End group removal and modification of RAFT polymers
Helen
Willcock
and
Rachel K.
O'Reilly
*
Department of Chemistry, University of Warwick, Coventry, UK CV4 7AL. E-mail: R.K.O-Reilly@warwick.ac.uk; Fax: +44 (0)247 7652 4112; Tel: +44 (0)247 652 3236
First published on 23rd December 2009
Abstract
This paper describes both well-established routes and recent advances in the end group modification of polymers synthesised by reversible addition–fragmentation chain transfer (RAFT) polymerisation. The lability of the thiocarbonylthio group, which facilitates the RAFT mechanism, allows for ready post-polymerisation functionalisation of RAFT polymers by a number of techniques. In particular, end group thermolysis, radical induced reduction, hetero-Diels–Alder reactions and reaction with nucleophiles are discussed as are the applications and limitations of each method. The versatility of RAFT as a polymerisation tool for the synthesis of polymers with functional end groups for a range of applications is demonstrated.
 Helen Willcock | Dr Willcock graduated in 2003 from the University of Liverpool having studied with Drs Robin Whyman and Neil Winterton. After working at Apollo Scientific Ltd she returned to Liverpool to commence a PhD (2004) under the supervision of Professors Steve Rannard and Andy Cooper researching the control of dendrimer properties through surface functionality. She became a postdoctoral researcher in the Rannard group in 2008. She then moved to the University of Warwick to conduct further postdoctoral research with Dr O'Reilly, where she has worked on the use of RAFT polymerisation in the synthesis of higher order supramolecular structures from block copolymers. |
 Rachel K. O'Reilly | Dr O'Reilly is currently an EPSRC fellow in the Chemistry Department at the University of Warwick. She graduated from the University of Cambridge (1999) and completed her PhD at Imperial College in 2003. She then moved to the US to work at IBM Almaden and Washington University in Saint Louis, under the joint direction of Professors Craig Hawker and Karen Wooley. In 2004 she was awarded a Royal Commission 1851 fellowship and in 2005 she moved to the University of Cambridge with a Royal Society Dorothy Hodgkin Fellowship. Her current research focuses on bridging the interface between creative synthetic, polymer and catalysis chemistry. |
Introduction
There has been great interest in the use of controlled radical polymerisation (CRP) techniques over the past decade, especially in reversible addition–fragmentation chain transfer (RAFT) polymerisation. RAFT is one of the most versatile CRP techniques as it exhibits good tolerance to a diverse range of functional groups in monomers, solvents and initiators and offers control over a wide range of monomers through the use of different classes of chain transfer agents (CTAs). It can be used to form narrow polydispersity polymers and copolymers and it is often possible to take the polymerisations to high conversion. RAFT polymerisation has been used to synthesise complex architectures such as block copolymers,1 stars,2 hyperbranched polymers3 and higher order supramolecular structures.4–6Careful choice of RAFT agent, reaction conditions and monomer is imperative to achieve good control over the polymerisation and therefore well-defined polymeric products. Many of the problems that can be associated with RAFT polymerisation (poor control, retardation) can be avoided by careful consideration and alteration of reaction conditions. There are four classes of CTAs differing by the substituent group next to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) S functionality: dithioesters, trithiocarbonates, xanthates and dithiocarbamates (Fig. 1). In general, given an appropriate choice of R group, trithiocarbonates are effective RAFT agents for the polymerisation of styrenic and acrylic monomers (acrylates, methacrylates and acrylamides), whereas xanthates offer good control over less activated vinyl monomers such as vinyl acetate, N-vinyl carbazole and N-vinylpyrrolidone.8 Substituents around the CS group are labelled Z and R (Fig. 1) and can be tailored to suit the monomer used. The Z group should activate the CS towards radical addition and stabilise the intermediate radical formed, whereas the R group should be a good free-radical leaving group and be capable of reinitiating free-radical polymerisation.9 As the RAFT mechanism proceeds by insertion of monomer units into the C–S bond (Scheme 1), end-functionalised polymers can be easily achieved by incorporating the functional groups into the RAFT agent (groups R and Z). There are a number of versatile synthetic routes for the synthesis of RAFT agents which allow the incorporation of a wide range of functional groups.10–13
S functionality: dithioesters, trithiocarbonates, xanthates and dithiocarbamates (Fig. 1). In general, given an appropriate choice of R group, trithiocarbonates are effective RAFT agents for the polymerisation of styrenic and acrylic monomers (acrylates, methacrylates and acrylamides), whereas xanthates offer good control over less activated vinyl monomers such as vinyl acetate, N-vinyl carbazole and N-vinylpyrrolidone.8 Substituents around the CS group are labelled Z and R (Fig. 1) and can be tailored to suit the monomer used. The Z group should activate the CS towards radical addition and stabilise the intermediate radical formed, whereas the R group should be a good free-radical leaving group and be capable of reinitiating free-radical polymerisation.9 As the RAFT mechanism proceeds by insertion of monomer units into the C–S bond (Scheme 1), end-functionalised polymers can be easily achieved by incorporating the functional groups into the RAFT agent (groups R and Z). There are a number of versatile synthetic routes for the synthesis of RAFT agents which allow the incorporation of a wide range of functional groups.10–13
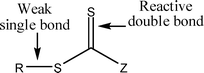 | ||
| Fig. 1 General RAFT agent structure (trithiocarbonate Z = SR, dithioester Z = alkyl or aryl, dithiocarbamate Z = NR2, xanthate Z = O-alkyl, R = alkyl or H).7 | ||
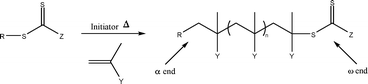 | ||
| Scheme 1 Schematic representation of RAFT polymerisation showing α- and ω-ends of the resulting polymer. | ||
The α-end group of RAFT polymers can be controlled in much the same way as polymers synthesised by other CRP methods (e.g. ATRP and NMP)—the incorporation of functionality into the R group of the initiating species results in end-functionalised polymers,14 with examples ranging from carboxylic acids10 to peptide15 and lipid groups,16 the in situ formation of protein–polymer conjugates via RAFT polymerisation using a bioconjugated RAFT agent,17 as well as many other examples.7,12,18–20 The synthesis of functionalised ATRP and NMP polymers has been studied extensively and there are a number of comprehensive reviews in this area.21,22 The end groups of both ATRP and NMP synthesised polymers can be modified post-polymerisation and the interconversion of NMP and RAFT initiating species as a bridge between different polymerisation techniques has also been described.23 Similarly ATRP initiating species have been converted to RAFT initiators,24 allowing the synthesis of diblock copolymers of both vinyl acetate and tert-butyl acrylate, monomers that are not easily controlled using one initiating species.24,25
One novel feature of RAFT synthesised polymers is the presence of a thiocarbonate at the ω-end of the polymer, which can be utilised to introduce functionality into the ω-end of the polymer post-polymerisation. The thiocarbonyl group can be thought of as a masked thiol or alkene, or a dienophile for hetero-Diels–Alder reactions. The nature of the labile C–S bond that facilitates the RAFT mechanism means that the retention of the RAFT end group in the polymer may be a problem for many commercial applications, therefore initial reasons for removal of end groups from polymers synthesised by RAFT included the removal of colour and odour. Previous articles have therefore extensively discussed the various methods available for the removal of the thiocarbonyl end group from RAFT polymers,7,26 however, since these earlier reports the importance has shifted from removal to functionalisation and use of the thiocarbonyl end groups. There is a great current interest in the conjugation of biomolecules to a range of polymers such as PEG as the resultant biomaterials often have increased stability and improved pharmokinetics compared to the parent biomolecule.27 Another class of bioconjugates can be formed from “smart” polymers such as NIPAM, which can respond to external stimuli and offer the ability to modulate protein activity.28 The ability to remove and subsequently functionalise the end group of polymers is an important and widely applicable topic. This paper describes the main methods for ω-end group conversion of RAFT polymers, with the emphasis on recent advances and current applications. It aims to give a highlighted overview of the use of RAFT in the synthesis of end-functionalised polymers by post-polymerisation ω-end group modification. A wide range of groups can be introduced at the α-end of RAFT polymers and this subject is considered to be beyond the scope of this review. The reader is instead directed towards a chapter of the Handbook of RAFT Polymerisation which describes methods of both α- and ω-end group functionalisation of RAFT polymers29 and several detailed reviews on RAFT polymerisation for further information.30–34
ω-End modification
The chemistry of the thiocarbonylthio group is well-established from small molecule chemistry35 and many of the same methods can be applied to RAFT polymers. The main methods of removal of the RAFT group functionality from polymers have been discussed in two reports by Moad et al. in 20057 and 200726 and in the Handbook of RAFT Polymerisation.29 End group removal is an important issue as residual RAFT agent functionality in polymers can be a problem due to their inherent reactivity and the possibility of decomposition into malodorous sulfur-containing materials. There are a number of methods available to cleave thiocarbonylthio groups (summarised in Scheme 2).
Reaction of thiocarbonylthio compounds with nucleophiles and ionic reducing agents (e.g. amines,36 hydroxide,37 borohydride38) is one of the most well-established and widely reported methods of RAFT polymer end group modification. The reaction transforms the thiocarbonyl containing group into a thiol which can be utilised in further coupling reactions, examples of which include disulfide formation and Michael addition of an acrylate. However, these reactions may leave reactive end group functionality so may not be suitable for all applications. Thermolysis provides complete desulfurisation of the polymer yielding an unsaturated chain end, but requires the polymer and any desired functionality to be stable to the conditions required for thermolysis. Radical induced reduction can also yield a sulfur-free hydrocarbon end group by using hypophosphite salts as the H atom source and has been reported with the advantage that both the excess reagent and byproducts are water soluble and thus can be easily removed from the polymer.26 Radical induced reduction has also been used to end cap the polymer with the required group through the use of functionalised azo-initiators.39 There are also a number of examples of the use of the thiocarbonyl as a dienophile with both small molecule dienes40 and dienes at polymer chain ends41 in reversible hetero-Diels–Alder reactions. Such reactions have been used to synthesise diblock copolymers as well as more complex macromolecular structures such as stars. The oxidation of thiocarbonylthio groups to give sulfines that readily decompose to thioesters and elemental sulfur has been reported. Barner-Kowollik and co-workers demonstrated this method for the modification of the dithiobenzoate end group of PMA by reaction with tert-butyl hydroperoxide, transforming the CS group to CO.42 They have since expanded upon this demonstrating the conversion of thiocarbonylthio end groups of acrylate and methacrylate polymers into hydroxyl capped polymers. This occurs via the reaction of dithioester capped polymers with an azo-initiator in the presence of air, quantitatively yielding hydroperoxide end groups that could subsequently be reduced to hydroxyl groups. This method provides a simple route to sulfur-free hydroxyl ended capped polymers.43,44
Analysis of the end group removed polymers can be achieved using traditional polymer characterisation methods such as nuclear magnetic resonance (NMR) and gel permeation chromatography (GPC). UV spectroscopy can also be used as the thiocarbonylthio chromophore absorbs strongly in the UV at around 300–310 nm and this absorbance will not be present in polymers not displaying the RAFT end group. Visual comparison of the colour of the polymers before and after end group removal can also be used to confirm if the end group has successfully been removed as the RAFT groups are often coloured whereas end group removed polymers are not.
Radical induced end group removal
Free-radical sources have been used to modify the end groups of RAFT synthesised polymers via radical cross-coupling. This has been achieved by reacting the polymer with a radical species which adds to the reactive CS bond resulting in the formation of an intermediate radical that can either fragment or react with a trapping group (which can be the radical species if used in a large excess) and terminate (Scheme 3).26 Perrier et al. demonstrated this method for the removal of dithiobenzoate end groups from PMMA using 2,2′-azo(bis)isobutyronitrile (AIBN) as the initiating species, resulting in complete removal of the RAFT end group along with the recovery of the RAFT agent. The process was reported to be effective for a variety of polymers and end groups. The structure of the product was confirmed by 1H NMR spectroscopy and also upon inspection of the polymer colour change (Fig. 2).45 A subsequent report has demonstrated that while this method is effective for methacrylate polymers, it provides incomplete end group removal when applied to acrylate and styrenic polymers and instead a combination of AIBN and lauroyl peroxide was found to be more effective.46
 | ||
| Scheme 3 Mechanism of radical induced removal of thiocarbonyl end groups from RAFT polymers.26 | ||
 | ||
| Fig. 2 Picture of poly(methyl methacrylate) (PMMA) synthesised by the RAFT process (a) before and (b) after reaction with AIBN.45 | ||
Since these reports, functionalised azo-initiators have been used to exchange with the RAFT end group of polymers in a number of studies.47 A recent study by Maynard et al. demonstrated the use of radical cross-coupling in the synthesis of heterotelechelic polymers which were used to form polymer–protein conjugates (Scheme 4). The thiocarbonyl end group of the α-biotinylated RAFT polymer was reacted with an excess of the protected maleimide functionalised azo-initiator. Deprotection of the maleimide resulted in the α- and ω-ends of the polymer being selectively reactive with streptavidin (SAv) and bovine serum albumin (BSA) proteins respectively, to afford a heterodimer conjugate.39

If both a free-radical source and a hydrogen atom donor are used the thiocarbonylthio group can be replaced with hydrogen, however, in order to avoid side reactions this requires an efficient hydrogen atom donor. This reaction was first achieved using tributylstannane,48 and although the reaction was successful yielding almost quantitative end group transformation of the polymers, concerns over the toxicity of the stannane and difficulty with removal have hindered its widespread application. An alternative H source, tris(trimethyl)silane, was found to be less efficient resulting in the coupling of the propagating radicals.49 Hypophosphite salts have more recently been investigated as free-radical reducing agents and have been shown to be very efficient for both acrylate and styrenic polymers.26 Their effectiveness was demonstrated in the successful removal of a trithiocarbonate end group from poly-tert-butyl acrylate with a Pd-pincer ligand incorporated into the R group that was then used in the synthesis of supramolecular nanostructures. In this case, the trithiocarbonate group was found to bind Pd competitively with the pincer end group and thus the complete removal of the end group was required. GPC analysis of the polymer both before and after end group removal using UV detection at 309 nm (the absorbance maxima of trithiocarbonate groups) confirmed the complete removal of the RAFT group from the polymer. This work also highlighted that there is no detectable bimolecular termination which would be indicated by a high molecular weight peak in the GPC/RI trace.50
Radical induced end group modification has been shown to be amenable to a variety of polymers and can be used to form stable polymer–protein conjugates.39 The radical initiators must be in relatively high concentrations and require high temperatures to fragment, which must be taken into account when working with unstable monomers. However, a varied range of groups can be added to the polymer ends via this method, dependent upon the synthesis or commercial availability of the functionalised azo-initiator. The use of hypophosphite salts to replace the thiocarbonyl group with a H atom is an extremely efficient reaction with which an essentially unreactive sulfur-free end group can be formed.26
Thermal elimination
Thermal elimination of the RAFT end group from polymers is one of the two methods of end group removal that can yield a sulfur-free end group functionality. It has the advantage of proceeding with no chemical additions, therefore reducing the amount of purification steps and complexity. One drawback of this method is that the polymer and any functionality must be stable to the thermolysis conditions, which typically involve temperatures from 120–200 °C. Studies into the thermal stability of CTAs during polymerisation reveal the order of thermal stability to be dithiobenzoates > trithiocarbonates > xanthates and that when Z is an aromatic group the thermal stability is much improved when compared to analogous non-aromatic CTAs.51 The polymerisation of MMA using cumyl dithiobenzoate was found to be hindered by decomposition of the RAFT end group above 120 °C,52 however, the polymerisation of styrene at temperatures up to 180 °C exhibited no decomposition. This was attributed to the fast conversion of the initial CTA into the more stable polymeric CTA.53 Removal of the end group by thermolysis can be followed by thermogravimetric analysis (TGA) as well as traditional polymer characterisation methods. The weight loss profile observed by TGA and the mechanism of loss have been found to depend strongly on both the RAFT agent54 and polymer type and there are a number of proposed mechanistic pathways (Scheme 5).55 | ||
| Scheme 5 Mechanisms of thermolysis for RAFT polymer end group removal (Y = CO2C4H9).55 | ||
The first mechanism A has been observed in the case of trithiocarbonate ended polystyrene and involves concerted elimination resulting in an unsaturated end group. This mechanism has some precedence in ester and xanthate pyrolysis, i.e. the Chugaev reaction for the formation of olefins from alcohols.56 The GPC trace of the polymer after end group removal showed the molecular weight of the polymer to be essentially unchanged; 1H NMR proved that the trithiocarbonate group was quantitatively removed whilst the α-end group (a phthalimidomethyl group) was retained and electrospray ionisation mass spectrometry (ES+ MS) showed a series of peaks in accordance with the expected structure (Fig. 3).55
 | ||
| Fig. 3 ES+ MS of polystyrene after end group removal via thermolysis, 1326.7 = (10 × 104.06) + 160.04 + 103.05 + 22.99 (Na+), the interpeak distance corresponding to the mass of the styrene repeat unit (104.1).55 | ||
The second mechanism B (observed for poly(n-butyl acrylate) with a trithiocarbonate end group) involves initial C–S bond homolysis yielding the propagating radical and a byproduct containing the thiocarbonylthio group that decomposes into CS2 and butanethiol. The polymer radical then decays by backbiting and β-scission, resulting in lower molecular weight oligomers with either unsaturated or radical chain ends, the latter of which can couple to form higher molecular weight species. In the case of polymethylmethacrylate (PMMA) it has been shown that the decomposition of the thiocarbonythio groups depends strongly upon the CTA used during polymer synthesis. The trithiocarbonate group was found to decompose at around 180 °C by homolysis of the C–S bond followed by depropagation, whereas dithiobenzoate-ended PMMA was stable at higher temperatures and decomposes via concerted elimination.54 In a separate study carried out by Xu et al., the thermolysis of low molecular weight PMMA (Mn 3600) with a dithiobenzoate end group was reported to occur at much lower temperatures, an effect that was attributed to molecular weight effects.52 Although this was not substantiated by later studies,57,58 the reported mechanism of decomposition was the same and the difference in temperature reported could be due to experimental differences between laboratories—for example it is known that oxygen can trap the radicals (in much the same way that it will hinder the progress of polymerisations) and can interfere with the unzipping of the chains. In the case of polymers with xanthate end groups, the mechanism has been found to depend upon the polymer structure. The decomposition of both polystyrene and poly(tert-butyl acrylate) terminated with an O-isobutyl xanthate is reported to proceed via selective elimination to form thiol-ended polymers, whereas the decomposition of polyvinylacetate with an O-ethyl xanthate is triggered by the homolysis of the C–S bond.8 If the thermal decomposition of the RAFT end group occurs during polymerisation, the unsaturated end group can take part in the polymerisation leading to a broadening in molecular weight distribution,59 however, it has been demonstrated that thermolysis can be used to yield macromonomers with low polydispersities.8
The release of sometimes toxic and odorous thermolysis byproducts (thiols and carbon disulfide in the case of trithiocarbonates) during thermolysis is an issue so appropriate safety precautions must be implemented. However, thermolysis has been shown to be a simple and effective method of end group removal, usually resulting in unsaturated end groups and therefore can provide a route to relatively narrow polydispersity macromonomers.8
Reaction with nucleophiles/aminolysis
The kinetics and mechanism of the reaction of thiocarbonythio groups with excess amine were reported in 199060 and the method has since been used to cleave RAFT end groups from polymers. It is now one of the most widely used and versatile methods of RAFT end group conversion, resulting in the formation of a thiol end group that can be subsequently utilised in a number of reactions. This reaction must be taken into account when designing RAFT agent and monomer systems as any amines in the reaction media may cause unwanted decomposition of the RAFT moiety during polymerisation. Either primary or secondary amines acting as nucleophiles can convert a thiocarbonylthio group to a thiol. The exclusion of oxygen from the reaction is vital as the thiols formed can be readily oxidised to disulfides resulting in higher molecular weight coupled species, a problem that has been in part avoided by the use of reducing agents in the reaction mixture.60,61 Examples include the use of Zn and acetic acid in the aminolysis of branched polymers formed by RAFT polymerisation of a monomer containing both a dithioester moiety and a double bond.36 The dithioesters at the branching points were cleaved by reaction with ethylamine, and after purification the product was treated with a Zn–acetic acid mixture to cleave the disulfide linkages formed. In the same study linear PS with dithioester end groups was subjected to the same treatment, the GPC analysis of which revealed that the thiol-ended species had indeed coupled to form a polymer with double the molecular weight of the starting polymer resulting in a bimodal peak. After treatment with the reducing agent this bimodality was removed and a polymer with molecular weight similar to the original was reformed, confirming the cleavage of the disulfide bond.36Other reports demonstrate the addition of other reducing agents (such as PBu3,61,62 tris(2-carboxyethyl)phosphine (TCEP)38) to the reaction during aminolysis to prevent the formation of unwanted disulfide-coupled species and while in most cases the intensity of higher molecular weight species can be reduced, they cannot always be completely eliminated.61 One example in which the formation of disulfide bonds between the polymer chains has been exploited describes the formation of telechelic and cyclic polymers via aminolysis of polymers with RAFT agent functionality at both ends. Polystyrene was formed using bifunctional RAFT agents, which when treated with excess hexylamine formed polymers with thiol groups at each end. Disulfide formation between the thiol groups in a relatively highly concentrated solution resulted in linear multiblock polystyrene, whereas in more dilute conditions monocyclic species were obtained (Scheme 6).63 A similar method was applied to the synthesis of telechelic multiblock copolymers of PMMA and PBA.64
 | ||
| Scheme 6 Synthesis of telechelic and cyclic polymers by a combination of RAFT polymerisation and aminolysis.63 | ||
An elegant one-pot route to metal nanoparticles stabilised by thiol-terminated polymers was described in 2002; aqueous solutions of RAFT synthesised polymers with dithioester end groups and transition metal salts were simultaneously reduced using NaBH4 as shown in Scheme 7. When the reduction of the transition metal complex was carried out in the absence of the polymer the reduced metal species flocculated, however, the polymer-stabilised particles were shown to be stable for months.65 Subsequent studies have shown that the RAFT agent can be used directly without reducing to a thiol to stabilise metal nanoparticles.47
 | ||
| Scheme 7 Preparation of polymer-stabilised transition metal nanoparticles.47 | ||
There is some evidence that the thiol formed during the aminolysis of PMMA and other methacrylate polymers can backbite to form stable thiolactone rings. The products resulting from the aminolysis of both dithiobenzoate-ended PMMA and PS were compared and whilst the PS formed the expected thiol end group with some disulfide formation, the PMMA thiol was shown to attack the penultimate group in the chain forming the thiolactone species. The driving force for this cyclisation is thought to be the stability of the five-membered ring formed.52 Other groups have observed the similar results from PMMA66 and PAA,37 however, in these studies the lactone terminus was only present in a small fraction of the sample.
As the isolation of the thiol-ended polymers is hindered by their inherent reactivity, recent reports demonstrate the trapping of the thiol by addition of activated alkenes (Michael acceptors) such as maleimides, acrylates and vinyl sulfones67 yielding thioether linkages, or thiols to give disulfide bonds. In these cases the thiol is reacted immediately as it is formed by the addition of both the nucleophile and the reactive species to the polymer solution, thus reducing the occurrence of unwanted side reactions. The addition of Michael acceptors is an efficient method of introducing desired groups into the polymer end. The thiol–ene reaction has been favourably compared to “click” reactions, demonstrating some advantages over the traditional copper catalysed alkyne–azide reactions.68 Namely, thiol–ene reactions use readily available starting materials, occur under mild conditions and often give quantitative results over period of seconds, compared to the extended reaction times and elevated temperatures that can be needed for alkyne–azide reactions.68 Common catalysts that have been used for the thiol–ene reaction between RAFT synthesised polymers and Michael acceptors include dimethylphenylphosphine (DMPP)69 and triethylamine (TEA).62 Li et al. reported the end group activation of PNIPAM prepared by RAFT polymerisation by the conversion of the trithiocarbonate end groups to thiols, followed by the subsequent triethylamine catalysed reaction with a bis-maleimide. The resulting maleimide group was both coupled with a model diene (9-anthracenemethanol) in a Diels–Alder reaction and importantly, was activated to addition with other macromolecular thiols, providing a route to block copolymers by the convenient coupling of two homopolymers.62
The maleimide–thiol coupling reaction has also been used for the fluorescent labelling of RAFT synthesised PNIPAM by reaction with maleimide functionalised pyrene, a synthesis that highlights the potential use of RAFT in biotechnology and drug delivery applications.38 DMPP was used as a catalyst in the synthesis of three-arm star polymers from RAFT prepared poly(N,N-diethylacrylamide) and a triacrylate core molecule (Scheme 8). This reaction was quantified by FTIR and NMR spectroscopic analysis and was found to be both rapid and facile and is the first demonstration of the convergent synthesis of star RAFT polymers via the thiol–ene reaction.69
 | ||
| Scheme 8 Star polymers via aminolysis of dithiobenzoate terminated poly(N,N-diethylacrylamide) and DMPP mediated thiol–ene reaction.69 | ||
The use of thiosulfonates as a trapping agent for the thiols formed during aminolysis of RAFT synthesised polymers has been demonstrated by Roth et al. In the first instance methyl methanethiosulfonate (MMTS) was used as it reacts quickly and selectively with thiols to form methyl disulfides, which were found to form well-defined self-assembled monolayers on gold surfaces.70 The use of an alkyne functionalised thiosulfonate was demonstrated as a route to polymers with “click” end functionality. This was reported for a variety of methacrylate polymers as well as polyacrylamide and polystyrene.71 In a systematic study by Li et al., thiol-ended poly(N,N-diethylacrylamide) synthesised by RAFT polymerisation was aminolysed in the presence of TCEP and the resultant reactive thiol was coupled to a host of commercially available isocyanates. FTIR spectroscopy revealed that the reaction was rapid with ∼95% conversion being reached within 15 min, yielding a wide range of end groups.72 Vinyl sulfone end-functionalised PEGylated polymers have been synthesised by a combination of RAFT polymerisation and aminolysis in the presence of divinyl sulfone. These functionalised Michael acceptor polymers were conjugated with bovine serum albumin (BSA) and esterase activity studies revealed that the conjugated protein retained 92% of the free BSA activity.67
A comprehensive study of the modification of RAFT polymers via thiol–ene reactions has been recently reported, demonstrating the synthesis of novel architectures and biofunctionalisation of polymers. The reactions studied are summarised in Scheme 9.73 The first synthetic route involves simultaneous aminolysis and TEA mediated thiol–ene reaction with the diene species 1,6-hexanediol diacrylate, yielding macromolecular monomers with defined structures and low polydispersities. This reaction has been proved successful for a variety of polymers; PMMA, PHPMA and PNIPAM and the resulting macromonomers have been used both in polymerisations with styrene yielding copolymers and in thiol–ene reactions with both PEG–thiol and DNA–thiol species.
 | ||
| Scheme 9 A schematic overview of the synthesis of new architectures and biofunctionalisation of polymers by thiol–ene reactions. (See ref. 73 for further details.) | ||
The second synthetic route demonstrates the biofunctionalisation of RAFT polymers achieved by the coupling of both trithiocarbonate ended PNIPAM and dithiobenzoate-ended PHPMA with a methacrylate-modified mannose and a maleimide-modified biotin. Simultaneous aminolysis and thiol–ene reactions were carried out in a one-pot procedure with product yields above 85% and the presence of the alkene compounds during aminolysis was found to prevent the formation of interchain disulfides.73 The biofunctionalisation of glycopolymers has been achieved using a similar method: poly(pentafluorophenyl acrylate) synthesised via RAFT polymerisation was modified by reaction with amine functionalised sugars yielding a trithiocarbonate ended glycopolymer. Subsequent aminolysis and addition of the resulting thiol onto the biotin-modified maleimide yielded biotin-functionalised glycopolymers in high yields (>95%).74 A further example of biofunctionalisation via a one-pot aminolysis and thiol–ene reaction has been reported for trithiocarbonate ended PNIPAM and 6-O-methacryloyl mannose forming non-reversible thioether linkages again in high yield. The same report details reversible disulfide formation between thiol containing oligonucleotide and peptide groups and the thiol end group of the polymers. In this case 2,2′-dithiopyridine (DTP) was used to eliminate unwanted side reactions by forming a stable pyridyl disulfide (PDS) group that could be displaced by the free thiol-bearing molecules in the next step. PDS–thiol exchange is widely used for efficient and site-selective conjugation of biological molecules under mild conditions.75 When the aminolysis was carried out in the absence of DTP both thiol-terminated and disulfide-coupled polymer populations were observed.76 DTP has also been used to introduce biodegradable linkers in the synthesis of biodegradable hyperbranched3 and star77 polymers, demonstrating the versatility of this method of functional group incorporation and it's compatibility with RAFT polymerisation.
In addition to the nucleophilic reactions of thiols with activated alkenes described so far, radical thiol–ene reactions can be used to couple thiols to unactivated alkenes. This has been demonstrated by Yu et al. in the synthesis of well-defined end-functionalised PNIPAM via sequential thiol–ene/thiol–ene and thiol–ene/thiol–yne reactions. The dithiobenzoate RAFT end groups were modified in a one-pot process involving amine cleavage followed by phosphine-mediated thiol–ene reactions with allyl methacrylate or propargyl acrylate. Commercially available thiols were used in a photoinitiated radical reaction with the resulting end groups, yielding mono and bis end-functionalised PNIPAM (Scheme 10).78
 | ||
| Scheme 10 PNIPAM end group modifications via a thiol–ene/radical thiol–ene and thiol–yne reactions.72 | ||
The mild conditions and commercially available catalysts used for the aminolysis of RAFT ended polymers make it an attractive and extremely versatile method of end group modification. The thiol end group resulting from this reaction can be used in a multitude of reactions, from the introduction of fluorescently labelled groups via the thiol–ene reaction38 to conjugation to biomolecules via disulfide coupling,76 demonstrating that these are robust, versatile reactions for the functionalisation of RAFT synthesised polymers.
Hetero-Diels–Alder reactions
It has been shown that thiocarbonylthio groups can be readily removed and the polymers subsequently modified using the remaining end functionality, however, an alternative method of end functionalisation is the direct use of the RAFT agent. The electron deficiency of thiocarbonylthio groups facilitates their use as dienophiles in hetero-Diels–Alder reactions. This is well known from small molecule chemistry79 and has since been demonstrated for polymer RAFT end groups.41 This method has been used to synthesise block copolymers from RAFT synthesised polystyrene and diene-functionalised ROP synthesised polycaprolactone. The reaction was found to be highly selective giving quantitative conversion and was carried out under moderate conditions with non-toxic catalysts.41 Subsequent reports have shown that hetero-Diels–Alder linkages can be utilised in the synthesis of more complex macromolecular architectures such as stars (Scheme 11),40 again with high reactivity and efficiency, allowing analogies to “click” chemistries80 to be drawn. | ||
| Scheme 11 Synthesis of polystyrene stars via a combination of RAFT and hetero-Diels–Alder cycloaddition.40 | ||
The surface modification of divinylbenzene microspheres with surface expressed RAFT groups by hetero-Diels–Alder reactions has been demonstrated, also using mild reaction conditions. Diene-functionalised polycaprolactone was again used to react with the thiocarbonylthio group and the successful grafting was evident by the colour change of the microspheres from purple to white.81 The same group has used ab initio calculations to design strongly electron deficient C-sulfonylthioformate based RAFT agents. These have been shown to offer control over the polymerisation of a limited range of monomers, and can be used for extremely efficient post-polymerisation reactions with electron rich dienes.82 Furthermore, a rapid and extremely efficient room temperature conjugation strategy that can be used to access block copolymers from RAFT and ATRP synthesised polymers has been demonstrated. Conversion of the ATRP synthesised block into a cyclopentadienyl terminated polymer was achieved followed by the rapid and catalyst free conjugation with the dithioester group of the RAFT synthesised block.83 This has since been proven to be effective for the coupling of styrene and isobornyl acrylate blocks to form diblock copolymers with molecular weights from 34![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 to over 100000 g mol−1, demonstrating for the first time that hetero-Diels–Alder click chemistry can be used to access high molecular weight block copolymers (Scheme 12).84
000 to over 100000 g mol−1, demonstrating for the first time that hetero-Diels–Alder click chemistry can be used to access high molecular weight block copolymers (Scheme 12).84
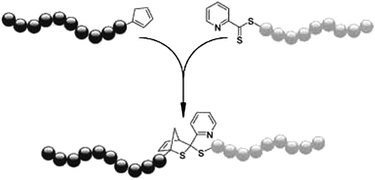 | ||
| Scheme 12 General synthetic strategy for producing well-defined high Mw block copolymers via the RAFT–HDA click reaction (TFA, CHCl3, RT).84 | ||
Conclusions
RAFT polymerisation can offer polymers that are readily functionalised at both the α- and ω-ends. The thiocarbonylthio end group that facilitates the RAFT mechanism also offers a functional handle for efficient post-polymerisation functionalisation of the ω-end. Early work was focussed on the removal of the sulfur-containing groups to prevent problems with reactivity and degradation into odorous compounds, but more recently the focus has switched to the use of this functional end group. This has been shown to be achieved by a number of methods including hetero-Diels–Alder reactions, thermolysis, radical induced substitution and the most widely demonstrated: reaction with a nucleophile. The latter method proceeds under mild conditions (room temperature) and yields a reactive thiol end group that can then be utilised in many reactions, from disulfide coupling to thiol–ene reactions. Initial problems with this method (disproportionation by homo-disulfide formation) have been overcome and the one-pot modification of polymers with a variety of activated alkene compounds has been demonstrated.Notes and references
- A. M. Bivigou-Koumba, J. Kristen, A. Laschewsky, P. Müller-Buschbaum and C. M. Papadakis, Macromol. Chem. Phys., 2009, 210, 565–578 CrossRef CAS.
- J. F. Quinn, R. P. Chaplin and T. P. Davis, J. Polym. Sci., Part A: Polym. Chem., 2002, 40, 2956–2966 CrossRef CAS.
- J. Xu, L. Tao, J. Liu, V. Bulmus and T. P. Davis, Macromolecules, 2009, 42, 6893–6901 CrossRef CAS.
- J. Bernard, F. Lortie and B. Fenet, Macromol. Rapid Commun., 2009, 30, 83–88 CrossRef CAS.
- M. H. Stenzel, Macromol. Rapid Commun., 2009, 30, 1603–1624 CrossRef CAS.
- M. H. Stenzel, Chem. Commun., 2008, 3486–3503 RSC.
- G. Moad, Y. K. Chong, A. Postma, E. Rizzardo and S. H. Thang, Polymer, 2005, 46, 8458–8468 CrossRef CAS.
- A. Postma, T. P. Davis, G. Li, G. Moad and M. S. O'Shea, Macromolecules, 2006, 39, 5307–5318 CrossRef CAS.
- J. Chiefari, Y. K. Chong, F. Ercole, J. Krstina, J. Jeffery, T. P. T. Le, R. T. A. Mayadunne, G. F. Meijs, C. L. Moad, G. Moad, E. Rizzardo and S. H. Thang, Macromolecules, 1998, 31, 5559–5562 CrossRef CAS.
- J. Skey and R. K. O'Reilly, Chem. Commun., 2008, 4183–4185 RSC.
- M. R. Wood, D. J. Duncalf, S. P. Rannard and S. Perrier, Org. Lett., 2006, 8, 553–556 CrossRef CAS.
- D. J. Coady, B. C. Norris, V. M. Lynch and C. W. Bielawski, Macromolecules, 2008, 41, 3775–3778 CrossRef CAS.
- M. Benaglia, J. Chiefari, Y. K. Chong, G. Moad, E. Rizzardo and S. H. Thang, J. Am. Chem. Soc., 2009, 131, 6914–6915 CrossRef CAS.
- V. Coessens, T. Pintauer and K. Matyjaszewski, Prog. Polym. Sci., 2001, 26, 337–377 CrossRef CAS.
- J. Hentschel, K. Bleek, O. Ernst, J. F. Lutz and H. G. Borner, Macromolecules, 2008, 41, 1073–1075 CrossRef CAS.
- M. Bathfield, D. Daviot, F. D'Agosto, R. Spitz, C. Ladaviere, M. T. Charreyre and T. Delair, Macromolecules, 2008, 41, 8346–8353 CrossRef CAS.
- J. Liu, V. Bulmus, D. L. Herlambang, C. Barner-Kowollik, M. H. Stenzel and T. P. Davis, Angew. Chem., Int. Ed., 2007, 46, 3099–3103 CrossRef CAS.
- G. Zhou and I. I. Harruna, Macromolecules, 2005, 38, 4114–4123 CrossRef CAS.
- A. Samakande, R. Chaghi, G. Derrien, C. Charnay and P. C. Hartmann, J. Colloid Interface Sci., 2008, 320, 315–320 CrossRef CAS.
- P. De, M. Li, S. R. Gondi and B. S. Sumerlin, J. Am. Chem. Soc., 2008, 130, 11288–11289 CrossRef CAS.
- K. Matyjaszewski and J. Xia, Chem. Rev., 2001, 101, 2921–2990 CrossRef CAS.
- C. J. Hawker, A. W. Bosman and E. Harth, Chem. Rev., 2001, 101, 3661–3688 CrossRef CAS.
- A. Favier, B. Luneau, J. Vinas, N. Laissaoui, D. Gigmes and D. Bertin, Macromolecules, 2009, 42, 5953–5964 CrossRef CAS.
- C. M. Wager, D. M. Haddleton and S. A. F. Bon, Eur. Polym. J., 2004, 40, 641–645 CrossRef CAS.
- C. D. Petruczok, R. F. Barlow and D. A. Shipp, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 7200–7206 CrossRef CAS.
- Y. K. Chong, G. Moad, E. Rizzardo and S. H. Thang, Macromolecules, 2007, 40, 4446–4455 CrossRef CAS.
- P. Caliceti and F. M. Veronese, Adv. Drug Delivery Rev., 2003, 55, 1261–1277 CrossRef CAS.
- A. S. Hoffman and P. S. Stayton, Macromol. Symp., 2004, 207, 139–152 CrossRef CAS.
- L. Barner and S. Perrier, in Handbook of RAFT Polymerisation, ed. C. Barner-Kowollik, Wiley-VCH, Wenheim, 2008, pp. 455–482 Search PubMed.
- G. Moad, E. Rizzardo and S. H. Thang, Polymer, 2008, 49, 1079–1131 CrossRef CAS.
- G. Moad, E. Rizzardo and S. H. Thang, Aust. J. Chem., 2005, 58, 379–410 CrossRef CAS.
- S. Perrier and P. Takolpuckdee, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 5347–5393 CrossRef CAS.
- C. Barner-Kowollik and S. Perrier, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 5715–5723 CrossRef CAS.
- A. B. Lowe and C. L. McCormick, Prog. Polym. Sci., 2007, 32, 283–351 CrossRef CAS.
- D. Crich and L. Quintero, Chem. Rev., 1989, 89, 1413–1432 CrossRef CAS.
- Z. Wang, J. He, Y. Tao, L. Yang, H. Jiang and Y. Yang, Macromolecules, 2003, 36, 7446–7452 CrossRef CAS.
- M. F. Llauro, J. Loiseau, F. Boisson, F. Delolme, C. Ladavière and J. Claverie, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 5439–5462 CrossRef CAS.
- C. W. Scales, A. J. Convertine and C. L. McCormick, Biomacromolecules, 2006, 7, 1389–1392 CrossRef CAS.
- K. L. Heredia, G. N. Grover, L. Tao and H. D. Maynard, Macromolecules, 2009, 42, 2360–2367 CrossRef CAS.
- A. J. Inglis, S. Sinnwell, T. P. Davis, C. Barner-Kowollik and M. H. Stenzel, Macromolecules, 2008, 41, 4120–4126 CrossRef CAS.
- S. Sinnwell, A. J. Inglis, T. P. Davis, M. H. Stenzel and C. Barner-Kowollik, Chem. Commun., 2008, 2052–2054 RSC.
- P. Vana, L. Albertin, L. Barner, T. P. Davis and C. Barner-Kowollik, J. Polym. Sci., Part A: Polym. Chem., 2002, 40, 4032–4037 CrossRef CAS.
- T. Gruendling, M. Dietrich and C. Barner-Kowollik, Aust. J. Chem., 2009, 62, 806–812 CrossRef CAS.
- T. Gruendling, R. Pickford, M. Guilhaus and C. Barner-Kowollik, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 7447–7461 CrossRef CAS.
- S. Perrier, P. Takolpuckdee and C. A. Mars, Macromolecules, 2005, 38, 2033–2036 CrossRef CAS.
- M. Chen, G. Moad and E. Rizzardo, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 6704–6714 CrossRef CAS.
- B. J. Kim, S. Given-Beck, J. Bang, C. J. Hawker and E. J. Kramer, Macromolecules, 2007, 40, 1796–1798 CrossRef CAS.
- M. Chen, K. P. Ghiggino, T. A. Smith, S. H. Thang and G. J. Wilson, Aust. J. Chem., 2004, 57, 1175–1177 CrossRef CAS.
- A. Postma, T. P. Davis, R. A. Evans, G. Li, G. Moad and M. S. O'Shea, Macromolecules, 2006, 39, 5293–5306 CrossRef CAS.
- A. O. Moughton, K. Stubenrauch and R. K. O'Reilly, Soft Matter, 2009, 5, 2361–2370 RSC.
- T. M. Legge, A. T. Slark and S. Perrier, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 6980–6987 CrossRef.
- J. Xu, J. He, D. Fan, X. Wang and Y. Yang, Macromolecules, 2006, 39, 8616–8624 CrossRef CAS.
- Y. Liu, J. He, J. Xu, D. Fan, W. Tang and Y. Yang, Macromolecules, 2005, 38, 10332–10335 CrossRef CAS.
- B. Chong, G. Moad, E. Rizzardo, M. Skidmore and S. H. Thang, Aust. J. Chem., 2006, 59, 755–762 CrossRef CAS.
- A. Postma, T. P. Davis, G. Moad and M. S. O'Shea, Macromolecules, 2005, 38, 5371–5374 CrossRef CAS.
- L. Chugaev, Chem. Ber., 1899, 32, 3332 CrossRef.
- V. Lima, H. Jiang, J. Brokken-Zijp, P. J. Schoenmakers, B. Klumperman and R. V. D. Linde, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 959–973 CrossRef CAS.
- D. L. Patton, M. Mullings, T. Fulghum and R. C. Advincula, Macromolecules, 2005, 38, 8597–8602 CrossRef CAS.
- J. Xu, J. He, D. Fan, W. Tang and Y. Yang, Macromolecules, 2006, 39, 3753–3759 CrossRef CAS.
- M. Deletre and G. Levesque, Macromolecules, 1990, 23, 4733–4741 CrossRef CAS.
- J. M. Spruell, B. A. Levy, A. Sutherland, W. R. Dichtel, J. Y. Cheng, F. J. Stoddart and A. Nelson, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 346–356 CrossRef CAS.
- M. Li, P. De, S. R. Gondi and B. S. Sumerlin, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 5093–5100 CrossRef CAS.
- M. R. Whittaker, Y. K. Goh, H. Gemici, T. M. Legge, S. Perrier and M. J. Monteiro, Macromolecules, 2006, 39, 9028–9034 CrossRef CAS.
- H. Gemici, T. M. Leggs, M. Whittaker, M. J. Monterio and S. Perrier, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 2334–2340 CrossRef CAS.
- A. B. Lowe, B. S. Sumerlin, M. S. Donovan and C. L. McCormick, J. Am. Chem. Soc., 2002, 124, 11562–11563 CrossRef CAS.
- X. Jiang, P. J. Schoenmakers, J. L. J. van Dongen, X. Lou, V. Lima and J. Brokken-Zijp, Anal. Chem., 2003, 75, 5517–5524 CrossRef CAS.
- G. N. Grover, S. N. S. Alconcel, N. M. Matsumoto and H. D. Maynard, Macromolecules, 2009, 42, 7657–7663 CrossRef CAS.
- W. H. Binder and R. Sachsenhofer, Macromol. Rapid Commun., 2007, 28, 15–54 CrossRef CAS.
- J. W. Chan, B. Yu, C. E. Hoyle and A. B. Lowe, Chem. Commun., 2008, 4959–4961 RSC.
- P. J. Roth, D. Kessler, R. Zentel and P. Theato, Macromolecules, 2008, 41, 8316–8319 CrossRef CAS.
- P. J. Roth, D. Kessler, R. Zentel and P. Theato, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 3118–3130 CrossRef CAS.
- H. Li, B. Yu, H. Matsushima, C. E. Hoyle and A. B. Lowe, Macromolecules, 2009, 42, 6537–6542 CrossRef CAS.
- C. Boyer, A. Granville, T. P. Davis and V. Bulmus, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 3773–3794 CrossRef CAS.
- C. Boyer and T. P. Davis, Chem. Commun., 2009, 6029–6031 RSC.
- G. H. Hsiue, H. Z. Chiang, C. H. Wang and T. M. Juang, Bioconjugate Chem., 2006, 17, 781–786 CrossRef CAS.
- C. Boyer, V. Bulmus and T. P. Davis, Macromol. Rapid Commun., 2009, 30, 493–497 CrossRef.
- E. Setijadi, L. Tao, J. Liu, Z. Jia, C. Boyer and T. P. Davis, Biomacromolecules, 2009, 10, 2699–2707 CrossRef CAS.
- B. Yu, J. W. Chan, C. E. Hoyle and A. B. Lowe, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 3544–3557 CrossRef CAS.
- B. Heuzé, R. Gasparova, M. Heras and S. Masson, Tetrahedron Lett., 2000, 41, 7327–7331 CrossRef CAS.
- C. K. Hartmuth, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS.
- L. Nebhani, S. Sinnwell, A. J. Inglis, M. H. Stenzel, C. Barner-Kowollik and L. Barner, Macromol. Rapid Commun., 2008, 29, 1431–1437 CrossRef CAS.
- L. Nebhani, S. Sinnwell, C. Y. Lin, M. L. Coote, M. H. Stenzel and C. Barner-Kowollik, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 6053–6071 CrossRef CAS.
- A. J. Inglis, S. Sinnwell, M. H. Stenzel and C. Barner-Kowollik, Angew. Chem., Int. Ed., 2009, 48, 2411–2414 CAS.
- A. J. Inglis, M. H. Stenzel and C. Barner-Kowollik, Macromol. Rapid Commun., 2009, 30, 1792–1798 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2010 |