
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe interaction of surface electron distribution-polarized Fe/polyimide hybrid nanosheets with organic pollutants driving a sustainable Fenton-like process†
Lai
Lyu
 ,
Kanglan
Deng
,
Junrong
Liang
,
Chao
Lu
,
Tingting
Gao
,
Wenrui
Cao
and
Chun
Hu
*
,
Kanglan
Deng
,
Junrong
Liang
,
Chao
Lu
,
Tingting
Gao
,
Wenrui
Cao
and
Chun
Hu
*
Institute of Environmental Research at Greater Bay Area, Key Laboratory for Water Quality and Conservation of the Pearl River Delta, Ministry of Education, Guangzhou University, Guangzhou 510006, China. E-mail: huchun@gzhu.edu.cn; Tel: +86-20-39346609
First published on 16th July 2020
Abstract
Many organic pollutants are rich in electrons, which are not used in the current pollutant removal technology, resulting in high energy consumption and low efficiency. To achieve a sustainable conversion of pollutants using Fenton-like technology, surface electron distribution-polarized lotus flower-like Fe-doped polyimide hybrid nanosheets (lf-Fe/PI HNs), a Fenton-like catalyst, are first developed for electron transfer and storage between pollutants and H2O2. Fe2+/Fe3+ ions are found in the framework of lf-Fe/PI HNs and directly bond with the C and N species of polyimide to form C–Fe–N–C in a closed loop. The experimental results and theoretical calculations demonstrate that electron-rich Fe and electron-deficient C areas are produced in the C–Fe–N–C of lf-Fe/PI HNs. This novel structure type in Fe-based materials leads to an excellent Fenton-like catalytic performance and efficiency for the refractory pollutant degradation under neutral conditions. The reaction rate is found to be 7–10 times higher than that of the conventional Fe-based Fenton-like catalyst. The reaction mechanism revealed that H2O2 is reduced to ˙OH radicals (further attacking pollutants) in the electron-rich Fe areas, and pollutants are mainly oxidized and degraded by providing electrons for the electron-deficient C areas. Fe–N–C acts as a bridge for electron transfer and electron storage carriers between pollutants and H2O2. This process enables the sustainable use of the electrons of pollutants, thereby greatly reducing the energy consumption for water treatment.
Introduction
The classical Fenton reaction based on the reaction of hydrogen peroxide (H2O2) with ferrous ions (Fe2+),1–3 which was first discovered over 120 years ago, is still of special significance for water treatment today.4 The most attractive part is the generation of highly aggressive hydroxyl radicals (˙OH, E0![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) = 2.80 V/NHE), which have been proved to be one of the most powerful and environment-friendly species to attack organic pollutants.5–8 Nevertheless, the widespread use of the Fenton reaction is being limited by its bottleneck problems, including the narrow pH range (pH 2–3) for normal reactions,9 and the Fe-containing sludge generation due to the rate limitation of electron recycling,10 which is considered secondary pollution and requires additional treatment processes. To address these problems, various types of heterogeneous Fenton-like materials such as zero-valent transition metals,11 transition metal oxides,12,13 supported/doped transition metal solids,14–16 and single atom catalysts17 were developed. These catalysts have demonstrated to be useful alternatives due to their good separability and the avoidance of Fe-containing sludge production.18 However, the heterogeneous Fenton-like process does not deviate from the basic principles of the classical Fenton reaction, and so the rate-limiting step upon the reduction of the stationary
= 2.80 V/NHE), which have been proved to be one of the most powerful and environment-friendly species to attack organic pollutants.5–8 Nevertheless, the widespread use of the Fenton reaction is being limited by its bottleneck problems, including the narrow pH range (pH 2–3) for normal reactions,9 and the Fe-containing sludge generation due to the rate limitation of electron recycling,10 which is considered secondary pollution and requires additional treatment processes. To address these problems, various types of heterogeneous Fenton-like materials such as zero-valent transition metals,11 transition metal oxides,12,13 supported/doped transition metal solids,14–16 and single atom catalysts17 were developed. These catalysts have demonstrated to be useful alternatives due to their good separability and the avoidance of Fe-containing sludge production.18 However, the heterogeneous Fenton-like process does not deviate from the basic principles of the classical Fenton reaction, and so the rate-limiting step upon the reduction of the stationary ![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) Fe3+ to Fe2+ still exists in the reaction process,19 which results in poor activity under neutral conditions and the excessive consumption of H2O2 as electron donors with a low H2O2 utilization efficiency.18 These problems have seriously hindered the application of heterogeneous Fenton catalysts.20
Fe3+ to Fe2+ still exists in the reaction process,19 which results in poor activity under neutral conditions and the excessive consumption of H2O2 as electron donors with a low H2O2 utilization efficiency.18 These problems have seriously hindered the application of heterogeneous Fenton catalysts.20
Our former research on Cu-based heterogeneous Fenton-like systems19–21 has shown that these problems could be overcome by forming σ-Cu2+-complexes on the surface of catalysts for aromatic pollutant degradation. On this basis, we developed a series of metal Cu–organic catalysts to construct the C–O–Cu bonding bridge and induce the generation of dual-reaction-centers (DRCs) with electron-rich and electron-deficient areas,22–24 which resulted in a significant increase in the activity of the catalyst and a great increase in the utilization efficiency of H2O2 (up to ∼90%). However, the toxicity of Cu is much stronger than that of Fe, resulting in the uneasy application of the Cu-based catalyst for water treatment. The development of Fe-based DRC catalysts is the most appropriate choice for sustainable water treatment.
In addition to our work on Cu-based systems, surface complexation25 and organometallic polymerization26,27 have also been reported to promote the activities of Fe-based Fenton-like catalytic systems. These studies imply the key roles of the interactions of Fe and N species. We have also explored the complexation of Fe with N-graphene and g-C3N4. Unfortunately, their Fenton activities for pollutant degradation are far from satisfactory under neutral conditions due to their inability to form the key bonding bridge to induce the generation of surface electron-rich/deficient areas. Polyimide (PI) is considered as an attractive high-performance polymer, which contains many benzene rings with π electrons and imide rings (–CO–NH–CO–) in the main chains. Furthermore, the precursor of PI can be transformed into aqueous solution by facile chemical modification.28–30 The special complexation may be easier to achieve before Fe and N-containing substrates.
In this work, we report for the first time a new Fe-containing catalyst consisting of lotus flower-like Fe-doped polyimide hybrid nanosheets (lf-Fe/PI HNs) prepared by a hydrothermal one-pot polymerization process. lf-Fe/PI HNs exhibit high Fenton-like catalytic efficiency for organic pollutant removal by inhibiting H2O2 overoxidation under neutral conditions. X-ray photoelectron spectroscopy (XPS), extended X-ray absorption fine structure (EXAFS) and Fourier-transform infrared spectroscopy (FTIR) analyses reveal the microstructure information of lf-Fe/PI HNs and prove the bonding of Fe with C and N of polyimide to form C–Fe–N–C bridges. Electron paramagnetic resonance (EPR) and density functional theory (DFT) calculations reveal that electron-rich Fe and electron-deficient C areas are produced in the C–Fe–N–C of lf-Fe/PI HNs, which is the key factor for the enhancement of the sustainable Fenton-like reaction. Fe–N–C acts as the bridge for the electron transfer and electron storage carrier between pollutants and H2O2. An effort to uncover the novel reaction mechanism has also been undertaken in detail.
Results and discussion
Fabrication and fine microstructure of lf-Fe/PI HNs
In this study, lotus flower-like Fe-doped polyimide hybrid nanosheets (lf-Fe/PI HNs) are fabricated by a hydrothermal polymerization process (see the Methods section for more details). As illustrated in Fig. 1, ethylenediamine (EDA) and pyromellitic dianhydride (PMDA) are used to react with N,N-dimethylformamide (DMF) to form linear polyamic acid polymer chains (LPAPCs). The iron source is introduced into LPAPCs under the protection and dispersion of glucose to form the precursor of the as-prepared polymer, which undergoes the dehydration–condensation reaction through a hydrothermal process. After further washing and drying, lf-Fe/PI HNs are fabricated. As a reference, the pure PI sample was also prepared using the same method as described above without adding the iron source and glucose.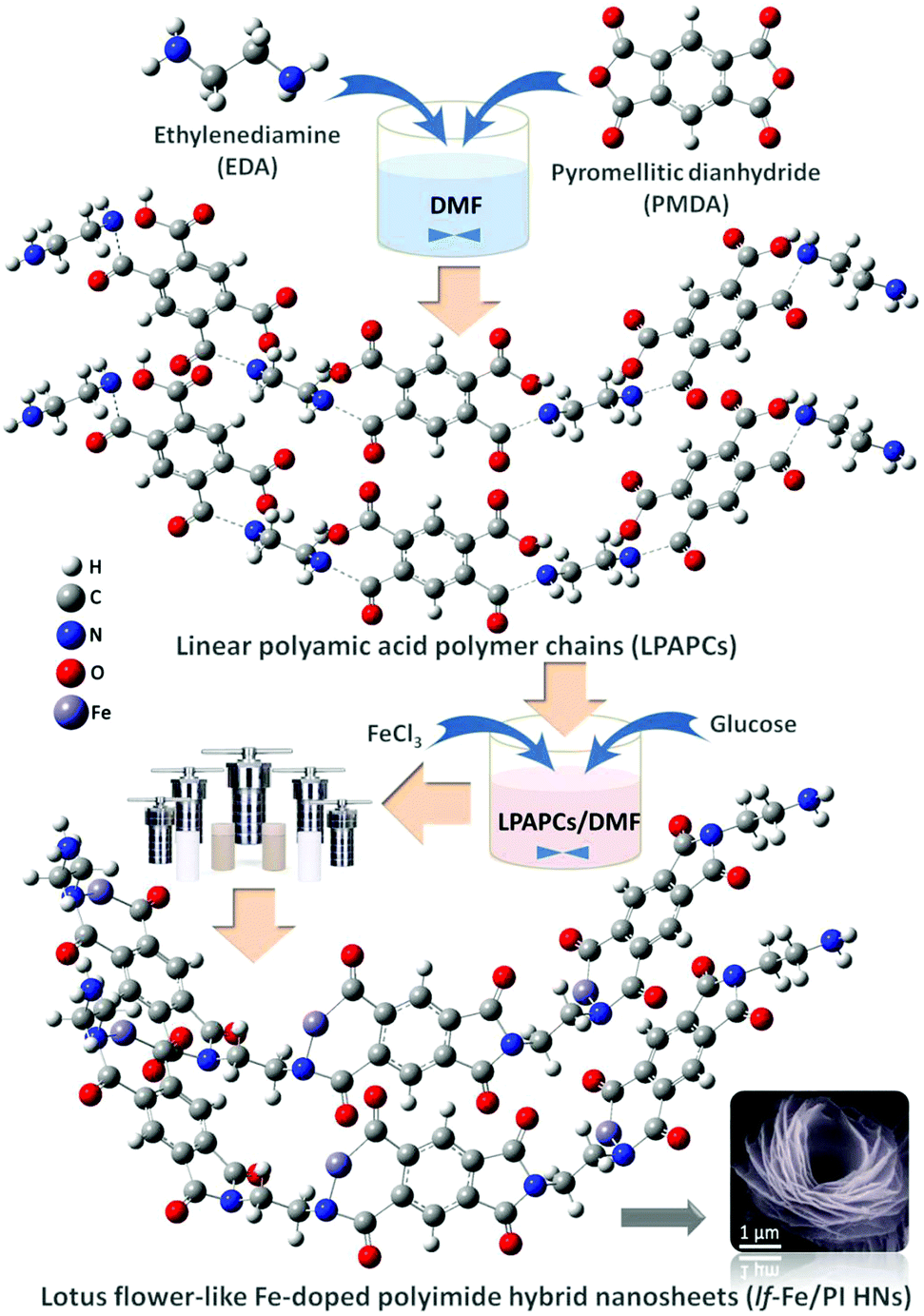 | ||
| Fig. 1 Schematic illustration of the synthesis route of lf-Fe/PI HNs. | ||
Scanning electron microscopy (SEM) images (Fig. 2a) show that lf-Fe/PI HNs clearly exhibit polymeric 3D architectures constructed using 2D nanosheets with a diameter of ∼2.0 μm and a thickness of ∼25 nm. The great number of stacking nanosheets with a highly ordered arrangement enabled lf-Fe/PI HNs to exhibit a lotus flower-like morphology. Transmission electron microscopy (TEM) images of lf-Fe/PI HNs (Fig. 2b) further confirm the neatness and orderliness of the self-arranged nanosheets and disclose the presence of numerous micropores and apertures between the nanosheets in lf-Fe/PI HNs. Moreover, no impurity clusters were found even at the edge of the nanosheets, indicating the well crystalized framework of lf-Fe/PI HNs. Fig. 2c shows the elemental mapping analyses of lf-Fe/PI HNs from SEM, which demonstrates the existence of the C, N, O and Fe elements in the structure of the catalyst. The mapping of Fe further shows that the doped Fe species are uniformly dispersed in lf-Fe/PI HNs. Fig. S1 (ESI†) shows the N2 adsorption–desorption isotherm of lf-Fe/PI HNs as well as its and BJH pore size distribution. The adsorption–desorption isotherm of lf-Fe/PI HNs has a hysteresis loop, belonging to a typical IV-type isotherm. In addition, the surface area of lf-Fe/PI HNs obtained is 19.8 m2 g−1 and the most distributed pore diameter is ∼3.62 nm, indicating that lf-Fe/PI HNs possess a typical mesoporous structure. Fig. S2 (ESI†) shows the powder X-ray diffraction (XRD) patterns of PI and lf-Fe/PI HNs. It is obvious that lf-Fe/PI HNs exhibit similar XRD patterns to the pure PI sample, suggesting that the skeleton structure of the substrate is well preserved with the introduction of Fe. However, all of the diffraction peaks of lf-Fe/PI HNs are slightly shifted compared to those of the pure PI sample, indicating that the incorporated Fe has affected the crystalline interplanar distance of the substrate.
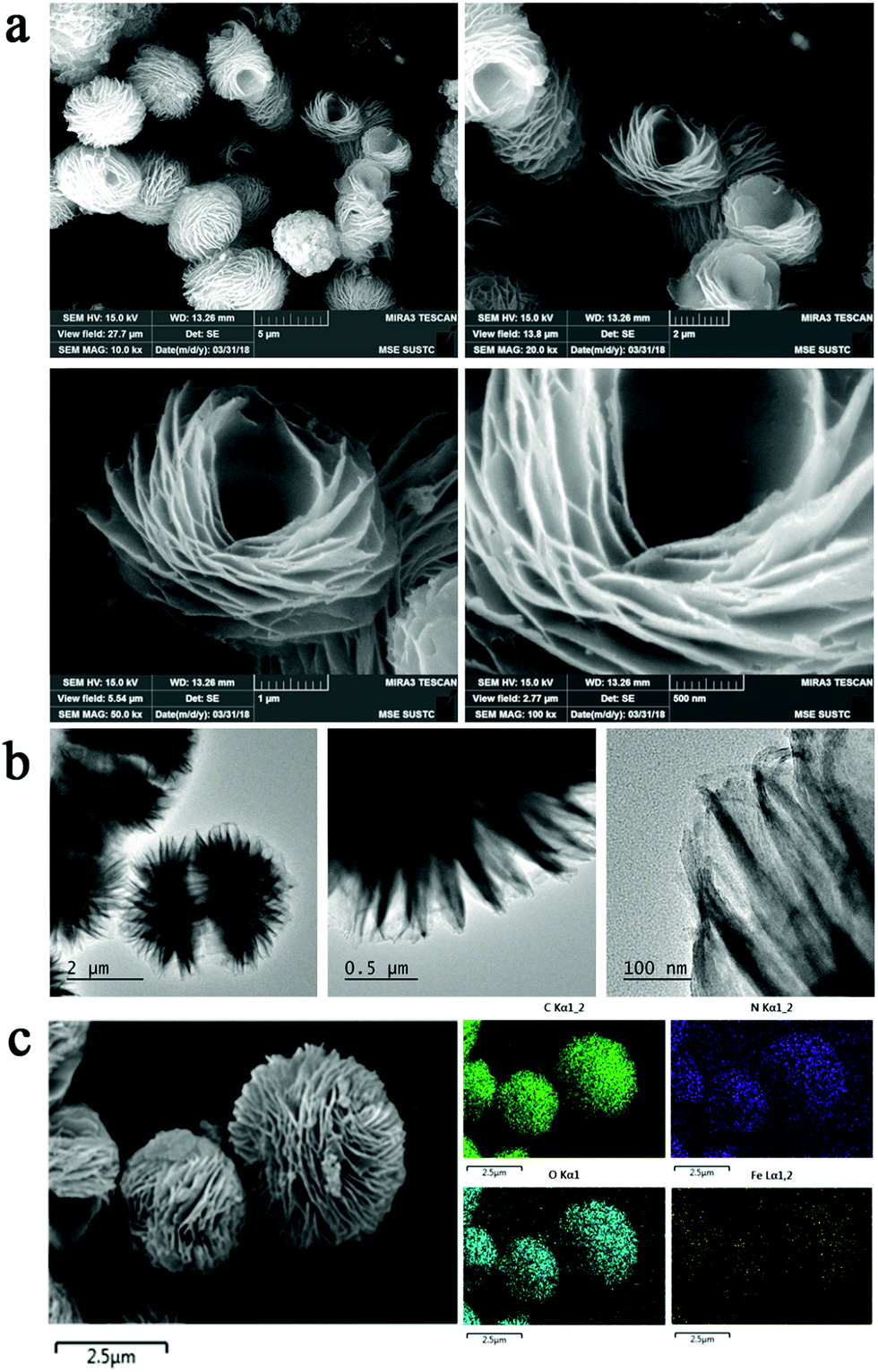 | ||
| Fig. 2 Morphology and structure of lf-Fe/PI HNs. (a) SEM images with different amplifications of lf-Fe/PI HNs. (b) TEM images with different amplifications of lf-Fe/PI HNs. (c) Corresponding SEM elemental mapping analysis for C, N, O and Fe of lf-Fe/PI HNs. | ||
The XPS spectrum in the Fe 2p orbital of lf-Fe/PI HNs is shown in Fig. 3a. The binding energy peaks located at 710.6 and 724.4 eV are attributed to the formation of surface Fe2+ species.31 The weaker peaks at 712.7 and 726.8 eV correspond to the surface Fe3+ species, which is also verified by the occurrence of the satellite peak at 717.2 eV.32 The curve fittings of the XPS spectrum showed that both Fe2+ and Fe3+ species were generated on the surface of lf-Fe/PI HNs with the atomic ratio of Fe2+/Fe3+ of 1.5:1. Fig. S3 (ESI†) exhibits the C 1s XPS spectrum of lf-Fe/PI HNs. The binding energy peaks located at 284.4, 285.4, 286.3 and 288.5 eV are attributed to the sp2 C–C/C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C, C–N, C–O– (from the adsorbing surface –OH) and CO bands in lf-Fe/PI HNs, respectively. The ratio of C–N to C–O is 1:0.36. lf-Fe/PI HNs almost preserve all the C 1s XPS characteristic peaks of PI (Fig. S4, ESI†), indicating the stable carbon skeleton even after introducing Fe. Differently, the binding energy corresponding to C–N in C 1s shifted to 285.4 from 285.8 eV and the C–N/C–O ratio decreased after the incorporation of Fe species, indicating that the binding sites of Fe species are relevant to the N atoms in lf-Fe/PI HNs. Fig. 3b presents the N 1s spectra of PI and lf-Fe/PI HNs. The strong peak in the PI spectrum centered at 400.5 eV originates from the pyrrolic N based on the intrinsic structure characteristics of PI. For lf-Fe/PI HNs, the binding energy peak shifted to 400.2 eV, which is largely due to the ligand N atoms of PI coordinating to Fe, forming the pyridine-like Fe–N bonds in lf-Fe/PI HNs.33 As expected, no obvious change was observed in the peak intensity and the binding energy of O 1s corresponding to CO after the introduction of Fe (Fig. S5, ESI†), which suggested that the binding sites of Fe species were independent of the framework O atoms in lf-Fe/PI HNs.
C, C–N, C–O– (from the adsorbing surface –OH) and CO bands in lf-Fe/PI HNs, respectively. The ratio of C–N to C–O is 1:0.36. lf-Fe/PI HNs almost preserve all the C 1s XPS characteristic peaks of PI (Fig. S4, ESI†), indicating the stable carbon skeleton even after introducing Fe. Differently, the binding energy corresponding to C–N in C 1s shifted to 285.4 from 285.8 eV and the C–N/C–O ratio decreased after the incorporation of Fe species, indicating that the binding sites of Fe species are relevant to the N atoms in lf-Fe/PI HNs. Fig. 3b presents the N 1s spectra of PI and lf-Fe/PI HNs. The strong peak in the PI spectrum centered at 400.5 eV originates from the pyrrolic N based on the intrinsic structure characteristics of PI. For lf-Fe/PI HNs, the binding energy peak shifted to 400.2 eV, which is largely due to the ligand N atoms of PI coordinating to Fe, forming the pyridine-like Fe–N bonds in lf-Fe/PI HNs.33 As expected, no obvious change was observed in the peak intensity and the binding energy of O 1s corresponding to CO after the introduction of Fe (Fig. S5, ESI†), which suggested that the binding sites of Fe species were independent of the framework O atoms in lf-Fe/PI HNs.
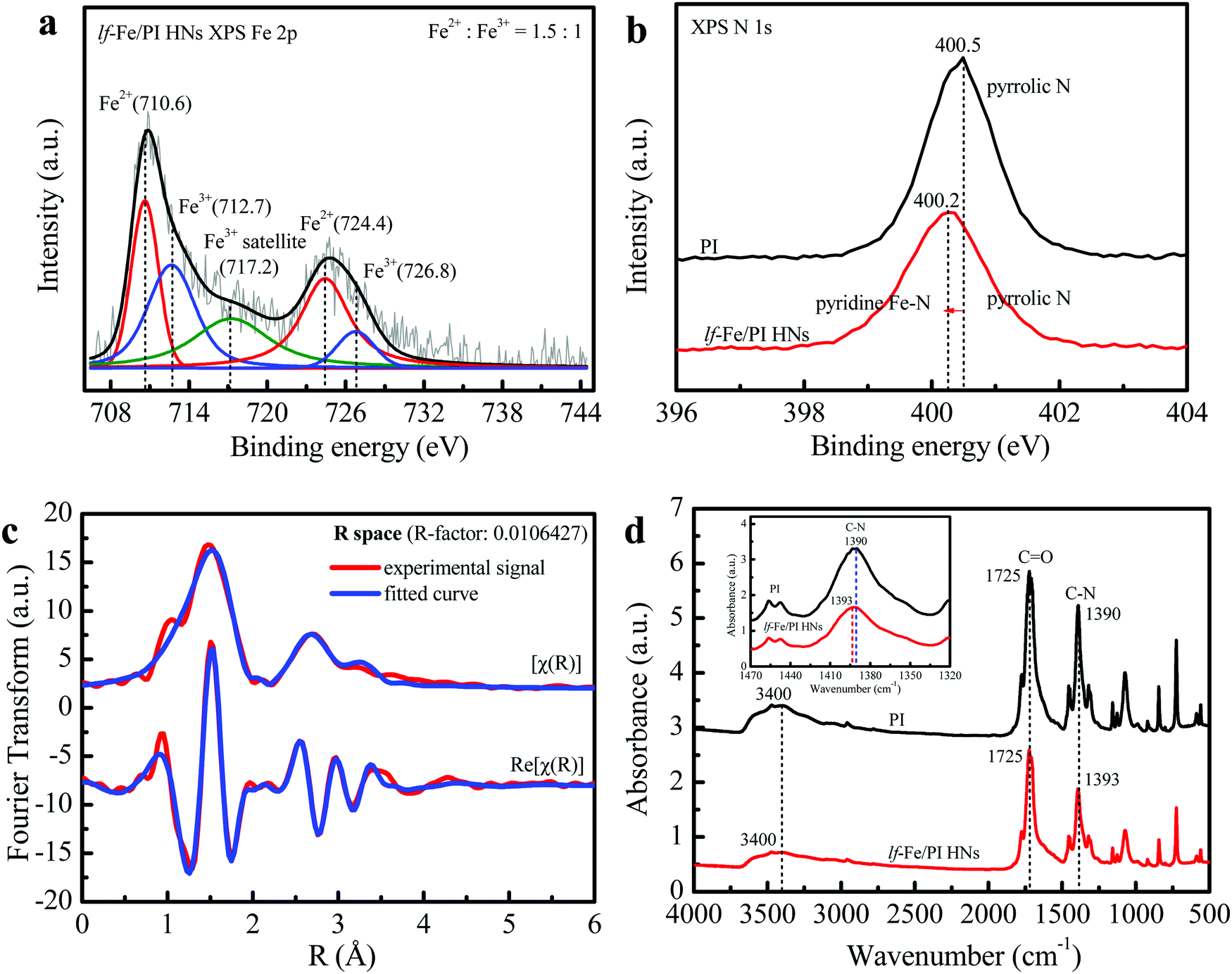 | ||
| Fig. 3 Local chemical environment and fine structure of surface elements. (a) Fe 2p XPS spectrum of lf-Fe/PI HNs. (b) N 1s XPS spectra of PI and lf-Fe/PI HNs. (c) Fe K-edge EXAFS oscillation and its curve fitting of lf-Fe/PI HNs. (d) FTIR spectra of PI and lf-Fe/PI HNs. | ||
To reveal the fine microstructure of lf-Fe/PI HNs and the coordination environment of Fe, the Fourier-transformed EXAFS signal magnitude curves at the Fe K-edge of lf-Fe/PI HNs were collected and the fitting curves were calculated using Artemis (Fig. 3c). The advanced fitting curves of the sample on [χ(R)] and Re[χ(R)] spaces with several shells almost completely coincide with those of the corresponding experimental signals of lf-Fe/PI HNs and the R-factor in the fitting process is less than 0.02, demonstrating the fitting credibility and certainty. Table 1 presents the finally obtained fitting information. The Fe–C shell with a bond distance of (R) =1.68 Å and a coordination number of (CN) = 1.0 and the Fe–N shell with R = 2.01 Å and CN = 18.1 are observed on the first-shells, which are due to the direct backscattering from C and N atoms, confirming the formation of C–Fe–N bonds on lf-Fe/PI HNs. In addition to the two first-shells, three other shells including the Fe–Fe shell (R = 3.05 Å, CN = 3.9), Fe–C shell (R = 3.46 Å, CN = 16.3) and Fe–O shell (R = 3.77 Å, CN = 11.0) are also observed. Obviously, the Fe–Fe shell is attributed to the backscattering from the close Fe atoms. The Fe–O shell is attributed to the Fe–CO bonds on lf-Fe/PI HNs. The Fe–C shell with the maximum coordination number (16.3) is due to the backscattering from multiple types of C atoms, which include the Fe–C–C bonds and Fe–N–C bonds on the formed six-membered rings and chains of lf-Fe/PI HNs. The EXAFS results clearly reveal the coordination and bonding structures (Fe–N, Fe–C, C–Fe–N, Fe–C–C, Fe–N–C and Fe–CO) of lf-Fe/PI HNs.
| Sample | Shell | R (Å) | CNb | σ 2 (Å2) | ΔE0d (eV) | R f |
|---|---|---|---|---|---|---|
| a Bond distance. b Coordination number. c Debye–Waller factor. d Inner potential correction. e Residual factor. | ||||||
| lf-Fe/PI HNs | Fe–C | 1.68 | 1.0 | 0.0121 | −5.477 | 0.0106 |
| Fe–N | 2.01 | 18.1 | 0.0121 | −5.477 | 0.0106 | |
| Fe–Fe | 3.05 | 3.9 | 0.0121 | −5.477 | 0.0106 | |
| Fe–C | 3.46 | 16.3 | 0.0121 | −5.477 | 0.0106 | |
| Fe–O | 3.77 | 11.0 | 0.0121 | −5.477 | 0.0106 | |
The FTIR spectra of the prepared PI and lf-Fe/PI HNs samples are shown in Fig. 3d. It is clear that both PI and lf-Fe/PI HNs exhibit a series of characteristic peaks in the range of 500–1800 cm−1, which are caused by the characteristic vibration of the basic group of the PI substrate. Typically, the strong stretching vibration of the CO bond is located at 1725 cm−1 and the asymmetric and symmetric stretching vibrations of the C–N bond are located at ∼1390 cm−1 for the PI substrate.34 After the introduction of Fe species, the CO characteristic peak does not change, while the wavenumber of the C–N vibration peak evidently shifts to 1393 cm−1 from 1390 cm−1, which confirms that the introduced Fe does not rely on the O site but connects with the N site, forming C–N–Fe bonds in lf-Fe/PI HNs. The two spectra of PI and lf-Fe/PI HNs display a broad absorption peak at 3400 cm−1 ascribing to the stretching vibrations of surface –OH [ν(OH)]. There is no change in the centered wavenumber for ν(OH) after introducing Fe, indicating no coordination occurrence between metal species and the surface –OH in lf-Fe/PI HNs. Thus, all the evidence mentioned above reveal that the Fe species was incorporated into the PI substrate framework and connected to N to form Fe–N in a closed loop, rather than Fe–O–C bonds.
Formation of electron-rich Fe and electron-deficient C areas on the lf-Fe/PI HN surface
Fig. 4a shows the EPR spectra of the prepared PI and lf-Fe/PI HN solid samples, which are used to investigate the electronic structural information of the catalysts. Only a very weak signal is observed for the PI substrate, indicating that there are almost no unpaired electrons on the PI structure, which confirms the inertness of the delocalized π electrons in the PI structure. However, a very strong and symmetrical signal appears on the spectra of lf-Fe/PI HNs. The g value of this EPR signal is 2.004, which is very close to the value of the free electrons (∼2.0023).35 This result indicates that the formed Fe–N draws a lot of free electrons around the Fe species from the organic ligand, forming electron-rich Fe areas in lf-Fe/PI HNs.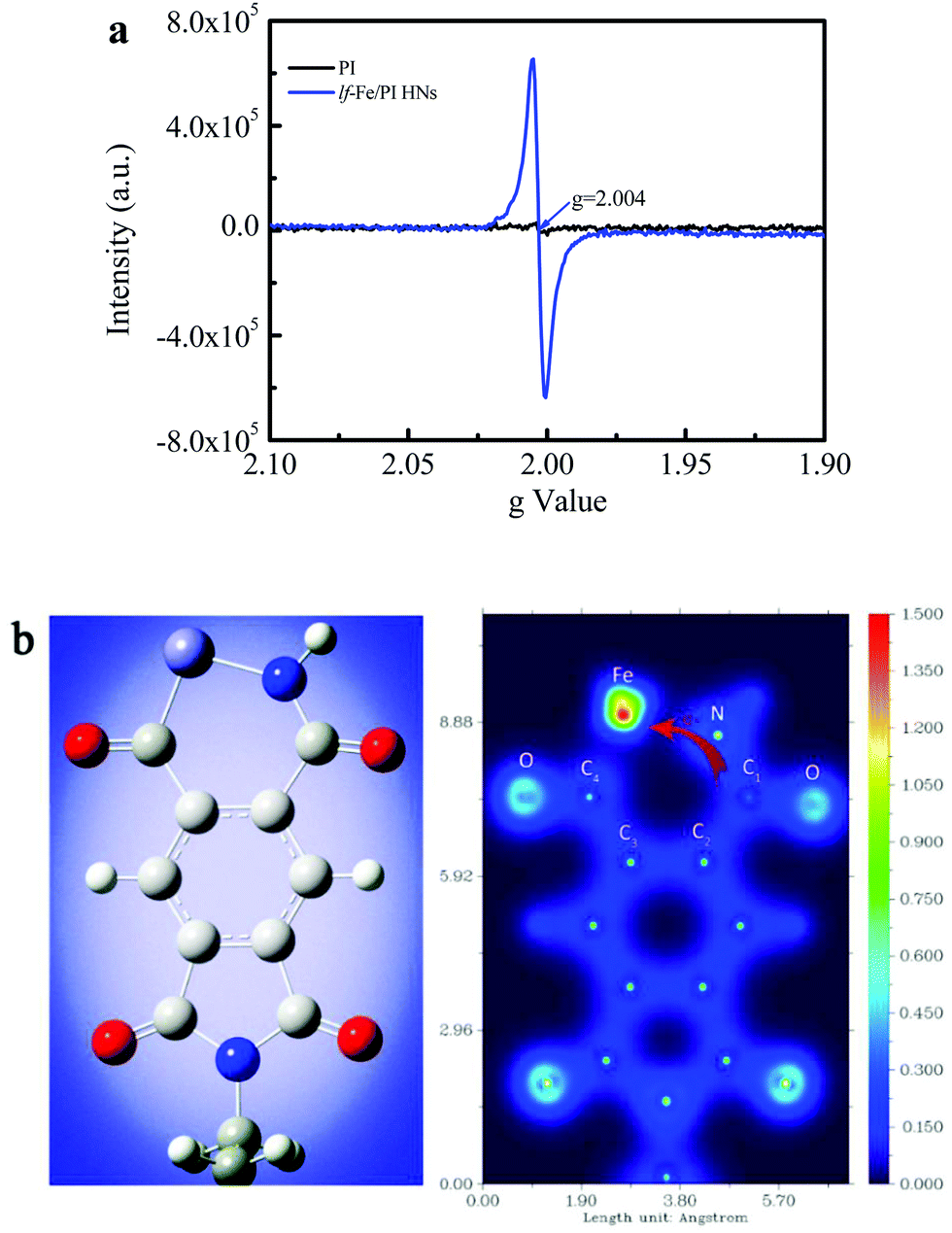 | ||
| Fig. 4 Non-uniform distribution of surface electrons on lf-Fe/PI HNs. (a) EPR spectra of PI and lf-Fe/PI HNs. (b) DFT calculations for the optimized structure of lf-Fe/PI HNs in one unit (left) and the corresponding two-dimensional valence-electron density color-filled maps of the lf-Fe/PI HNs model (right). Purple, red, grey and blue circles denote Fe, O, C and N atoms, respectively. The valence-electron density is given in e Å−3. | ||
The valence-electron density (VED) distributions of the lf-Fe/PI HNs are investigated by DFT calculations to obtain the reactive site information. Fig. 4b (left) presents the optimum geometric structure of the lf-Fe/PI HN model fragments obtained with a Gaussian 09 package with a B3LYP functional. Fig. 4b (right) shows the corresponding two-dimensional VED color-filled map of the model obtained through Multiwfn package. In the lf-Fe/PI HN model fragment, the largest VED area occurs around the Fe species with the relative maximum value (1.5 e Å−3) for the VED. Obviously, the maximum VED value around Fe is much higher than those around the C and N atoms in the six-element rings, demonstrating that the electron-rich micro-center was formed around Fe theoretically, which is the same as the experimental results. It is worth noting that the VED distribution area around the carbon atom (C1) connecting with the N atom on the formed Fe–N–C1 bonding bridge is much narrower than that around the other C atoms (C2, C3 and C4) and the N atom, and the valence-electron density of C1 with the maximum VED of ∼0.5 e Å−3, is also evidently less than that of the N atom (∼1.3 e Å−3) on Fe–N–C1 as well as the other C atoms (0.7–1.0 e Å−3) on lf-Fe/PI HNs, confirming the electron transfer from C1 to the electron-rich Fe area through the Fe–N–C1 bonding bridge. These results reveal that the formed Fe–N–C on lf-Fe/PI HNs can draw lots of free electrons around the Fe species from the organic ligand, forming electron-rich Fe areas and electron-deficient C sites connecting with N atoms in lf-Fe/PI HNs.
The chemical reactivity and kinetic stability of the catalysts were also theoretically evaluated through DFT calculations based on the HOMO (highest occupied molecular orbital)–LUMO (lowest unoccupied molecular orbital) gap.36 For the pure PI, the HOMO–LUMO gap is 4.40 eV (the values are equivalent for α and β electrons). For lf-Fe/PI HNs, the energy gap for α electrons decreases to 3.54, and the gap for β electrons decreases to 2.24 eV. These results indicate that the electrons are activated and the chemical reactivity of lf-Fe/PI HNs is significantly enhanced with the introduction of Fe species and the formation of electron-rich areas.
Catalytic performance for refractory pollutant conversion in the lf-Fe/PI HN system
The Fenton-catalytic activity of the synthesized catalysts was first evaluated by the carcinogenic dyestuff Rhodamine B (RhB) removal at natural pH (∼7) with H2O2. As presented in Fig. 5a, only 30–40% of RhB could be decolorized within 120 min in the conventional Fenton catalyst Fe2O3 and Fe3O4 (with equivalent Fe content) systems. Under the same conditions, RhB decolorization was only 24.7% in the pure PI system. Astonishingly, the RhB decolorization reached 90.0% within only 60 min and the degradation rate reached 98.3% at 120 min in the lf-Fe/PI HNs/H2O2 suspension. The kinetic curves for RhB decolorization in various suspensions (Fig. 5a inset) show that the reaction rates in the lf-Fe/PI HNs/H2O2 suspension was ∼14, ∼10 and ∼7 times faster than that in the suspensions of PI/H2O2, Fe2O3/H2O2 and Fe3O4/H2O2, respectively. Furthermore, within 120 min, the removal rate of TOC could reach 50.1% in the lf-Fe/PI HNs/H2O2 suspension. This removal rate was substantially higher than those in the PI, Fe2O3 and Fe3O4 suspensions (TOC removal rate less than 10%). To explore the adaptability of lf-Fe/PI HNs for removing different pollutants, other refractory organic compounds including the azo dyestuff acid orange 7 (AO7), pesticide 2-chlorophenol (2-CP), endocrine disruptor bisphenol A (BPA) and pharmaceutical ciprofloxacin (CIP) were used for the degradation in the lf-Fe/PI HNs/H2O2 systems under natural conditions (pH ∼ 7). As shown in Fig. 5b, all of the selected pollutants (AO7, 2-CP, BPA and CIP) could be substantially removed in the lf-Fe/PI HN Fenton-like process and the conversion rates were all higher than 80% in 120 min, in particular for CIP, which could be completely eliminated within 60 min under the neutral conditions.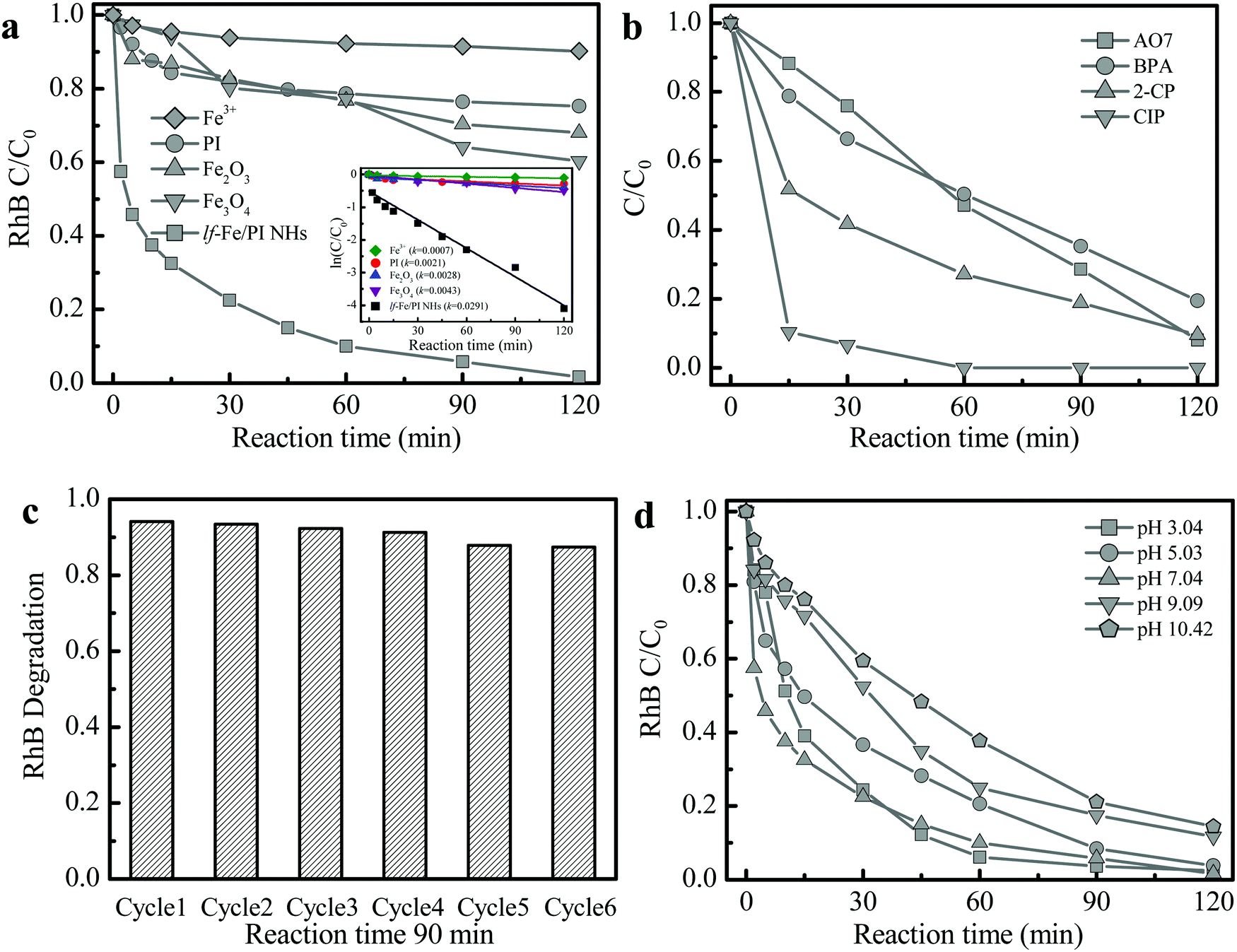 | ||
| Fig. 5 Performance of lf-Fe/PI HNs for pollutant removal. (a) RhB degradation curves in various suspensions with H2O2 (inset shows the corresponding kinetic curves). (b) Decomposition curves of other different pollutants in the lf-Fe/PI HNs suspensions with H2O2. (c) Reusability of lf-Fe/PI HNs for RhB degradation. (d) Effect of initial pH values for RhB degradation. Reaction conditions: natural initial pH (pH 6–7, except (d)), initial pollutants 10 mg L−1, initial H2O2 10 mM, catalyst 1.0 g L−1 (Fe ions 0.15 mg L−1 in (a)). | ||
After the reactions, the metal concentration released from the lf-Fe/PI HN sample was detected. It was found that the concentration of Fe ions was only ∼0.15 mg L−1. A homogeneous test using 0.15 mg L−1 Fe ions and 10 mM H2O2 was conducted to investigate the contribution of the released Fe for RhB removal (Fig. 5a), from which only ∼9.8% RhB decolorization and ∼1.7% TOC elimination within 120 min were achieved, showing that the homogeneous contribution was negligible. Fig. 5c shows the reusability of lf-Fe/PI HNs. The activity of the catalyst only decreased by 6.8% and the RhB decolorization rate could still reach ∼87.4% within 90 min after six successive cycles, and the catalyst became more and more stable in the later cycles due to the impurities on the catalyst surface being washed away.
The effect of initial pH on the catalyst activity is shown in Fig. 5d. lf-Fe/PI HNs exhibited the highest reactivity for RhB degradation under neutral conditions (pH ∼ 7.04). Although the activity of the lf-Fe/PI HN Fenton-like system decreased slightly after deviating neutrality, it was not obviously changed in a broad pH range (3.04–10.42), revealing the wide applicable range of pH for lf-Fe/PI HNs. The metal-based material activity often depends on the acid–base properties of the aqueous solution due to the generation of surface hydroxyl groups between the water molecules and the metal sites through the dissociative chemisorption,37 while lf-Fe/PI HNs overcame this barrier effect due to the generation of the electron-rich reaction areas, causing the metal species not to oxidize H2O2 directly. So the sensitivity of the catalyst to the change of pH decreased.
Insight into the interaction mechanism of lf-Fe/PI HNs and organic pollutants driving sustainable Fenton-like reaction
Fig. 6a shows the H2O2 utilization during the reaction process in the lf-Fe/PI HN system (see the Notes S1 of ESI† for the specific calculation process). The actual H2O2 consumption was close to the theoretical value in the first 30 min, which indicated a very low useless decomposition of H2O2 over lf-Fe/PI HNs. The calculated utilization efficiency of H2O2 was over 80% at this stage, and this value remained at a relatively high level during the whole Fenton-like process (reaching 62.6% at 120 min), which was significantly greater than the value (∼5%) of the conventional Fe3O4 Fenton systems.38 These results suggest the excellent catalytic performance and efficiency of lf-Fe/PI HNs with the formation of the electron-rich reaction areas.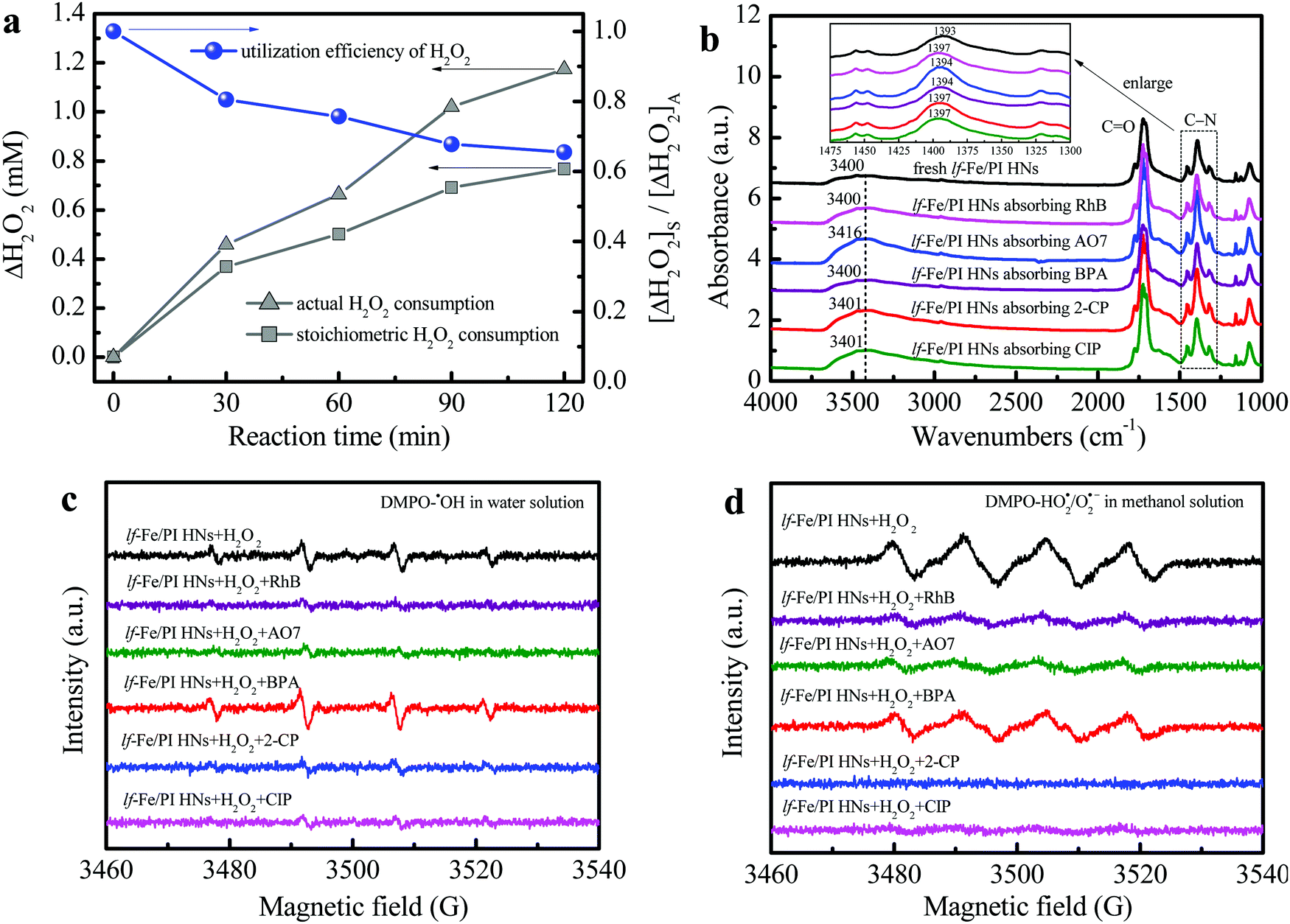 | ||
| Fig. 6 H2O2 utilization efficiency and free radical change during the reaction. (a) Actual and stoichiometric H2O2 consumptions and the utilization efficiency of H2O2 during the Fenton-like process in the lf-Fe/PI HN suspension (reaction conditions: natural initial pH, initial pollutants 10 mg L−1, initial H2O2 10 mM, catalyst 1.0 g L−1). (b) FTIR spectra of the lf-Fe/PI HN samples before/after absorbing different pollutants. DMPO spin-trapping EPR spectra for (c) ˙OH in the lf-Fe/PI HN aqueous suspensions before/after adding different pollutants and (d) HO2˙/O2˙− in lf-Fe/PI HN aqueous suspensions before/after adding different pollutants. | ||
FTIR analysis was used to investigate the adsorption and complexation sites of pollutants on the catalyst surface. The complexation of organic compounds with metal species on the catalyst surface often leads to a shift of the surface hydroxyl ν(OH) band to a low wavenumber according to previous reports.19,22 As shown in Fig. 6b, the ν(OH) bands (∼3400 cm−1) of lf-Fe/PI HNs did not redshift with the pollutants absorbing on the surface and even blue-shifted to 3416, 3401 and 3401 cm−1 after absorbing the pollutants AO7, 2-CP and CIP, respectively, which indicated that the pollutants did not complex with the metal Fe species on the surface of lf-Fe/PI HNs during the reaction process. It was also worth noting that the FTIR absorption peak ascribed to the stretching vibration of the C–N bond (1393 cm−1) on lf-Fe/PI HNs shifted to higher wavenumbers (1394–1397 cm−1) after absorbing pollutants due to the interaction of the pollutants and the C sites of the C–N bonds on lf-Fe/PI HNs, implying the adsorption of pollutants on the electron-deficient C sites (C-rings), which made it possible for pollutants to donate electrons directly.
The DMPO-trapped EPR technique was used to analyze the interfacial electron transfer process between lf-Fe/PI HNs, H2O2 and pollutants through free radical detection. As shown in Fig. S6a and b (ESI†), no signals were detected in the pure water/methanol and lf-Fe/PI HN solutions. However, four centrosymmetrical characteristic peaks corresponding to the DMPO–˙OH signals with an intensity ratio of ∼1:2:2:1 and a magnetic field spacing of ∼14 G were observed (Fig. S6a, ESI†) after adding H2O2 into the lf-Fe/PI HN suspensions (without pollutants), which suggested that H2O2 was quickly reduced by the electrons around the electron-rich Fe areas of lf-Fe/PI HNs to produce ˙OH radicals. At the same time, four evident characteristic peaks corresponding to DMPO–HO2˙/O2˙− were also observed in the lf-Fe/PI HN methanol-aqueous dispersion (Fig. S6b, ESI†), which is due to the H2O2 oxidation in the electron-deficient C areas.
After introducing pollutants into the reaction process, the electron transfer pathways in the lf-Fe/PI HN Fenton-like system have changed. As shown in Fig. 6c, after adding various pollutants to the lf-Fe/PI HNs/H2O2 systems, most of the signal intensities of DMPO–˙OH weakened due to the consumption of the produced ˙OH radicals by the pollutants, implying the fast reactivity of the lf-Fe/PI HNs/H2O2 systems. Differently, the DMPO–˙OH signal intensity increased after adding BPA, suggesting that the electron-rich BPA could promote the reduction of H2O2 and affect the consumption of ˙OH. The change of the DMPO–HO2˙/O2˙− signals give more important information about the electron transfers (Fig. 6d). After adding pollutants in the lf-Fe/PI HNs/H2O2 systems, all of the DMPO–HO2˙/O2˙− signals were significantly weakened, which revealed that the organic pollutants directly acted as electron-donors on the electron-deficient C sites, greatly avoiding H2O2 oxidation to generate HO2˙/O2˙− and accelerating the electron transfer cycles that from pollutants to electron-deficient areas. In this process, Fe–N–C acted as an electron transfer bridge and electron storage carrier from pollutants to H2O2, resulting in the efficient oxidation of pollutants in electron-deficient C areas and efficient reduction (rather than oxidation) of H2O2 in electron-rich Fe areas. Especially in the CIP and 2-CP systems, almost no HO2˙/O2˙− signals were observed, indicating that the ineffective oxidation and decomposition of H2O2 have been completely suppressed, and thus the lf-Fe/PI HNs/H2O2 Fenton-like system exhibits excellent activity for the degradation of CIP and 2-CP.
Conclusions
In summary, we have developed a new Fe-containing catalyst (lf-Fe/PI HNs) consisting of lotus flower-like Fe-doped polyimide hybrid nanosheets prepared by a hydrothermal one-pot polymerization process in this work. lf-Fe/PI HNs exhibited high Fenton-like catalytic efficiency for organic pollutant removal by inhibiting H2O2 overoxidation under neutral conditions. The reaction rate is found to be 7–10 times higher than that of the conventional Fe-based Fenton-like catalyst. This work greatly resolved the bottleneck breakthrough of the Fe-based Fenton reaction by constructing surface electron distribution-polarized areas on the catalyst, using the electron compensation effect of pollutants, and utilizing the Fe–N–C electron transfer bridge and electron storage carrier during the reaction, which significantly saved the oxidative consumption of H2O2, and improved the utilization rate of H2O2 and degradation efficiency for pollutants under neutral conditions. Obviously, the lf-Fe/PI HNs/H2O2 Fenton-like system is of importance environmentally and energetically for solving the bottleneck in the application of the Fe-based Fenton reaction for water treatment, which also has significance to the development of sustainable environmental remediation technology by utilizing the energy of pollutants.Methods
Catalyst preparation
lf-Fe/PI HNs were synthesized by a hydrothermal polymerization process. Typically, 7.2 mmol EDA and 7.2 mmol PMDA were dissolved in 150 mL of DMF, which was then stirred at 45 °C for 2 h to form LPAPCs. After that, 7.2 mmol FeCl3 and 2.4 mmol glucose were added to the above mixture with stirring for 2 h, which could form the precursor of the as-prepared polymer. The precursor was then transferred into a hydrothermal autoclave with Teflon-lined stainless steel and subjected to a dehydration–condensation reaction at 180 °C for 15 h. After naturally cooling to ambient temperature, the obtained solid sample was washed several times using DMF and deionized water and dried in an oven (70 °C) to yield lf-Fe/PI HNs. As a reference, the pure PI sample was also prepared using the same method as described above without adding FeCl3 and glucose. The chemicals and reagents used in this work are shown in the Notes S2 of ESI.†Characterization methods
TEM and SEM images were observed through a FEI Tecnai G2 F20 TEM and a TESCAN MIRA 3 GMU FESEM instrument, respectively. The Brunner–Emmett–Teller (BET) specific surface area and pore diameter were obtained using a gas sorption analyzer (ASAP2460) cooling by liquid nitrogen. Powder XRD patterns were obtained using a Bruker D8 ADVANCE diffractometer (40 kV, 40 mM) with Cu Kα radiation λ = 1.540598 Å. The XPS data of the prepared samples were recorded using an ESCALAB 250Xi XPS system (Thermo Fisher Scientific Inc., USA) with monochromatic Al Kα radiation as well as low-energy electron flooding for charge compensation. The FTIR spectra of the solid samples were recorded on an IS10 FTIR spectrophotometer (Thermo Fisher Scientific Inc., USA) after pressing potassium bromide tablets. Fe K-edge EXAFS data of lf-Fe/PI HNs were recorded at the 1W1B-XAFS beamline of the Beijing Synchrotron Radiation Facility (BSRF) using a Si(111) double-crystal monochromator under natural conditions. The EXAFS data were pre-treated using the Athena program. The advanced fitting of EXAFS spectra was performed using Artemis interfaces of IFFEFIT 7.0 package. The solid EPR spectra of the samples were recorded on a Bruker A300-10/12 electron paramagnetic resonance (EPR) spectrometer.Procedures and analysis
RhB, AO7, BPA, 2-CP and CIP, as various types of typical refractory organic pollutants, were selected as the representative refractory organic pollutants in different industries to evaluate the Fenton-like reaction performance of the prepared catalysts. Typically, 0.05 g of the catalyst powder and 50 mL of the pollutant solution (10 mg L−1, natural pH) were first mixed in a glass beaker of appropriate capacity with consecutive stirring for approximately 20 min to achieve adsorption–desorption equilibrium of pollutants on the catalyst surface (∼50% RhB could be adsorbed on the catalyst surface). Then, the Fenton-like reaction was triggered by adding H2O2 (10 mM) under magnetic stirring. Approximately 2 mL of the sample of the reaction suspension was collected with a syringe and filtered with a filter (0.45 μm) at certain intervals for follow-up analysis. To investigate the repeatability of lf-Fe/PI HNs, the used sample was filtered and washed with deionized water. The obtained solid was dried in the oven and recycled in the subsequent reaction. All the information and methods for HPLC, ICP-MS, TOC and EPR measurements are shown in Notes S3 of ESI.† The computational methods of DFT are shown in the Notes S4 of ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the Introduced Innovative R&D Team Project under the “Pearl River Talent Recruitment Program” of Guangdong Province (2019ZT08L387), the National Natural Science Foundation of China (51838005, 51808140 and 51538013), the National Key Research and Development Plan of China (2016YFA0203200), the Natural Science Foundation of Guangdong Province (2018A030313487), the Young Innovative Talent Project in Higher Education of Guangdong Province (2017KQNCX150), the Guangdong Province Universities and Colleges Pearl River Scholar Funded Scheme (Young Scholar), and the National College Students’ Innovation and Entrepreneurship Training Program of China (201911078037). The authors also appreciate the support from the 1W1B-XAFS beamline of the Beijing Synchrotron Radiation Facility (BSRF, China).References
- C. Vilcheze, T. Hartman, B. Weinrick and W. R. Jacobs, Nat. Commun., 2013, 4, 1881 CrossRef PubMed.
- C. Liu, Y. Cao, Y. Cheng, D. Wang, T. Xu, L. Su, X. Zhang and H. Dong, Nat. Commun., 2020, 11, 1735 CrossRef CAS PubMed.
- M. F. Huo, L. Y. Wang, Y. Chen and J. L. Shi, Nat. Commun., 2017, 8, 357 CrossRef PubMed.
- L. Lyu and C. Hu, Prog. Chem., 2017, 29, 981–999 Search PubMed.
- S. G. Huling and S. Hwang, Water Res., 2010, 44, 2663–2671 CrossRef CAS PubMed.
- L. Lyu, G. F. Yu, L. L. Zhang, C. Hu and Y. Sun, Environ. Sci. Technol., 2018, 52, 747–756 CrossRef CAS PubMed.
- Y. Ren, M. Shi, W. Zhang, D. D. Dionysiou, J. Lu, C. Shan, Y. Zhang, L. Lv and B. Pan, Environ. Sci. Technol., 2020, 54, 5258–5267 CrossRef CAS PubMed.
- Z. Ye, J. A. Padilla, E. Xuriguera, J. L. Beltran, F. Alcaide, E. Brillas and I. Sirés, Environ. Sci. Technol., 2020, 54, 4664–4674 CrossRef CAS PubMed.
- N. N. Tusar, D. Maucec, M. Rangus, I. Arcon, M. Mazaj, M. Cotman, A. Pintar and V. Kaucic, Adv. Funct. Mater., 2012, 22, 820–826 CrossRef CAS.
- S. H. Yoo, D. Jang, H. I. Joh and S. Lee, J. Mater. Chem. A, 2017, 5, 748–755 RSC.
- Y. W. Pan, M. H. Zhou, X. Li, L. T. Xu, Z. X. Tang and M. M. Liu, Sep. Purif. Technol., 2016, 169, 83–92 CrossRef CAS.
- S. T. Jiang, J. Z. Zhu, Y. Ding, S. L. Bai, Y. J. Guan and J. Wang, Ozone: Sci. Eng., 2016, 38, 225–232 CrossRef CAS.
- M. A. Fontecha-Camara, C. Moreno-Castilla, M. V. Lopez-Ramon and M. A. Alvarez, Appl. Catal., B, 2016, 196, 207–215 CrossRef CAS.
- X. J. Yang, P. F. Tian, C. X. Zhang, Y. Q. Deng, J. Xu, J. L. Gong and Y. F. Han, Appl. Catal., B, 2013, 134, 145–152 CrossRef.
- Y. T. Zhang, C. Liu, B. B. Xu, F. Qi and W. Chu, Appl. Catal., B, 2016, 199, 447–457 CrossRef CAS.
- X. R. Wang, W. Z. Yang, Y. Ji, X. S. Yin, Y. Liu, X. Z. Liu, F. Y. Zhang, B. H. Chen and N. Yang, RSC Adv., 2016, 6, 26155–26162 RSC.
- Y. Yin, L. Shi, W. L. Li, X. N. Li, H. Wu, Z. M. Ao, W. J. Tian, S. M. Liu, S. B. Wang and H. Q. Sun, Environ. Sci. Technol., 2019, 53, 11391–11400 CrossRef CAS PubMed.
- L. Lyu, L. L. Zhang and C. Hu, Environ. Sci.: Nano, 2016, 3, 1483–1492 RSC.
- L. Lyu, L. L. Zhang, Q. Y. Wang, Y. L. Nie and C. Hu, Environ. Sci. Technol., 2015, 49, 8639–8647 CrossRef PubMed.
- L. Lyu, L. L. Zhang, C. Hu and M. Yang, J. Mater. Chem. A, 2016, 4, 8610–8619 RSC.
- L. Lyu, L. L. Zhang and C. Hu, Chem. Eng. J., 2015, 274, 298–306 CrossRef.
- L. Lyu, L. L. Zhang, G. Z. He, H. He and C. Hu, J. Mater. Chem. A, 2017, 5, 7153–7164 RSC.
- L. Lyu, D. B. Yan, G. F. Yu, W. R. Cao and C. Hu, Environ. Sci. Technol., 2018, 52, 4294–4304 CrossRef CAS PubMed.
- L. Lyu, W. Cao, G. Yu, D. Yan, K. Deng, C. Lu and C. Hu, J. Hazard. Mater., 2020, 383, 121182 CrossRef PubMed.
- X. J. Hou, X. P. Huang, F. L. Jia, Z. H. Ai, J. C. Zhao and L. Z. Zhang, Environ. Sci. Technol., 2017, 51, 5118–5126 CrossRef CAS PubMed.
- J. Q. Ma, Q. F. Yang, Y. Z. Wen and W. P. Liu, Appl. Catal., B, 2017, 201, 232–240 CrossRef CAS.
- J. Q. Ma, L. L. Xu, C. S. Shen, C. Hu, W. P. Liu and Y. Z. Wen, Environ. Sci. Technol., 2018, 52, 3608–3614 CrossRef CAS PubMed.
- Y. Y. Qin, Q. Y. Peng, Y. J. Ding, Z. S. Lin, C. H. Wang, Y. Li, J. J. Li, Y. Yuan, X. D. He and Y. B. Li, ACS Nano, 2015, 9, 8933–8941 CrossRef CAS PubMed.
- L. Lyu, M. E. Han, W. R. Cao, Y. W. Gao, Q. Y. Zeng, G. F. Yu, X. Huang and C. Hu, Environ. Sci.: Nano, 2019, 6, 798–808 RSC.
- W. R. Cao, M. E. Han, L. Lyu, C. Hu and F. Xiao, ACS Appl. Mater. Interfaces, 2019, 11, 16496–16505 CrossRef CAS PubMed.
- B. B. Xing, N. Graham and W. Z. Yu, Commun. Chem., 2020, 3, 38 CrossRef CAS.
- H. Jin, X. K. Tian, Y. L. Nie, Z. X. Zhou, C. Yang, Y. Li and L. Q. Lu, Environ. Sci. Technol., 2017, 51, 12699–12706 CrossRef CAS PubMed.
- R. V. Jagadeesh, A. E. Surkus, H. Junge, M. M. Pohl, J. Radnik, J. Rabeah, H. M. Huan, V. Schunemann, A. Bruckner and M. Beller, Science, 2013, 342, 1073–1076 CrossRef CAS PubMed.
- Q. Wu, J. Q. Liu, C. P. Yuan, Q. Li and H. G. Wang, Appl. Surf. Sci., 2017, 425, 1082–1088 CrossRef CAS.
- R. R. Cheng, Z. L. Wu, Y. L. Hou, J. Dong, J. Z. Cui and B. Zhao, Inorg. Chem. Commun., 2015, 51, 95–98 CrossRef CAS.
- L. P. Zhang and Z. H. Xia, J. Phys. Chem. C, 2011, 115, 11170–11176 CrossRef CAS.
- H. Tamura, K. Mita, A. Tanaka and M. Ito, J. Colloid Interface Sci., 2001, 243, 202–207 CrossRef CAS.
- X. Y. Zhang, Y. B. Ding, H. Q. Tang, X. Y. Han, L. H. Zhu and N. Wang, Chem. Eng. J., 2014, 236, 251–262 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Supplementary Fig. 1–6 and supplementary Notes 1–4. See DOI: 10.1039/d0ma00374c |
| This journal is © The Royal Society of Chemistry 2020 |