
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Effect of glycerol concentration on rate and product speciation for Ni and Au-based catalysts†
Lauren C.
Harris
a,
Rachel N.
Gaines
b,
Qi
Hua
a,
Gavin S.
Lindsay
a,
James J.
Griebler
b,
Paul J. A.
Kenis
b and
Andrew A.
Gewirth
 *a
*a
aDepartment of Chemistry, University of Illinois Urbana-Champaign, Illinois, USA. E-mail: agewirth@illinois.edu
bDepartment of Chemical and Biomolecular Engineering, University of Illinois Urbana-Champaign, Illinois, USA
First published on 28th April 2025
Abstract
In this paper, we investigate the glycerol electrooxidation reaction (GEOR) on Au and Ni catalysts, specifically the effect of glycerol concentration on electrochemical activity and product speciation for GEOR in an electrochemical flow cell system. With Au foil, cyclic voltammogram behavior shifted from hysteretic to near-linear by increasing the concentration of glycerol from 0.1 M to 1 M. As a result, glycerol electrooxidation increased up to 1.4 V vs. RHE with a higher glycerol concentration. The major products were formic acid and glycolic acid, yet minor products of value-added glyceric acid, lactic acid, and dihydroxyacetone were observed at a higher glycerol concentration. Competition between glycerol and the Au surface for hydroxide inhibits the formation of poisoning Au oxide (AuOx) species and enables the formation of low degree oxidation products. With Ni foil, the GEOR peak current density in cyclic voltammetry increased with glycerol concentration, however, formation of the major product, formic acid, decreased. This study examines and utilizes differences in GEOR mechanism on Ni vs. Au catalysts to vary product speciation in flow cell systems.
1. Introduction
Over the next few decades, global anthropogenic CO2 emissions need to be reduced drastically to combat climate change and its associated detrimental effects. While transportation and electricity generation represent two-thirds of these emissions, the chemical industry is also a major emitter at 23–25%.1,2 One way to diminish the emissions of the chemical industry is to transition to electrified processes that can be driven by renewable energy sources and use renewable feedstocks such as biomass.2–6 As such, the electrocatalytic transformation of glycerol, a byproduct of biodiesel production, to value-added products has been widely investigated.7,8 Electrochemical technologies are promising for glycerol conversion as they are environmentally-friendly and can be performed under ambient temperature and pressure.7–9 Glycerol can be converted into a wide variety of products, including dihydroxyacetone, glyceraldehyde, acetic acid, mesoxalic acid, oxalic acid, tartronic acid, glyceric acid, glycolic acid, lactic acid, and formic acid. Some of these products are value-added chemicals with commercial applications. For example, dihydroxyacetone and glycolic acid have been utilized in the cosmetics industry, whereas lactic acid has applications in the food industry and in the development of biodegradable polylactic acid.7,10–12Significant effort has focused on the development of different metal catalysts for glycerol electrooxidation (GEOR) in alkaline conditions, including Ni, Au, Pt, and Pd.6,8 On Ni-based catalysts, the major product generated is formic acid, with glycolic acid, glyceric acid, and oxalic acid formed as minor products.13–15 On Au materials, the generation of GEOR products is heavily dependent on applied potential. At potentials greater than 1 V vs. RHE, the major observed products are glycolic acid, glyceric acid, and formic acid.16,17 However, at lower potentials, the major products are dihydroxyacetone and lactic acid.18
Both flow cell and batch cell configurations have been used to study glycerol oxidation with the intent to control product speciation. In recirculating flow conditions at elevated temperatures, Zhang et al. observed glycolic acid and glyceric acid to be the major products for GEOR on Au/C.19 In recirculating flow conditions at ambient temperatures, Morales et al. observed formic acid as the major product on NiOx/MWCNTs-Ox, with increased production of oxalic acid at high potentials and high KOH concentrations.14
Recently, we showed that methanol oxidation behavior could be altered by changing the concentration of methanol at the electrode surface.20 In the presence of higher methanol concentrations (>1 M on a rough Pt surface), methanol oxidation no longer exhibited the characteristic poisoning behavior, but rather showed a linear response up until the 1.4 V vs. RHE positive limit.20 The origin of this behavior is in the competition between the methanol and the Pt substrate for OH− equivalents (H2O in the case of acid) at positive potentials.21
After observing changes in voltammetric behavior of methanol oxidation due to variation in reactant concentrations, we wondered what the effect of changing glycerol concentration would be on the products and voltammetry associated with glycerol oxidation under flow conditions. Oliveira et al. observed increased current density and a delay of the oxygen evolution reaction with higher glycerol concentrations on Ni/C in a batch configuration.22 Zhang et al. observed increased production of tartronic acid and mesoxalic acid with increased concentrations of both glycerol and KOH for GEOR on Au/C in a flow configuration.19 A direct comparison regarding the effect of glycerol concentration on product speciation and voltammetry in a flow cell on both Au and Ni has not been reported. Consequently, in this paper, we investigate the effect of glycerol concentration with Ni and Au foil catalysts in a single-pass flow electrolytic system.
2. Experimental
2.1 Electrochemical measurements
A two-channel electrolyzer flow cell was utilized for electrochemical measurements by using a modification of a system previously described (Fig. S1, ESI†).23 The cell consisted of two stainless steel current collectors between which was sandwiched the anode, a channel layer made from polyetheretherketone (PEEK), an anion exchange membrane (Fumasep FAA-3-PK-130), another PEEK channel layer, and the cathode. Spacers (Marian HT6210) between the current collector and PEEK layer accommodated the thickness of the electrodes while gaskets (Marian HT6210) were utilized between the PEEK layer and the anion exchange membrane. PEEK screws and a C-clamp provided pressure on the cell to prevent leaks. The anode and cathode each had an area of 1 cm2. Anolyte and catholyte electrolyte streams were introduced to the cell by using a syringe pump (Harvard apparatus) connected to the cell with polyethylene tubing (BD Intramedic). Typical flow rates were 2 mL min−1. The catholyte consisted of 1 M KOH (VWR ≥ 85%) while the anolyte contained glycerol (Sigma-Aldrich > 95%) + 1 M KOH. The Hg/HgO reference electrode (BASI) was placed upstream from the flow cell. Potentials are reported with respect to RHE, unless otherwise stated. The cathode electrode was a Pt foil. Anode electrodes were Ni foil (Alfa Aesar 99%) or Au foil (Alfa Aesar 99.9975%) and cleaned prior to use. Pt foil and Au foil were flamed-cleaned with a hydrogen torch between runs to remove surface contaminants. Ni foil was cleaned by successive sonication for 10 minutes in acetone, 0.1 M NaOH, 0.1 M HNO3, and 5 minutes in Milli-Q water.24 Au foil was cleaned by electrochemical cycling in 50 mM H2SO4 from −0.4 V to 1.4 V vs. Ag/AgCl (3.4 M KCl) prior to experimentation.25–27 Voltammetry obtained from these foils (Fig. 1A and 6A) was consistent with published reports.13,28–31 Anodes were pretreated electrochemically in the flow cell with 10 cycles in 1 M KOH conditions and 2 cycles in glycerol + 1 M KOH conditions prior to use. A CHI 6002E or Biologic SP-150 potentiostat was used to control the potential or current with 85% IR compensation consistent with recommendations.32,33 | ||
| Fig. 1 Cyclic voltammograms for glycerol electrooxidation on Ni foil with 1 M KOH in (A) absence of glycerol and (B) presence of 0.1 M glycerol (red) or 1 M glycerol (blue) in the flow cell (2 mL min−1) at a scan rate of 50 mV s−1. | ||
Batch measurements of GEOR activity were obtained by using a 5 mm diameter polycrystalline Au disk (Aupoly) in a three-electrode round-bottom flask. Hg/HgO was the reference electrode and Pt mesh was used for the cathode. All experiments were carried out at 21 °C. A Biologic SP-150 potentiostat was used to control the potential or current with 85% IR compensation.
2.2 Product characterization
The anolyte product stream was collected in a vial attached to the outlet of the flow cell. A 0.5 mL aliquot of the product stream was neutralized immediately after collection with 0.5 mL of 0.5 M H2SO4, and analyzed by using high-performance liquid chromatography (HPLC) (Shimadzu Nexera LC-40D with an LC-40D pump, CTO-40C column oven, SPD-40 UV detector, and RID-20A refractive index detector). The HPLC column was an Aminex HPX-87H (Bio-Rad) ion exclusion column and diluted sulfuric acid (5 mM) at a flow rate of 0.6 mL min−1 was used as the eluent to separate compounds. Column temperature was maintained at 60 °C and products were detected with a UV-vis detector (210 nm) and/or a refractive index detector (RID). Calibration curves for specific glycerol oxidation productions were constructed using commercially available compounds (Table S1 and Fig. S5, S6, and S11, ESI†). Oxalic acid, glycolic acid, formic acid, lactic acid, and dihydroxyacetone were acquired from Sigma Aldrich. Glyceric acid, glyceraldehyde, and tartronic acid were acquired from Thermo Fisher.Faradaic efficiencies were calculated by using the following equation:

![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 485 C mol−1), and Q is charge (C) passed.14,15,34 These calculations typically have a 10% error from multiple trials, which results in some faradaic efficiency values greater than 100%.35,36
485 C mol−1), and Q is charge (C) passed.14,15,34 These calculations typically have a 10% error from multiple trials, which results in some faradaic efficiency values greater than 100%.35,36
3. Results
3.1 Glycerol oxidation on Ni
Fig. 1A shows a CV obtained in the flow cell showing the response of the Ni foil electrode in a solution containing 1 M KOH flowing at a rate of 2 mL min−1. The voltammetry shows the presence of the well-known Ni(OH)2/NiOOH oxidation and reduction waves at 1.35 V and 1.3 V, respectively.22 The magnitude of the wave was consistent to ca. 10% between different foils and runs.Fig. 1B shows the voltammetry in the flow cell obtained following addition of either 0.1 M glycerol (red) or 1 M glycerol (blue) to the 1 M KOH solution. The current density was normalized to the coloumbs passed to form NiOOH, observed in the oxidation wave in Fig. 1A. The figure shows the presence of an oxidative wave starting at ca. 1.3 V and rising at 1.4 V, consistent with prior flow cell reports.14 The lack of substantial current associated with glycerol oxidation at potentials negative of 1.3 V suggests that the active surface is made from NiOOH.22 Prior reports suggest that the more cathodic form of Ni, Ni(OH)2, is not active for glycerol oxidation because Ni(OH)2 is a wide band gap (3–3.5 eV) semiconductor.37,38 The magnitude of the glycerol-associated oxidative wave, normalized by the magnitude of the Ni(OH)2/NiOOH wave, is increased by ca. 150% between the different glycerol concentrations. This result is consistent with prior work in a static cell, showing that an increase in glycerol concentration at a Ni electrode led to an increase in peak current associated with oxidation.22 At higher concentrations, however, the peak current did not increase linearly with concentration. The asymptotic response of the peak current with glycerol concentration is associated with saturation of NiOOH sites by glycerol.22 Interestingly, no change is observed in either the onset of oxidation or the CV behavior with increased glycerol concentration on the NiOOH electrode.
Fig. 2 reports the average current density during chronoamperometry obtained in the flow cell in 1 M KOH with 0.1 M glycerol and 1 M glycerol. The figure shows a lack of oxidative current at 1.3 V, consistent with the CV results. No oxidative current was found between 0.9 V and 1.2 V as well. Following a step to 1.4 V, the Ni surface oxidizes, NiOOH is formed, and glycerol oxidation current is seen. Interestingly, the onset of oxidation differs only marginally between the two different concentrations of glycerol used here. The average oxidative current is slightly higher with higher glycerol concentration. Fig. S2 (ESI†) shows that following an initial oxidative spike (associated with surface oxidation) the oxidative current becomes stable, decreasing by ∼9% between 120 s and 240 s, where product quantification was obtained.
 | ||
| Fig. 2 Average current density from chronoamperometry for glycerol electrooxidation on Ni foil in 0.1 M glycerol + 1 M KOH (■) and 1 M glycerol + 1 M KOH (▶) in a flow cell. | ||
Fig. 3 reports the potential dependence of oxidation products obtained for glycerol oxidation on Ni foil. Only formic acid was observed as a product at 1.4 V, although trace (<0.05 mM) amounts of glyceric, glycolic, and oxalic acid are evident at this potential as well (Fig. S7 and S8, ESI†). This result is consistent with prior reports.14 Interestingly, the concentration of formic acid was found to be lower in the product stream with the higher concentration of glycerol relative to that with the lower glycerol concentration.
 | ||
| Fig. 3 Formic acid product concentration of glycerol electrooxidation on Ni foil with 0.1 M glycerol + 1 M KOH (□) and 1 M glycerol + 1 M KOH (▷) in a flow cell. | ||
The concentration of product and the current passed during oxidation are used to calculate a faradaic efficiency (FE) for formic acid production, the values for which are reported in Fig. S4 (ESI†). The FE for formic acid production was 54% from the 0.1 M glycerol solution, but that number decreased to 37% with the use of the higher 1 M glycerol feed. FEs for formic acid production on Ni and Ni-alloys range from 30–98% for Ni nanoparticles deposited on carbon in a multipass or batch cell configuration.14,15,23,39,40 Single-pass FEs are expected to be lower relative to those obtained from multipass configurations.
Perhaps the most interesting result from these measurements is the drop in FE with the higher glycerol concentration. The origin of this drop may be due to additional oxidative processes occurring with the higher glycerol concentration. We noted only trace amounts of other oxidation products in the HPLC and these did not appreciably change between the two concentrations. One possibility is the oxidation of glycerol to CO2. Indeed, prior reports suggest that CO2 is a product of glycerol oxidation.13,22 We were not able to measure CO2 production from our cell. One reason that CO2 production might be enhanced with increased glycerol concentration is greater residence time of the glycerol at the electrode surface, possibly due to the surface saturation discussed previously.22
3.2 Glycerol oxidation on Au
Fig. 4 shows a representative set of CVs obtained from a Aupoly disk electrode in 1 M KOH with increasing concentrations of glycerol in batch cell conditions. The inset to the figure shows the CV obtained absent glycerol which exhibits the features expected for Au(poly) in aerated basic solution.29,41 At 0.1 M glycerol, the CV shows the expected hysteretic behavior for glycerol oxidation on Au exhibiting current maxima at 1.04 and 1.26 V during the anodic scan.16,29,41,42 These two maxima are consistent with prior literature.43 Following an increase in current up to ca. 1.22 V, the current density drops to nearly 0 mA cm−2, consistent with prior literature.16,29 This drop in current is associated with oxidation of the Au electrode at potentials where Au oxide (AuOx) is formed on the electrode surface.29,43 As the potential is reversed, the AuOx is reduced at ca. 1.25 V, and glycerol oxidation commences again. | ||
| Fig. 4 (A) Cyclic voltammetry obtained from a polycrystalline Au disk in 1 M KOH + x M glycerol at a scan rate of 20 mV s−1 with an inset blank voltammetry in 1 M KOH. (B) Potential of Au catalyst poisoning as a function of glycerol concentration. | ||
In the presence of higher glycerol concentrations, the CV changes. Fig. 4 shows that as the glycerol concentration is increased, the potential at which the current density achieves a maximum and then drops (associated with Au electrode oxidation) moves to more positive values. At a concentration of 5 M glycerol, the CV only evinces a quasi-linear increase going to positive potentials.
Fig. 4B shows the concentration dependence of the poisoning potential (the potential at which the oxidation current achieves a maximum and then drops). The figure shows a sharp increase in the potential of Au poisoning up to 1 M glycerol, after which the poisoning potential slowly approaches the upper limit of 1.42 V.
Fig. 5A shows a plot of the log of the oxidation current as a function of the log of the glycerol concentration obtained at different potentials. The order of the glycerol oxidation reaction is m = ∂[logi]/∂[log[glycerol]].18 The figure shows a nearly linear increase in the log of current for the different potentials considered up until the concentration of glycerol reached 1 M. At that point, the current is roughly constant with increased glycerol concentration, indicating that m is small. Fig. 5B plots m as a function of potential for these two different concentration regimes. At low glycerol concentrations, m ranges from ca. 1 to ca. 0.6, indicating participation of glycerol in the GEOR. At high concentrations, however, m is approximately = 0, which suggests that solution glycerol does not participate in the GEOR. This observation that m ≈ 0 suggests that the surface is saturated with glycerol as further changes in glycerol concentration do not significantly change the reaction rate. In turn, this saturation suggests that OH− participation in the GEOR will be restricted.
 | ||
| Fig. 5 (A) Dependence of log of current on log of glycerol concentration. (B) Potential dependence of reaction order (m) for [Glycerol] ≤ 1 M (orange) and [Glycerol] > 1 M (blue) concentrations (B). | ||
Fig. 6A shows a CV obtained from a Au foil in a flow cell in 1 M KOH. The characteristic gold oxide oxidation and reduction peaks are observed at 1.3 V and 1.1 V, respectively.30Fig. 6B shows the CV in the presence of two different concentrations of glycerol. In the presence of 0.1 M glycerol, the CV shows hysteretic behavior similar to that described above. In this case, however, the current does not decay to below 15 mA cm−2 in the positive sweep until 1.6 V. The somewhat diminished poisoning of the Au surface in the flow cell may reflect current inhomogeneities in the flowing KOH. The drop in current at 1.6 V indicates that the Au surface is oxidized and that glycerol oxidation activity has ceased. On the reverse scan, oxide begins to be reduced at ca. 1.2 V and glycerol oxidation activity recommences.29 In the presence of 1 M glycerol, however, the CV does not show the same hysteretic behavior seen with lower glycerol concentrations. Rather, the current increases up to 1.2 V and is sustained before diminishing after 1.4 V. In contrast to the case with lower glycerol concentration, the current does not decrease to below 100 mA cm−2 until the sweep is reversed. The noise evident in the CV at higher current densities is due to bubble formation at the cathode.
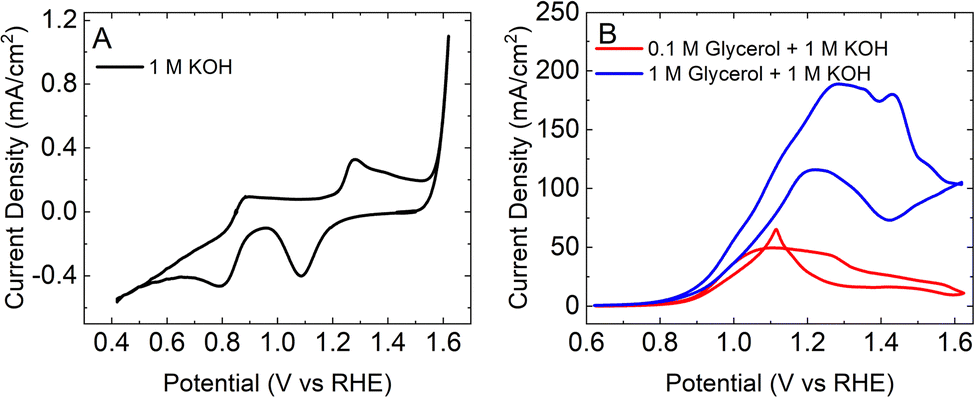 | ||
| Fig. 6 Cyclic voltammogram obtained from a Au foil in a flow cell (2 mL min−1) at a scan rate of 50 mV s−1 in (A) 1 M KOH and (B) 1 M KOH + 0.1 M glycerol (red) and 1 M KOH + 1 M glycerol (blue). | ||
Fig. 7 reports the results of chronoamperometry in the flow cell obtained at different potentials with low and high glycerol concentrations. At low concentrations, the current begins to increase at 0.8 V and achieves a maximum of 28 mA cm−2 at 1.0 and 1.2 V. At 1.4 V, the current decays. This behavior is consistent with that found in the batch cell in Fig. 4. At high concentration in the flow cell, the current profile is somewhat different. Fig. 7 shows the current begins to increase at 0.8 V as before, but achieves an average maximum of 96 mA cm−2 at 1.2 V and maintains this value at 1.4 V. There is no evidence of electrode poisoning with the higher glycerol concentration. Fig. S3 (ESI†) shows that the oxidative current from chronoamperometry exhibits noise but is stable between 120 s and 240 s, where product quantification was obtained.
 | ||
| Fig. 7 Average current density from chronoamperometry of glycerol electrooxidation on Au foil in 0.1 M glycerol + 1 M KOH (red) and 1 M glycerol + 1 M KOH (blue) in a flow cell. | ||
Fig. 8 reports the potential dependence of the product distribution obtained from a Au foil in the flow cell. Fig. 8A shows the result for an electrolyte containing 0.1 M glycerol + 1 M KOH. The figure highlights the lack of significant oxidation products at either 0.6 V or 0.8 V, consistent with the lack of oxidation current at these potentials. At 1.0 V, Fig. 8A shows that the major oxidation products are formic acid and glycolic acid, with lower concentrations of glyceric acid also found (Fig. S9, ESI†). Formic acid and glycolic acid are also found at 1.2 V, but are diminished at 1.4 V, consistent with the decreased oxidation current seen in the voltammetry. The formic acid and glycolic acid products and their potential dependence are identical with that found in prior work.16
 | ||
| Fig. 8 Product distribution and faradaic efficiency for glycerol electrooxidation on Au foil. Product concentrations are displayed for GEOR in (A) 0.1 M glycerol + 1 M KOH and (B) 1 M glycerol + 1 M KOH in a flow cell. Faradaic efficiency as a function of potential is shown for GEOR in (C) 0.1 M glycerol + 1 M KOH and (D) 1 M glycerol + 1 M KOH. | ||
Fig. 8B shows the potential dependence of oxidation products obtained from a solution containing 1 M glycerol + 1 M KOH. The figure shows that significant oxidation starts at 1.0 V with the appearance of formic and glycolic acid, as well as trace amounts of glyceric acid (Fig. S10, ESI†). The product concentration, however, is substantially lower than that found in 0.1 M glycerol. At 1.2 V the amount of product increases and this increase is maintained at 1.4 V. The appearance of lactic acid and dihydroxyacetone is also observed at 1.2 and 1.4 V. Thus, a consequence of the increased glycerol concentration is the presence of oxidation products at 1.4 V, due to continued glycerol oxidation at this potential.
Fig. 8C and D report FEs obtained for oxidation products on Au from solutions containing either 0.1 M glycerol (Fig. 8C) or 1 M glycerol (Fig. 8D). Fig. 8C shows the FE for glyceric acid is ca. 40% at 0.8 V, and then decreases at higher potentials where formic acid and glycolic acid are produced. The figure shows relatively little change in FE for these products between 1 V and 1.2 V.
Fig. 8D reports the FEs on Au obtained from a 1 M glycerol solution. The figure shows that the FEs are relatively smaller at 0.8 V and 1 V, but increase at 1.2 V and 1.4 V. Interestingly, the figure shows the presence of lower oxidation products such as lactic acid and dihydroxyacetone at these more positive potentials. The lower oxidation products were not observed at any glycerol concentration on Ni.
3.3 Flow rate dependence
Fig. 9 reports the flow rate dependence of the glycerol oxidation current. For the Ni electrode (Fig. 9A), very little dependence on solution flow rate is observed, consistent with the lack of rotation rate dependence found in Ni RDE studies during the oxidation of other organic substrates, such as urea.44 Alternatively, Fig. 9B shows there is a weak dependence of oxidation current with flow rate on Au, as higher flow rates give higher oxidation currents. | ||
| Fig. 9 Cyclic voltammograms for glycerol electrooxidation in 0.1 M glycerol + 1 M KOH with Ni foil (A) and Au foil (B) at various flow rates. | ||
In order to evaluate electrode kinetics in the flow cell, we used the method developed by Cooper et al. to calculate the kinetic current (ik) with flow rates between 1 and 4 mL min−1.45,46 Here,
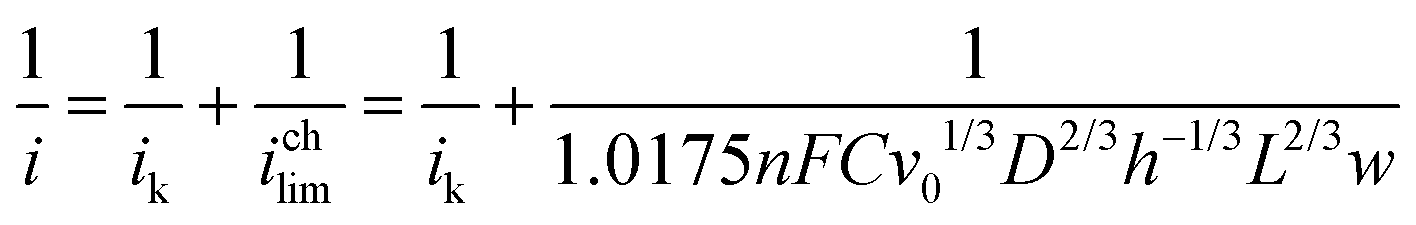
Fig. 10 reports the results of this analysis. At low potentials, the change in 1/i with corrected flow rate is relatively shallow, while this slope becomes steeper at higher potentials. Table 1 reports the kinetic currents obtained from this analysis. These kinetic currents are similar (within a factor of five) to those obtained in other GEOR analyses by using RDE41 and also within a factor of four of ik obtained for other electrooxidations on Au, such as glucose oxidation.47
 | ||
| Fig. 10 Plots of the dependence of reciprocal current density on the negative cubic root of flow rate-containing parameter, v0. | ||
| Potential (V vs. RHE) | i k (mA cm−2) |
|---|---|
| 1.0 | 61.3 |
| 1.1 | 64.8 |
| 1.2 | 56.5 |
| 1.3 | 50.6 |
| 1.4 | 55.0 |
| 1.5 | 49.1 |
| 1.6 | 51.8 |
Fig. 11 reports products obtained from GEOR on Au at different flow rates. The figure shows that – at least at the flow rates chosen here – product speciation does not change. In particular, formic acid, glycolic acid, and glyceric acid are the major products at all flow rates. There are greater amounts of all products formed at higher flow rates (Fig. 11D) consistent with the higher currents measured in the CV obtained in the flow cell. The amount of glyceric acid product formed increases steadily from 0.5 mL min−1 to 4 mL min−1, while formic acid and glycolic acid production changes very little until the fastest flow rate, 4 mL min−1, is reached. Formic acid and glycolic acid are higher degree oxidation products, formed by cleavage of a C–C bond in glyceric acid. Formation of these products likely requires a higher surface concentration of hydroxide relative to what is needed for less oxidized products, such as glyceric acid. The higher flow rate could be associated with the higher surface hydroxide concentration.
 | ||
| Fig. 11 Molar product distribution for glycerol electrooxidation on Au foil in a flow cell with 0.1 M glycerol + 1 M KOH at (A) 0.5 mL min−1, (B) 1 mL min−1, (C) 2 mL min−1, and (D) 4 mL min−1 flow rates. | ||
4. Discussion
The results from this paper show that the concentration of glycerol in a flow cell using a Au catalyst changes both the appearance of the voltammetry and the product speciation. Alternatively, using different concentrations of glycerol with a Ni catalyst yielded only minor changes in both the voltammetry and product speciation.4.1 Oxidation on Au
Glycerol oxidation in alkaline solution on Au is thought to proceed via the following mechanism:31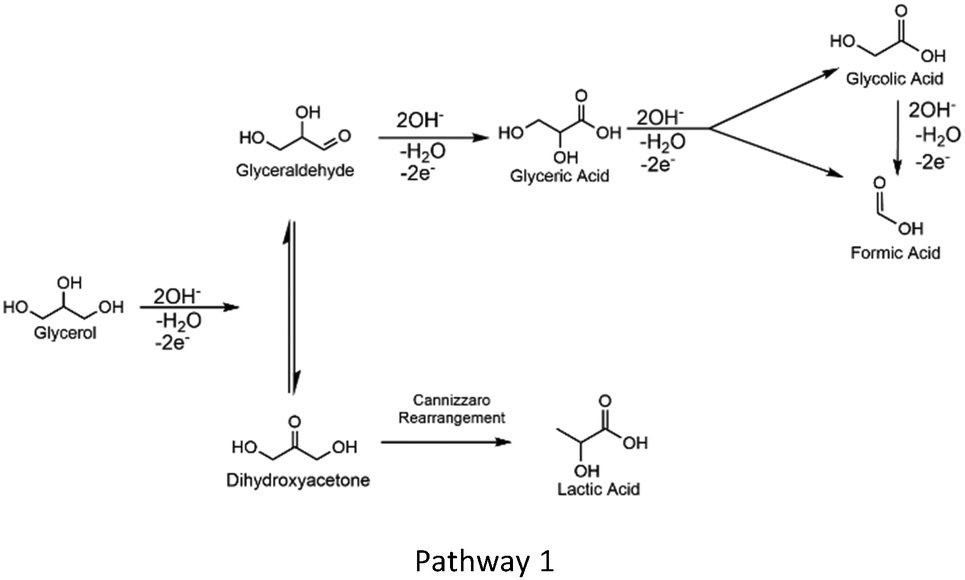
Pathway 1 shows that on Au catalysts, glycerol oxidation utilizes hydroxide, which is present as OH− in solution and OH adsorbed onto the Au surface.48,49 Different intermediates, such as glyceraldehyde, glyceric acid, and dihydroxyacetone can either be released from the surface or be oxidized or altered further to make products such as lactic acid, formic acid, and glycolic acid. The mechanism here is similar in concept with that involved with methanol and ethanol oxidation in that there is direct participation of surface or near-surface OH− in this oxidation.50
The base-catalyzed deprotonation of glycerol in solution is suggested to be the first step in GEOR on Au, and subsequent abstraction of hydrogens to form intermediates is also performed by OH−. As a result, OH− is vital to catalyst activity and product formation. Glycerol is adsorbed to Au,49,51,52 while base in solution deprotonates glycerol to make an active species. The Au surface accepts electrons and adsorbed surface hydroxide lowers barriers for activation of C–H and C–O bonds.48,53 Adsorbed OH− on Au engages in hydrogen bonding with adsorbed glycerol.52
The results presented here show that both the CV and product speciation associated with glycerol oxidation are dependent on the glycerol concentration. In particular, increasing the concentration of glycerol results in the formation of less oxidized products. At the same time, the CV obtained with higher glycerol concentrations does not exhibit the poisoning behavior seen on the Au surface with lower glycerol concentrations. The origin of this behavior is likely found in the competition between glycerol and the Au surface for OH− at the positive potentials associated with glycerol oxidation. As the glycerol concentration is increased, there is less OH− available to oxidize the Au surface to AuOx and thus the CV does not exhibit the poisoning behavior found at lower glycerol concentrations.
An additional consequence of the increased glycerol concentration is the change in product speciation toward less oxidized species. Alternatively, more oxidized products (which require more OH−) are found with a lower glycerol concentration. We suggest that this change is associated with the diminished presence of OH− and consequently AuOH and AuOx on the electrode surface with the higher glycerol concentrations. Further, this behavior suggests that the interplay of glycerol and OH− is responsible for product speciation on Au. Increasing the glycerol concentration induces competition between OH− and glycerol availability at the Au surface, consequently limiting the degree of glycerol oxidation. This is the reason behind the increased presence of dihydroxyacetone, lactic acid, and glyceric acid in 1 M glycerol. We show here that this interplay can be accomplished by changing the glycerol concentration on a surface that becomes oxidized in the region where glycerol oxidation occurs. Indeed, decreasing OH− availability by lowering the pH also leads to the formation of less oxidized products.15
An interesting feature associated with changing glycerol concentration is the change in the order, m, of the glycerol oxidation reaction. We showed that at low glycerol concentrations ([glycerol] ≤ 1 M), m ranges from 1 to 0.6, consistent with m values for other electrooxidations on Au and Pt surfaces.54 Alternatively, at high concentrations ([glycerol] > 1 M), m approaches 0. This behavior suggests that the Au surface is saturated with glycerol at concentrations above 1 M. Consequently, hardly any surface is available for adsorbed OH− or O. This is the reason for the increased poisoning potential observed in Fig. 4B.
The concentration dependence for glycerol oxidation shown here is similar to that shown previously on Pt for methanol oxidation.20 In particular, we showed that increasing methanol concentration inhibited surface oxidation on Pt leading to linear or near linear oxidation CVs. In our previous work, methanol concentration at the surface was enhanced by the presence of surface roughness. In this case, induced surface roughness was not required to observe the concentration effect, possibly because glycerol associates more strongly with Au than methanol does with Pt.52,55
Analysis shows that the ik values obtained by using the flow rate dependence in the flow cell are similar to those obtained by using RDE, thus validating the technique. The flow rate dependence of speciation shows that speciation of the less oxidized minor products can be incrementally altered at low flow rates, but privileging of the more oxidized products is observed at higher flow rates. The origin of this behavior is likely the higher effective OH− concentration at the solution/electrode interface at high flow rates, which favors oxidative C–C cleavage.56
4.2 Oxidation on Ni
In contrast to the behavior seen with Au, GEOR on Ni exhibited little dependence on glycerol concentration. The CVs obtained in 0.1 M and 1 M glycerol were almost identical, consistent with prior work22 and product speciation also did not change. Additionally, only the high oxidation product formic acid was observed in both cases.The origin of the different behavior on Ni must be related to the mechanism of glycerol oxidation on this surface. On Ni, glycerol oxidation between 1.2 V and 1.4 V is thought to proceed primarily by an indirect mechanism shown in Pathway 2.15,57,58

In this mechanism, Ni(OH)2 is present in the potential range between 0.9 V to ca. 1.35 V vs. RHE.13 At further increased potentials, Ni(OH)2 is oxidized to the nickel oxyhydroxide, NiOOH, species.22,59 The indirect mechanism uses Ni(OH)2 and NiOOH as a mediator for glycerol oxidation. Glycerol is oxidized on NiOOH to form oxidized intermediates, such as a geminal diol58,60 and NiOOH is reduced to make Ni(OH)2. In the presence of solution OH−, the Ni(OH)2 surface is reoxidized to form NiOOH. The NiOOH then mediates additional intermediate oxidation.
The predominance of formic acid as the product under these conditions is ascribed to facile C–C bond cleavage due to the formation of the geminal diol which weakens the C–C bond.58,60 In addition, studies suggest that higher energy barriers for desorption on Ni will cause reaction products/intermediates to remain on the surface after formation and undergo further oxidation to formic acid.61,62
In this indirect mechanism, OH− does not participate in the oxidation of glycerol directly, but is only involved in reforming the active NiOOH surface. Variations of this mechanism suggest that NiOOH accepts a hydrogen atom from carbon via a hydrogen atom transfer, subsequently producing a radical on glycerol and this radical induces dehydrogenation of the adsorbed glycerol molecule.15 The presence of the indirect mechanism explains the relative glycerol concentration invariance seen in this paper on Ni. The role of solution OH− is only to participate in the oxidation of the Ni(OH)2 surface. It does not incorporate directly into glycerol. Thus, surface oxidation and glycerol oxidation do not compete anymore, as was the case for Au.
5. Conclusions
We showed that glycerol oxidation proceeds via different mechanisms on Au and Ni. In the former, near surface OH− participates directly in glycerol oxidation. At the same time, near surface OH− can also participate in Au surface oxidation. Consequently, Au and glycerol compete for near surface OH−. Changing the concentration of glycerol changes the relative amount of OH− that is utilized for glycerol and surface oxidation. If the glycerol concentration is high, then relatively little surface oxidation occurs. At the same time, the relative scarcity of OH− at high glycerol concentrations yields less oxidized products during glycerol electrooxidation.On Ni, glycerol oxidation occurs via an indirect mechanism featuring the use of the Ni(OH)2/NiOOH surface as a redox mediator. Consequently, OH− consumption does not change with a change in glycerol concentration. Likewise, product speciation is also not a function of glycerol concentration.
This work shows that even with the same catalyst and potential, product speciation during glycerol oxidation can be altered by changing glycerol concentration on Au, which participates in a direct glycerol oxidation mechanism. In contrast, little change occurs on Ni.
Author contributions
Lauren Harris: conceptualization, formal analysis, methodology, investigation, validation, visualization, and writing – original draft. Rachel Gaines: software, methodology, and writing – review & editing. Qi Hua: methodology, writing – review & editing. Gavin Lindsay: software. James Griebler: software. Prof. Paul Kenis: resources and writing – review & editing. Prof. Andrew Gewirth: conceptualization, supervision, funding acquisition, and writing – review & editing.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by the NSF 2029326 which is gratefully acknowledged. LCH acknowledges the John and Margaret Witt Fellowship, John C. Bailar Fellowship, 3 M Fellowship, and University of Illinois Urbana Champaign Graduate College Fellowship.References
- Inventory of U.S. Greenhouse Gas Emissions and Sinks: 1990-2021, https://www.epa.gov/ghgemissions/inventory-us-greenhouse-gas-emissions-and-sinks-1990-2021, (accessed January 2024).
- CO2 Emissions in 2022, https://www.iea.org/reports/co2-emissions-in-2022, (accessed November 2023).
- Annual Energy Outlook 2022, https://www.eia.gov/outlooks/aeo/pdf/AEO2022_ReleasePresentation.pdf, (accessed November 2023).
- T. Noël, Y. Cao and G. Laudadio, Acc. Chem. Res., 2019, 52, 2858–2869 CrossRef PubMed.
- M. S. Ahmad, M. H. Ab Rahim, T. M. Alqahtani, T. Witoon, J.-W. Lim and C. K. Cheng, Chemosphere, 2021, 276, 130128 CrossRef CAS PubMed.
- G. Dodekatos, S. Schünemann and H. Tüysüz, ACS Catal., 2018, 8, 6301–6333 Search PubMed.
- L. F. Fan, B. W. Liu, X. Liu, N. Senthilkumar, G. X. Wang and Z. H. Wen, Energy Technol., 2021, 9, 2000804 Search PubMed.
- T. Li and D. A. Harrington, ChemSusChem, 2021, 14, 1472–1495 CrossRef CAS PubMed.
- M. Guschakowski and U. Schröder, ChemSusChem, 2021, 14, 5216–5225 CrossRef CAS PubMed.
- M. Simoes, S. Baranton and C. Coutanceau, ChemSusChem, 2012, 5, 2106–2124 CrossRef CAS PubMed.
- N. Razali and A. Z. Abdullah, Appl. Catal., A, 2017, 543, 234–246 CrossRef CAS.
- C. Dai, L. Sun, H. Liao, B. Khezri, R. D. Webster, A. C. Fisher and Z. J. Xu, J. Catal., 2017, 356, 14–21 Search PubMed.
- V. L. Oliveira, C. Morais, K. Servat, T. W. Napporn, G. Tremiliosi-Filho and K. B. Kokoh, J. Electroanal. Chem., 2013, 703, 56–62 CrossRef CAS.
- D. M. Morales, D. Jambrec, M. A. Kazakova, M. Braun, N. Sikdar, A. Koul, A. C. Brix, S. Seisel, C. Andronescu and W. Schuhmann, ACS Catal., 2022, 12, 982–992 CrossRef CAS.
- M. K. Goetz, M. T. Bender and K.-S. Choi, Nat. Commun., 2022, 13, 5848 CrossRef CAS PubMed.
- Y. Kwon, K. J. P. Schouten and M. T. M. Koper, ChemCatChem, 2011, 3, 1176–1185 Search PubMed.
- Y. A. Xie, L. Z. Sun, X. Pan, Z. Y. Zhou, Y. C. Zheng, X. F. Yang and G. H. Zhao, Carbon, 2023, 203, 88–96 Search PubMed.
- Y. Yan, H. Zhou, S.-M. Xu, J. Yang, P. Hao, X. Cai, Y. Ren, M. Xu, X. Kong, M. Shao, Z. Li and H. Duan, J. Am. Chem. Soc., 2023, 145, 6144–6155 CrossRef CAS PubMed.
- Z. Zhang, L. Xin, J. Qi, D. J. Chadderdon, K. Sun, K. M. Warsko and W. Li, Appl. Catal., B, 2014, 147, 871–878 CrossRef CAS.
- Q. Hua, N. M. Alghoraibi, X. Chen and A. A. Gewirth, ACS Catal., 2023, 13, 10683–10693 CrossRef CAS.
- P. V. B. Santiago, R. A. G. Oliveira, J. M. Roquetto, N. Akiba, I. Gaubeur, C. A. Angelucci, J. Souza-Garcia and J. M. Feliu, Electrochim. Acta, 2019, 317, 694–700 CrossRef CAS.
- V. L. Oliveira, C. Morais, K. Servat, T. W. Napporn, P. Olivi, K. B. Kokoh and G. Tremiliosi-Filho, Electrocatalysis, 2015, 6, 447–454 CrossRef CAS.
- M. S. E. Houache, R. Safari, U. O. Nwabara, T. Rafaïdeen, G. A. Botton, P. J. A. Kenis, S. Baranton, C. Coutanceau and E. A. Baranova, ACS Appl. Energy Mater., 2020, 3, 8725–8738 CrossRef CAS.
- N. Singer, R. G. Pillai, A. I. D. Johnson, K. D. Harris and A. B. Jemere, Microchim. Acta, 2020, 187, 196 CrossRef CAS PubMed.
- L. M. Fischer, M. Tenje, A. R. Heiskanen, N. Masuda, J. Castillo, A. Bentien, J. Émneus, M. H. Jakobsen and A. Boisen, Microelectron. Eng., 2009, 86, 1282–1285 CrossRef CAS.
- A. L. Gui, G. Z. Liu, M. Chockalingam, G. Le Saux, J. B. Harper and J. J. Gooding, Electroanalysis, 2010, 22, 1283–1289 CrossRef CAS.
- B. R. Lydon, A. Germann and J. Y. Yang, Inorg. Chem. Front., 2016, 3, 836–841 RSC.
- T. T. H. Hoang and A. A. Gewirth, ACS Catal., 2016, 6, 1159–1164 CrossRef CAS.
- L. Pérez-Martínez, L. Balke and A. Cuesta, J. Catal., 2021, 394, 1–7 CrossRef.
- S. Yang and D. G. H. Hetterscheid, ACS Catal., 2020, 10, 12582–12589 CrossRef CAS.
- Y. Zhou, Y. Shen and X. Luo, J. Catal., 2020, 381, 130–138 CrossRef CAS.
- J. White, L. Peters, D. Martín-Yerga, I. Terekhina, A. Anil, H. Lundberg, M. Johnsson, G. Salazar-Alvarez, G. Henriksson and A. Cornell, J. Electrochem. Soc., 2023, 170 Search PubMed.
- X. Yu, E. C. dos Santos, J. White, G. Salazar-Alvarez, L. G. M. Pettersson, A. Cornell and M. Johnsson, Small, 2021, 17, 2104288 CrossRef CAS PubMed.
- X. Han, H. Sheng, C. Yu, T. W. Walker, G. W. Huber, J. Qiu and S. Jin, ACS Catal., 2020, 10, 6741–6752 CrossRef CAS.
- D. Liu, J.-C. Liu, W. Cai, J. Ma, H. B. Yang, H. Xiao, J. Li, Y. Xiong, Y. Huang and B. Liu, Nat. Commun., 2019, 10, 1779 CrossRef PubMed.
- W. Luo, H. Tian, Q. Li, G. Meng, Z. Chang, C. Chen, R. Shen, X. Yu, L. Zhu, F. Kong, X. Cui and J. Shi, Adv. Funct. Mater., 2024, 34, 2306995 CrossRef CAS.
- Z. H. Zhao, P. S. Lamoureux, A. Kulkarni and M. Bajdich, ChemCatChem, 2019, 11, 3423–3431 CrossRef CAS.
- K. Abe and M. Shimura, Semicond. Sci. Technol., 2022, 37 Search PubMed.
- M. S. E. Houache, E. Cossar, S. Ntais and E. A. Baranova, J. Power Sources, 2018, 375, 310–319 CrossRef CAS.
- T. Andreu, M. Mallafre, M. Molera, M. Sarret, R. Oriol and I. Sires, ChemElectroChem, 2022, 9 Search PubMed.
- X. Shi, D. E. Simpson and D. Roy, Phys. Chem. Chem. Phys., 2015, 17, 11432–11444 Search PubMed.
- N. E. de Souza, J. F. Gomes and G. Tremiliosi-Filho, J. Electroanal. Chem., 2017, 800, 106–113 Search PubMed.
- A. T. Marshall, V. Golovko and D. Padayachee, Electrochim. Acta, 2015, 153, 370–378 CrossRef CAS.
- V. Vedharathinam and G. G. Botte, Electrochim. Acta, 2012, 81, 292–300 Search PubMed.
- J. A. Cooper and R. G. Compton, Electroanalysis, 1998, 10, 141–155 CrossRef CAS.
- D. A. Scherson, Y. V. Tolmachev, Z. H. Wang, J. Wang and A. Palencsar, Electrochem. Solid-State Lett., 2008, 11, F1–F4 CrossRef CAS.
- N. Schlegel, G. K. H. Wiberg and M. Arenz, Electrochim. Acta, 2022, 410, 140023 CrossRef CAS.
- B. N. Zope, D. D. Hibbitts, M. Neurock and R. J. Davis, Science, 2010, 330, 74–78 CrossRef CAS PubMed.
- A. M. Verma, L. Laverdure, M. M. Melander and K. Honkala, ACS Catal., 2022, 12, 662–675 CrossRef CAS.
- P. Rodriguez and M. T. M. Koper, Phys. Chem. Chem. Phys., 2014, 16, 13583–13594 RSC.
- E. C. dos Santos, R. B. Araujo, M. Valter, G. Salazar-Alvarez, M. Johnsson, M. Bajdich, F. Abild-Pedersen and L. G. M. Pettersson, Electrochim. Acta, 2021, 398, 139283 CrossRef CAS.
- F. C. Calaza, M. A. Baltanás, M. Sterrer and H. J. Freund, Top. Catal., 2019, 62, 1053–1066 CrossRef CAS.
- Y. Kwon, S. C. S. Lai, P. Rodriguez and M. T. M. Koper, J. Am. Chem. Soc., 2011, 133, 6914–6917 CrossRef CAS PubMed.
- C. Alonso and J. Gonzalez-Velsasco, Z. Phys. Chem., 1990, 2710, 799–822 CrossRef.
- C. Panja, N. Saliba and B. E. Koel, Surf. Sci., 1998, 395, 248–259 CrossRef CAS.
- M. K. Goetz, M. T. Bender and K. S. Choi, Nat. Commun., 2022, 13 Search PubMed.
- M. Fleischmann, K. Korinek and D. Pletcher, J. Electroanal. Chem. Interfacial Electrochem., 1971, 31, 39–49 CrossRef CAS.
- M. K. Goetz, E. Usman and K.-S. Choi, ACS Catal., 2023, 15758–15769, DOI:10.1021/acscatal.3c03365.
- S. L. Medway, C. A. Lucas, A. Kowal, R. J. Nichols and D. Johnson, J. Electroanal. Chem., 2006, 587, 172–181 Search PubMed.
- M. T. Bender, Y. C. Lam, S. Hammes-Schiffer and K.-S. Choi, J. Am. Chem. Soc., 2020, 142, 21538–21547 CrossRef CAS PubMed.
- Z. He, J. Hwang, Z. Gong, M. Zhou, N. Zhang, X. Kang, J. W. Han and Y. Chen, Nat. Commun., 2022, 13, 3777 CrossRef CAS PubMed.
- L. A. Calderón, A. Montoya, A. Soon and C. Stampfl, Mol. Catal., 2019, 474, 110412 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cp04013a |
| This journal is © the Owner Societies 2025 |