
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Indications for a universal hydrogen catalysis mechanism in [FeFe]-hydrogenases of different phylogenetic groups†
Moritz
Senger
 *ab,
Conrad
Schumann
a,
Princess R.
Cabotaje
a,
Afridi
Zamader‡
a,
Ping
Huang
a,
Henrik
Land
a and
Gustav
Berggren
*a
*ab,
Conrad
Schumann
a,
Princess R.
Cabotaje
a,
Afridi
Zamader‡
a,
Ping
Huang
a,
Henrik
Land
a and
Gustav
Berggren
*a
aDepartment of Chemistry – Ångström Laboratory, Molecular Biomimetics, Uppsala University, 75120 Uppsala, Sweden. E-mail: moritz.senger@kemi.uu.se; gustav.berggren@kemi.uu.se
bDepartment of Chemistry – BMC, Biochemistry, Uppsala University, 75120 Uppsala, Sweden
First published on 16th April 2025
Abstract
[FeFe]-hydrogenases are metalloenzymes catalysing bidirectional hydrogen (H2) turnover. These enzymes are generally considered to be extremely efficient and fast catalysts. However, [FeFe]-hydrogenases constitute a very diverse enzyme family that can be divided into several distinct phylogenetic groups, denoted as groups A–G. Very little is known about the properties of [FeFe]-hydrogenases outside of the intensively studied group A, but recent studies on putatively sensory group C and D enzymes have revealed distinct differences in reactivity. The variation in structure, reactivity and physiological function observed between phylogenetic groups raises the question if all [FeFe]-hydrogenases follow the same mechanism for H2 turnover. Here, we provide the first detailed spectroscopic investigation of a slow-acting putatively sensory group D [FeFe]-hydrogenase from Thermoanaerobacter mathranii (TamHydS). Photo-reduction enabled us to characterize redox states in group D [FeFe]-hydrogenase via infrared spectroscopy under catalytic conditions. The sequential population of redox states similar to group A [FeFe]-hydrogenases supports the notion that group A and D [FeFe]-hydrogenases follow a universal catalytic mechanism. However, clear differences between enzymes from different phylogenetic groups become evident when comparing the relative stability and protonation state of suggested key catalytic intermediates. Moreover, the spectroscopic data collected on TamHydS provides new insight into the structure of the reduced active site, lending further support for the notion of a retained bridging CO ligand throughout the entire catalytic cycle.
Introduction
[FeFe]-hydrogenases are metalloenzymes involved in hydrogen metabolism. They are a structurally and functionally highly diverse enzyme family and can be classified into phylogenetic groups (denoted group A–G).1 Outside of group A, there is limited data available on [FeFe]-hydrogenases. Still, group C and D [FeFe]-hydrogenases stand out as they are proposed to serve a H2 sensory function rather than a catalytic one.2,3 The different phylogenetic groups likely share the same organometallic catalytic cofactor but differ in the active-site pocket and substrate transport pathways.2,4 The active site consists of a unique diiron site with a terminal CO and CN− ligand at each iron ion. The iron ions are further coordinated by two bridging ligands, a bidentate azadithiolate (–SCH2NCH2S–, ADT) and an additional CO molecule (μCO). The diiron site is linked via a cysteine residue to a [4Fe–4S] cluster ([4Fe–4S]H) to form the so-called H-cluster (Fig. 1A). Many [FeFe]-hydrogenases harbour additional [4Fe–4S] clusters in the N terminal domain involved in electron transfer, commonly denoted as F-clusters. In addition, group C and D enzymes often harbour an additional FeS cluster in the C-terminal domain called C-clusters. Due to their outstanding catalytic properties, [FeFe]-hydrogenases and the H-cluster have received significant attention from the bioinorganic chemistry and solar fuel research communities.5–8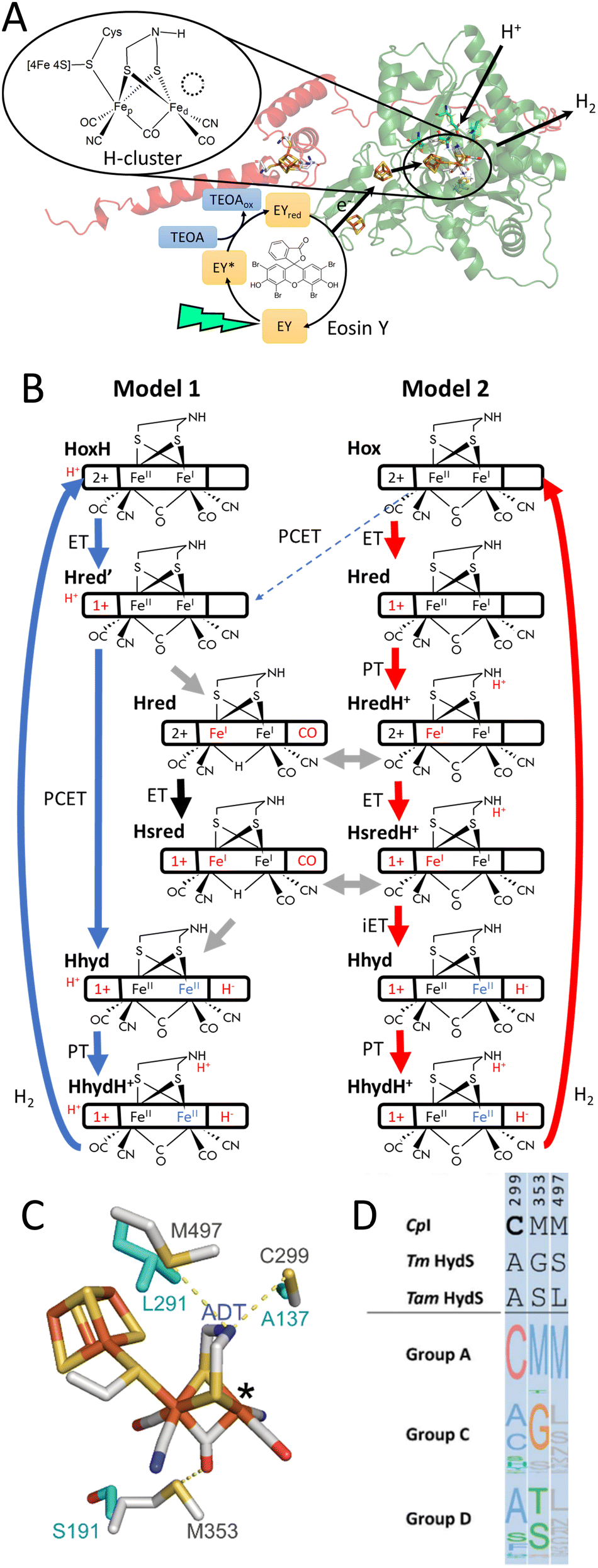 | ||
| Fig. 1 (A) TamHydS protein structure, H-cluster and photo-reduction scheme. YASARA-generated homology model (green cartoon) of TamHydS9,10 based on the crystal structure of CpI (PDB ID 4XDC11). The [4Fe–4S] clusters and the H-cluster are shown as sticks (C: gray, Fe: orange, S: yellow, N: blue, O: red) The additional C-terminal domain was predicted with Alphafold (red cartoon) and the associated [4Fe–4S] cluster inserted manually. Details are described in the TamHydS structure section in the ESI.† Eosin Y (EY) is excited by visible light (EY*) and reductively quenched by triethanolamine (TEOA). The reduced Eosin Y (EYred) can donate electrons to TamHydS resulting in population of reduced states and eventually H2 production. (B) Two main catalytic cycle models. Model 1 blue arrows, model 2 red arrows, grey arrows indicate ligand rotation. The CO ligand occupying the apical vacancy and bound protons are indicated in red. Electron transfer (ET), internal electron transfer (iET), proton transfer (PT) and proton coupled electron transfer (PCET) steps are indicated at the arrows. Red letters indicate the site of reduction, blue letters the site of oxidation relative to the oxidized state (Hox). Note that for model 2 the nomenclature introduced by Sommer et al.12 is used which leads to different redox states having the same abbreviation. (C) Group A and D cofactor second coordination sphere differences. Amino acids of CpI interacting with the H-cluster (C299, M353, and M497) are shown in sticks (grey: C, yellow: S). The azadithiolate (ADT) bridgehead is labelled as blue text. Hydrogen turnover is catalysed at the distal iron denoted as Fed (*). The corresponding residues from the homology model of TamHydS are overlaid (cyan: C, red: O). (D) Normalized consensus logos of groups A, C, and D [FeFe]-hydrogenase generated in Jalview using ClustalΩ13 sequence alignment of sequences retrieved from Greening et al.14 The numbering is based on the sequence of CpI. The conservation of the amino acid in a position is proportional to its font size. | ||
To-date, mechanistic investigations have focused almost exclusively on group A [FeFe]-hydrogenases, for which several protonation and oxidation states of the H-cluster have been characterized. Albeit the mechanism remains debated, all currently proposed catalytic cycles include oxidized, single and double reduced H-cluster states.15–17 Multiple spectroscopic signals associated with such states have been identified by Fourier-transform infrared (FTIR) and electron paramagnetic resonance (EPR) spectroscopy. The oxidized state (Hox) features a [4Fe–4S]H2+ cluster and a Fe(II)Fe(I) diiron site. The Hox state and its potentially protonated equivalent HoxH, feature the aforementioned bridging CO ligand.18 This ligation results in an open coordination site in an apical position at the distal iron atom (Fed), a structure defining the “rotated” diiron site geometry.19–22 The first reduction occurs at the [4Fe–4S]H cluster forming the  state.12,18,23 Protonation coupled to intramolecular electron transfer from the [4Fe–4S]H cluster to the diiron site has been proposed to give rise to a diiron site reduced state denoted Hred (sometimes HredH+). Addition of a second electron to the H-cluster gives rise to the “super-reduced” state Hsred (or HsredH+), with a reduced [4Fe–4S]H+ cluster. The hydride state, Hhyd, is a tautomer of Hsred featuring a terminal hydride at the former apical vacancy giving a formally di-ferrous diiron site.24–26 Protonation of Hhyd at the amine of the ADT ligand is proposed to yield the still only partially characterized state, HhydH+.27,28 The hydride and the proton may form H2, returning the H-cluster to the oxidized state, Hox. The exact structure and catalytic relevance of these states are subject to ongoing discussion, which has resulted in competing mechanistic models, at least two of which are extensively reviewed in recent literature.2,5,16,29–31
state.12,18,23 Protonation coupled to intramolecular electron transfer from the [4Fe–4S]H cluster to the diiron site has been proposed to give rise to a diiron site reduced state denoted Hred (sometimes HredH+). Addition of a second electron to the H-cluster gives rise to the “super-reduced” state Hsred (or HsredH+), with a reduced [4Fe–4S]H+ cluster. The hydride state, Hhyd, is a tautomer of Hsred featuring a terminal hydride at the former apical vacancy giving a formally di-ferrous diiron site.24–26 Protonation of Hhyd at the amine of the ADT ligand is proposed to yield the still only partially characterized state, HhydH+.27,28 The hydride and the proton may form H2, returning the H-cluster to the oxidized state, Hox. The exact structure and catalytic relevance of these states are subject to ongoing discussion, which has resulted in competing mechanistic models, at least two of which are extensively reviewed in recent literature.2,5,16,29–31
In short, the two mechanistic models diverge in the interpretation of the spectroscopic data, both with regards to ligand geometry of the diiron subsite as well as protonation status of the H-cluster (Fig. 1B). In model 1, two of the aforementioned reduced states, Hred and Hsred, are interpreted as inhibited states adopting an “inverted” diiron site geometry. Critically, here protonation of the diiron subsite is proposed to occur at the Fe ions yielding a bridging hydride (μH), while the former μCO ligand shifts to a terminal position that otherwise provides the apical vacancy. The expected thermodynamic stability of the μH geometry,32,33 and the additional ligand rotation necessary to form a terminal hydride disfavour their involvement in fast H2 catalysis.30,34–36 Conversely, in model 2, the same spectroscopic signals are attributed to species protonated at the ADT-amine instead of the iron ions, and commonly denoted HredH+ and HsredH+. As a consequence, HredH+ and HsredH+ are proposed to retain the rotated geometry of the Hox state and to be catalytically relevant.12,16 In favour of model 1, the absence of a μCO band for Hred has been noted in numerous independent studies. An observation which, coupled with comprehensive DFT calculations and isotope editing, supports the notion of an inverted geometry in Hred.34,35 However, low temperature experiments provide support for model 2. Albeit the μCO ligand band expected for the HredH+ state has proven challenging to observe at room temperature it has been detected via FTIR spectroscopy at cryogenic temperatures.37–39
In the following, we will use the  /Hred/Hsred nomenclature to define specific spectroscopic fingerprint signals, without implying a specific ligand geometry and/or protonation status.
/Hred/Hsred nomenclature to define specific spectroscopic fingerprint signals, without implying a specific ligand geometry and/or protonation status.
It is well-established that the protein environment has a large effect on the H-cluster.17,40–42 In group A [FeFe]-hydrogenases both the active-site pocket and a proton transfer pathway (PTP), composed of conserved amino acids and water molecules, are supposed to play a key role in fast catalysis.43–45 The H-bonding partners of the CN− ligands of the H-cluster are conserved in the vast majority of groups of [FeFe]-hydrogenases.1,46 Beyond these immediate interactions, several outer-coordination sphere amino acid residues are strictly conserved in the active-site pocket of group A [FeFe]-hydrogenases. Variation of these conserved residues through site directed mutagenesis has been shown to significantly impair turnover rates and in some cases even prevent H-cluster assembly.40 However, the putatively sensory enzymes in groups C and D display variations of several conserved amino acids constituting the outer-coordination sphere in group A [FeFe]-hydrogenases (Fig. 1C and D).3,9 Moreover, the PTP of group A is not conserved in either group C or group D [FeFe]-hydrogenases, and a distinct alternative PTP has been proposed for group D.47
As expected from these structural differences, the reactivity of group C and D enzymes is clearly different from the well-known group A [FeFe]-hydrogenases. Activity assays have shown that group C and group D enzymes are significantly slower than any reported group A enzyme.3,9 Introducing the PTP and active-site of group A [FeFe]-hydrogenase into a group D enzyme has been shown to increase the catalytic rate more than 100-fold, underscoring the impact of the protein scaffold on H-cluster reactivity.42 Moreover, an alternative diiron site reduced state Fe(I)Fe(I), denoted  , has so far been detected exclusively in spectro-electrochemistry studies of the group C [FeFe]-hydrogenase from Thermotoga maritima (TmHydS).3,48 Compared to HredH+,
, has so far been detected exclusively in spectro-electrochemistry studies of the group C [FeFe]-hydrogenase from Thermotoga maritima (TmHydS).3,48 Compared to HredH+,  is proposed to lack the additional protonation at the ADT while featuring the rotated, μCO, geometry (compare Fig. S1, ESI†). In the only characterized group D [FeFe]-hydrogenase, from Thermoanaerobacter mathranii (TamHydS), catalysis is bidirectional but thermodynamically irreversible.9 Kinetic modelling of cyclic voltammetry traces recorded under varied pH and H2 pressure supports the notion that TamHydS nevertheless follows the same mechanism as group A hydrogenases.49,50 However, there is currently very limited spectroscopic data available for TamHydS under turnover conditions to support this hypothesis. With regards to possibly catalytically active states, previously observed in group A enzymes, only oxidized (Hox/HoxH) and a one-electron reduced redox state (Hred) of the diiron site have been reported to-date.9,10
is proposed to lack the additional protonation at the ADT while featuring the rotated, μCO, geometry (compare Fig. S1, ESI†). In the only characterized group D [FeFe]-hydrogenase, from Thermoanaerobacter mathranii (TamHydS), catalysis is bidirectional but thermodynamically irreversible.9 Kinetic modelling of cyclic voltammetry traces recorded under varied pH and H2 pressure supports the notion that TamHydS nevertheless follows the same mechanism as group A hydrogenases.49,50 However, there is currently very limited spectroscopic data available for TamHydS under turnover conditions to support this hypothesis. With regards to possibly catalytically active states, previously observed in group A enzymes, only oxidized (Hox/HoxH) and a one-electron reduced redox state (Hred) of the diiron site have been reported to-date.9,10
[FeFe]-hydrogenases serve as model systems for probing general concepts such as thermodynamically reversible vs. irreversible catalysis.51 Thus, spectroscopic insight into the mechanism of these “non-prototypical” [FeFe]-hydrogenases is required to support or refute the suggestion arising from kinetic modelling work that the irreversible group D enzyme TamHydS still follows the same mechanism as the reversible group A enzymes.50 Furthermore, considering how the active site of [FeFe]-hydrogenases inspire the design of synthetic catalysts for sustainable H2 catalysis,5,6 a more detailed characterization of the group C and D enzymes is arguably critical to improve our understanding of how the outer-coordination sphere modulates the reactivity of the H-cluster.
Our initial efforts using spectro-electrochemistry and chemical reductants to probe the H-cluster of TamHydS have only revealed interconversion between the Hred and Hox states.9 To gain further insight into the H-cluster chemistry under turn-over conditions we instead applied photo-reduction. [FeFe]-hydrogenases are not intrinsically photoactive, but catalysis can be photo-triggered via a suitable photosensitizer (Fig. 1A). Such photo-reduction has been shown to induce H2 catalysis in vitro52–55 and in living cells.56,57 Moreover, this technique has facilitated investigations into the population of the reduced state (Hred) of the H-cluster as well as associated changes in the proton transfer pathway (PTP) of group A [FeFe]-hydrogenases under both steady-state and time-resolved conditions.45,57,58
Here, we investigate the group D [FeFe]-hydrogenase TamHydS via attenuated total reflection Fourier-transform infrared (ATR-FTIR) spectroscopy, complemented by electron paramagnetic resonance (EPR) spectroscopy. Through photo-reduction, we now have detected a variety of additional states that feature similar redox levels as in group A [FeFe]-hydrogenases. We observe the accumulation of four distinct diiron site reduced states that most likely differ in protonation chemistry, of which two were exclusively observed in the group C [FeFe]-hydrogenase TmHydS before.3 In turnover experiments, we can confirm a sequential population of redox states, namely from the oxidized state to one-electron reduced to two-electron reduced and putatively terminal hydride bound intermediates. Despite notable differences in active site cofactor environment, PTP, as well as catalytic activity rates, our findings provide support for a conserved catalytic mechanism in group A and D [FeFe]-hydrogenases. Additionally, the study sheds new light on the contested Hred intermediate.16,29 The detection of a potential bridging CO ligand in the spectroscopic signature of the Hred state indicates that the catalytically important rotated cofactor geometry can be conserved for the reduced diiron site intermediate at ambient conditions.
Results and discussion
Spectroscopic investigation of group D [FeFe]-hydrogenase TamHydSPDT
We initially studied an artificially matured variant of TamHydS where the secondary amine of the ADT bridgehead is replaced by a methylene group (PDT) denoted TamHydSPDT to avoid the complexity arising from protonation of the diiron subsite. For PDT cofactor variants of group A [FeFe]-hydrogenases, Hox and are the main redox states reported.59,60 A mixture of TamHydSPDT [FeFe]-hydrogenase, photosensitizer (Eosin Y), and sacrificial electron donor (triethanolamine (TEOA)) was applied to the surface of the ATR crystal, dried, and subsequently rehydrated with pH 8 Tris buffer. During continuous white light illumination, the photo-reduction of TamHydSPDT was followed in real time by ATR-FTIR difference spectroscopy. Fig. 2A (top spectrum) displays the “light minus dark” difference spectrum in the region of the CO and CN− ligands of the diiron site. Before illumination, the sample adopted the oxidized resting state, Hox, with its characteristic bands at 2084, 2073, 1971, 1949 and 1789 cm−1 (negative grey bands Fig. 2A). Upon photo-reduction, Hox depopulates, converting into the commonly observed [4Fe–4S]H cluster reduced state,
are the main redox states reported.59,60 A mixture of TamHydSPDT [FeFe]-hydrogenase, photosensitizer (Eosin Y), and sacrificial electron donor (triethanolamine (TEOA)) was applied to the surface of the ATR crystal, dried, and subsequently rehydrated with pH 8 Tris buffer. During continuous white light illumination, the photo-reduction of TamHydSPDT was followed in real time by ATR-FTIR difference spectroscopy. Fig. 2A (top spectrum) displays the “light minus dark” difference spectrum in the region of the CO and CN− ligands of the diiron site. Before illumination, the sample adopted the oxidized resting state, Hox, with its characteristic bands at 2084, 2073, 1971, 1949 and 1789 cm−1 (negative grey bands Fig. 2A). Upon photo-reduction, Hox depopulates, converting into the commonly observed [4Fe–4S]H cluster reduced state,  , with bands at 2079, 2064, 1969, 1939 and 1778 cm−1 (positive orange bands Fig. 2A). The absolute spectra indicate that the transition is nearly quantitative (Fig. S2, ESI†). However, minor unassigned peaks in the difference spectrum indicate the formation of additional reduced species (marked by an * in Fig. 2A, top).
, with bands at 2079, 2064, 1969, 1939 and 1778 cm−1 (positive orange bands Fig. 2A). The absolute spectra indicate that the transition is nearly quantitative (Fig. S2, ESI†). However, minor unassigned peaks in the difference spectrum indicate the formation of additional reduced species (marked by an * in Fig. 2A, top).
 | ||
Fig. 2 (A) ATR-FTIR difference spectra of the TamHydSPDT/Eosin Y/TEOA mixture. Reduction upon illumination (66 s of illumination minus the initial pre-illumination “0 s” spectrum, top spectrum) and auto-oxidation after illumination was stopped (480 seconds minus the last illuminated spectrum at 455 seconds, bottom spectrum). The peaks of terminal cyanide (CN−), terminal (tCO) and bridging carbon monoxide (μCO) ligands for different redox states are colour coded and the band patterns indicated by coloured bars. (Top spectrum) Upon photo-reduction the oxidized state (Hox, 2084, 2073, 1971, 1949 and 1789 cm−1) depopulates, predominantly in favour of the one-electron reduced state ( , 2079, 2064, 1969, 1939 and 1778 cm−1). Potential bands indicated by # do not correlate with any of the band patterns observed here. (Bottom spectrum) The 10-fold magnified difference spectrum displays the changes during re-oxidation after illumination. The minor redox species indicated by an * in the top spectrum can be assigned to , 2079, 2064, 1969, 1939 and 1778 cm−1). Potential bands indicated by # do not correlate with any of the band patterns observed here. (Bottom spectrum) The 10-fold magnified difference spectrum displays the changes during re-oxidation after illumination. The minor redox species indicated by an * in the top spectrum can be assigned to  (2050, 2018, 1903 and 1868 cm−1) and (2050, 2018, 1903 and 1868 cm−1) and  (2040, 2005, 1910 and 1855 cm−1). Compare with (B) for the kinetics of (de)-population during and after photo-reduction. (B) Redox state populations over time during and after photo-reduction. The time traces represent the sum of the difference peak area associated with a given redox state. To facilitate comparison the population of (2040, 2005, 1910 and 1855 cm−1). Compare with (B) for the kinetics of (de)-population during and after photo-reduction. (B) Redox state populations over time during and after photo-reduction. The time traces represent the sum of the difference peak area associated with a given redox state. To facilitate comparison the population of  and and  are multiplied by a factor of 3. The yellow box indicates the illumination period. Within 22 seconds most of the Hox population (black) is converted into are multiplied by a factor of 3. The yellow box indicates the illumination period. Within 22 seconds most of the Hox population (black) is converted into 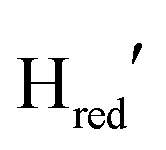 (orange). Half the (orange). Half the  population (red) is formed on the same timescale. Subsequently, population (red) is formed on the same timescale. Subsequently, 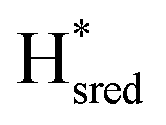 (blue) starts to accumulate. Directly after illumination Hox and (blue) starts to accumulate. Directly after illumination Hox and  appear to remain constant while appear to remain constant while  decays and decays and  gets transiently populated. On a longer timescale > 150 seconds after illumination, all reduced species convert into Hox. Compare Fig. S4 (ESI†) for zoom-ins on the direct changes upon illumination and after illumination. gets transiently populated. On a longer timescale > 150 seconds after illumination, all reduced species convert into Hox. Compare Fig. S4 (ESI†) for zoom-ins on the direct changes upon illumination and after illumination. | ||
The origin of these minor contributions becomes clearer when studying the auto-oxidation of the enzyme, spontaneously occurring once illumination was stopped. The difference spectrum computed from a dark spectrum collected 55 s after stopping illumination and the last illuminated spectrum (timepoints indicated by dashed lines in Fig. 2B) clearly shows that the unassigned features can be attributed to two additional interconverting states (Fig. 2A bottom spectrum). We observe positive bands at 2050, 2018, 1903 and 1868 cm−1 and negative bands at 2040, 2005, 1910 and 1855 cm−1. In both band patterns, the lowest wavenumber band is the most intense, which is a characteristic of diiron site reduced states.12,18,19 Since the PDT ligand cannot be protonated and the cofactor cannot yield Hred and Hsred, we assign the minor species with a pronounced absorbance feature at 1868 cm−1 to  (Fig. 2A, bottom spectrum, positive bands), in agreement with data reported for the group C [FeFe] hydrogenase TmHydSPDT.3 It follows that the second minor component, which is lost upon incubation in darkness, is attributable to
(Fig. 2A, bottom spectrum, positive bands), in agreement with data reported for the group C [FeFe] hydrogenase TmHydSPDT.3 It follows that the second minor component, which is lost upon incubation in darkness, is attributable to  (compare Fig. S1, ESI†) additionally reduced at the [4Fe–4S]H cluster (Fig. 2A, bottom spectrum, negative bands). We do not observe the photolysis of CO ligands during photo-reduction.61,62 Repeating the experiment with TamHydSPDT samples including sodium dithionite led to practically indistinguishable results (Fig. S3, ESI†). An overview of all TamHydS infrared signatures is given in Table 1 and Fig. S9 (ESI†). The detailed fit parameters for all infrared signatures can be found in the ESI.†
(compare Fig. S1, ESI†) additionally reduced at the [4Fe–4S]H cluster (Fig. 2A, bottom spectrum, negative bands). We do not observe the photolysis of CO ligands during photo-reduction.61,62 Repeating the experiment with TamHydSPDT samples including sodium dithionite led to practically indistinguishable results (Fig. S3, ESI†). An overview of all TamHydS infrared signatures is given in Table 1 and Fig. S9 (ESI†). The detailed fit parameters for all infrared signatures can be found in the ESI.†
Bands are indicated in the order CN, CN, tCO, tCO, μ/tCO. #Data reported for the group C enzyme TmHydS is listed for  and and  to compare the observed band positions since this is the only [FeFe]-hydrogenases previously reported to adopt these states. to compare the observed band positions since this is the only [FeFe]-hydrogenases previously reported to adopt these states. |
||
|---|---|---|
| TamHydS (group D) bands in cm−1 | CrHydA1 (group A) bands in cm−1 | |
| Hox | 2082, 2074, 1970, 1948, 17879 | 2088, 2072, 1964, 1940, 180025 |
| HoxH | n.d., n.d., 1978, 1954, 17959 | 2092, 2072, 1970, 1946, 181218 |
| Hred | 2062, 2030, 1961, 1922, 18959 | 2072, 2033, 1961, 1915, 189135 |
| 2062, 2032, 1922, 1896, 1803 (this study) | At 90 K, n.d, n.d., 1919, 1891, 181739 | |

|
2052, 2012, 1903, 1875, n.d. (this study) | #TmHydSADT (group C) 2055, 2022, 1894, 1871, 17633 |
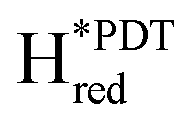
|
2050, 2018, 1903, 1868, n.d. (this study) | #TmHydSPDT (group C) 2048, 2012, 1883, 1862, n.d.3 |

|
n.d, n.d., 1960, 1939, 1782 (this study) | 2084, 2066, 1962, 1933, 179215 |
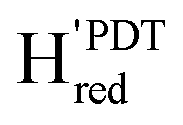
|
2079, 2064, 1969, 1939, 1778 (this study) | 2090, 2072, 1966, 1941, 181059 |
| Hsred | n.d., n.d., 1912, 1887, n.d. (this study) | 2068, 2026, 1953, 1918, 188235 |

|
n.d., n.d., n.d.,1865, n.d. (this study) | #TmHydSADT (group C) 2047, 2013, 1900, 1861, 17513 |

|
2040, 2005, 1910, 1855, n.d. (this study) | #TmHydSPDT (group C) 2042, 2007, 1894, 1853, n.d.3 |
| Hhyd | 2082, 2075, 1981, 1970, 1848 (this study) | 2082, 2068, 1978, 1960, 186024 |
Using the aforementioned redox state assignment, we can monitor the redox state population over time during and after photo-reduction (Fig. 2B). A rapid conversion of Hox primarily into  is evident during the first 22 seconds of illumination.
is evident during the first 22 seconds of illumination.  also starts to accumulate rapidly, reaching approximately half of the final
also starts to accumulate rapidly, reaching approximately half of the final  population on the same timescale (Fig. S4 for zoom in, ESI†). However, while the
population on the same timescale (Fig. S4 for zoom in, ESI†). However, while the 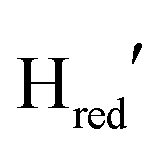 population appears to plateau, we observe further population of
population appears to plateau, we observe further population of 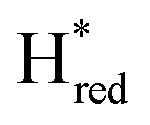 on a longer timescale. A lag-phase of about 20 seconds is observed for the population of
on a longer timescale. A lag-phase of about 20 seconds is observed for the population of  . The fact that
. The fact that  starts to accumulate when a large fraction of the enzyme has already converted to
starts to accumulate when a large fraction of the enzyme has already converted to  and
and  is in line with the proposed two-electron reduced nature of this species. Note that the redox state populations plotted in Fig. 2 are normalized, and that the
is in line with the proposed two-electron reduced nature of this species. Note that the redox state populations plotted in Fig. 2 are normalized, and that the  and
and 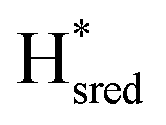 populations remain minor species when compared to the Hox to
populations remain minor species when compared to the Hox to  populations throughout the entire experiment (Fig. S5, ESI†). After photo-reduction, the
populations throughout the entire experiment (Fig. S5, ESI†). After photo-reduction, the 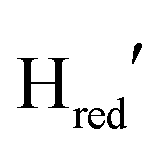 population appears to stay constant for ca. 150 seconds. The
population appears to stay constant for ca. 150 seconds. The  population decays fastest and most probably converts into
population decays fastest and most probably converts into  whose population is transiently increased before decaying as well (Fig. S4, ESI†). The Hox population remains constant for ca. 30 seconds after illumination and recovers subsequently. Similar relatively sluggish conversion from one-electron reduced states to Hox has been observed in group A [FeFe]-hydrogenases before.63 Thus, the study of TamHydSPDT has allowed us to observe three reduced states
whose population is transiently increased before decaying as well (Fig. S4, ESI†). The Hox population remains constant for ca. 30 seconds after illumination and recovers subsequently. Similar relatively sluggish conversion from one-electron reduced states to Hox has been observed in group A [FeFe]-hydrogenases before.63 Thus, the study of TamHydSPDT has allowed us to observe three reduced states  , complementing the observation of the Hred state previously reported for TamHydSADT.9,10
, complementing the observation of the Hred state previously reported for TamHydSADT.9,10
New redox states for group D [FeFe]-Hydrogenase TamHydSADT
Performing the analogous photo-reduction experiment on the catalytically active form TamHydSADT (in the following TamHydS) resulted in more complex spectral changes. In Fig. 3A, difference spectra reflecting changes after 44 seconds (44s–0s, top spectrum) and 220 seconds (220s–0s, middle spectrum) of continuous illumination relative to the starting “pre-illumination” (“0”) spectrum are displayed (absolute and full series of difference spectra in Fig. S6, ESI†). As expected, we observe a decrease of bands associated with the oxidized states, Hox and HoxH (grey and dark grey bands). The difference spectra also suggest the de-population of an additional minor species with small negative bands detected at 1960, 1939 and 1782 cm−1 (orange bands). These bands are reminiscent of when compared to TamHydSPDT (Fig. 2) and group A enzymes,12,15 implying that this reduced species is present in low amounts at pH 8.
when compared to TamHydSPDT (Fig. 2) and group A enzymes,12,15 implying that this reduced species is present in low amounts at pH 8.
 | ||
Fig. 3 (A) Light minus dark ATR-FTIR difference spectra of the TamHydS/Eosin Y/TEOA mixture after 44 seconds (top spectrum) and 220 seconds of illumination (middle spectrum). The bottom difference spectrum is calculated between 44 and 220 seconds of illumination (timepoints indicated by vertical dashed lines) where only small changes in oxidized redox states population occur (compare dashed lines in Fig. 3B) enabling the detection of the hydride state bands. The peaks of terminal cyanide (CN), terminal (tCO) and bridging carbon monoxide (μCO) ligands for each different redox state are colour coded, and the band patterns indicated by coloured bars. The difference spectrum is indicated in black and the fit by a dashed line. Upon photo-reduction the oxidized states (Hox, HoxH) and  depopulate in favour of the one-electron and two electron reduced states (Hred, Hsred, depopulate in favour of the one-electron and two electron reduced states (Hred, Hsred,  , ,  and Hhyd). Compare to Fig. 3B for the kinetics of (de)-population during and after photo-reduction. An unassigned band is indicated in magenta. (B) Redox state population kinetics during and after photo-reduction of TamHydS. The summed band area of each individual redox state is plotted for visual comparison of the (de)-population kinetics. Within the first 40–60 seconds we observe a depopulation of the oxidized states (Hox, HoxH) as well as the one-electron reduced state and Hhyd). Compare to Fig. 3B for the kinetics of (de)-population during and after photo-reduction. An unassigned band is indicated in magenta. (B) Redox state population kinetics during and after photo-reduction of TamHydS. The summed band area of each individual redox state is plotted for visual comparison of the (de)-population kinetics. Within the first 40–60 seconds we observe a depopulation of the oxidized states (Hox, HoxH) as well as the one-electron reduced state  . The population of Hred reaches its maximum after 44 seconds and depopulates subsequently in the expense of . The population of Hred reaches its maximum after 44 seconds and depopulates subsequently in the expense of 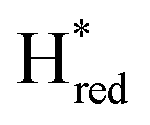 and the two electron reduced states (Hsred, and the two electron reduced states (Hsred,  , Hhyd). Hhyd seems to accumulate the slowest reaching its maximum around 200 seconds. After photo-reduction, the two-electron reduced protonated redox states Hhyd and Hsred depopulate at the same time scale as we observe an increase of Hred. , Hhyd). Hhyd seems to accumulate the slowest reaching its maximum around 200 seconds. After photo-reduction, the two-electron reduced protonated redox states Hhyd and Hsred depopulate at the same time scale as we observe an increase of Hred. 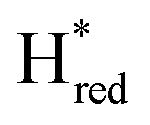 (and (and 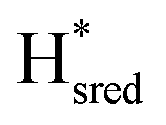 ) decay on a slower timescale. The population of Hox, HoxH and ) decay on a slower timescale. The population of Hox, HoxH and  remains nearly constant for 80 seconds after photo-reduction. The yellow box indicates the illumination period. Each point is extracted from a difference spectrum (x–0 seconds) (Fig. S6, ESI†) and difference spectra for 44–0, 220–0 and 220–44 seconds are shown in Fig. 3A. remains nearly constant for 80 seconds after photo-reduction. The yellow box indicates the illumination period. Each point is extracted from a difference spectrum (x–0 seconds) (Fig. S6, ESI†) and difference spectra for 44–0, 220–0 and 220–44 seconds are shown in Fig. 3A. | ||
In contrast to the reduction of TamHydS by H2 gas and electrochemistry as reported earlier,9,10 we detect two distinct terminal distal CO bands at 1895 and 1875 cm−1. To fit these bands, additional bands at 1887 and 1865 cm−1 were necessary, overall indicative of four separate diiron site reduced states. The assignment to four distinct H-cluster states is further strengthened by their independent (de-)population kinetics during the photo-reduction experiment (Fig. 3B). The highest wavenumber band at 1895 cm−1 is attributable to the previously observed protonated and reduced diiron site state, Hred, with an additional CO band detected at 1922 cm−1 and CN− bands at 2030 and 2062 cm−1. The band at 1887 cm−1, which increases together with a band at 1912 cm−1, is attributed to the two electron reduced state Hsred, based on its position relative to that of Hred at 1895 cm−1 and comparison to the infrared signatures of group A [FeFe]-hydrogenases.19,25 The possibility that the signals at 1887 and 1895 cm−1 instead reflect two Hred state populations with different redox states of neighbouring F-clusters is highly unlikely to rationalize the here observed shift of ca. 10 cm−1.64,65 The band at 1875 cm−1, which follows the same kinetics as the bands at 2052, 2012 and 1903 cm−1, can be assigned to 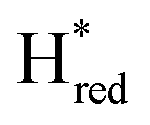 , based on the
, based on the  state signature observed in TamHydSPDT. Finally, based on the assignment of
state signature observed in TamHydSPDT. Finally, based on the assignment of  , we can assign the small band detected at 1865 cm−1 to
, we can assign the small band detected at 1865 cm−1 to 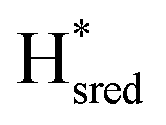 . We note that this implies a blue-shift of the band by about 8 cm−1, when the dithiolate ligand featured an amine bridgehead group relative to the methylene analogue. This would be in good agreement with what has been reported for the group C [FeFe]-hydrogenase TmHydS, for which the band pattern of
. We note that this implies a blue-shift of the band by about 8 cm−1, when the dithiolate ligand featured an amine bridgehead group relative to the methylene analogue. This would be in good agreement with what has been reported for the group C [FeFe]-hydrogenase TmHydS, for which the band pattern of  in TmHydSADT shifted to higher wavenumbers by ca. 10 cm−1 when compared to TmHydSPDT.3 Chongdar et al. attributed this shift to a change in electron density at the diiron core induced by the nature of the bridging dithiolate ligand.
in TmHydSADT shifted to higher wavenumbers by ca. 10 cm−1 when compared to TmHydSPDT.3 Chongdar et al. attributed this shift to a change in electron density at the diiron core induced by the nature of the bridging dithiolate ligand.
To test this latter hypothesis, regarding the influence of the bridgehead group on the FTIR signatures of the H-cluster, we studied a series of diiron site mimics (Fe(I)Fe(I)) featuring ADT and PDT ligands with different combinations of carbonyl, phosphine and CN− ligands (Fig. S7, ESI†). The effect of the bridgehead group of the dithiolate ligand was found to vary depending on the rest of the ligand sphere of the model complexes. It was most distinct in the dicyanide complexes, where the mimic featuring the PDT ligand displayed a shift of certain CO bands by 8 cm−1 relative to the ADT ligated analogue. When comparing the infrared signatures of TamHydSPDT and TamHydSADT, a clear effect by H-cluster redox state was found (overview of infrared signatures in Table 1 and Fig. S9, ESI†). The two TamHydS versions seem to be nearly identical for Hox with 0–2 cm−1 shifts of the cofactor ligand bands. Conversely, for  , we observe a 9 cm−1 shift on the proximal CO (pCO) band while the distal CO (dCO) band is found at an identical position. While for the
, we observe a 9 cm−1 shift on the proximal CO (pCO) band while the distal CO (dCO) band is found at an identical position. While for the  and
and  states, the dCO band is shifted by 7–10 cm−1 to higher energies in TamHydSADT relative to TamHydSPDT. In combination, these observations support the idea that the nature of the bridgehead can influence the FTIR signature of H-cluster states. However, evidently, the effect is difficult to predict as the effect varies both with oxidation state and the remaining ligand sphere.
states, the dCO band is shifted by 7–10 cm−1 to higher energies in TamHydSADT relative to TamHydSPDT. In combination, these observations support the idea that the nature of the bridgehead can influence the FTIR signature of H-cluster states. However, evidently, the effect is difficult to predict as the effect varies both with oxidation state and the remaining ligand sphere.
As the concentration of Hox states remains practically constant after 44 seconds (Fig. 3B) the difference spectrum calculated between 220 and 44 seconds of illumination (220s–44s, Fig. 3A bottom) has no significant negative contributions from the oxidized state bands between 1980–1948 cm−1. This allows assigning the additional positive bands at 2082, 2075, 1981, 1970 and 1848 cm−1 to the CN− and CO ligands of a redox state with a super-oxidized diiron site when compared to Hox. The inhibited states, Hinact and Htrans, feature a super-oxidized diiron site. However, reduction reactivates these inhibited states, thus they are highly unlikely to accumulate under photo-reduction.19,66 Instead we attribute the band pattern to the hydride state, Hhyd (positive green bands, Fig. 3 bottom spectrum), which exhibits similar band patterns in group A [FeFe]-hydrogenases.24,55 However, due to the low population of the putative Hhyd species when performing the experiment in D2O we refrain from a definite assignment.24,28 Attempts at enriching this proposed essential catalytic intermediate in TamHydS via our group A [FeFe]-hydrogenase specific protocols (low pH value, optional high reductant concentration, and exposure to H2 gas24,26) led to the population of Hred only, even though a significant fraction of HoxH was present.9
To conclude, we observe similar redox states in group A and D [FeFe]-hydrogenases. The H-cluster in TamHydS can clearly adopt the oxidized state (Hox), the one-electron reduced states  and Hred with the reducing equivalent on the [4Fe–4S]H or the diiron site respectively, the super reduced state (Hsred) with one reducing equivalent on the [4Fe–4S]H, and the diiron site each, and appears to form also the di-ferrous terminal hydride state (Hhyd).
and Hred with the reducing equivalent on the [4Fe–4S]H or the diiron site respectively, the super reduced state (Hsred) with one reducing equivalent on the [4Fe–4S]H, and the diiron site each, and appears to form also the di-ferrous terminal hydride state (Hhyd).
To further investigate the interconversion of these redox states during and after photo-reduction, we again analysed the redox state population changes as a function of time (Fig. 3B). The quantification was done through summation of the areas of the bands associated with each state. The oxidized states Hox and HoxH as well as for the one-electron reduced state, 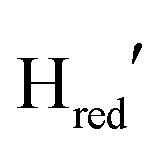 depopulate mainly during the first 44 seconds, and reach a steady concentration after 66 seconds. The protonated one-electron reduced state Hred reaches its maximum population after 44 seconds, after which time the population of this species clearly decreases again. The alternative one-electron reduced state
depopulate mainly during the first 44 seconds, and reach a steady concentration after 66 seconds. The protonated one-electron reduced state Hred reaches its maximum population after 44 seconds, after which time the population of this species clearly decreases again. The alternative one-electron reduced state 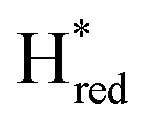 instead increases on a slower timescale (ca. 50% populated at 44 s) and reaches a steady-state population after about 220 seconds. As can be seen in Fig. 3A, Hred and
instead increases on a slower timescale (ca. 50% populated at 44 s) and reaches a steady-state population after about 220 seconds. As can be seen in Fig. 3A, Hred and  are the main reduced states formed during photo-reduction. The two-electron reduced state Hsred rise to a plateau population within 120 seconds, while the Hhyd population takes approximately double the time (220 seconds, compare as well bottom spectrum Fig. 3A). Notably, the rise of Hhyd coincides with the decay of Hred. The time-dependency of the
are the main reduced states formed during photo-reduction. The two-electron reduced state Hsred rise to a plateau population within 120 seconds, while the Hhyd population takes approximately double the time (220 seconds, compare as well bottom spectrum Fig. 3A). Notably, the rise of Hhyd coincides with the decay of Hred. The time-dependency of the  population is difficult to track with certainty due to the low signal intensity but seem to continuously increase slowly during illumination. Overall, these trends are in good agreement with an increasingly negative solution potential upon extended illumination. After illumination, we observe the converse transitions between H-cluster state populations. The protonated two-electron reduced states, Hsred and Hhyd, depopulate rapidly (negligible residual signal at t ≈ 420 s and 460 s, respectively; where illumination is stopped at t = 374 s). On a much longer timescale compared to the decay of Hsred and Hhyd the populations of
population is difficult to track with certainty due to the low signal intensity but seem to continuously increase slowly during illumination. Overall, these trends are in good agreement with an increasingly negative solution potential upon extended illumination. After illumination, we observe the converse transitions between H-cluster state populations. The protonated two-electron reduced states, Hsred and Hhyd, depopulate rapidly (negligible residual signal at t ≈ 420 s and 460 s, respectively; where illumination is stopped at t = 374 s). On a much longer timescale compared to the decay of Hsred and Hhyd the populations of 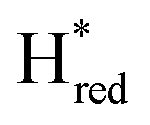 (50% de-populated at 500 s) and
(50% de-populated at 500 s) and 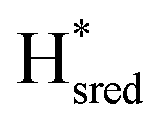 decrease. The population of Hred varies in a more complex fashion. Again, coinciding with the opposite kinetics of Hhyd, Hred increases shortly after illumination is arrested (maximum population at ca. 500 s) before it slowly starts to decrease. Similar to TamHydSPDT the population of the oxidized states, Hox and HoxH, remain nearly constant for ca. 80 seconds and repopulate slowly afterwards. The rapid appearance of Hred but more sluggish re-formation of the Hox/HoxH after illumination agrees with Hred as a quasi-stable resting state as previously noted in H2 flushing experiments.9
decrease. The population of Hred varies in a more complex fashion. Again, coinciding with the opposite kinetics of Hhyd, Hred increases shortly after illumination is arrested (maximum population at ca. 500 s) before it slowly starts to decrease. Similar to TamHydSPDT the population of the oxidized states, Hox and HoxH, remain nearly constant for ca. 80 seconds and repopulate slowly afterwards. The rapid appearance of Hred but more sluggish re-formation of the Hox/HoxH after illumination agrees with Hred as a quasi-stable resting state as previously noted in H2 flushing experiments.9
Considering the more recently identified 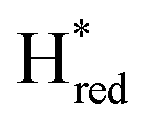 and
and 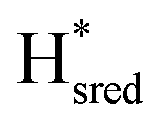 states, and assuming that the structural assignment of
states, and assuming that the structural assignment of  proposed by Chongdar et al. is correct,3 there are at least two possible scenarios that can explain the kinetics observed in this study.
proposed by Chongdar et al. is correct,3 there are at least two possible scenarios that can explain the kinetics observed in this study. 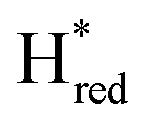 , and the associated
, and the associated  , state could reflect inhibited states, potentially related to the reductive inhibition phenomena previously noted by Léger and co-workers,67 or represent catalytically relevant states closely related to the
, state could reflect inhibited states, potentially related to the reductive inhibition phenomena previously noted by Léger and co-workers,67 or represent catalytically relevant states closely related to the  state.
state.
If 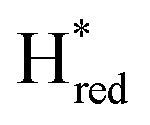 would represent an inhibited, off-pathway, species, Hred and Hhyd would be the main reduced catalytic species interconverting during and after photo reduction. Indeed, the decay of Hred during prolonged illumination (photo-reduction) coincides with the population of Hhyd. Similar but opposite behaviour is observed directly after photo-reduction; Hred gets populated while Hhyd de-populates on the same timescale. The kinetics of
would represent an inhibited, off-pathway, species, Hred and Hhyd would be the main reduced catalytic species interconverting during and after photo reduction. Indeed, the decay of Hred during prolonged illumination (photo-reduction) coincides with the population of Hhyd. Similar but opposite behaviour is observed directly after photo-reduction; Hred gets populated while Hhyd de-populates on the same timescale. The kinetics of  do not seem to be following the kinetics of either of these states besides getting populated alongside Hred at the beginning of the photo-reduction experiment. During the post-illumination phase,
do not seem to be following the kinetics of either of these states besides getting populated alongside Hred at the beginning of the photo-reduction experiment. During the post-illumination phase,  decays slowly, potentially interconverting into Hred after the Hhyd state already decayed (around 500 seconds). The accumulation of a large population of an inhibited state under reducing conditions could also partially rationalize the low catalytic activities observed for group C and D [FeFe]-hydrogenases.3,9 Still, we note that this model would require that the inhibition is reversible on the time-scale of the experiment, as otherwise the
decays slowly, potentially interconverting into Hred after the Hhyd state already decayed (around 500 seconds). The accumulation of a large population of an inhibited state under reducing conditions could also partially rationalize the low catalytic activities observed for group C and D [FeFe]-hydrogenases.3,9 Still, we note that this model would require that the inhibition is reversible on the time-scale of the experiment, as otherwise the  state population would be expected to continuously increase during the photo-reduction. Arguing against this scenario is the fact that no reductive inhibition has been reported from earlier electrochemical studies of TamHydS.9,49,50 The alternative rationale would be that
state population would be expected to continuously increase during the photo-reduction. Arguing against this scenario is the fact that no reductive inhibition has been reported from earlier electrochemical studies of TamHydS.9,49,50 The alternative rationale would be that  vs. Hred populations are governed by the relative rates of electron transfer and protonation. Here, pure electron transfer would yield
vs. Hred populations are governed by the relative rates of electron transfer and protonation. Here, pure electron transfer would yield  , while coupled (but not necessarily concerted) electron transfer and protonation yields Hred. During extended illumination we can expect electron transfer rates to increase as the population of photo-reduced EY increases, while proton transfer rates remain unchanged. This would potentially result in an increase in population of the reduced unprotonated state
, while coupled (but not necessarily concerted) electron transfer and protonation yields Hred. During extended illumination we can expect electron transfer rates to increase as the population of photo-reduced EY increases, while proton transfer rates remain unchanged. This would potentially result in an increase in population of the reduced unprotonated state  relative to Hred. As illumination is arrested, the solution potential decreases and the protonation step can now compete, resulting in a decrease of
relative to Hred. As illumination is arrested, the solution potential decreases and the protonation step can now compete, resulting in a decrease of  relative to Hred, as observed in our experiments on a longer timescale after illumination is stopped (starting around 500 seconds). This would instead make
relative to Hred, as observed in our experiments on a longer timescale after illumination is stopped (starting around 500 seconds). This would instead make  a catalytically relevant intermediate analogous but not identical to
a catalytically relevant intermediate analogous but not identical to 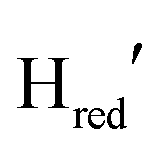 , with still unidentified structural aspects of TmHydS and TamHydS favoring reduction of the di-iron site instead of the [4Fe4S] cluster during the initial electron injection into the H-cluster. The currently available data does not allow us to strictly distinguish these two hypotheses.
, with still unidentified structural aspects of TmHydS and TamHydS favoring reduction of the di-iron site instead of the [4Fe4S] cluster during the initial electron injection into the H-cluster. The currently available data does not allow us to strictly distinguish these two hypotheses.
We note that when analysing the kinetics of the photo-reduction experiment in isolation, the population of the redox state assigned via spectroscopic analysis to  would be intuitively rather assigned to a two electron reduced species e.g. Hsred. However, such an assignment would imply a down shift of 20 cm−1 of the main band of Hred (1895 cm−1) to the putative Hsred main band (1875 cm−1), contradicting all previous studies involving these two redox states reporting a shift of ≈10 cm−1.12,19,23,25,35,37
would be intuitively rather assigned to a two electron reduced species e.g. Hsred. However, such an assignment would imply a down shift of 20 cm−1 of the main band of Hred (1895 cm−1) to the putative Hsred main band (1875 cm−1), contradicting all previous studies involving these two redox states reporting a shift of ≈10 cm−1.12,19,23,25,35,37
Altering the pH in these photo-reduction assays can significantly affect both the H-cluster kinetics as well as the photochemistry, and should thus be interpreted carefully. Still, to further probe the nature of the species giving rise to the 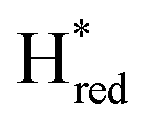 signature, we performed additional photo-reduction experiments at lower pH (Fig. S6, ESI†). Albeit this data should only be considered qualitative, reduction at lower pH did suppress the 1875 cm−1 signal, while the relative amplitude of the Hred signature increased, in good agreement with the expected behaviour of the
signature, we performed additional photo-reduction experiments at lower pH (Fig. S6, ESI†). Albeit this data should only be considered qualitative, reduction at lower pH did suppress the 1875 cm−1 signal, while the relative amplitude of the Hred signature increased, in good agreement with the expected behaviour of the  species.
species.
A bridging CO band in Hred observed at room temperature in TamHydSADT
The FTIR signature of the Hred species generated under photo-reduction was consistently shifted to slightly lower energies, relative to our earlier reported data for the H2 induced Hred state.9 To verify that this was not due to variations in enzyme preparations, we exposed films of TamHydS to a 10% H2 atmosphere. As expected, this treatment generated difference spectra displaying a nearly exclusive Hox/HoxH to Hred (2063, 2032, 1922 and 1896 cm−1) transition, with only residual traces of other redox states being formed (Fig. 4). This verified that the FTIR signature of Hred obtained in the photo-reduction experiment was indeed shifted relative to the Hred species enriched under H2, with the latter displaying shifts of the CO and CN− ligand bands of 1–2 cm−1 to higher wavenumbers. Such small but distinct shifts have previously been observed for group A [FeFe]-hydrogenase and have been attributed to differences in the redox states of surrounding F-clusters.64,65 | ||
| Fig. 4 H2 induced Hred in TamHydS exhibits a potential bridging CO ligand band at ambient conditions. A TamHydS film equilibrated under 2% H2 (pH 8) was exposed for 44 seconds to 10% H2 resulting in an increased population of Hred. The difference spectrum shows a nearly exclusive Hox/HoxH to Hred transition when compared to the rather crowded difference spectra obtained via photo-reduction (compare Fig. 3). A band in the bridging CO ligand region at 1803 cm−1 co-populates with the bands at 2063, 2032, 1922 and 1896 cm−1 and is thus assigned to Hred. When compared to the IR signature obtained via photo-reduction the bands of Hredvia H2 exposure are slightly shifted to higher wavenumbers indicative of oxidized F-clusters.64,65 The spectrum contains trances of State 1 and State 2, described earlier.47 Magenta peaks indicate residual unassigned species. | ||
To verify this hypothesis, we performed EPR spectroscopy on photo-reduced TamHydS samples. Our earlier EPR studies of TamHydS have shown that H2 gas exposure only results in partial reduction of the F-clusters. More specifically, H2 treatment gives rise to one broad rhombic feature, attributable to the reduction of a single [4Fe4S] cluster.9,47 The photo-reductant employed here resulted in a larger degree of [4Fe4S] cluster reduction, as evident from the appearance of at least two distinct rhombic signals in samples illuminated in the presence of Eosin Y and TEOA (Fig. S8, ESI†).
We note that attempts at fitting the Hred state observed during or after photo-reduction (Fig. 3) with two peaks at 1895 and 1896 cm−1 excludes the population of the H2 induced Hred state with potentially oxidized F-clusters (Fig. S6, ESI†). More importantly, these H2 induced Hred spectra also display an additional broad but distinct band at 1803 cm−1, which correlates in intensity with the other features of the Hred signature. The frequency of this band strongly supports the notion that it reflects a μCO ligand.
A similar μCO band was observed in photo-reduction experiments with TamHydS, showing a nearly exclusive transition from Hox to Hred.68 In these experiments, the μCO band of Hred was downshifted by about 1 cm−1 compared to the H2-induced Hred state seen here, further supporting our hypothesis of further reduced F-clusters. However, we did not include this band in the analysis in Fig. 3, as it did not improve the fit.
Surprisingly, the μCO band of the reduced diiron site (1803 cm−1) is found at higher wavenumbers when compared to the μCO band position of the oxidized state, Hox (1787 cm−1). This high wavenumber position seems counter intuitive for a μCO vibration at a more reduced diiron site, as that should instead induce a significant shift to lower wavenumbers due to increased π back-bonding. As a comparison, the μCO band of the formally super-oxidized state, Hhyd (Fe(II)Fe(II)), shifts by ca. 60 cm−1 to higher wavenumbers when compared to the oxidized state Hox (Fe(II)Fe(I)).24,55 A comparable up-shift relative to Hox is observed for the inhibited state, Hinact, as well.69 Furthermore, in group C [FeFe]-hydrogenase, TmHydS, the μCO band for the putatively unprotonated reduced state,  , has been reported red shifted by 40 cm−1, relative to Hox, to 1763 cm−1.3 The fact that Hred and
, has been reported red shifted by 40 cm−1, relative to Hox, to 1763 cm−1.3 The fact that Hred and 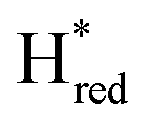 most likely vary in protonation state of the ADT-amine would only partially rationalize the higher energy of the μCO for Hred in TamHydS as compared to
most likely vary in protonation state of the ADT-amine would only partially rationalize the higher energy of the μCO for Hred in TamHydS as compared to  in TmHydS,70,71 as model complexes have consistently shown that protonation of the ADT amine cause a 16–20 cm−1 upshift of the CO bands. However, similar high μCO band positions for Hred have been observed at cryogenic temperatures for group A [FeFe]-hydrogenases,37–39 and correlation of this experimental data with Density Functional Theory calculations favour its attribution to a Fe(I)Fe(I) species with strict rotated μCO geometry and a protonated ADT ligand.38,72 Summing up, the fact that a band is now observed at this position provides compelling support for the notion that Hred can harbour a μCO ligand at room temperature. We further note that the absence of any μCO band in our spectra for
in TmHydS,70,71 as model complexes have consistently shown that protonation of the ADT amine cause a 16–20 cm−1 upshift of the CO bands. However, similar high μCO band positions for Hred have been observed at cryogenic temperatures for group A [FeFe]-hydrogenases,37–39 and correlation of this experimental data with Density Functional Theory calculations favour its attribution to a Fe(I)Fe(I) species with strict rotated μCO geometry and a protonated ADT ligand.38,72 Summing up, the fact that a band is now observed at this position provides compelling support for the notion that Hred can harbour a μCO ligand at room temperature. We further note that the absence of any μCO band in our spectra for  and
and  in both the ADT and PDT version of TamHydS differs somewhat from what has been reported for the group C representative TmHydS.3 In the latter case, the fully functional ADT version of the enzyme did show a distinct μCO band, while no corresponding band was observed for the PDT variant. Evidently, the relative amplitude of the μCO band is strongly dependent on a range of factors including temperature and nature of the bridgehead group.
in both the ADT and PDT version of TamHydS differs somewhat from what has been reported for the group C representative TmHydS.3 In the latter case, the fully functional ADT version of the enzyme did show a distinct μCO band, while no corresponding band was observed for the PDT variant. Evidently, the relative amplitude of the μCO band is strongly dependent on a range of factors including temperature and nature of the bridgehead group.
Conclusions
Besides Hox/HoxH and Hred, this study confirms that the H-cluster in TamHydS forms additional redox states analogous to the ones observed for group A [FeFe]-hydrogenases (Table 1 and Fig. S9, ESI†).5,72 This lends spectroscopy support to the notion that group A and group D [FeFe]-hydrogenases follow the same catalytic cycle, as previously proposed from kinetic fitting of electrochemical data.49 The idea of a shared catalytic mechanism is further supported by the observed sequential population of key redox states. In this scenario, the slow catalytic kinetics of TamHydS are attributable to an increased stability of Hred in the putatively sensory enzymes group C and D hydrogenases, relative to the group A. In the current study, this enhanced stability of Hred is reflected in its fast formation upon illumination and relatively slow conversion back to Hox once illumination is stopped (Fig. 3). The stability of the one-electron reduced intermediate over a wide potential range is further in agreement with the spectro-electrochemistry data reported for the group C [FeFe]-hydrogenase TmHydS.3,48However, different mechanisms operating in different groups of [FeFe]-hydrogenases cannot be fully excluded at this stage. The fact that  and
and  are found to accumulate in both group C and D [FeFe]-hydrogenases, while no corresponding states have been observed for group A enzymes, underscores that the protein environment induces distinct differences in the properties of the H-cluster between groups. As noted above, one possible explanation for the accumulation of
are found to accumulate in both group C and D [FeFe]-hydrogenases, while no corresponding states have been observed for group A enzymes, underscores that the protein environment induces distinct differences in the properties of the H-cluster between groups. As noted above, one possible explanation for the accumulation of  and
and  in these slow [FeFe]-hydrogenases could be that these are inhibited states, which would explain why mainly Hred/Hhyd populations interconvert during and after photo-reduction. On the other hand, the diiron site is clearly much more easily reduced in these two putatively sensory hydrogenases, as it accumulates without concomitant protonation and the slow interconversion of Hred/
in these slow [FeFe]-hydrogenases could be that these are inhibited states, which would explain why mainly Hred/Hhyd populations interconvert during and after photo-reduction. On the other hand, the diiron site is clearly much more easily reduced in these two putatively sensory hydrogenases, as it accumulates without concomitant protonation and the slow interconversion of Hred/ after photo-reduction would favour
after photo-reduction would favour  to be a potential catalytic relevant intermediate. When comparing group D and A enzymes the differences in H-cluster reactivity are further evidenced by the fact that still incompletely characterized spectroscopic features have been observed in TamHydS under H2 pressure.9,47 Beside reporting numerous H-cluster states previously unidentified in group D, our study sheds new light on the common Hred intermediate. Besides group C, this species appears to be shared across all [FeFe]-hydrogenases studied to date, but its structural details remain contested. The data reported herein does not allow for definite distinction between models 1 or 2, as direct experimental proof of either a μH ligand or a protonation of the ADT ligand (Fig. 1B) remains elusive. Still, when the diiron site reduced state, Hred, is induced via exposure to H2 gas in TamHydS, we now observe a band in the μCO band region that appears to be associated with Hred, indicating the existence of a bridging CO ligand in the Hred state at room temperature in line with the recent report of a μCO band for photo-reduced TamHydS.68 We note that in earlier reports of group A [FeFe]-hydrogenases traces of a μCO bands potentially attributable to the Hred state could be visible at room temperatures as well, but to the best of our knowledge this is the first reported instance where the band is so clearly discernible. We show that Hred can adopt different F-cluster redox configurations, and the retained rotated diiron site geometry implied from the μCO band at ambient temperature would render Hred a potential catalytic intermediate.
to be a potential catalytic relevant intermediate. When comparing group D and A enzymes the differences in H-cluster reactivity are further evidenced by the fact that still incompletely characterized spectroscopic features have been observed in TamHydS under H2 pressure.9,47 Beside reporting numerous H-cluster states previously unidentified in group D, our study sheds new light on the common Hred intermediate. Besides group C, this species appears to be shared across all [FeFe]-hydrogenases studied to date, but its structural details remain contested. The data reported herein does not allow for definite distinction between models 1 or 2, as direct experimental proof of either a μH ligand or a protonation of the ADT ligand (Fig. 1B) remains elusive. Still, when the diiron site reduced state, Hred, is induced via exposure to H2 gas in TamHydS, we now observe a band in the μCO band region that appears to be associated with Hred, indicating the existence of a bridging CO ligand in the Hred state at room temperature in line with the recent report of a μCO band for photo-reduced TamHydS.68 We note that in earlier reports of group A [FeFe]-hydrogenases traces of a μCO bands potentially attributable to the Hred state could be visible at room temperatures as well, but to the best of our knowledge this is the first reported instance where the band is so clearly discernible. We show that Hred can adopt different F-cluster redox configurations, and the retained rotated diiron site geometry implied from the μCO band at ambient temperature would render Hred a potential catalytic intermediate.
In closing, we stress that many details surrounding the catalytic mechanism(s) of [FeFe]-hydrogenase remain to be resolved. However, the fact that Hox,  , Hred, Hsred and Hhyd, have now all been observed in both group A and D [FeFe]-hydrogenase, arguably suggests a shared catalytic cycle. In addition, the here observed μCO ligand in Hred, favours a catalytic cycle involving diiron site reduced intermediates. Overall, these findings highlight how studies of [FeFe]-hydrogenases from alternative groups can provide new insight into the catalytic cycle, underscoring the need for continuous exploration of this diverse and fascinating enzyme family.
, Hred, Hsred and Hhyd, have now all been observed in both group A and D [FeFe]-hydrogenase, arguably suggests a shared catalytic cycle. In addition, the here observed μCO ligand in Hred, favours a catalytic cycle involving diiron site reduced intermediates. Overall, these findings highlight how studies of [FeFe]-hydrogenases from alternative groups can provide new insight into the catalytic cycle, underscoring the need for continuous exploration of this diverse and fascinating enzyme family.
Author contributions
M. S. designed experiments, performed the FTIR spectroscopy, analysed data and wrote the first draft of the manuscript. C. S. and P. H. performed EPR spectroscopy and analysed data. A. Z. performed the synthesis work. C. S., P. R. C. and H. L. isolated the proteins. G. B. contributed to the data analysis and manuscript preparation. All authors were involved in the analysis and revision of the manuscript and have given approval to the final version of the manuscript.Data availability
The methods and data supporting the findings of this study are available in the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank Leopold Fichet for his contributions to the FTIR spectroscopy experiments. The work presented in this article is supported by the Novo Nordisk Foundation (NNF23OC0085682 to MS) and by the Swedish Research Council (2023-04593 to MS, 2021-04471 to GB). The Swedish Energy Agency (STEM, grant no 48574-1 to GB), the Olle Engkvist stiftelse (grant no 220-0226 to GB and MS) and the European Union Horizon Europe - the Framework Programme for Research and Innovation (2021–2027) under the grant agreement number 101070948 (project PhotoSynH2 to GB) are gratefully acknowledged for funding.Notes and references
- C. Greening, P. R. Cabotaje, L. E. Valentin Alvarado, P. M. Leung, H. Land, T. Rodrigues-Oliveira, R. I. Ponce-Toledo, M. Senger, M. A. Klamke, M. Milton, R. Lappan, S. Mullen, J. West-Roberts, J. Mao, J. Song, M. Schoelmerich, C. W. Stairs, C. Schleper, R. Grinter, A. Spang, J. F. Banfield and G. Berggren, Minimal and hybrid hydrogenases are active from archaea, Cell, 2024, 187(3357–3372), e3319 Search PubMed.
- H. Land, M. Senger, G. Berggren and S. T. Stripp, Current State of [FeFe]-Hydrogenase Research: Biodiversity and Spectroscopic Investigations, ACS Catal., 2020, 10, 7069–7086 CrossRef CAS.
- N. Chongdar, J. A. Birrell, K. Pawlak, C. Sommer, E. J. Reijerse, O. Rudiger, W. Lubitz and H. Ogata, Unique Spectroscopic Properties of the H-Cluster in a Putative Sensory [FeFe] Hydrogenase, J. Am. Chem. Soc., 2018, 140, 1057–1068 CrossRef CAS PubMed.
- S. T. Stripp, B. R. Duffus, V. Fourmond, C. Leger, S. Leimkuhler, S. Hirota, Y. Hu, A. Jasniewski, H. Ogata and M. W. Ribbe, Second and Outer Coordination Sphere Effects in Nitrogenase, Hydrogenase, Formate Dehydrogenase, and CO Dehydrogenase, Chem. Rev., 2022, 122, 11900–11973 CrossRef CAS PubMed.
- J. T. Kleinhaus, F. Wittkamp, S. Yadav, D. Siegmund and U. P. Apfel, [FeFe]-Hydrogenases: maturation and reactivity of enzymatic systems and overview of biomimetic models, Chem. Soc. Rev., 2021, 50, 1668–1784 RSC.
- T. R. Simmons, G. Berggren, M. Bacchi, M. Fontecave and V. Artero, Mimicking hydrogenases: From biomimetics to artificial enzymes, Coordin. Chem. Rev., 2014, 270, 127–150 CrossRef.
- N. Kornienko, J. Z. Zhang, K. K. Sakimoto, P. Yang and E. Reisner, Interfacing nature's catalytic machinery with synthetic materials for semi-artificial photosynthesis, Nat. Nanotechnol., 2018, 13, 890–899 CrossRef CAS PubMed.
- W. Lubitz, H. Ogata, O. Rudiger and E. Reijerse, Hydrogenases, Chem. Rev., 2014, 114, 4081–4148 CrossRef CAS.
- H. Land, A. Sekretareva, P. Huang, H. J. Redman, B. Nemeth, N. Polidori, L. S. Meszaros, M. Senger, S. T. Stripp and G. Berggren, Characterization of a putative sensory [FeFe]-hydrogenase provides new insight into the role of the active site architecture, Chem. Sci., 2020, 11, 12789–12801 RSC.
- H. Land, P. Ceccaldi, L. S. Meszaros, M. Lorenzi, H. J. Redman, M. Senger, S. T. Stripp and G. Berggren, Discovery of novel [FeFe]-hydrogenases for biocatalytic H(2)-production, Chem. Sci., 2019, 10, 9941–9948 RSC.
- J. Esselborn, N. Muraki, K. Klein, V. Engelbrecht, N. Metzler-Nolte, U. P. Apfel, E. Hofmann, G. Kurisu and T. Happe, A structural view of synthetic cofactor integration into [FeFe]-hydrogenases, Chem. Sci., 2016, 7, 959–968 RSC.
- C. Sommer, A. Adamska-Venkatesh, K. Pawlak, J. A. Birrell, O. Rudiger, E. J. Reijerse and W. Lubitz, Proton Coupled Electronic Rearrangement within the H-Cluster as an Essential Step in the Catalytic Cycle of [FeFe] Hydrogenases, J. Am. Chem. Soc., 2017, 139, 1440–1443 CrossRef CAS PubMed.
- F. Sievers, A. Wilm, D. Dineen, T. J. Gibson, K. Karplus, W. Li, R. Lopez, H. McWilliam, M. Remmert, J. Soding, J. D. Thompson and D. G. Higgins, Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega, Mol. Syst. Biol., 2011, 7, 539 CrossRef PubMed.
- C. Greening, A. Biswas, C. R. Carere, C. J. Jackson, M. C. Taylor, M. B. Stott, G. M. Cook and S. E. Morales, Genomic and metagenomic surveys of hydrogenase distribution indicate H2 is a widely utilised energy source for microbial growth and survival, ISME J., 2016, 10, 761–777 CrossRef CAS PubMed.
- K. Laun, I. Baranova, J. Duan, L. Kertess, F. Wittkamp, U. P. Apfel, T. Happe, M. Senger and S. T. Stripp, Site-selective protonation of the one-electron reduced cofactor in [FeFe]-hydrogenase, Dalton Trans., 2021, 50, 3641–3650 RSC.
- J. A. Birrell, P. Rodríguez-Maciá, E. J. Reijerse, M. A. Martini and W. Lubitz, The catalytic cycle of [FeFe] hydrogenase: A tale of two sites, Coordin. Chem. Rev., 2021, 449, 214191 CrossRef CAS.
- J. H. Artz, O. A. Zadvornyy, D. W. Mulder, S. M. Keable, A. E. Cohen, M. W. Ratzloff, S. G. Williams, B. Ginovska, N. Kumar, J. Song, S. E. McPhillips, C. M. Davidson, A. Y. Lyubimov, N. Pence, G. J. Schut, A. K. Jones, S. M. Soltis, M. W. W. Adams, S. Raugei, P. W. King and J. W. Peters, Tuning Catalytic Bias of Hydrogen Gas Producing Hydrogenases, J. Am. Chem. Soc., 2020, 142, 1227–1235 CrossRef CAS PubMed.
- M. Senger, S. Mebs, J. Duan, O. Shulenina, K. Laun, L. Kertess, F. Wittkamp, U. P. Apfel, T. Happe, M. Winkler, M. Haumann and S. T. Stripp, Protonation/reduction dynamics at the [4Fe-4S] cluster of the hydrogen-forming cofactor in [FeFe]-hydrogenases, Phys. Chem. Chem. Phys., 2018, 20, 3128–3140 RSC.
- W. Roseboom, A. L. De Lacey, V. M. Fernandez, E. C. Hatchikian and S. P. Albracht, The active site of the [FeFe]-hydrogenase from Desulfovibrio desulfuricans. II. Redox properties, light sensitivity and CO-ligand exchange as observed by infrared spectroscopy, J. Biol. Inorg. Chem., 2006, 11, 102–118 CrossRef CAS PubMed.
- J. W. Peters, W. N. Lanzilotta, B. J. Lemon and L. C. Seefeldt, X-ray crystal structure of the Fe-only hydrogenase (CpI) from Clostridium pasteurianum to 1.8 angstrom resolution, Science, 1998, 282, 1853–1858 CrossRef CAS PubMed.
- Y. Nicolet, C. Piras, P. Legrand, C. E. Hatchikian and J. C. Fontecilla-Camps, Desulfovibrio desulfuricans iron hydrogenase: the structure shows unusual coordination to an active site Fe binuclear center, Structure, 1999, 7, 13–23 CrossRef CAS PubMed.
- M. L. Singleton, N. Bhuvanesh, J. H. Reibenspies and M. Y. Darensbourg, Synthetic support of de novo design: sterically bulky [FeFe]-hydrogenase models, Angew. Chem., Int. Ed., 2008, 47, 9492–9495 CrossRef CAS PubMed.
- S. Katz, J. Noth, M. Horch, H. S. Shafaat, T. Happe, P. Hildebrandt and I. Zebger, Vibrational spectroscopy reveals the initial steps of biological hydrogen evolution, Chem. Sci., 2016, 7, 6746–6752 RSC.
- M. Winkler, M. Senger, J. Duan, J. Esselborn, F. Wittkamp, E. Hofmann, U. P. Apfel, S. T. Stripp and T. Happe, Accumulating the hydride state in the catalytic cycle of [FeFe]-hydrogenases, Nat. Commun., 2017, 8, 16115 CrossRef CAS PubMed.
- A. Silakov, C. Kamp, E. Reijerse, T. Happe and W. Lubitz, Spectroelectrochemical characterization of the active site of the [FeFe] hydrogenase HydA1 from Chlamydomonas reinhardtii, Biochemistry, 2009, 48, 7780–7786 CrossRef CAS PubMed.
- M. Senger, T. Kernmayr, M. Lorenzi, H. J. Redman and G. Berggren, Hydride state accumulation in native [FeFe]-hydrogenase with the physiological reductant H(2) supports its catalytic relevance, Chem. Commun., 2022, 58, 7184–7187 RSC.
- L. S. Meszaros, P. Ceccaldi, M. Lorenzi, H. J. Redman, E. Pfitzner, J. Heberle, M. Senger, S. T. Stripp and G. Berggren, Spectroscopic investigations under whole-cell conditions provide new insight into the metal hydride chemistry of [FeFe]-hydrogenase, Chem. Sci., 2020, 11, 4608–4617 RSC.
- D. W. Mulder, M. W. Ratzloff, M. Bruschi, C. Greco, E. Koonce, J. W. Peters and P. W. King, Investigations on the role of proton-coupled electron transfer in hydrogen activation by [FeFe]-hydrogenase, J. Am. Chem. Soc., 2014, 136, 15394–15402 CrossRef CAS PubMed.
- M. Haumann and S. T. Stripp, The Molecular Proceedings of Biological Hydrogen Turnover, Acc. Chem. Res., 2018, 51, 1755–1763 CrossRef CAS PubMed.
- F. Wittkamp, M. Senger, S. T. Stripp and U. P. Apfel, [FeFe]-Hydrogenases: recent developments and future perspectives, Chem. Commun., 2018, 54, 5934–5942 RSC.
- J. W. Sidabras and S. T. Stripp, A personal account on 25 years of scientific literature on [FeFe]-hydrogenase, J. Biol. Inorg. Chem., 2023, 28, 355–378 CrossRef CAS PubMed.
- M. Bruschi, C. Greco, M. Kaukonen, P. Fantucci, U. Ryde and L. De Gioia, Influence of the [2Fe]H subcluster environment on the properties of key intermediates in the catalytic cycle of [FeFe] hydrogenases: hints for the rational design of synthetic catalysts, Angew. Chem., Int. Ed., 2009, 48, 3503–3506 CrossRef CAS PubMed.
- G. Filippi, F. Arrigoni, L. Bertini, L. De Gioia and G. Zampella, DFT dissection of the reduction step in H2 catalytic production by [FeFe]-hydrogenase-inspired models: can the bridging hydride become more reactive than the terminal isomer?, Inorg. Chem., 2015, 54, 9529–9542 CrossRef CAS PubMed.
- S. Mebs, R. Kositzki, J. Duan, L. Kertess, M. Senger, F. Wittkamp, U. P. Apfel, T. Happe, S. T. Stripp, M. Winkler and M. Haumann, Hydrogen and oxygen trapping at the H-cluster of [FeFe]-hydrogenase revealed by site-selective spectroscopy and QM/MM calculations, Biochim. Biophys. Acta, Bioenerg., 2018, 1859, 28–41 CrossRef CAS PubMed.
- S. Mebs, M. Senger, J. Duan, F. Wittkamp, U. P. Apfel, T. Happe, M. Winkler, S. T. Stripp and M. Haumann, Bridging Hydride at Reduced H-Cluster Species in [FeFe]-Hydrogenases Revealed by Infrared Spectroscopy, Isotope Editing, and Quantum Chemistry, J. Am. Chem. Soc., 2017, 139, 12157–12160 CrossRef CAS PubMed.
- P. Chernev, C. Lambertz, N. Leidel, K. Sigfridsson, R. Kositzki, T. Happe and M. Haumann, Hydride binding to the active-site H-cluster of [FeFe]-hydrogenase, J. Biol. Inorg. Chem., 2014, 19, S758 Search PubMed.
- M. W. Ratzloff, J. H. Artz, D. W. Mulder, R. T. Collins, T. E. Furtak and P. W. King, CO-Bridged H-Cluster Intermediates in the Catalytic Mechanism of [FeFe]-Hydrogenase CaI, J. Am. Chem. Soc., 2018, 140, 7623–7628 CrossRef CAS PubMed.
- J. A. Birrell, V. Pelmenschikov, N. Mishra, H. Wang, Y. Yoda, K. Tamasaku, T. B. Rauchfuss, S. P. Cramer, W. Lubitz and S. DeBeer, Spectroscopic and Computational Evidence that [FeFe] Hydrogenases Operate Exclusively with CO-Bridged Intermediates, J. Am. Chem. Soc., 2020, 142, 222–232 CrossRef CAS PubMed.
- C. Lorent, S. Katz, J. Duan, C. J. Kulka, G. Caserta, C. Teutloff, S. Yadav, U. P. Apfel, M. Winkler, T. Happe, M. Horch and I. Zebger, Shedding Light on Proton and Electron Dynamics in [FeFe] Hydrogenases, J. Am. Chem. Soc., 2020, 142, 5493–5497 CrossRef CAS PubMed.
- M. Winkler, J. Esselborn and T. Happe, Molecular basis of [FeFe]-hydrogenase function: an insight into the complex interplay between protein and catalytic cofactor, Biochim. Biophys. Acta, 2013, 1827, 974–985 CrossRef CAS PubMed.
- H. J. Redman, P. Huang, M. Haumann, M. H. Cheah and G. Berggren, Lewis acid protection turns cyanide containing [FeFe]-hydrogenase mimics into proton reduction catalysts, Dalton Trans., 2022, 51, 4634–4643 RSC.
- P. R. Cabotaje, A. Sekretareva, M. Senger, P. Huang, K. Walter, H. J. Redman, N. Croy, S. T. Stripp, H. Land and G. Berggren, Probing the Influence of the Protein Scaffold on H-Cluster Reactivity via Gain-of-Function Studies horizontal line Improved H(2) Evolution and O(2) Tolerance through Rational Design of [FeFe] Hydrogenase, J. Am. Chem. Soc., 2025, 147, 4654–4666 CrossRef CAS PubMed.
- O. Lampret, A. Adamska-Venkatesh, H. Konegger, F. Wittkamp, U. P. Apfel, E. J. Reijerse, W. Lubitz, O. Rudiger, T. Happe and M. Winkler, Interplay between CN(-) Ligands and the Secondary Coordination Sphere of the H-Cluster in [FeFe]-Hydrogenases, J. Am. Chem. Soc., 2017, 139, 18222–18230 CrossRef CAS PubMed.
- J. Duan, M. Senger, J. Esselborn, V. Engelbrecht, F. Wittkamp, U. P. Apfel, E. Hofmann, S. T. Stripp, T. Happe and M. Winkler, Crystallographic and spectroscopic assignment of the proton transfer pathway in [FeFe]-hydrogenases, Nat. Commun., 2018, 9, 4726 CrossRef PubMed.
- M. Senger, V. Eichmann, K. Laun, J. Duan, F. Wittkamp, G. Knor, U. P. Apfel, T. Happe, M. Winkler, J. Heberle and S. T. Stripp, How [FeFe]-Hydrogenase Facilitates Bidirectional Proton Transfer, J. Am. Chem. Soc., 2019, 141, 17394–17403 CrossRef CAS PubMed.
- L. S. Mészáros, H. Land, H. J. Redman and G. Berggren, Semi-synthetic hydrogenases—in vitro and in vivo applications, Curr. Opin. Green Sustainable Chem., 2021, 32, 100521 CrossRef.
- P. R. Cabotaje, K. Walter, A. Zamader, P. Huang, F. Ho, H. Land, M. Senger and G. Berggren, Probing Substrate Transport Effects on Enzymatic Hydrogen Catalysis: An Alternative Proton Transfer Pathway in Putatively Sensory [FeFe] Hydrogenase, ACS Catal., 2023, 13, 10435–10446 CrossRef CAS PubMed.
- N. Chongdar, P. Rodriguez-Macia, E. J. Reijerse, W. Lubitz, H. Ogata and J. A. Birrell, Redox tuning of the H-cluster by second coordination sphere amino acids in the sensory [FeFe] hydrogenase from Thermotoga maritima, Chem. Sci., 2023, 14, 3682–3692 RSC.
- A. Fasano, C. Baffert, C. Schumann, G. Berggren, J. A. Birrell, V. Fourmond and C. Leger, Kinetic Modeling of the Reversible or Irreversible Electrochemical Responses of FeFe-Hydrogenases, J. Am. Chem. Soc., 2024, 146, 1455–1466 CrossRef CAS PubMed.
- A. Fasano, H. Land, V. Fourmond, G. Berggren and C. Leger, Reversible or Irreversible Catalysis of H(+)/H(2) Conversion by FeFe Hydrogenases, J. Am. Chem. Soc., 2021, 143, 20320–20325 CrossRef CAS PubMed.
- F. Vincent, P. Nicolas and L. Christophe, Reversible catalysis, Nat. Rev. Chem., 2021, 5, 348–360 CrossRef PubMed.
- K. Holá, M. V. Pavliuk, B. Németh, P. Huang, L. Zdrazil, H. Land, G. Berggren and H. N. Tian, Carbon Dots and [FeFe] Hydrogenase Biohybrid Assemblies for Efficient Light-Driven Hydrogen Evolution, ACS Catal., 2020, 10, 9943–9952 CrossRef.
- D. Adam, L. Bosche, L. Castaneda-Losada, M. Winkler, U. P. Apfel and T. Happe, Sunlight-Dependent Hydrogen Production by Photosensitizer/Hydrogenase Systems, ChemSusChem, 2017, 10, 894–902 CrossRef CAS PubMed.
- M. V. Pavliuk, M. Lorenzi, D. R. Morado, L. Gedda, S. Wrede, S. H. Mejias, A. Liu, M. Senger, S. Glover, K. Edwards, G. Berggren and H. Tian, Polymer Dots as Photoactive Membrane Vesicles for [FeFe]-Hydrogenase Self-Assembly and Solar-Driven Hydrogen Evolution, J. Am. Chem. Soc., 2022, 144, 13600–13611 CrossRef CAS PubMed.
- M. W. Ratzloff, M. B. Wilker, D. W. Mulder, C. E. Lubner, H. Hamby, K. A. Brown, G. Dukovic and P. W. King, Activation Thermodynamics and H/D Kinetic Isotope Effect of the H(ox) to H(red)H(+) Transition in [FeFe] Hydrogenase, J. Am. Chem. Soc., 2017, 139, 12879–12882 CrossRef CAS PubMed.
- Y. Honda, Y. Shinohara and H. Fujii, Visible light-driven, external mediator-free H2 production by a combination of a photosensitizer and a whole-cell biocatalyst: Escherichia coli expressing [FeFe]-hydrogenase and maturase genes, Catal. Sci. Technol., 2020, 10, 6006–6012 RSC.
- M. Lorenzi, M. T. Gamache, H. J. Redman, H. Land, M. Senger and G. Berggren, Light-Driven [FeFe] Hydrogenase Based H(2) Production in E. coli: A Model Reaction for Exploring E. coli Based Semiartificial Photosynthetic Systems, ACS Sustainable Chem. Eng., 2022, 10, 10760–10767 CrossRef CAS PubMed.
- M. L. K. Sanchez, C. Sommer, E. Reijerse, J. A. Birrell, W. Lubitz and R. B. Dyer, Investigating the Kinetic Competency of CrHydA1 [FeFe] Hydrogenase Intermediate States via Time-Resolved Infrared Spectroscopy, J. Am. Chem. Soc., 2019, 141, 16064–16070 CrossRef CAS PubMed.
- A. Adamska-Venkatesh, D. Krawietz, J. Siebel, K. Weber, T. Happe, E. Reijerse and W. Lubitz, New redox states observed in [FeFe] hydrogenases reveal redox coupling within the H-cluster, J. Am. Chem. Soc., 2014, 136, 11339–11346 CrossRef CAS PubMed.
- M. Senger, K. Laun, F. Wittkamp, J. Duan, M. Haumann, T. Happe, M. Winkler, U. P. Apfel and S. T. Stripp, Proton-Coupled Reduction of the Catalytic [4Fe-4S] Cluster in [FeFe]-Hydrogenases, Angew. Chem., Int. Ed., 2017, 56, 16503–16506 CrossRef CAS PubMed.
- M. Sensi, C. Baffert, V. Fourmond, L. de Gioia, L. Bertini and C. Léger, Photochemistry and photoinhibition of the H-cluster of FeFe hydrogenases, Sustainable Energy Fuels, 2021, 5, 4248–4260 RSC.
- M. Senger, S. Mebs, J. Duan, F. Wittkamp, U. P. Apfel, J. Heberle, M. Haumann and S. T. Stripp, Stepwise isotope editing of [FeFe]-hydrogenases exposes cofactor dynamics, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 8454–8459 CrossRef CAS PubMed.
- J. F. Duan, S. Mebs, K. Laun, F. Wittkamp, J. Heberle, T. Happe, E. Hofmann, U. P. Apfel, M. Winkler, M. Senger, M. Haumann and S. T. Stripp, Geometry of the Catalytic Active Site in [FeFe]-Hydrogenase Is Determined by Hydrogen Bonding and Proton Transfer, ACS Catal., 2019, 9, 9140–9149 CrossRef CAS.
- P. Rodríguez-Maciá, N. Breuer, S. DeBeer and J. A. Birrell, Insight into the Redox Behavior of the [4Fe–4S] Subcluster in [FeFe] Hydrogenases, ACS Catal., 2020, 10, 13084–13095 CrossRef.
- P. Rodriguez-Macia, K. Pawlak, O. Rudiger, E. J. Reijerse, W. Lubitz and J. A. Birrell, Intercluster Redox Coupling Influences Protonation at the H-cluster in [FeFe] Hydrogenases, J. Am. Chem. Soc., 2017, 139, 15122–15134 CrossRef CAS PubMed.
- P. Rodriguez-Macia, E. J. Reijerse, M. van Gastel, S. DeBeer, W. Lubitz, O. Rudiger and J. A. Birrell, Sulfide Protects [FeFe] Hydrogenases From O(2), J. Am. Chem. Soc., 2018, 140, 9346–9350 CrossRef CAS PubMed.
- V. Hajj, C. Baffert, K. Sybirna, I. Meynial-Salles, P. Soucaille, H. Bottin, V. Fourmond and C. Léger, FeFe hydrogenase reductive inactivation and implication for catalysis, Energy Environ. Sci., 2014, 7, 715–719 RSC.
- I. Voloshyn, C. Schumann, P. R. Cabotaje, A. Zamader, H. Land and M. Senger, Secondary structure changes as the potential H(2) sensing mechanism of group D [FeFe]-hydrogenases, Chem. Commun., 2024, 60, 10914–10917 RSC.
- P. Rodriguez-Macia, L. M. Galle, R. Bjornsson, C. Lorent, I. Zebger, Y. Yoda, S. P. Cramer, S. DeBeer, I. Span and J. A. Birrell, Caught in the H(inact): Crystal Structure and Spectroscopy Reveal a Sulfur Bound to the Active Site of an O(2) -stable State of [FeFe] Hydrogenase, Angew. Chem., Int. Ed., 2020, 59, 16786–16794 CrossRef CAS PubMed.
- G. Eilers, L. Schwartz, M. Stein, G. Zampella, L. de Gioia, S. Ott and R. Lomoth, Ligand versus metal protonation of an iron hydrogenase active site mimic, Chemistry, 2007, 13, 7075–7084 CrossRef CAS PubMed.
- J. D. Lawrence, H. Li, T. B. Rauchfuss, M. Benard and M. M. Rohmer, Diiron Azadithiolates as Models for the Iron-Only Hydrogenase Active Site: Synthesis, Structure, and Stereoelectronics This research was supported by the NIH and the Centre Universitaire et Regional de Ressources Informatiques of ULP and CNRS, Angew. Chem., Int. Ed., 2001, 40, 1768–1771 CrossRef CAS PubMed.
- S. T. Stripp, S. Mebs and M. Haumann, Temperature Dependence of Structural Dynamics at the Catalytic Cofactor of [FeFe]-hydrogenase, Inorg. Chem., 2020, 59, 16474–16488 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5cp00412h |
| ‡ Current address: Laboratoire d’Electrochimie Moléculaire (LEM), Université Paris Cité, CNRS, F-75006, Paris, France. |
| This journal is © the Owner Societies 2025 |