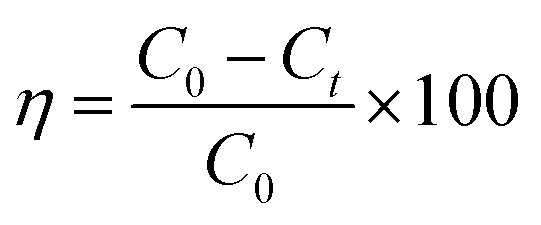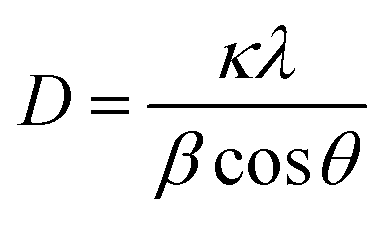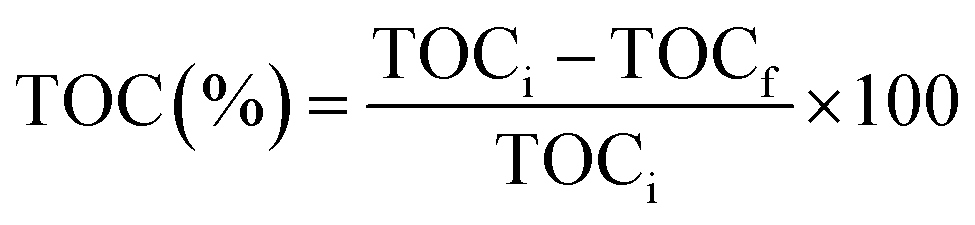DOI:
10.1039/D0RA02062A
(Paper)
RSC Adv., 2020,
10, 20494-20503
Reduced graphene oxide supported ZnO quantum dots for visible light-induced simultaneous removal of tetracycline and hexavalent chromium†
Received
4th March 2020
, Accepted 22nd May 2020
First published on 28th May 2020
Abstract
Semiconducting nanomaterials play an important role in the photocatalytic removal of aqueous pollutants like heavy metals, organic compounds, pathogens and antibiotics. In this study, we prepared ZnO quantum dots (QD) by the precipitation method and ZnO/rGO materials with varying percentages (0.5–2 wt%) of ZnO were prepared by the hydrothermal method. The synthesized catalysts were characterized by various physicochemical techniques, such as powder X-ray diffraction (XRD), Raman spectroscopy, ultraviolet-visible-diffuse reflectance spectroscopy (UV-VIS-DRS), Transmission Electron Microscopy (TEM), Fourier-transform infrared spectroscopy (FT-IR) and Brunauer–Emmett–Teller (BET) analysis to determine the structural as well as textural and surface properties. The photocatalytic activity of the prepared catalysts was analyzed during the individual and simultaneous removal of tetracycline (TC) and hexavalent chromium [Cr(VI)] in aqueous medium. Among all the catalysts, 1.5 wt% ZnO/rGO showed the highest visible light activity, where 68% tetracycline and 84% Cr(VI) abatement was observed after 120 min irradiation time. Moreover, tetracycline showed 70% mineralization. The photocatalytic activity is explained based on the photo-generated electron transfer from the conduction band (CB) of ZnO to the surface of rGO which prevents the recombination of excitons and produces OH˙ and O2−˙; these radicals play an important role in degrading the TC and Cr(VI). The mechanism suggested that the co-existence of oxidizable and reducible species such as TC and Cr(VI) ensured the effective use of the photo-generated electrons and holes that leads to the efficient oxidation of TC and Cr(VI) reduction.
1. Introduction
Industrial wastewater may contain heavy metals, toxic organic compounds, pathogens, etc. and removal of these contaminants is a challenge. The presence of heavy metals in industrial wastewater is dangerous to human health and the environment. In general, antibiotics discharged in water are resistant to biodegradation. There are several antibiotics like sulfonamides, trimethoprim, fluoroquinolones, etc. present in effluent steams.1–3 Tetracycline is one of the antibiotics in wastewater and it is obtained from the fermentation of the bacteria Streptomyces aureofaciens and Streptomyces rimosus with efficient hepatic excretion.4 The presence of tetracycline in water causes renal failure, diarrhea, vomiting and nausea, and teeth yellowing ensues in humans and animals like dogs and cats.5,6
Chromium metal existing as two stable oxidation states such as Cr(VI) and Cr(III). The hexavalent chromium is mainly discharged from the leather industry wastes, paint pigments and easily soluble in water, which creates carcinogenic and mutagenic effects on humans and by inhalation of chromium cause anemia, vertigo, ulcers, gastrointestinal illness and failure in kidneys and liver.7,8 At the same time, the trivalent chromium is useful to human health and it is available in vegetables, yeast, and meat, etc.9
The conventional techniques like reverse osmosis, electrochemical treatment, precipitation, ion exchange, adsorption, chemical oxidation, membrane technology, ultrafiltration, ozonization, and anaerobic biological treatment have been used to treat industrial wastewater.10–15 The main disadvantage of these methods is the production of toxic byproducts, high energy consumption and operating cost16–18 Hence, there is a need to develop methods that are inexpensive, clean, time saving and environmentally benign, such as photocatalysis based advance oxidation process (AOP).
In recent years, various semiconductors like ZnO, TiO2, WO3 etc., have been used as photocatalysts for the removal of tetracycline and Cr(VI).19–23 However, among all, ZnO is has shown good photocatalytic activity for the removal of different pollutants due to its unique properties like non-toxic, high surface energy and electron mobility and large bandgap (3.2 eV).24 The activity of ZnO was highlighted by various researchers for the removal of tetracycline and Cr(VI) under sunlight irradiation.25–27 Being a large bandgap semiconductor, ZnO shows activity only under UV light irradiation. To shift the absorption edge of ZnO into visible region various attempts like either doping with transitional metals/noble metals or creation of heterojunction by combining with small bandgap semiconductors was widely tested.28–33 In addition, carbonaceous materials like g-C3N4, carbon nanotubes, reduced graphene oxide, carbon nanofibers were also combined with ZnO.34–36 Due to the higher conductivity, chemical durability, and specific surface area, rGO is a preferred choice for ZnO to improve its photocatalytic activity. A variety of synthetic approaches like hydrothermal, co-precipitation, combustion synthesis, etc. were followed for the preparation of ZnO/rGO composite and hydrothermal method is known produce crystalline materials with large surface area.37–40
Recently, Ghorbani et al., Haldorai et al., Teh et al., Zhang et al., Pant et al., Xu et al., Darwish et al., and Sandhya et al., synthesized ZnO/rGO composite by different methods and used for photocatalytic water splitting, reduction of CO2 and removal of various organic pollutants and textile dyes.41–48 However, to date, scientists focused on individual removal of pollutants but in this manuscript, our study reports the use of modified ZnO for the simultaneous removal of pollutants. We prepared ZnO and rGO supported ZnO (0.5–2 wt%) catalysts for the individual and simultaneous removal of tetracycline and hexavalent chromium from the wastewater. Also, we characterized all the catalysts by using various physicochemical techniques.
2. Materials and methods
2.1. Materials
The materials used in this project were zinc acetate dihydrate (Sigma-Aldrich), potassium hydroxide (Sigma-Aldrich), tetraethyl orthosilicate (Alfa-Aeser), methanol (Alfa-Aeser), graphite powder (Sigma-Aldrich), sodium nitrate (Sigma-Aldrich), conc. H2SO4 (Alfa-Aeser), potassium permanganate (Sigma-Aldrich), 30% H2O2 (Sigma-Aldrich), tetracycline (Sigma-Aldrich), potassium dichromate (Alfa-Aeser). All materials used without purification and Millipore water used throughout the analysis.
2.2. Synthesis of photocatalysts
2.2.1 Synthesis of ZnO QD. The ZnO QD prepared by the simple chemical precipitation method. Firstly, 0.1 M (0.55 g) of zinc acetate dissolved in 25 mL methanol and after that 1 M (2.8 g) of KOH also dissolved in 50 mL methanol separately. The reaction was carried out at room temperature by dropwise addition of KOH–methanol solution to the zinc solution with constant stirring and maintain the final pH at ∼10 for one hour. On this point, 0.25 mL tetraethyl orthosilicate and 0.5 mL of DI water were added immediately and continued the stirring for the other 3 h. Finally, the obtained white colloidal solution was centrifuged and washed with DI water and methanol several times and dispersed this white suspension in water for further analysis.
2.2.2 Synthesis of ZnO QD/rGO nanocomposite. The various weight percentages of ZnO (0.5 to 2 wt%) on reduced graphene oxide were prepared by the in situ hydrothermal method. To a round bottom flask, we took 50 mg of graphene oxide (GO), 20 mL of DI water and 10 mL of ethanol were added and then sonicated for 30 min. To this mixture, different weight percentages (0.5–2 wt%) of ZnO QDs were added and continued the sonication for 15 min. After the sonication, the reaction was stirred for 3 h at room temperature. The resultant mixture was transferred into the 100 mL Teflon lined autoclave and heated at 120 °C for 12 h. Finally, the autoclave cooled to room temperature and then the resultant solution was centrifuged, washed with water and ethanol several times and dried at 60 °C for overnight and then resultant powders used for further analysis. The various weight percentages of ZnO on rGO named as 0.5 ZnO/rGO (P), 1 ZnO/rGO (Q), 1.5 ZnO/rGO (R) and 2 ZnO/rGO (S) for our identifications.
2.3. Characterization techniques
The phase purity and crystallite sizes of ZnO QD and ZnO/rGO nanocomposites were analyzed by Rigaku Ultima IV powder X-ray diffractometer with Cu-Kα radiation with wavelength 1.54 Å and Cu used as a filter. The applied voltage and applied current are sustained at 40 kV and 40 mA h respectively. The absorption effects of synthesized samples were analyzed by Shimadzu-3600 UV-vis-diffuse reflectance spectrophotometer with BaSO4 as a reference sample. The crystal defects present in the prepared samples were evaluated by Bruker Senterra dispersive Raman microscopy with a laser excitation wavelength of 532 nm. The JEM-2100 transmission electron microscope examined the morphology and average particle sizes of ZnO QD and ZnO/rGO composite with accelerating voltage 200 kV. For recording the images, samples were dispersed in ethanol and coated on a carbon-coated copper grid. The functional groups present in the synthesized materials were detected by Bruker Alpha-E Fourier transformation infrared spectrometer with a resolution of 4 cm−1. The total organic carbon existing in the pollutants was studied by Shimadzu TOC analyzer and surface area of ZnO and ZnO/rGO composite was measured by NOVA 2200e surface analyzer at liquid nitrogen temperature.
2.4. Photocatalytic activity measurements
The visible light activity of synthesized materials was tested by specially designed photoreactors which consist of two non-halogen lamps (24 V, 250 W) from both sides of the reactor. The photoactivity of ZnO and ZnO–rGO composite measured by the simultaneous and individual removal of Cr(VI) and TC, one is undergoing oxidation and other undergoes reduction during the photocatalytic process. For photo activity tests, 50 mg L−1 of catalyst and 50 mL of 20 ppm of tetracycline and Cr(VI) has taken in 100 mL round bottom flask and placed in between two lights by maintaining the distance 10 cm. Before light irradiation, the pollutant with catalyst kept in the dark for 30 min to attain the adsorption–desorption equilibrium between catalyst and pollutant. When started the light irradiation, the sample was collected every 30 min and centrifuged then analyzed by using a UV-visible spectrophotometer. In the UV-visible spectrophotometer, the tetracycline peak attributed to 358 nm. The removal percentage of tetracycline can be observed with a decreasing intensity of the peak at 358 nm. Whereas in the case of Cr(VI) reduction, initially, we made Cr(VI) complex (purple color) via the addition of 1 mL of 10% H2SO4 and 1 mL of 1,5-diphenyl carbazide solutions into the 2 mL of Cr(VI) solution.49 We observed the complex peak at 542 nm in the UV-visible spectrophotometer. The percentage of removal/degradation can calculate using eqn (1).| |
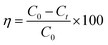 | (1) |
Here, C0 is the concentration at 0 min, Ct is the concentration at time t min, and η is the degradation efficiency of tetracycline and Cr(VI).28,29 The surface area of ZnO QD was 60 m2 g−1 with average pore size 43.1 Å, and surface area of 1.5 ZnO/rGO (R) was 135 m2 g−1 with average pore size 37.9 Å.
3. Results and discussions
3.1. Powder-XRD analysis
The phase purity and crystallite size of ZnO QDs and ZnO/rGO nanocomposites was examined by using powder XRD analysis and results are presented in Fig. 1. ZnO QDs displayed diffraction peaks at d-spacing values of 2.563 Å, 1.566 Å, 1.3548 Å corresponding to the lattice planes (101), (110), (112) and. All the above diffraction peaks of ZnO reliable with the wurtzite phase structure (JC-PDS number: 79-2205). We observed two broad peaks at a 2θ values of 24.9° and 43.5° with lattice planes (002), (100) and d-spacing values of 3.69 Å, 2.10 Å approves the formation of reduced graphene oxide (rGO). In all composites, we detected the diffraction peaks related to ZnO and rGO which indicate the formation of ZnO/rGO composites. The intensity of diffraction peaks of ZnO was very less compared to rGO due to the presence of less amount of ZnO.50–52 The crystallite sizes of ZnO and ZnO/rGO composites were measured by the Debye–Scherer formula exposed in eqn (2):| |
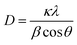 | (2) |
Here, ‘D’ is crystallite size, κ is a unit factor (0.9), λ is the wavelength of X-ray irradiation (1.54 Å), β is full width at half maximum of the peak in radians and θ is the angle of X-ray radiation. The average crystallite size of pristine ZnO was ∼6 nm which indicates the formation of ZnO quantum dot53 and catalysts P–S showed average crystallite sizes 4.5 nm, 4.2 nm, 3.5 nm, and 3.2 nm respectively.
 |
| | Fig. 1 Powder-XRD patterns of ZnO QD and ZnO/rGO composites. | |
3.2. Raman analysis
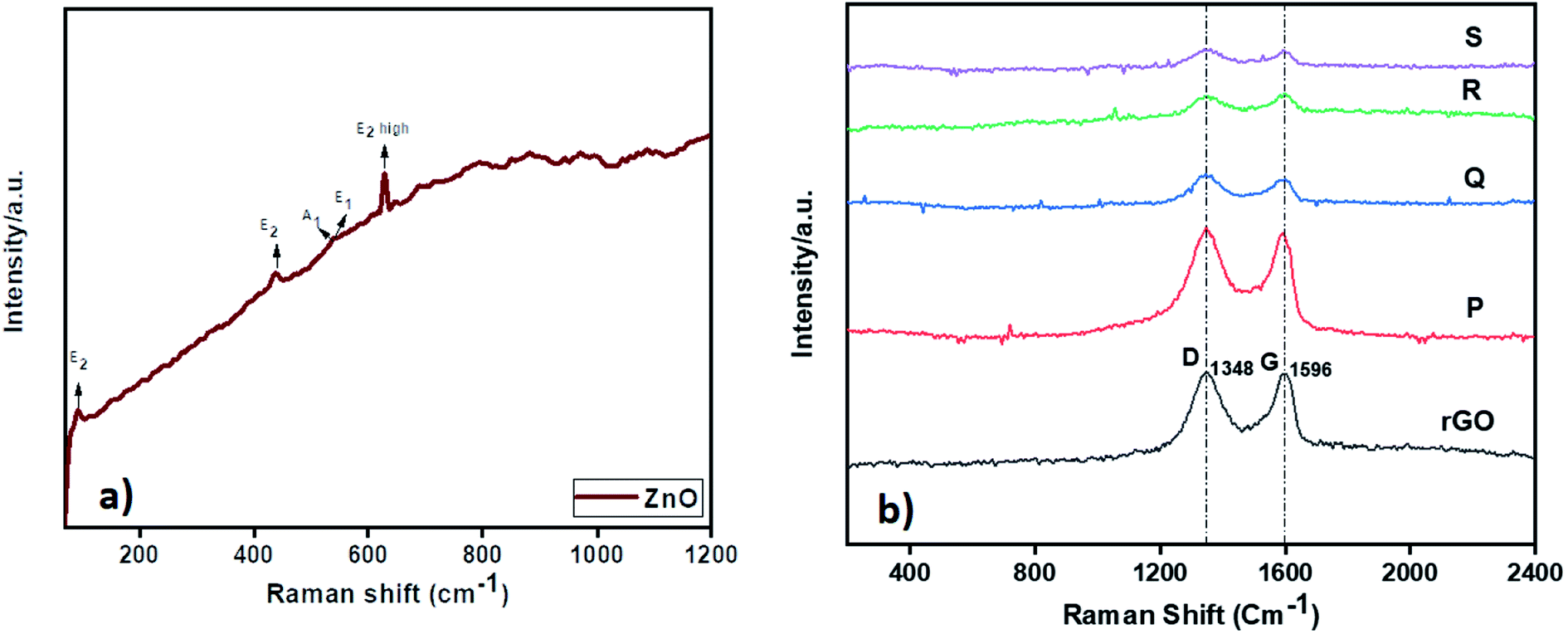
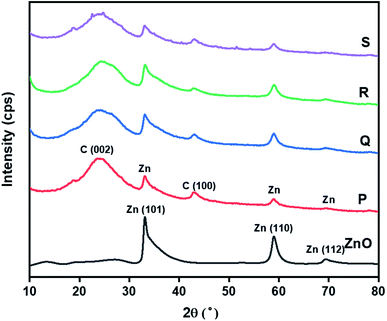
The Raman analysis of ZnO and ZnO–rGO nanocomposite at 532 nm laser excitation is displayed in Fig. 2. From Fig. 2a, ZnO showed prominent peaks at 93, 436, 540, and 630 cm−1 were designates the E2, A1, E1, and E2 (high) vibrational modes of ZnO QD with P63mc symmetry respectively and the peaks between 1000 to 1200 cm−1 correlated to multiphonon process in Raman spectra.28,54 In Fig. 2b, the ZnO–rGO showed two broad peaks at 1348 cm−1 and 1596 cm−1 associated with D band and G band in graphene materials where D band represents to measure the disorders and defects in materials and G band is E2g symmetric stretching of sp2 hybridized C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond in graphene materials. Each composite has solid D and G bands along with very weak peaks of ZnO due to the presence of less amount of ZnO. Compare with ID/IG ratio of rGO i.e., 0.98, ZnO/rGO showed lower ID/IG ratio values55 The decrease in ID/IG ratios indicates the formation of ZnO/rGO composites.
C bond in graphene materials. Each composite has solid D and G bands along with very weak peaks of ZnO due to the presence of less amount of ZnO. Compare with ID/IG ratio of rGO i.e., 0.98, ZnO/rGO showed lower ID/IG ratio values55 The decrease in ID/IG ratios indicates the formation of ZnO/rGO composites.
 |
| | Fig. 2 Raman spectrum of (a) ZnO and (b) ZnO/rGO composites. | |
3.3. UV-VIS-DRS analysis
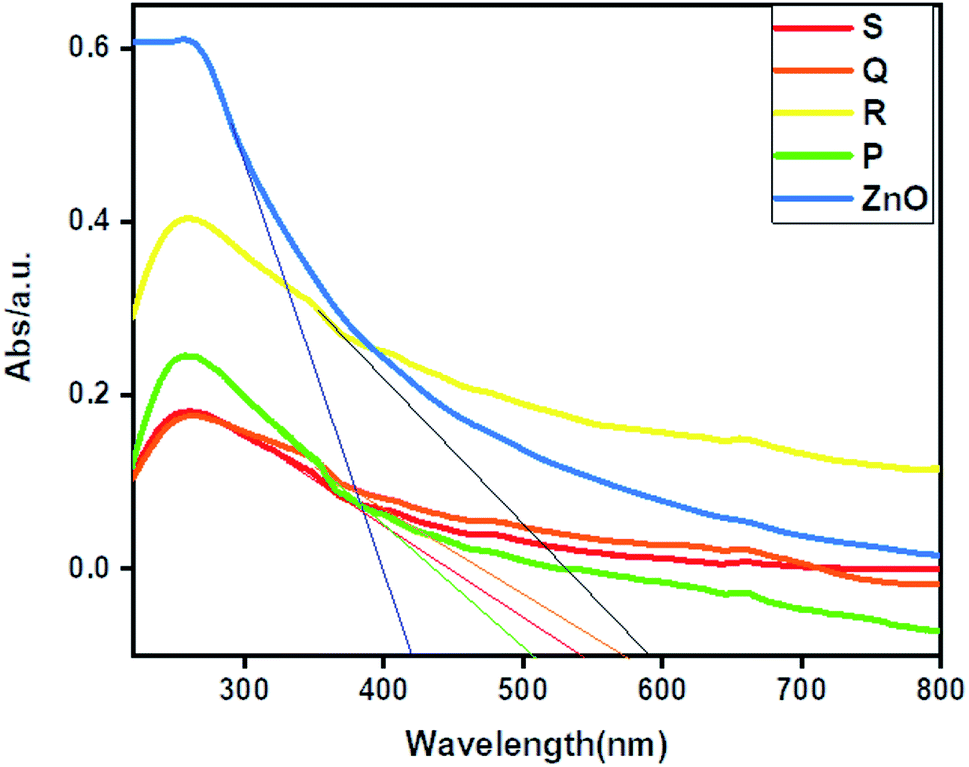
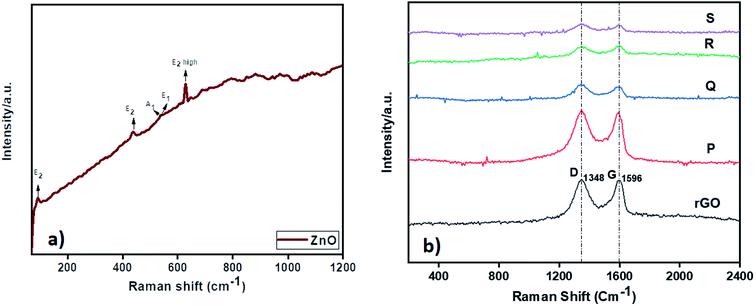
The UV-DRS spectrum of ZnO and ZnO–rGO composites shown in Fig. 3. From Fig. 3, ZnO showed an absorption edge at 420 nm. Whereas ZnO/rGO composites showed absorption edge values at 590 nm for R, 542 nm for S, 570 nm for Q and 504 nm for P.56 Compare with ZnO, composite ZnO/rGO, showed redshift which indicates that there is an interaction between ZnO and rGO.57 We calculated the bandgap energies of ZnO (3 eV) and ZnO/rGO catalysts showed the bandgap values 2.5, 2.3, 2.2 and 2.1 eV respectively for P, S, Q and R photocatalysts.58 From UV-visible spectrum analysis, we confirm the catalyst R has less bandgap energy. Therefore, which (catalyst R) leads to showed higher photocatalytic activity.59,60
 |
| | Fig. 3 UV-VIS-DRS spectrum of ZnO and ZnO/rGO composites. | |
3.4. Transmission electron microscopy analysis
The morphology of ZnO, rGO, and 1.5 ZnO/rGO composite was determined by using TEM analysis and the obtained results are depicted in Fig. 4. From Fig. 4a, dots type morphology observed for pure ZnO with an average particle size of ∼6 nm. Whereas, rGO showed folded sheet morphology (Fig. 4b). From Fig. 4c, the ZnO QD's agglomerated on the rGO sheets. The lattice fringes of ZnO QD's (inset in Fig. 4a) represents the lattice plane (101) with an interplanar distance of 0.23 nm and these results competed with the XRD observations.61,62
 |
| | Fig. 4 TEM images of (a) ZnO, (b) rGO and (c) ZnO/rGO composite (inset: lattice fringes of ZnO). | |
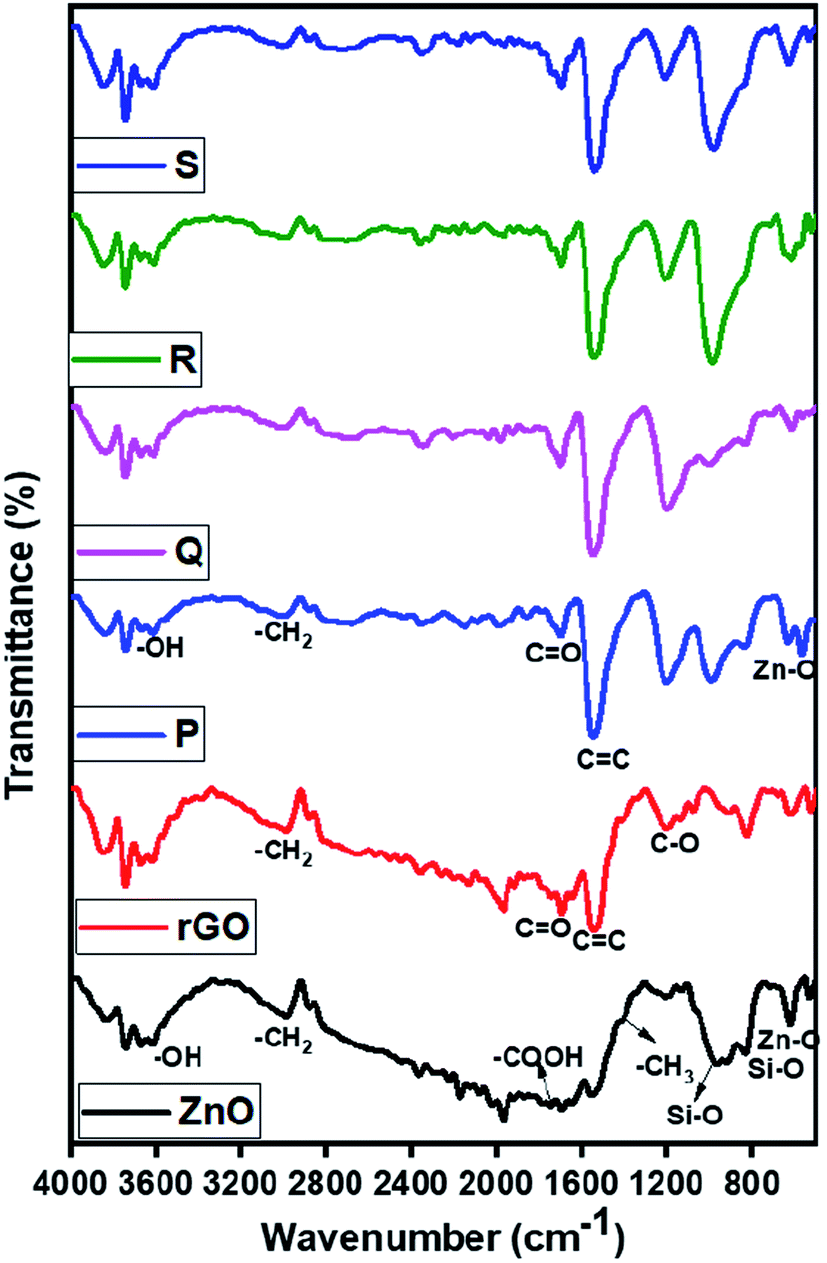
3.5. FTIR analysis
The nature of functional groups presents in the ZnO and ZnO/rGO nanocomposites were evaluated by FTIR analysis and results are showed in Fig. 5. In the ZnO FTIR spectrum, the observed peaks at 620 cm−1 ascribed to Zn–O stretching vibration and peaks at 1400, 1730, 2978 cm−1 were related to bending and stretching vibrations of methyl and carboxyl group (–COO) due to usage of acetate in the synthetic procedure. However, rGO showed a peak at 1545 cm−1 which indicates the presence of the CC functional group and peaks at 1220 cm−1 belongs to the C–OH band and we didn't observe any peak related to –OH it confirmed the formation of rGO after hydrothermal treatment.63 In ZnO/rGO composites, we observed the peaks of both ZnO and rGO indicates the formation of ZnO/rGO nanocomposite. Moreover, when increasing the weight percentages of ZnO, all peaks of ZnO/rGO composites slightly shifted to higher wavenumber side (redshift).64–66
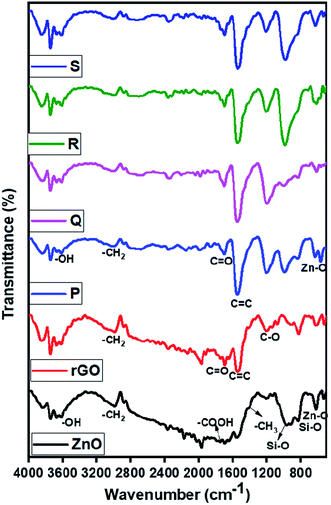 |
| | Fig. 5 FTIR spectrum of ZnO, rGO, and ZnO/rGO composites. | |
3.6. Photocatalytic experiments
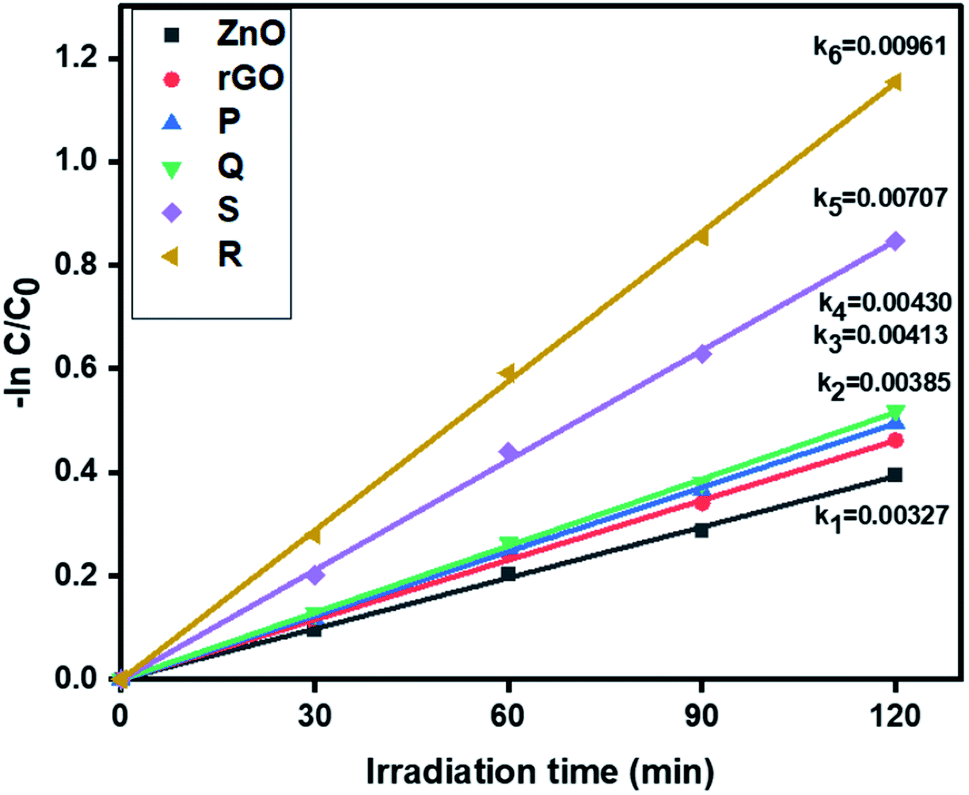
3.6.1. Photocatalytic removal of tetracycline. The photocatalytic activity of ZnO, rGO and ZnO–rGO nanocomposites (P, Q, R, and S) were examined by the removal of antibiotic tetracycline at pH = 5 from wastewater under light irradiation and the outcomes are displayed in Fig. 6. The pseudo-first-order rate constants of ZnO, rGO and ZnO–rGO composite are 0.00327, 0.00385, 0.00413, 0.00430, 0.00707 and 0.00961 min−1 respectively. Among all catalysts, we observed 1.5 ZnO/rGO (R) showed a high rate constant value (0.00961 min−1) and showed higher activity. The treatment time (120 min) and catalyst quantity (50 mg L−1) were the same for the entire reaction. The degradation profiles of tetracycline are shown in Fig. S1.†
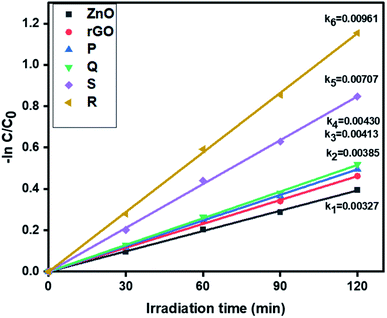 |
| | Fig. 6 The kinetics data of ZnO, rGO and ZnO/rGO composites for tetracycline oxidation (120 min of irradiation time, 50 mg L−1 of catalyst, 20 ppm of TC). | |
Prior to the experiment, under dark conditions, TC and photocatalysts were stirred for 30 min to allow adsorption of the TC on the catalyst. Under visible light irradiation, only 33% TC removal was observed with pure ZnO, whereas, photocatalyst R shows the best removal efficiency within 120 min irradiation. The degradation percentage of tetracycline is shown in Fig. S2† and photocatalyst R shows higher degradation percentage (68%) than remaining ZnO (33%), P (39%), Q (41%), and S (57%). The percentage mineralization of tetracycline was determined by the total organic carbon analyzer (TOC) and TOC percentages were calculated by the eqn (3):
| |
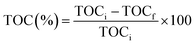 | (3) |
Here, TOC
i is the initial total organic carbon and TOC
f is the final total organic carbon present in the tetracycline antibiotic. Comparatively, catalyst
R showed higher mineralization
i.e., 70%.
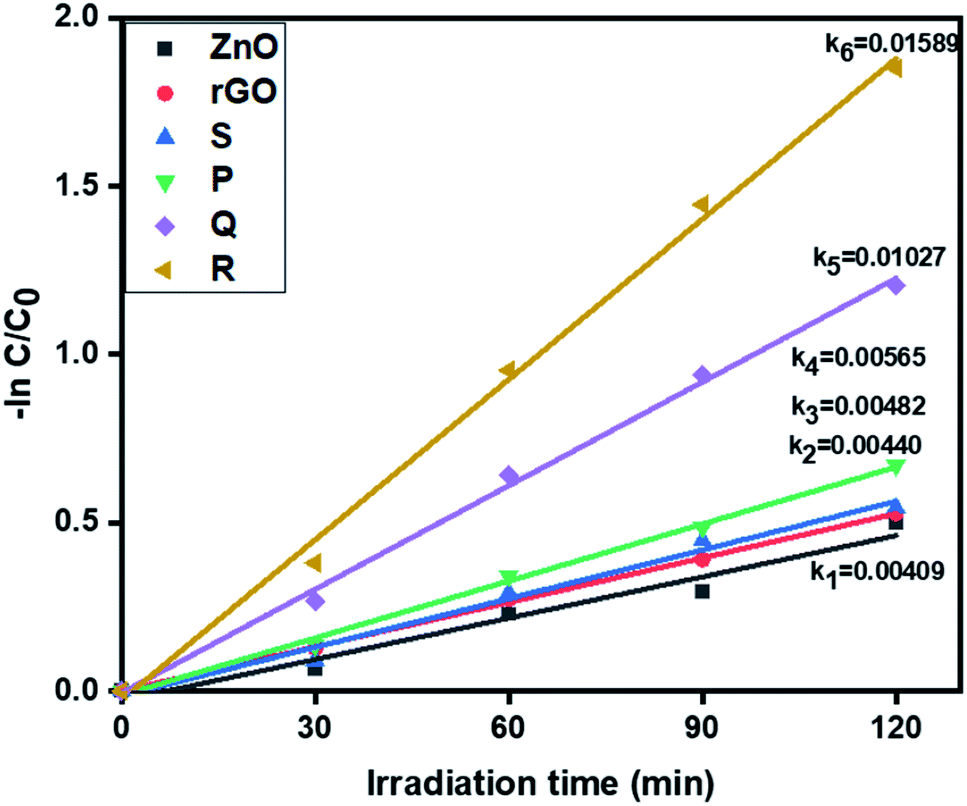
3.6.2. Photocatalytic removal of hexavalent chromium. Further, the visible light activity of ZnO, rGO and ZnO–rGO composites (P, Q, R, and S) were studied by the reduction of hexavalent chromium at pH = 4 and corresponding results are depicted in Fig. 7. From Fig. 7, the catalyst R showed the higher rate constant value (0.01589 min−1) than remaining photocatalysts ZnO (0.00409 min−1), rGO (0.00440 min−1), P (0.00565 min−1), Q (0.01027 min−1) and S (0.00482 min−1).
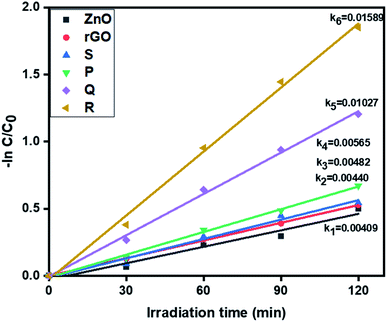 |
| | Fig. 7 The kinetic data of ZnO, rGO and ZnO/rGO composites for Cr(VI) reduction (120 min of irradiation time, 50 mg L−1 of catalyst, 20 ppm of Cr(VI)). | |
The degradation profiles of Cr(VI) reduction are shown in Fig. S3.† Pure ZnO shows very less reduction efficiency of Cr(VI) and it was less compared to ZnO/rGO composites. Also, comparatively, the catalyst R showed the higher Cr(VI) removal i.e. 84%. The conversion percentages of Cr(VI) were revealed in Fig. S4.†
3.6.3. Simultaneous removal of tetracycline and Cr(VI). From the above results, we optimized the catalyst R showed higher activity for the individual removal of tetracycline and Cr(VI). Further, we tried with the optimized catalyst R for the simultaneous removal of tetracycline and Cr(VI). In the experiment, we took 20 ppm Cr(VI) and 20 ppm tetracycline for the simultaneous removal analysis. In this, Cr(VI) improves the oxidizing capacity of tetracycline by tricking the hydrogen radicals in the pollutant solution and similarly, tetracycline traps the hydroxyl radicals and generates more hydrogen radicals for reduction of hexavalent chromium. The pseudo-first-order rate constant of tetracycline and Cr(VI) are 0.01433 min−1 and 0.02693 min−1 respectively and the results showed in Fig. 8. The degradation profiles for the simultaneous removal of TC and Cr(VI) is represented in Fig. S5† which shows a slight absorption under dark condition within 30 min and after 120 min light irradiation the photo-degradation efficiency of the simultaneous removal of pollutants was more than the individual pollutants. Compared to the individual abatement, the simultaneous removal percentages of tetracycline and Cr(VI) by catalyst R showed better activity and the removal percentages are displayed in Fig. S6.†
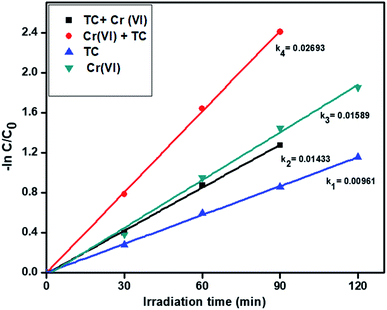 |
| | Fig. 8 The kinetics data for the simultaneous removal of TC oxidation and Cr(VI) reduction (120 min of irradiation time, 50 mg L−1 of catalyst R, 20 ppm of TC, 20 ppm of Cr(VI), pH = 5 of TC, pH = 4 of Cr(VI)). | |
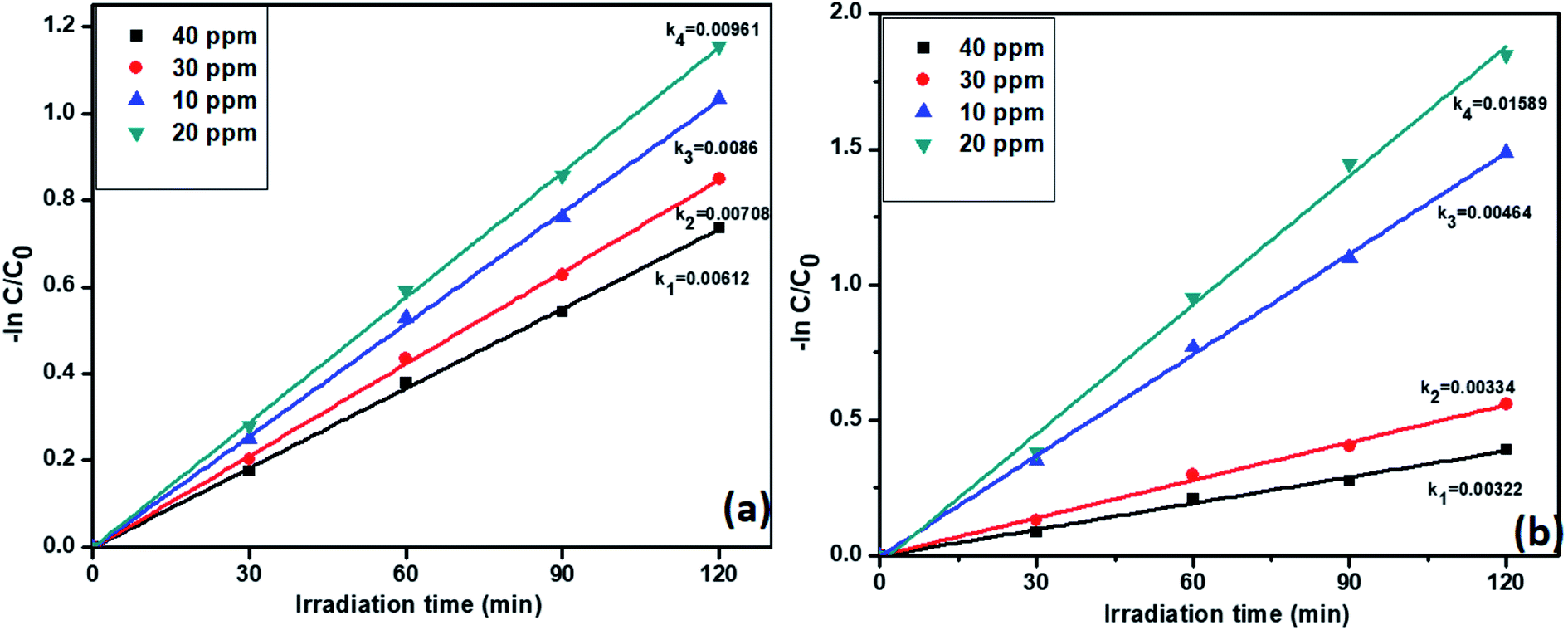
3.6.4. Effect of concentrations of pollutants on catalyst R activity. Previous studies confirmed that the catalyst R showed highest activity for both individual and simultaneous removal of the pollutants. Further, we want to study the effect of pollutants concentration on the catalyst R photocatalytic activity. The degradation profiles during the variation of the concentration of TC and Cr(VI) is displayed in Fig. S7,† which confirmed the decreasing efficiency of the removal of the pollutant with increasing concentration of TC. For this reason, we varied the concentrations of tetracycline and Cr(VI) from 10 to 40 ppm. Notably, the pseudo-first-order rate constants of tetracycline and Cr(VI) were decreased with increasing concentration. However, at 20 ppm, both the pollutants showed high rate constant values 0.00961 and 0.01589 min−1 respectively. The results are displayed in Fig. 9a and b. At higher concentrations i.e., 30 and 40 ppm, catalyst R showed lesser activity due to a large number of pollutant molecules situated on the surface of the catalyst and it condenses the light absorption from the light source and diminishes the generation of electron–hole pairs on the surface of photocatalyst R. From these results we optimized that at 20 ppm of both the pollutants with 50 mg of the catalyst R showed higher activity. The removal percentage of pollutants with the catalyst R showed in Fig. S8†
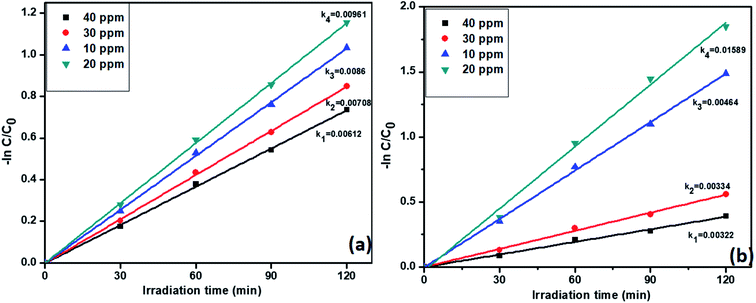 |
| | Fig. 9 The kinetic data for the effect of concentration on (a) TC oxidation and (b) Cr(VI) reduction with photocatalyst R (2 h of irradiation time, 50 mg L−1 of catalyst, pH = 5 of TC, pH = 4 of Cr(VI)). | |
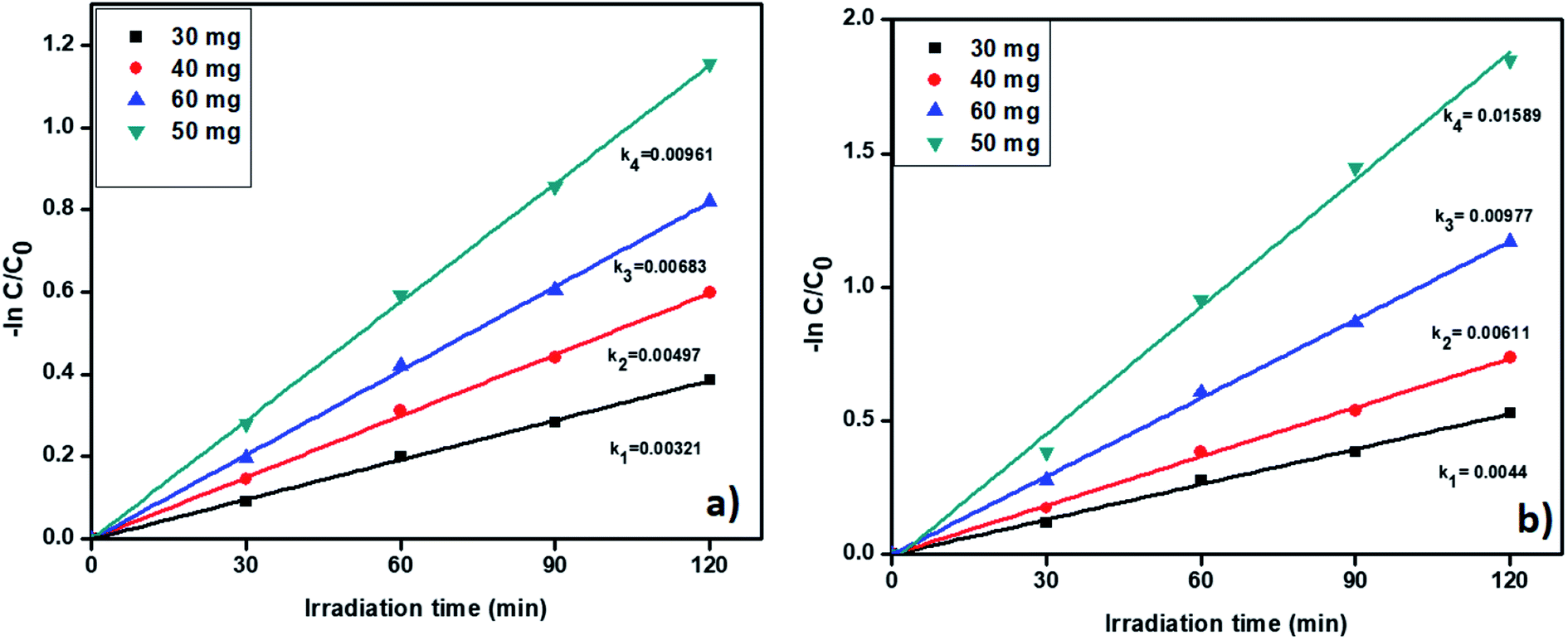
3.6.5. Catalyst amount effect on removal of tetracycline and Cr(VI). Further, we studied the effect of catalyst quantity on the pollutant removal. The degradation profiles of TC and Cr(VI) by changing the catalyst amount is shown in Fig. S9† which confirmed that the degradation efficiency increases with increasing the catalysts amount. The catalyst amount was varied from 30 to 60 mg and corresponding results displayed in Fig. 10a and b. From Fig. 10, the pseudo-first-order rate constants were increased with increasing the catalyst amount up to 50 mg. Whereas, when we used 60 mg of catalyst R, the rate of constant value was decreased. In general, a higher amount of the catalyst provides a large number of active sites. At high catalyst amount a large number of active sites available for removal of pollutants. Nevertheless, while using 60 mg of catalyst R, catalyst particles were agglomerated and develop an obstacle between catalyst surface and pollutant molecules and gradually reduced the activity. As per the results, we confirm the 50 mg of catalyst is showed better activity for the removal of the pollutants. The degradation percentage also increases up to 50 mg and results are included in Fig. S10.†
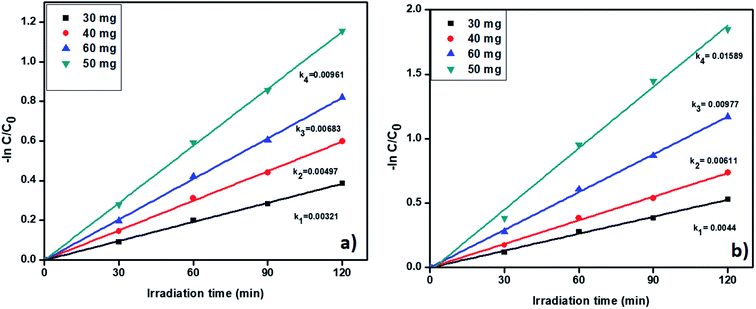 |
| | Fig. 10 The kinetic data for effect of catalyst on (a) TC oxidation and (b) Cr(VI) reduction (2 h of irradiation time, 20 ppm of TC, 20 ppm of Cr(VI), pH = 5 of TC, pH = 4 of Cr(VI)). | |
3.6.6. pH effect on photocatalytic removal of tetracycline and Cr(VI). The pH impacts the removal of tetracycline and Cr(VI) in the wastewater with the photocatalyst ‘R’ and results shown in Fig. 11. The removal of tetracycline was examined by varying the pH values from 3 to 11. The degradation profiles of TC and Cr(VI) at various pH values is shown in Fig. S11,† which confirms that at pH = 11 and pH = 2 the highest removal efficiency of TC and Cr(VI) was obtained. Generally, OH˙ plays a vital role in the oxidation of antibiotics. Therefore, a large amount of OH˙ required to enhanced the tetracycline removal from the wastewater. In neutral and basic conditions, removal percentage of tetracycline showed higher than the acidic condition due to in basic condition presence of more number of OH radicals (OH˙). The results are displayed in the Fig. 11a. Whereas in acidic conditions many H+ ions are available and these ions converted into H˙ after visible light irradiation and it suppresses the OH˙ formation via recombining with OH˙ and forms water. In experiment, at pH = 11 we observed the high pseudo-first-order rate constant (0.01356 min−1) compare to pH = 3, 5, 7 and 9 (0.00502 min−1, 0.00961 min−1, 0.01008 min−1 and 0.01156 min−1). In the same way, the degradation percentage of tetracycline showed higher in basic conditions (80%). Whereas, in acidic conditions, showed only 45% abatement and the results showed in Fig. S12a.† To reduce Cr(VI), a large number of H+ ions are required which are highly available in acidic medium. Whereas in basic medium, the presence of a large number of OH− ions which leads to the formation of Cr(OH)3. Moreover, which forms a layer on the catalyst surface and causes decreasing the catalyst efficiency. Therefore, we observed the highest removal of Cr(VI) in acidic conditions. In this study, we alter the pH values from 2 to 11 and the results showed in Fig. 11b. The experimental results reveal that the highest rate constant (0.0192 min−1) and conversion efficiency (90%) showed at pH = 2. Whereas, in basic conditions showed comparatively lesser conversion. The results are shown in Fig. S12b.†
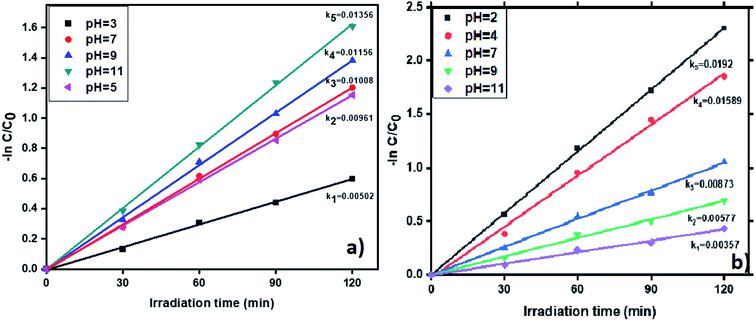 |
| | Fig. 11 The kinetic data for effect of pH on (a) TC and (b) Cr(VI) with photocatalyst R (2 h of irradiation time, 50 mg L−1 of catalyst, 20 ppm of TC, 20 ppm of Cr(VI)). | |
3.7. The photocatalytic mechanism for individual removal of pollutants
The mechanism involved in the deduction of tetracycline and Cr(VI) under visible light irradiation was represented as follows. When the catalyst (ZnO/rGO composite) irradiated with visible light produces a hole (h+) in VB and electron (e−) in CB shown in eqn (4). The photo-excited electron in CB moves on surface of rGO (eqn (5)) to suppress the recombination of excitons and electrons present on rGO further react with adsorbed/dissolved oxygen forms a superoxide radical anion (O2˙−) (eqn (6)) and this radical react with photons (H+) to build a new peroxy radical (HOO˙) further react with electron in CB and H+ to produce a hydrogen peroxide (eqn (7) and (8)). The formed H2O2 attacked by the CB electron forms hydroxyl radicals (OH˙) and hydroxyl ions (OH−) and holes in VB react with adsorbed water and hydroxyl ions yield hydroxyl radical (OH˙) and this radical decomposes the tetracycline molecules and converts into non-toxic products (CO2 & H2O) (eqn (9)–(12)).| | |
ZnO/rGO + hν → h+(VB) + e−(CB)
| (4) |
| | |
e−(CB) + rGO → rGO(e−)
| (5) |
| | |
rGO(e−) + O2(ads) → O2−˙ + rGO
| (6) |
| | |
HOO˙ + e−(CB) + H+ → H2O2
| (8) |
| | |
H2O2 + e−(CB) → OH˙ + OH−
| (9) |
| | |
H2O(ads) + h+(VB) → OH˙ + H+
| (10) |
| | |
h+(VB) + OH−(ads) → OH˙
| (11) |
| | |
OH˙ + tetracycline → degradation products (CO2 + H2O)
| (12) |
Similarly, the mechanism involved in Cr(VI) reduction was explained below: upon light irradiation on catalyst produces an electron–hole pairs (excitons) and these excitons further react with Cr(VI) and water form a Cr(V) and H+ ions and this Cr(V) also reduced to Cr(III) by photoelectron present in CB (eqn (13) and (14)).
| | |
Cr(VI) + e−(CB) → Cr(V) + e−(CB) → Cr(IV) + e−(CB) → Cr(III)
| (13) |
| | |
2H2O + 4h+(VB) → O2 + 4H+
| (14) |
3.8. The photocatalytic mechanism for simultaneous removal of tetracycline and Cr(VI)
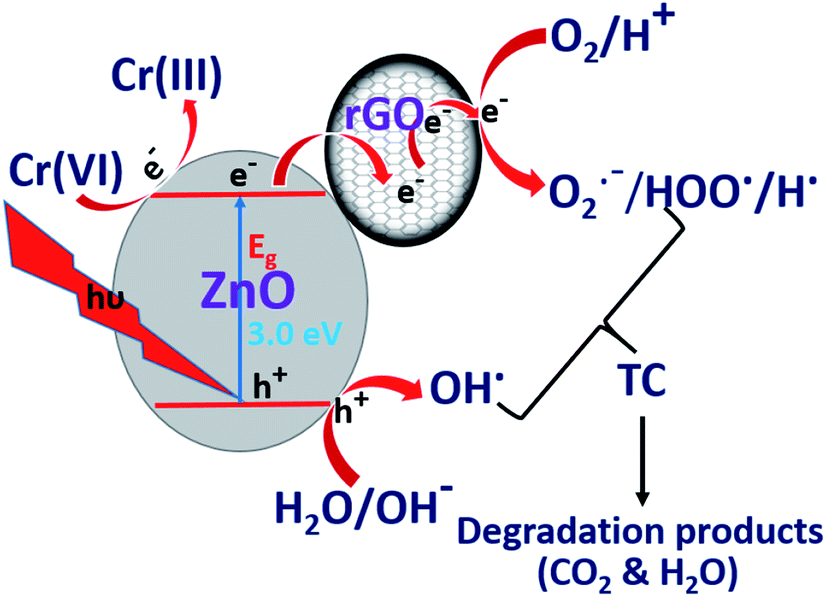
Under light irradiation, ZnO/rGO catalyst generates electrons in the CB and holes in the VB respectively. The electrons in the CB are quickly transferred to rGO and these electrons reduce Cr(VI) to Cr(III), whereas, holes present in the VB produce hydroxyl radicals, which oxidize TC, as shown in eqn (10) and (11). The facile utilization of electrons by Cr(VI) suppresses the recombination of the excitons and holes oxidize tetracycline. This synergetic effect provides better results during the simultaneous removal of the pollutants than for the removal of the individual pollutants. The schematic representation of the mechanism is shown in Fig. 12.67–69
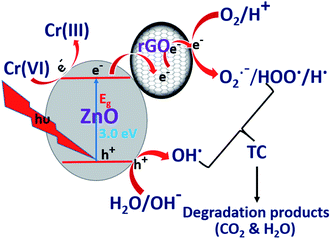 |
| | Fig. 12 Synergetic promotion effect for simultaneous removal of TC and Cr(VI). | |
4. Conclusions
In this context, we synthesized ZnO QDs and ZnO/rGO composites with varying wt% of ZnO (0.5–2 wt%) are prepared by via simple chemical precipitation and hydrothermal methods respectively. Also, we explored these catalysts for the photocatalytic individual and simultaneous removal of antibiotic tetracycline and hexavalent chromium. Among all catalyst, we found the catalyst 1.5 ZnO/rGO (R) showed highest photocatalytic performance for the removal of tetracycline (k = 0.00961 min−1, % of removal = 68%) and Cr(VI) (k = 0.01589 min−1, % of removal = 84%) and mineralization percentage of tetracycline was 70%. Finally, we concluded that both tetracycline and Cr(VI) helped each other for better degradation.
Conflicts of interest
There are no conflicts of interest.
Acknowledgements
KVA is thankful to MHRD-India and Dr B. L. is thankful to UAY/CHY/F019/2017-18/G119 for financial support.
References
- R. H. Lindberg, K. Björklund, P. Rendahl, M. I. Johansson, M. Tysklind and B. A. Andersson, Water Res., 2007, 41, 613 CrossRef CAS PubMed.
- K. G. Karthikeyan and M. T. Meyer, Sci. Total Environ., 2006, 361, 196 CrossRef CAS PubMed.
- A. J. Watkinson, E. J. Murby, D. W. Kolpin and S. D. Costanzo, Sci. Total Environ., 2009, 407, 2711 CrossRef CAS PubMed.
- A. A. Borghi and M. S. A. Palma, Braz. J. Pharm. Sci., 2014, 50, 25 CrossRef CAS.
- K. W. Peden, Gene, 1983, 22, 277 CrossRef CAS PubMed.
- Z. Chi and R. Liu, Biomacromolecules, 2010, 12, 203 CrossRef PubMed.
- M. Tripathi, S. K. Upadhyay, M. Kaur and K. Kaur, J. Biotechnol. Bioeng., 2018, 2, 40 Search PubMed.
- A. Linos, A. Petralias, C. A. Christophi, E. Christoforidou, P. Kouroutou, M. Stoltidis, A. Veloudaki, E. Tzala, K. C. Makris and M. R. Karagas, Environ. Health, 2011, 10, 50 CrossRef CAS PubMed.
- A. Zhitkovich, Chem. Res. Toxicol., 2011, 24, 1617 Search PubMed.
- C. A. Kozlowski and W. Walkowiak, Water Res., 2002, 36, 4870 CrossRef CAS PubMed.
- A. Di Nardo, M. Di Natale, A. Erto, D. Musmarra and I. Bortonea, Comput.-Aided Chem. Eng., 2010, 28, 1015 CAS.
- S. Habibi, A. Nematollahzadeh and S. A. Mousavi, Chem. Eng. J., 2015, 267, 306 CrossRef CAS.
- F. Pomati, A. G. Netting, D. Calamari and B. A. Neilan, Aquat. Toxicol., 2004, 67, 387 CrossRef CAS PubMed.
- A. B. A. Boxall, L. A. Fogg, P. A. Blackwell, P. Blackwell, P. Kay, E. J. Pemberton and A. Croxford, Rev. Environ. Contam. Toxicol., 2004, 180, 1 CrossRef CAS PubMed.
- X. Q. Cheng, C. Zhang, Z. X. Wang and L. Shao, J. Membr. Sci., 2016, 499, 326 CrossRef CAS.
- B. Li and T. Zhang, Environ. Sci. Technol., 2010, 44, 3468 CrossRef CAS PubMed.
- Z. Wang, X. Yu, B. Pan and B. Xing, Environ. Sci. Technol., 2009, 44, 978 CrossRef PubMed.
- R. Ding, P. Zhang, M. Seredych and T. J. Bandosz, Water Res., 2012, 46, 4081 CrossRef CAS PubMed.
- X. D. Zhu, Y. J. Wang, R. J. Sun and D. M. Zhou, Chemosphere, 2013, 92, 925 CrossRef CAS PubMed.
- V. Loryuenyong, N. Jarunsak, T. Chuangchai and A. Buasri, Adv. Mater. Sci. Eng., 2014, 348427 Search PubMed.
- G. Zhang, W. Guan, H. Shen, X. Zhang, W. Fan, C. Lu, H. Bai, L. Xiao, W. Gu and W. Shi, Ind. Eng. Chem. Res., 2014, 53, 5443 CrossRef CAS.
- V. M. Jerin, R. Remya, M. Thomas and J. T. Varkey, Mater. Today: Proc., 2019, 9, 27 Search PubMed.
- G. Zhang, X. Zhang, Y. Wu, W. Shi and W. Guan, Micro Nano Lett., 2013, 8, 177 CrossRef CAS.
- G. H. Munshi, A. M. Ibrahim and L. M. Al-Harbi, Int. J. Photoenergy, 2018, 5094741 Search PubMed.
- J. Choina, A. Bagabas, C. Fischer, G. U. Flechsig, H. Kosslick, A. Alshammari and A. Schulz, Catal. Today, 2015, 241, 47 CrossRef CAS.
- H. Wang, H. Yao, J. Pei, F. Liu and D. Li, Desalin. Water Treat., 2016, 57, 19981 CrossRef CAS.
- Y. Yang, Y. Yang, H. Wu and S. Guo, CrystEngComm, 2013, 15, 2608 RSC.
- K. A. Kumar, S. R. Amanchi, B. Sreedhar, P. Ghosal and C. Subrahmanyam, RSC Adv., 2017, 7, 43030 RSC.
- K. A. Kumar, B. Lakshminarayana, T. Vinodkumar and C. Subrahmanyam, J. Environ. Chem. Eng., 2019, 7, 103057 CrossRef CAS.
- S. Faure, T. Guillet, P. Lefebvre, T. Bretagnon and B. Gil, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 78, 235323 CrossRef.
- S. Hotchandani and P. V. Kamat, J. Phys. Chem., 1992, 96, 6834 CrossRef CAS.
- S. M. Hosseini, I. A. Sarsari, P. Kameli and H. Salamati, J. Alloys Compd., 2015, 640, 408 CrossRef CAS.
- J. J. M. Mesa, J. A. G. Arias, H. A. Rojas and O. E. C. Espinosa, Rev. Fac. Ing., Univ. Antioquia, 2020, 94, 24 Search PubMed.
- P. G. Ramos, E. Flores, C. Luyo, L. A. Sánchez and J. Rodriguez, Mater. Today Commun., 2019, 19, 407 CrossRef CAS.
- M. Salem, S. Akir, I. Massoudi, Y. Litaiem, M. Gaidi and K. Khirouni, J. Alloys Compd., 2018, 767, 982 CrossRef CAS.
- S. Kumar, V. Pandit, K. Bhattacharyya and V. Krishnan, Mater. Chem. Phys., 2018, 214, 364 CrossRef CAS.
- Z. K. Bolaghi, S. M. Masoudpanah and M. Hasheminiasari, Mater. Res. Bull., 2019, 115, 191 CrossRef.
- P. Molaei, M. Cheraghizade and R. Yousefi, Mater. Res. Express, 2018, 6, 025051 CrossRef.
- T. Xu, J. Hu, Y. Yang, W. Que, X. Yin, H. Wu and L. Chen, J. Mater. Sci.: Mater. Electron., 2018, 29, 4888 CrossRef CAS.
- T. T. Chen, I. C. Chang, M. H. Yang, H. T. Chiu and C. Y. Lee, Appl. Catal., B, 2013, 142, 442–449 CrossRef.
- M. Ghorbani, H. Abdizadeh, M. Taheri and M. R. Golobostanfard, Int. J. Hydrogen Energy, 2018, 43, 7754 CrossRef CAS.
- Y. Haldorai and J. J. Shim, Mater. Lett., 2013, 133, 24 CrossRef.
- S. J. Teh, C. W. Lai and S. B. A. Hamid, J. Energy Chem., 2016, 25, 336 CrossRef.
- L. Zhang, N. Li, H. Jiu, G. Qi and Y. Huang, Ceram. Int., 2015, 41, 6256 CrossRef CAS.
- H. R. Pant, C. H. Park, P. Pokharel, L. D. Tijing and C. S. Kim, Powder Technol., 2013, 235, 853 CrossRef CAS.
- S. Xu, L. Fu, T. S. H. Pham, A. Yu, F. Han and L. Chen, Ceram. Int., 2015, 41, 4007 CrossRef CAS.
- M. Darwish, A. Mohammadi, N. Assi, Q. S. Manuchehri, Y. Alahmad and S. Abuzerr, J. Alloys Compd., 2017, 703, 396–406 CrossRef CAS.
- P. K. Sandhya, J. Jose, M. S. Sreekala, M. Padmanabhan, N. Kalarikkal and S. Thomas, Ceram. Int., 2018, 44, 15092 CrossRef CAS.
- L. Chandana, B. Lakshminarayana and C. Subrahmanyam, J. Environ. Chem. Eng., 2015, 3, 2760 CrossRef CAS.
- S. Gayathri, P. Jayabal, M. Kottaisamy and V. Ramakrishnan, J. Appl. Phys., 2014, 115, 173504 CrossRef.
- M. Zubair, M. Mustafa, A. Ali, Y. H. Doh and K. H. Choi, J. Mater. Sci.: Mater. Electron., 2015, 26, 3344 CrossRef CAS.
- X. Zhou, T. Shi and H. Zhou, Appl. Surf. Sci., 2012, 258, 6204 CrossRef CAS.
- V. Muşat, A. Tăbăcaru, B. S. Vasile and V. A. Surdu, RSC Adv., 2014, 4, 63128 RSC.
- D. N. Montenegro, V. Hortelano, O. Martínez, M. C. Martínez-Tomas, V. Sallet, V. Muñoz-Sanjosé and J. Jiménez, J. Phys. D: Appl. Phys., 2013, 46, 235302 CrossRef.
- R. Paul, R. N. Gayen, S. Biswas, S. V. Bhat and R. Bhunia, RSC Adv., 2016, 6, 61661 RSC.
- W. Liu, Y. Li, F. Liu, W. Jiang, D. Zhang and J. Liang, Water Res., 2019, 151, 8 CrossRef CAS PubMed.
- Y. Feng, N. Feng, Y. Wei and G. Zhang, RSC Adv., 2014, 4, 7933 RSC.
- Q. Chen, L. Chen, J. Qi, Y. Tong, Y. Lv, C. Xu, J. Ni and W. Liu, Chin. Chem. Lett., 2019, 30, 1214 CrossRef CAS.
- L. Zhang, N. Li, H. Jiu, G. Qi and Y. Huang, Ceram. Int., 2015, 41, 6256 CrossRef CAS.
- X. Pan, M. Q. Yang and Y. J. Xu, Phys. Chem. Chem. Phys., 2014, 16, 5589 RSC.
- S. K. Mandal, K. Dutta, S. Pal, S. Mandal, A. Naskar, P. K. Pal, T. S. Bhattacharya, A. Singha, R. Saikh, S. De and D. Jana, Mater. Chem. Phys., 2019, 223, 456 CrossRef CAS.
- M. Raja, A. K. Kumar, N. Arora and J. Subha, Fullerenes, Nanotubes, Carbon Nanostruct., 2015, 23, 691 CrossRef CAS.
- Y. Gong, D. Li, Q. Fu and C. Pan, Prog. Nat. Sci.: Mater. Int., 2015, 25, 379 CrossRef CAS.
- M. Strankowski, D. Włodarczyk, L. Piszczyk and J. Strankowska, J. Spectrosc., 2016, 1 Search PubMed.
- K. Ravi, B. Sathish Mohan, G. Satya Sree, I. Manga Raju, K. Basavaiah and B. Venkateswara Rao, Int. J. Chem. Stud., 2018, 6, 20 CAS.
- S. Liu, B. Li, H. Kan, H. Liu, B. Xie, X. Zhu, Y. Hu and S. Jiang, J. Mater. Sci.: Mater. Electron., 2017, 28, 9403 CrossRef CAS.
- W. Liu, J. Ni and X. Yin, Water Res., 2014, 53, 12 CrossRef CAS PubMed.
- W. Liu, W. Sun, A. G. Borthwick, T. Wang, F. Li and Y. Guan, J. Hazard. Mater., 2016, 317, 385 CrossRef CAS PubMed.
- J. Wan, P. Xue, R. Wang, L. Liu, E. Liu, X. Bai, J. Fan and X. Hu, Appl. Surf. Sci., 2019, 483, 677 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra02062a |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a
*a