
Oxidant free conversion of alcohols to nitriles over Ni-based catalysts†
Yunzhu
Wang
 a,
Shinya
Furukawa
bc,
Zhang
Zhang
a,
Laura
Torrente-Murciano
d,
Saif A.
Khan
a and
Ning
Yan
*a
a,
Shinya
Furukawa
bc,
Zhang
Zhang
a,
Laura
Torrente-Murciano
d,
Saif A.
Khan
a and
Ning
Yan
*a
aDepartment of Chemical and Biomolecular Engineering, National University of Singapore, 4 Engineering Drive 4, Singapore 117585, Singapore. E-mail: ning.yan@nus.edu.sg
bInstitute for Catalysis, Hokkaido University, N-21, W-10, Sapporo 001-0021, Japan
cElementary Strategy Initiative for Catalysis and Battery, Kyoto University, Kyoto Daigaku Katsura, Nishikyo-ku, Kyoto, Japan 615-8510
dDepartment of Chemical Engineering and Biotechnology, University of Cambridge, Philippa Fawcett Drive, Cambridge CB3 0AS, UK
First published on 2nd November 2018
Abstract
Organic nitriles are significant and versatile industrial feedstocks, but their conventional synthetic protocols require hazardous starting materials and/or harsh reaction conditions posing environmental and health risks. Herein, we established a Ni-based catalytic system to convert primary alcohols to nitriles with ammonia gas as the sole nitrogen source under oxidant-free conditions at merely 190–230 °C. Based on isotope labelling experiments, in situ DRIFTS and control experiments, the reaction pathway was identified to follow a dehydrogenation–imination–dehydrogenation sequence, with α-carbon C–H bond breakage as the rate determining step. Ni is superior to all noble metal catalysts tested, due to its excellent dehydrogenation ability that is not inhibited by NH3. The support plays an auxiliary role, promoting the reaction between aldehyde and ammonia to form imine as a critical intermediate. Ni/Al2O3 catalyst prepared via a deposition–precipitation method, featuring both excellent dispersion of metallic Ni and suitable acid sites, enabled alcohol transformation into nitrile under unprecedented low temperature. Various alcohols were converted into their corresponding nitriles in high conversions and yields (both up to 99%), while the catalyst kept 90% of its original activity after 48 hours in the stability test, highlighting the wide applicability and the robustness of the catalytic system.
1. Introduction
Organic nitriles find wide applications as significant structural motifs in pharmaceuticals, fine chemicals, biological materials and high performance polymer materials.1,2 Nitriles are conventionally synthesized via the Sandmeyer reaction3 and the Rosenmund–von Braun reaction,4 and more recently via the nucleophilic substitution of cyanides to alkyl and aryl halides.5 These methods suffer from a number of drawbacks including the employment of toxic or hazardous starting materials, generation of large amounts of chemical wastes, and the requirement of harsh reaction conditions. Both metal based cyanides, such as KCN,6 NaCN,7 K4[Fe(CN)6],8 and Zn(CN)2,9 and non-metallic cyanides, such as trimethylsilyl cyanide,10 are highly toxic starting materials. Moreover, stoichiometric waste metal salts are produced, and HCN gas is often released as a side product. These factors raise environmental and health problems, requesting the development of alternative, more sustainable synthetic routes.Alcohol represents a better starting material for organic nitrile compounds that avoid the use of cyanides/halides. However, alcohols are not reactive enough and normally require in situ conversion into ketones/aldehydes prior to further transformation.11–14 Currently, production of nitriles from alcohols is dominated by the ammoxidation reaction, i.e., OH group is oxidized into a carbonyl group before reacting with ammonia.15 Strong oxidants such as iodine,16,17 1,3-diiodo-5,5-dimethylhydantoin,16,18 K2S2O8,19 MnO2,20 and (Bu4N)2S2O8 (ref. 21) have been utilized to produce nitriles from their corresponding alcohols. More recently, catalytic systems based on Cu,22–25 Ru(OH)x,26 Mn and NiO,27 Fe2O3 and Co3O4,28,29 and Pd (ref. 30) catalysts employing oxygen gas as the oxidant have been developed, but over-oxidation is difficult to avoid, and side reactions easily occur for multi-functionalized substrates. In addition, the current ammoxidation systems were mainly applied on aromatic substrates, and only limited cases demonstrated the possibility to transform aliphatic alcohols, in which long reaction time was necessary.22,23,27,28 For example, 26 to 30 hours were required to achieve 65% to 82% nitrile yields from aliphatic alcohols over Co3O4-NGr/C catalyst.28
In fact, the conversion of hydroxyl group to carbonyl group can be achieved through dehydrogenation rather than oxidation, enabling nitrile formation without any oxidant. Metal catalysts based on Cu,31,32 Co–Ni,33 FeS,34 Zn–Cr (ref. 35 and 36) etc. have been investigated to transform alcohol substrates to corresponding nitriles, among which only Cu/m-ZrO2 (ref. 31) and FeS (ref. 34) demonstrated its applicability in more than one substrate while others were only applied in converting one specific starting material. Moreover, the reported catalysts were not efficient, requiring high temperature to function properly (280–500 °C). These conditions result in a high energy consumption, product decomposition and severe catalyst deactivation. For instance, the Zn30Cr4.5/γ-Al2O3 catalyst afforded 61% selectivity towards propionitrile at 420 °C, producing considerable amounts of side products such as 3-picoline.36
Thermodynamically, dehydrogenation of alcohols does not require such a high temperature when the reaction is conducted in a continuous flow reactor where hydrogen is constantly taken away from the system. For example, ethanol underwent dehydrogenation at 200 °C on ZnZrOx supported Au catalyst,37 whereas cyclohexanol dehydrogenation occurred at the same temperature on Cu–MgO–Cr2O3 catalyst.38 The dehydrogenation of amine was also achieved at 200 °C employing a Ru complex.39 Recently, Ni catalysts have been widely used in many catalytic reactions,40 including efficiently catalysing the conversion of alcohols to amines.41–45 However, Ni based heterogeneous catalysts for various nitrile synthesis under mild conditions have not yet been reported. Herein, we developed a low-temperature, oxidant-free system based on a non-noble Ni catalyst to transform a series of aliphatic and aromatic alcohols to corresponding nitriles in the presence of ammonia gas. The reaction pathway was verified with a set of control experiments, reaction kinetics were investigated with deuterated substrates, while reactive surface species were monitored by in situ diffuse reflectance infrared Fourier-transform spectroscopy (DRIFTS) technique. Cleavage of C–H bond at α-carbon in the dehydrogenation of alcohol is the rate determining step in this reaction.
2. Experimental
2.1 Materials
1-Hexanol (99%), tert-butanol (99%), hexanal (98%), hexylamine (99%), 1-butanol (99%), 1-heptanol (98%), 1-octanol (99%), 1-nonanol (98%), cyclopentanemethanol (98%), cyclohexanemethanol (99%), benzyl alcohol (99%), 2-phenylethanol (99%), 3-phenyl-1-propanol (98%), 4-isopropylbenzyl alcohol (97%), tetrahydrofurfuryl alcohol (99%), furfuryl alcohol (98%), 3-pyridinemethanol (98%), 4-nitrobenzyl alcohol (99%), 3-pyridinemethanol (98%), nickel(II) nitrate hexahydrate, silica fumed (powder, 0.2–0.3 um avg. part. size (aggregate)), aluminum oxide (activated, neutral, Brockmann I), calcium silicate (purum, 12–22% Ca (as CaO) basis, ≥87% SiO2 basis), titanium(IV) oxide (nanopowder, 21 nm particle size, ≥99.5% trace metals basis), urea (99%), platinum on carbon (10 wt%), palladium on carbon (10 wt%) were purchased from Sigma-Aldrich Pte. Ltd. (Singapore). Ruthenium on carbon (10 wt%) and rhodium on carbon (10 wt%) were supplied by Shanghai Kaida Chemical Co., Ltd. (China). Deuterated compounds were supplied by Wuhan Livika Technology Co., Ltd. (China). Commercially available organic chemicals, metal precursors, and carbon supported noble metal catalysts were used without further purification. Al2O3-Nanorods and Al2O3-nanoplates were prepared via hydrothermal treatment method.462.2 Catalysts preparation
Ni-Based catalysts were prepared by three methods, including deposition–precipitation (DP) method, wet-impregnation (WI) method and ion-exchange method. DP method was modified from a literature.47 Ni(NO3)2·6H2O (2.1 g) was dissolved in H2O (50 mL), and the solution was divided into two portions, 10 mL and 40 mL, respectively. Into the 40 mL portion, support (1.0 g) was added (mixture A), after which the mixture was stirred and heated at 80 °C. Urea (3.3 g) was added into the 10 mL portion and the solution was added dropwise into mixture A. The final mixture was stirred and heated at 90 °C overnight. The solid was washed with deionized (DI) water and centrifuged for separation, and freeze-dried to obtain dry sample. Al2O3, SiO2 and TiO2 were used as supports, and the as-prepared catalysts were named as Ni/Al2O3-DP, Ni/SiO2-DP and Ni/TiO2-DP, respectively, with metal loading of ca. 20 wt%.Wet-impregnation method was also used to prepare Al2O3, SiO2, and TiO2 supported Ni catalysts. Ni(NO3)2·6H2O (1.2 g) was dissolved in H2O (50 mL), and support (1.0 g) was added to the solution. The mixture was stirred at room temperature overnight. After that, the mixture was freeze-dried. The catalysts based on different supports were named as Ni/Al2O3-WI, Ni/SiO2-WI and Ni/TiO2-WI, respectively, with metal loading of ca. 20 wt%.
Ni/CaSiO3 was prepared by an ion-exchange method, in which Ni2+ exchanged with Ca2+ in the CaSiO3 support.48 Ni(NO3)2·6H2O (1.0 g) was dissolved in DI water (50 mL), in which CaSiO3 (1.0 g) was added. The mixture was stirred at room temperature for 12 hours. After that, the solid part was washed with DI water for three times. The solid was dried in an oven at 90 °C for 12 hours and calcined at 350 °C for 4 hours. The as-prepared catalyst was named as Ni/CaSiO3, with metal loading of ca. 20 wt%.
2.3 General procedure for catalytic reactions
All experiments were conducted in a vertical, fixed-bed tube reactor (SS316), with length of 0.3 m and diameter of 3/8 inch (9.5 mm). A furnace (Yuanbang Furnace, 1 KW, 220 V, up to 1000 °C) was used to heat the reactor and a temperature probe was placed at reactor outer surface. The furnace temperature was controlled by a temperature controller. The catalysts were placed at the middle part of the reactor supported by a sieve and quartz wool. Liquid substrates were pumped into the reactor at the top part by a glass syringe (Hamilton 81620) with a syringe pump (Harvard PHD 2000 Infusion) and solid substrates were dissolved in the solvent before charged into the syringe. Flow rates of ammonia gas and nitrogen gas were controlled by a gas flow meter, and they were mixed before entering the reactor and carrying the substrate through the catalyst bed (Scheme 1).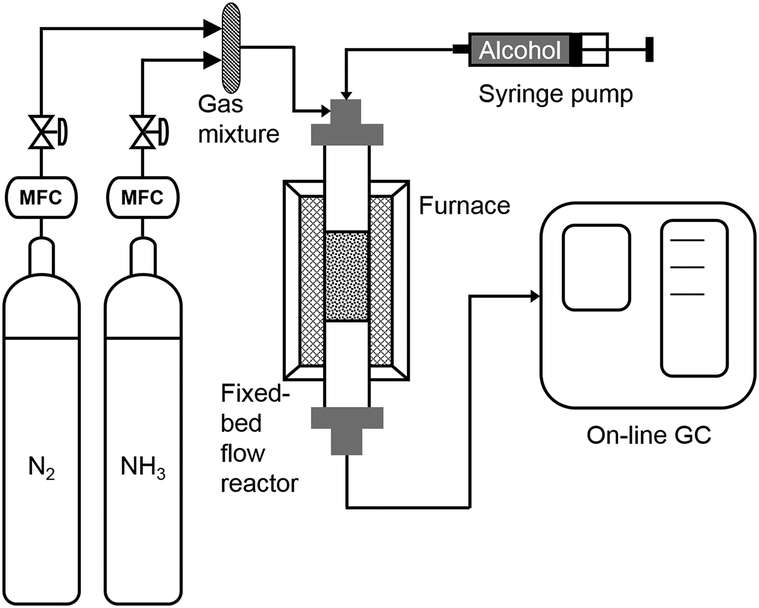 | ||
| Scheme 1 Experimental set-up for the conversion of alcohols to nitriles. | ||
Ni-Based catalysts or noble metal catalysts (equal-mole of metals) were added in the reactor. All the Ni-based catalysts, except Ni/CaSiO3, were pre-reduced with H2 (40 mL min−1) at 460 °C for 1 hour. Ni/CaSiO3 was pre-reduced with H2 (40 mL min−1) at 600 °C for 0.5 hour. All the noble metal catalysts, including Ru/C, Pt/C, Rh/C, and Pd/C, were treated at 190 °C under H2 (40 mL min−1) for 0.5 hour to remove moisture. After reduction, the temperature was decreased to reaction temperature under hydrogen flow followed by a nitrogen purge of the system. In this manner, there will be some hydrides adsorbed on metal surface unless a high-temperature desorption is conducted, since the reaction temperature (130–230 °C) is below the H2 desorption temperature on metallic Ni.49 Then, substrates together with ammonia gas and nitrogen gas were supplied to the reactor at desired flow rates. The products were detected with an online gas chromatography (GC)-flame ionization detector (FID) system equipped with an Agilent HP-5 capillary column with He as the carrier gas. In case off-line GC or GC-mass spectrometry (MS) analysis is needed, the outlet stream was passed through a bottle of ethanol in ice bath to trap the organic products, with 40 μL dodecane as the internal standard.
2.4 Catalyst characterization
Brunauer–Emmett–Teller (BET) surface analysis was carried out by N2 adsorption and desorption at 77 K in a Micromeritics ASAP 2020 surface area and pore size analyzer. H2 chemisorption were conducted over a ChemBET Pulsar TPR/TPD automated chemisorption analyzer. For H2 chemisorption experiments, the catalyst (100 mg) was pre-reduced under H2 at 460 °C for 1 h. The temperature was reduced to 400 °C, and the system was purged with N2 for 1 hour before decreasing to room temperature under N2 flow. After signal stabilization, H2 pulses (159 μL per pulse) were injected every 10 min. The crystallographic information was analysed by X-ray diffraction (XRD, Bruker D8 Advance) equipped with a Cu Kα radiation source. To maintain the metallic state of Ni-based catalysts, passivation was conducted before XRD analysis, using 1% O2/N2 at room temperature for an hour after catalyst reduction. Thermogravimetric (TG) analysis was conducted on a DTG-60A thermogravimetry analyser (Shimadzu) in air atmosphere. Electrospray ionization time-of-flight mass (ESI-TOF-MS) spectra were obtained from a Bruker MicroTOF-Q system. The samples were directly injected into the chamber at 200 μL h−1. Typical instrument parameters: capillary voltage, 4 kV; nebulizer, 0.4 bar; dry gas, 2 L min−1 at 120 °C; m/z range, 50–3000.IR spectroscopy of adsorbed pyridine (py-IR) was performed on a Thermo Fisher Nicolet 5700 IR spectrometer at a resolution of 4 cm−1 with 120 scans in the range of 1000–4000 cm−1 equipped with CaF2 windows. For the adsorption measurements, 20 mg sample was pressed into a self-supporting disc (wafer, diameter of 6.15 mm) and mounted on the sample holder. The samples were activated under vacuum (p = 10−6 mbar) at 450 °C for 1 h. As soon as the sample cooled to 150 °C, a spectrum of the activated sample was taken as the background. Subsequently, adsorption of pyridine was performed at 0.1 mbar for 30 min until saturation/equilibration of the surface was reached and the peak area of the IR signal remained constant. After physisorbed pyridine was removed by evacuation (p = 10−6 mbar) at 150 °C, another spectrum was recorded. Finally, spectra were taken after desorption at 200 °C, 300 °C, and 450 °C for 30 min (p = 10−6 mbar). The IR spectra of adsorbed pyridine were obtained by subtracting the spectrum of the activated sample.
2.5 Fourier transform infrared (FT-IR) spectroscopy experiment
The in situ DRIFTS spectra were recorded with a Thermo Scientific Nicolet iS50 FT-IR, with a liquid nitrogen cooled MCT-A detector with ZnS window. All the experiments were carried out under atmospheric pressure. Catalysts were loaded into the DRIFTS cell and reduced at 460 °C under H2 flow for one hour and then the cell temperature was decreased to the desired one. Ammonia was switched on until it was fully saturated on the surface of catalyst. After that, N2 was turned on to remove the gaseous ammonia as well as physically adsorbed ammonia. Background was taken under N2 atmosphere, and substrate (10 μL) was injected and passed through the catalyst cell. The spectra were recorded by accumulating 32 scans with a resolution of 4 cm−1. Attenuated total reflectance infrared (ATR-IR) spectroscopy analysis was carried out on a Thermo Scientific Nicolet iS50 FT-IR spectrometer integrated with a diamond ATR accessory. The IR spectra were collected in the spectral range 4000–500 cm−1 with a resolution of 4 cm−1 and scan number of 128.3. Results and discussion
Transformation of 1-hexanol into hexanenitrile in the presence of NH3 gas was selected as a model reaction. The reaction is endothermic and its ΔrG value becomes negative at around 200 °C and above (see page S2 in the ESI†). It is a low pressure favoured reaction so when conducted under low alcohol partial pressure, the reaction is thermodynamically favourable at even lower temperature (i.e., 190 °C). In addition to the desired product hexanenitrile, a small amount of 1-hexylamine (<10%) and trace amount of hexene and di-hexylamine (<2%) were observed as the main side products.3.1 Catalysts screening and optimization
A series of catalysts were screened under the same reaction conditions. As presented in Table 1, commercial noble metal catalysts (Ru, Rh, Pt, and Pd, ca. 10 wt%) supported on active carbon did not exhibit good performance (Table 1, entry 1–4), with the highest hexanenitrile yield of 37% on Ru/C (Table 1, entry 2). This suggests the activities of most noble metal catalysts are limited in the presence of ammonia, even though they are widely used in dehydrogenation reactions. Ni-Based catalysts, in sharp contrast, showed significantly higher activity than noble metal catalysts. Nickel supported on SiO2 and Al2O3 by deposition–precipitation (DP) method (Table 1, entry 5 and 6) gave the highest yields of hexanenitrile, reaching 69% and 66%, respectively, superior to the catalysts prepared by wet-impregnation (WI) method (entry 8 and 9). Ni catalysts supported on TiO2 (entry 7 and 10) and CaSiO3 (entry 11) were less active, regardless of preparation methods. A control experiment employing Al2O3 support only was conducted and no activity was observed even at a higher temperature (entry 20), demonstrating the active role of Ni species in the transformation.|
|
||||||
|---|---|---|---|---|---|---|
| Catalyst | Catalyst amount (mg) | GHSV (× 104 h−1) | Conv. (%) | Yield (%) | Selectivity (%) | |
Reaction conditions: 1 μL min−1 1-hexanol, 10 mL min−1 N2, 4 mL min−1 NH3, 190 °C.
a Same mole of metal with nickel in other entries.
b 210 °C.
c 230 °C.
d Mixing catalyst (25 v/v%) with quartz sand (75 v/v%). NH3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1-hexanol = 22:1. :1-hexanol = 22:1.
|
||||||
| 1 | Rh/Ca | — | — | 92 | 23 | 25 |
| 2 | Ru/Ca | — | — | 96 | 37 | 39 |
| 3 | Pt/Ca | — | — | 72 | 14 | 19 |
| 4 | Pd/Ca | — | — | 33 | 13 | 39 |
| 5 | Ni/SiO2-DP | 50 | 2.24 | 94 | 69 | 73 |
| 6 | Ni/Al2O3-DP | 50 | 2.24 | 82 | 66 | 80 |
| 7 | Ni/TiO2-DP | 50 | 2.24 | 41 | 32 | 78 |
| 8 | Ni/SiO2-WI | 50 | 2.24 | 50 | 43 | 86 |
| 9 | Ni/Al2O3-WI | 50 | 2.24 | 31 | 26 | 84 |
| 10 | Ni/TiO2-WI | 50 | 2.24 | 29 | 26 | 90 |
| 11 | Ni/CaSiO3 | 50 | 2.24 | 61 | 51 | 84 |
| 12 | Ni/Al2O3-Plate-DP | 50 | 2.24 | 24 | 21 | 88 |
| 13 | Ni/Al2O3-Rod-DP | 50 | 2.24 | 26 | 11 | 42 |
| 14 | Ni/Al2O3-Plate-WI | 50 | 2.24 | 49 | 11 | 22 |
| 15 | Ni/Al2O3-Rod-WI | 50 | 2.24 | 41 | 12 | 29 |
| 16 | Ni/Al2O3-DP | 100 | 1.12 | 87 | 71 | 82 |
| 17b | Ni/Al2O3-DP | 100 | 1.12 | 97 | 85 | 88 |
| 18c | Ni/Al2O3-DP | 100 | 1.12 | >99 | >99 | >99 |
| 19d | Ni/Al2O3-DP | 100 | 0.281 | 82 | 64 | 78 |
| 20b | Al2O3 | 100 | 1.12 | 0 | 0 | — |

To understand different catalytic activities among various Ni-based catalysts, we first measured metal dispersion by H2 chemisorption, assuming that one surface Ni atom adsorbs one hydrogen atom. Nickel dispersion was 9.9% and 8.3% on Ni/SiO2-DP and Ni/Al2O3-DP, respectively (Table S1†), higher than the catalysts prepared by WI methods. Ni/CaSiO3 also afforded a lower Ni dispersion of 3.1%. The higher dispersion of Ni/SiO2-DP and Ni/Al2O3-DP may result in their higher activity. The chemisorption results are consistent with transmission electron microscopy (TEM) results obtained, where catalysts prepared by DP methods have smaller particle sizes than the others (Fig. S1†). Other Al2O3 supports with different morphology were prepared, including Al2O3-nanoplates and Al2O3-nanorods.46 The same preparation methods, i.e., DP and WI, were utilized to prepare the catalysts. The four catalysts (Table 1, entry 12–15) did not outperform commercial Al2O3, presumably because of the different acid sites produced on Al2O3-nanoplates and Al2O3-nanorods and the presence of Na residue after their base hydrothermal synthesis.50 Three peaks at 44.2°, 51.6°, and 76.2° were observed in the XRD pattern of reduced Ni/Al2O3-DP catalyst (Fig. S2†), ascribable to (111), (200) and (220) crystal phases of Ni nanoparticles respectively,51 while no peak for NiO was observed,52 indicating very small amount of NiO existing, if any, under treatment conditions.
Reaction parameters were further scrutinized using the Ni/Al2O3-DP catalyst. The product yield increased linearly with increased catalyst loading from 15 mg to 50 mg (Fig. S3†). Further increasing the catalyst amount did not lead to a proportional increase of product yield, presumably due to the reaction was approaching equilibrium. When the reaction temperature increased from 150 °C to 230 °C (Fig. S4†), a monotonic increase of hexanenitrile yield from 26% to 82% was observed. This is exactly as expected, because dehydrogenation of the substrate is favored at high temperature both thermodynamically (see page S2 in the ESI†) and kinetically. Decreasing gas hourly space velocity (GHSV) by mixing catalyst with quartz sands did not induce substantial change of reactivity (Table 1, entry 19). With 100 mg catalyst, increased temperature also induced increased activity, and at 230 °C, quantitative formation of hexanenitrile was observed at a GHSV of 1.12 × 104 h−1 (Table 1, entry 16–18), mainly due to the elimination of 1-hexylamine via further dehydrogenation into nitrile product. Close to 100% selectivity can be achieved under optimized conditions, highlighting the feasibility of continuous production of organic nitrile compounds from alcohols without complex post-reaction treatment.
3.2 Reaction pathway
Two reaction pathways from alcohol to nitrile are considered, as shown in Scheme 2. Pathway I follows an SN2 mechanism, where a NH3 molecule directly attacks the α-carbon, inducing simultaneous C–O bond cleavage and C–N bond formation. This leads to the formation of a primary amine, which further undergoes dehydrogenation to produce a nitrile product together with two hydrogen molecules. In pathway II, alcohols undergo dehydrogenation to form aldehydes, and then the aldehydes react with ammonia to produce the imine intermediates.53 Imines undergo either a dehydrogenation step to form nitriles directly, or a hydrogenation step to the amine intermediates. The transformations between imine and amine are reversible so that the selectivity towards nitrile is not compromised. In the latter case, the hydrogen comes from the dehydrogenated substrate, which is reminiscent of the pathway in “hydrogen borrowing mechanism” that is well-established in homogeneous catalysis.11,54,55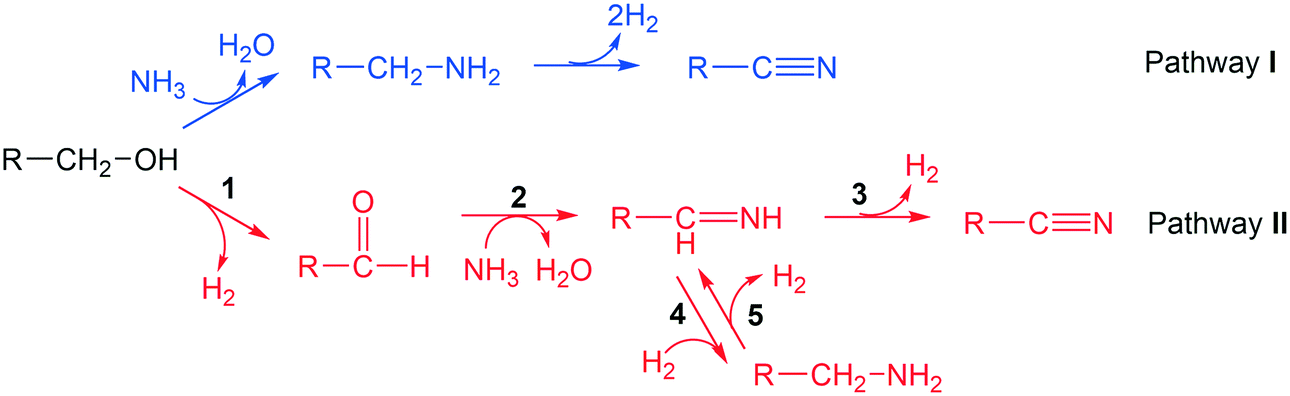 | ||
| Scheme 2 Two possible reaction schemes from alcohols to nitriles. | ||
To gather evidence to identify which pathway is dominant, deuterated butanol (CH3(CH2)2–CD2–OH, 1,1-dideuteriobutan-1-ol) was converted to butylamine with ammonia and hydrogen at 150 °C. A low temperature was chosen and excess hydrogen was supplied to make sure dehydrogenation of butylamine to butanenitrile was unfavoured. If the reaction follows pathway I, the major product should be 1-butan-1,1-d2-amine (CH3(CH2)2–CD2–NH2, m/z = 75), with deuteration of both hydrogen at α-carbon. If the reaction follows pathway II, where dehydrogenation and re-hydrogenation on α-carbon occurs, 1-butan-1-d-amine (CH3(CH2)2–CDH–NH2, m/z = 74) and 1-butylamine (CH3(CH2)2–CH2–NH2, m/z = 73) should be the major products. According to GC-MS spectra (Fig. S5†), the major molecular ion peaks were 73 and 74, corresponding to none- and mono-deuterated butylamine, respectively. This strongly supports that pathway II is dominant in the reaction. A small peak at m/z = 75 was observed, bearing a height of 4.1% compared with the peak at m/z = 74, in excellent agreement with the predicted abundance of 13C isotope peak for CH3(CH2)2–CDH–NH2 (4.4%, considering 4 carbons with 13C abundance of 1.1% each). In another word, no CH3(CH2)2–CD2–NH2 was detected in the product. Therefore, direct substitution of hydroxyl group by NH3 was not occurring at an appreciable level in the reaction system. In addition to this, tert-butanol, which is unable to be dehydrogenated, was also applied as the substrate to identify the validity of pathway I. Under N2 and NH3, 0% conversion was observed at 190 °C and 210 °C on Ni/Al2O3-DP catalyst. No tert-butylamine was observed by GC-MS analysis even with increased temperature at 290 °C, further suggesting that direct amination followed by dehydrogenation was not the major reaction pathway in our system. We have to point out, that while both experiments suggest pathway II is more likely to be the dominant pathway, neither of them are conclusive.
To investigate the validity of pathway II, reaction rate (mmol of substrate converted per gram of catalyst per hour) of each step was evaluated. When ammonia was not supplied (Table 2, entry 1), Ni/Al2O3-DP catalyst afforded aldehyde as the main product with 4% conversion. This observation matches pathway II, which comprises multiple reaction steps starting from alcohol dehydrogenation into an aldehyde. Consequently, reaction intermediates suggested in pathway II were used as the starting materials to further probe its feasibility. Hexanal was highly reactive in the system, reaching much higher conversion (96%) under the same reaction conditions (Table 2, entry 2). Interestingly, 3,5-dibutyl-2-pentyl-pyridine (compound X, Fig. 1(a) and S6†) was detected by GC-MS in the presence of Al2O3 support only, which was formed by one molecule of 1-hexanimine and two molecules of hexanal, due to the instability of imine under reaction conditions. This provided strong evidence for the formation of imine intermediate, as predicted by pathway II.
| Substrate | NH3 (mL min−1) | Ni/Al2O3-DP | Al2O3 | |||
|---|---|---|---|---|---|---|
| Product | Conversion (%) | Reaction rate (mmol g−1 hour−1) | Product | |||
| Reaction conditions: 1 μL min−1 substrate, 20 mg catalyst, 190 °C, 8 mL min−1 NH3 with 20 mL min−1 N2 or 28 mL min−1 N2 if no NH3 was supplied. GHSV = 1.12 × 105 h−1. N.D. = not detected. | ||||||
| 1 | 1-Hexanol | 0 | Hexanal | 4 | 0.98 | N.D. |
| 2 | Hexanal | 8 | Hexanenitrile | 96 | 22 | X |
| 1-Hexylamine | ||||||
| 3 | 1-Hexylamine | 8 | Hexanenitrile | 19 | 4.4 | N.D. |
| 4 | 1-Hexanol | 8 | Hexanenitrile | 14 | 3.1 | N.D. |
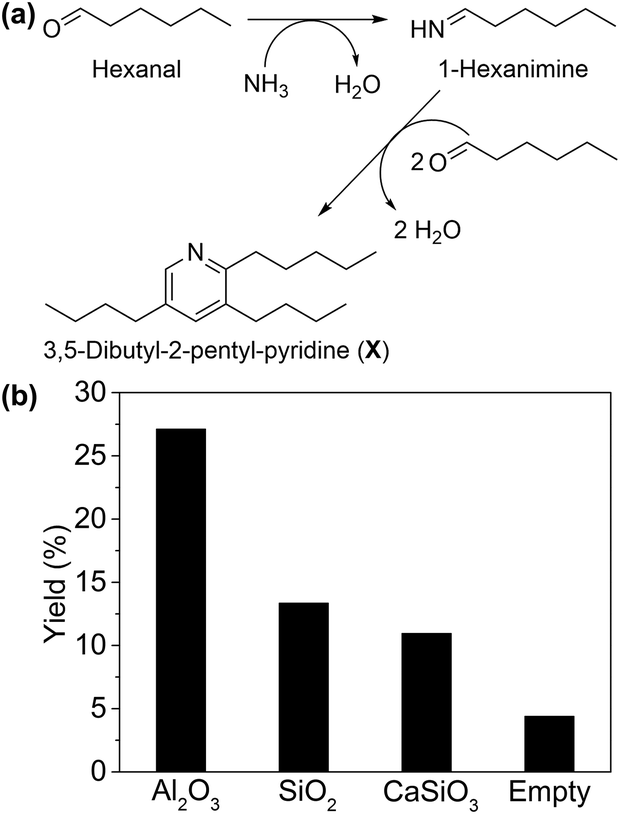 | ||
| Fig. 1 (a) Scheme for formation of 3,5-dibutyl-2-pentyl-pyridine (compound X) and (b) yield of compound X with different supports. Reaction conditions: 1 μL min−1 hexanal, 200 mg catalyst, 190 °C, 8 mL min−1 NH3, 20 mL min−1 N2. GHSV = 1.12 × 104 h−1. | ||
The formation of X from hexanal and ammonia was utilized as a descriptor of the ability of the support to promote imine formation. As shown in Fig. 1(b), Al2O3 presented much higher yield of X than SiO2, CaSiO3 and the blank control, indicating its promotional effect in this step. This is plausibly due to the weak Lewis acid sites (LAS) on Al2O3,56 suggested by Py-IR on Ni/Al2O3-DP (Fig. S7†). The peaks at 1447 cm−1 and 1606 cm−1 were ascribed to the LAS on Ni/Al2O3-DP catalyst,57 which disappeared as temperature increased to 450 °C. No obvious Brønsted acid sites (BAS) were observed on Ni/Al2O3-DP surface.58 Starting with hexanal, both hexanenitrile and 1-hexylamine were produced on Ni/Al2O3-DP catalyst, indicating the hydrogenation and dehydrogenation of imine were catalysed by Ni species. Hexanenitrile was also formed on Ni/Al2O3-DP when 1-hexylamine was injected into the reactor (Table 2, entry 3), suggesting the feasibility of step 5. The dehydrogenation of 1-hexylamine to hexanenitrile was faster than the dehydrogenation of hexanol (step 1), but much slower than step 2. Both dehydrogenation steps and the overall reaction (Table 2, entry 4) were slower than the step 2 in one order of magnitude, indicating the dehydrogenation limited the whole reaction rate.
Combining these control experiments, the following important insights are obtained: 1) the major pathway for nitrile compound formation is an indirect route, following dehydrogenation–imination–dehydrogenation reaction sequence; 2) imination reaction (step 2) is catalysed by the Al2O3 support, whereas other steps are catalysed by metallic Ni; 3) the dehydrogenation from alcohol to aldehyde has the smallest reaction rate, and therefore is likely to be the rate-determining step.
3.3 Reaction kinetics
Since the alcohol is vaporized under reaction temperature, the reaction is approximated as a gas–gas reaction on a solid catalyst surface. Therefore, the pseudo reaction rate can be represented as:| r = k × [alcohol]α × [NH3]β |
The values of α and β were measured for 1-hexanol conversion to hexanenitrile on Ni/Al2O3-DP catalyst. A high GHSV (1.28 × 105 h−1) is used to maintain low conversion of the substrate. When 1-hexanol flow rate was varied while the concentration of ammonia was kept constant, a positive correlation between the reaction rate (mmol of substrate converted per gram of catalyst per hour) and the substrate flow rate was observed.
k × [NH3]β was treated as a constant and α was determined as 0.6 by fitting the experimental data (Fig. 2(a)). Similarly, β was determined by maintaining 1-hexanol flow rate as a constant, while altering ammonia concentration. The reaction order of ammonia was close to 0 (Fig. 2(b)). The experiments were repeated under varied conditions, while the results obtained were essentially the same (Fig. S8†). The kinetic measurements provided further evidence to support pathway II, in which only the alcohol is involved in the rate-determining step.
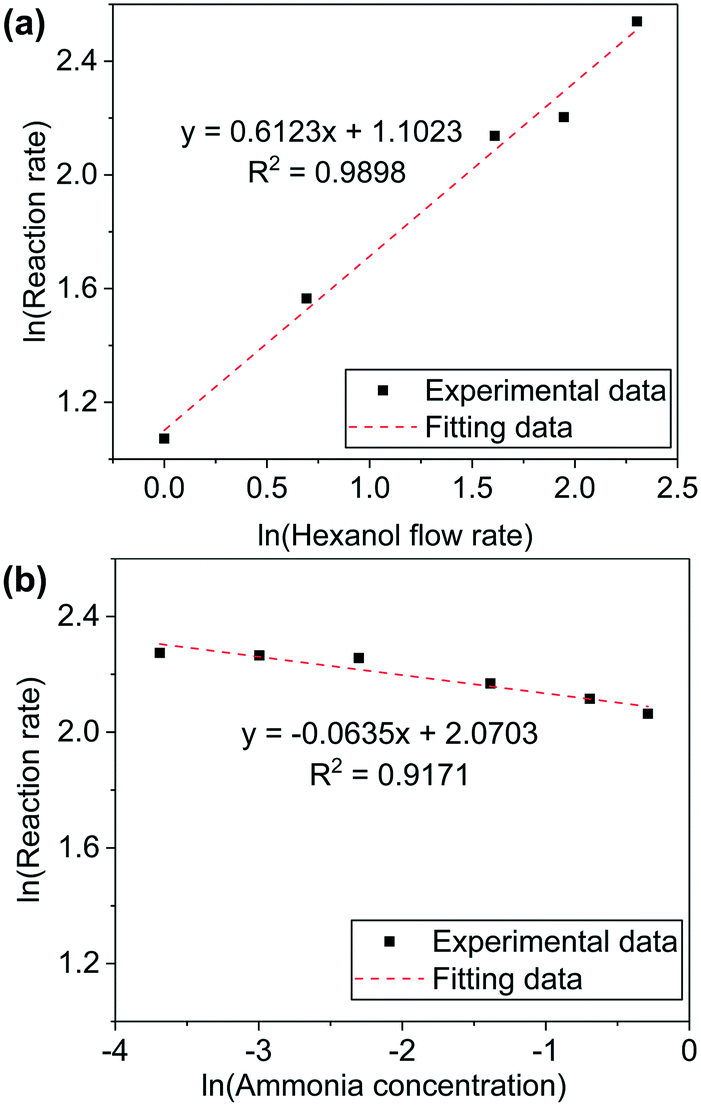 | ||
| Fig. 2 Plots of ln(reaction rate) v.s. (a) ln(hexanol flow rate), hexanol flow rate was in unit of μL min−1 and flow rate of NH3 and N2 were 4 mL min−1 and 76 mL min−1; and (b) ln(ammonia concentration), flow rate of 1-hexanol was 5 μL min−1 and total gas flow rate was 80 mL min−1 by supplementing N2. Reaction rate was calculated as mmol of substrate converted per gram of catalyst per hour. 190 °C, 50 mg Ni/Al2O3-DP catalyst. GHSV = 1.28 × 105 h−1. | ||
We further conducted the density functional theory (DFT) calculations to investigate the adsorption energies of possible species on Ni surface (see page S3 and Fig. S9 in the ESI†). Adsorption on a Ni top site was considered for each adsorbate to avoid any steric repulsion between molecule and surface. However, for unsaturated molecules such as aldehyde, imine, and nitrile, side-on conformations on hollow sites were possible (Fig. S9†), and aldehyde and nitrile were found to prefer side-on conformations. As shown in Table 3, N-containing species, including ammonia, generally showed much more negative adsorption energies than O-contacting species, reflecting the intrinsic azophilic character of Ni. As such, it is plausible that Ni surface is covered by ammonia, while alcohol be adsorbed on the surface of Al2O3 support (also suggested by DRIFTS, Fig. 3, in section 3.4) and the interface between Ni and Al2O3. This gives an explanation of the reaction orders of ammonia and alcohol as observed in the kinetic study.
| Adsorbate | Conformation | E ad (kJ mol−1) |
|---|---|---|
| 1-Hexanol | Top | −20.5 |
| Hexanal | Top | −25.9 |
| Hollow | −34.9 | |
| 1-Hexylamine | Top | −71.7 |
| 1-Hexanimine | Top | −86.9 |
| Hollow | −72.3 | |
| Hexanenitrile | Top | −70.6 |
| Hollow | −97.3 | |
| Ammonia | Top | −75.8 |
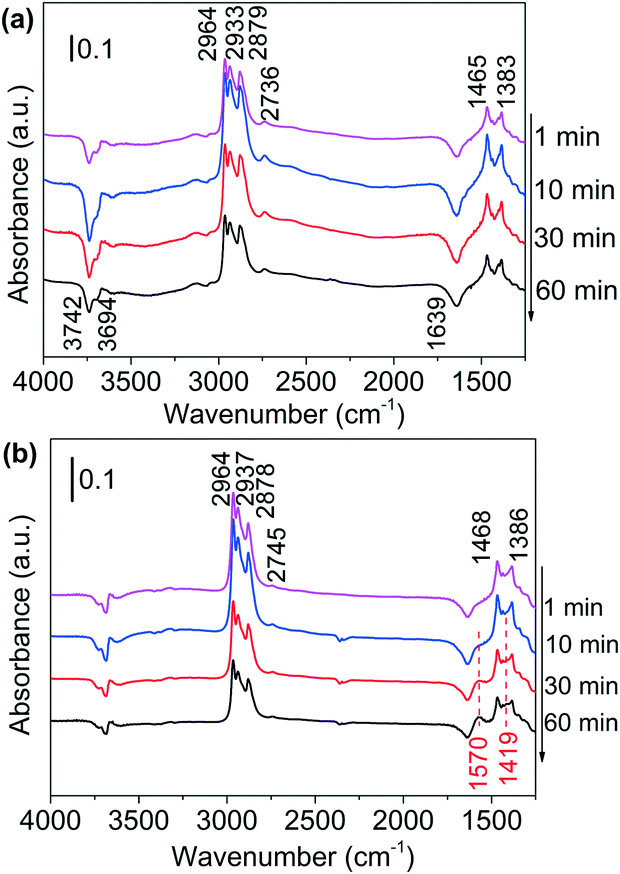 | ||
| Fig. 3 DRIFTS of adsorbed species on (a) Al2O3 surface and (b) Ni/Al2O3-DP surface as a function of time under N2 flow at 130 °C. At t = 0 min, 1-butanol was injected. | ||
In alcohol dehydrogenation, O–H bond cleavage in hydroxyl group and hydride abstraction via α-carbon C–H bond breakage are the two key steps. To investigate the kinetic relevance of each elementary step, isotope labelled substrates were used to test the reaction activity. Undeuterated 1-butanol (CH3(CH2)3–OH), 1-butanol-d (CH3(CH2)3–OD, deuteration at the hydroxyl group of 1-butanol) and CH3(CH2)2–CD2–OH (deuteration of both H at the α-carbon of 1-butanol) were converted under the same reaction conditions at 160 °C. Turnover frequency (TOF, calculated by molar amount of substrate converted per molar amount of surface Ni atom per hour) of CH3(CH2)2–CD2–OH was smaller than undeuterated 1-butanol, affording a normal KIE of 1.52 (Table 4), while no normal KIE was observed with CH3(CH2)3–OD (Table S2†). The results suggest that C–H bond cleavage at α-carbon determines the reaction rate. In the literature, the KIE of dehydrogenation of (CH3)2CH–OH over (CH3)2CD–OH was determined to be 2.0,59 which was higher than our result likely due to the lower reaction temperature in that study (60 °C).
| Alcohol | TOF (h−1) | KIE | |
|---|---|---|---|
| Reaction conditions: 160 °C, 1 μL min−1 substrate, 200 mg Ni/Al2O3-DP, 76 mL min−1 N2, 4 mL min−1 NH3. GHSV = 3.21 × 104 h−1. | |||
| 1 | 1-CH3(CH2)3–OH | 1.88 | |
| 2 | 1-CH3(CH2)2–CD2–OH | 1.24 | |
| 3 | k 1-CH3(CH2)3–OH/k-CH3(CH2)2–CD2–OH | 1.52 | |
3.4 In situ DRIFTS investigation
In situ DRIFTS, an efficient surface species analysis technique,60 is employed to monitor the reaction between alcohols and ammonia over Al2O3 support and supported Ni catalysts. To slow down the reaction and observe possible intermediates, the DRIFTS analysis was conducted at 130 °C. Adsorption of 1-butanol on the Al2O3 support was shown in Fig. 3(a). The four bands between 2964 cm−1 and 2736 cm−1 are attributed to C–H stretching of methyl (CH3–) and methylene (–CH2–) groups, while the bands at 1465 cm−1 and 1383 cm−1 belong to methyl C–H deformation and α-C–H deformation.59 The negative bands at 3742 cm−1, 3694 cm−1 and 1639 cm−1 are due to the consumption of surface O–H groups (Al–OH) of Al2O3 by hydroxyl group of butanol.61–63 The spectra intensity did not change with time, suggesting that Al2O3 could not catalyse the reaction of alcohol. Adsorption of 1-butanol on Ni/Al2O3-DP catalyst was performed at the same temperature (Fig. 3(b)). The six bands indicating C–H stretching and deformation (2964, 2937, 2878, 2745, 1468 and 1386 cm−1) were observed at the same positions with Al2O3, suggesting that butanol was mainly adsorbed on the surface of Al2O3. After 30 minutes, two new bands at 1570 cm−1 and 1419 cm−1, attributed to asymmetric and symmetric vibration of surface-bound carboxylate species (–COO–),64,65 started to increase with time, which was not observed on Al2O3 surface. The surface-bound carboxyl species were plausibly formed by the condensation reaction between aldehyde groups and surface hydroxyl groups (Fig. S10(a)†),62 providing indirect evidence for the formation of aldehydes via dehydrogenation. The observation suggested that Ni species provided the active sites for dehydrogenation of alcohols.DRIFTS were also used to study the adsorption of NH3 on Al2O3 and Ni/Al2O3-DP (Fig. S11†). Ammonia could be adsorbed on both LAS (1626, 1611, 1580 and 1268 cm−1) and BAS (1688 and 1454 cm−1) on the Al2O3 surface.66 With loading of Ni species, the peaks representing adsorbed ammonia on LAS (1626 and 1611 cm−1) maintained, and a new peak at 1219 cm−1, which may be ascribed to ammonia adsorbed on Ni surface,67 appeared. The BAS of Al2O3 were largely suppressed after loading of Ni species, therefore inhibiting undesired dehydration reaction catalysed by BAS. This observation is consistent with the Py-IR results (Fig. S7†).
1-Butanol was subsequently added to the NH3 saturated catalysts. On NH3 saturated Al2O3, the adsorption bands of butanol did not change with time, suggesting Al2O3 could not initiate the reaction between alcohol and ammonia (Fig. S12(a)†). As the alcohol was injected to the NH3 saturated Ni/Al2O3-DP (Fig. 4(a)), the two bands assigned to adsorbed ammonia at 1219 cm−1 and 1626 cm−1 started to decrease, indicating consumption of ammonia. The new peaks at 1606 cm−1 and 1097 cm−1 which could be ascribed as the N–H deformation and C–N stretching in amine species (Fig. S10(b)†),68 appeared quickly after 1 min, which further increased and then decreased with time. This observation confirmed the existence of step 4 in proposed pathway II. Within the first 10 min, no peak relevant to aldehyde was observed, indicating the dehydrogenation of alcohol was slow (rate-determining step) and the produced aldehyde reacted immediately with ammonia. After 20 min, the peaks for vibration of surface-bound carboxylate species (1562 cm−1 and 1411 cm−1) started to grow, probably because the amine species can condense on the catalyst surface as well (Fig. S10(b)†).
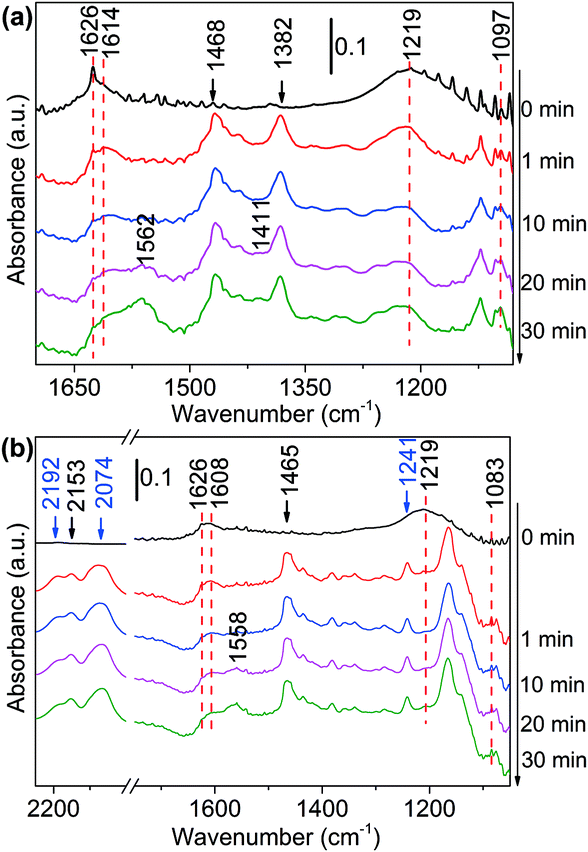 | ||
| Fig. 4 DRIFTS of adsorbed species on NH3 saturated Ni/Al2O3-DP surface as a function of time under N2 flow at 130 °C. At t = 0 min, (a) 1-butanol and (b) 1,1-dideuteriobutan-1-ol was injected. | ||
The deuterated substrates were also studied via in situ DRIFTS. Fig. 4(b) presented the reaction of CH3(CH2)2–CD2–OH with ammonia on the surface of Ni/Al2O3-DP. The two peaks at 2192 cm−1 and 2074 cm−1 were ascribed to α-C–D stretching, the peak at 1241 cm−1 represented α-C–D deformation.59 The cleavage of α-C–D bond was relatively slow, indicating this step determined the overall reaction rate. As the peak at 2192 cm−1 decreased, a new peak at 2153 cm−1 increased accordingly, which might be ascribed to vibration of C–D bond in deuterated imine/amine group. In addition, the peak assigned to ammonia adsorbed on Ni at 1219 cm−1 disappeared rapidly at 1 min. This is probably due to the proton exchange between ammonia and deuterium from the substrate, and thus the peak for deuterated ammonia moved to a lower wavenumber. This was also observed with CH3(CH2)3–OD adsorbed on ammonia saturated Ni/Al2O3-DP surface (Fig. S12(b)†). Similarly, the bands for amine (1606 cm−1 and 1083 cm−1) and carboxylate species (1558 cm−1 and 1417 cm−1) were observed. In Fig. S12(b),† the broad peak at 2589 cm−1 represented the vibration of deuterated O–D bond, and its rapid decrease suggested fast cleavage of O–D bond of hydroxyl group. With deuteration at the hydroxyl group, the reaction rate was fast that only carboxyl species (1566 cm−1) were observed.
Finally, the adsorption of the product (using butanenitrile as an example) has been conducted on Ni/Al2O3-DP. At merely 100 °C, the intensity of C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N peak at 2250 cm−1 decreased with time, suggesting either desorption or conversion of butanenitrile under mild condition (Fig. 5).69 This explains why the characteristic peak for CN was not observed in in situ DRIFTS analysis at 130 °C. The two peaks at 1665 cm−1 and 1608 cm−1 could be assigned to C
N peak at 2250 cm−1 decreased with time, suggesting either desorption or conversion of butanenitrile under mild condition (Fig. 5).69 This explains why the characteristic peak for CN was not observed in in situ DRIFTS analysis at 130 °C. The two peaks at 1665 cm−1 and 1608 cm−1 could be assigned to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N stretching and N–H bending.70 The hydrogenation of nitrile to imine was observed probably because the hydride adsorbed during catalyst reduction step or produced in dehydrogenation step remained on the catalyst was added to the nitrile group.
N stretching and N–H bending.70 The hydrogenation of nitrile to imine was observed probably because the hydride adsorbed during catalyst reduction step or produced in dehydrogenation step remained on the catalyst was added to the nitrile group.
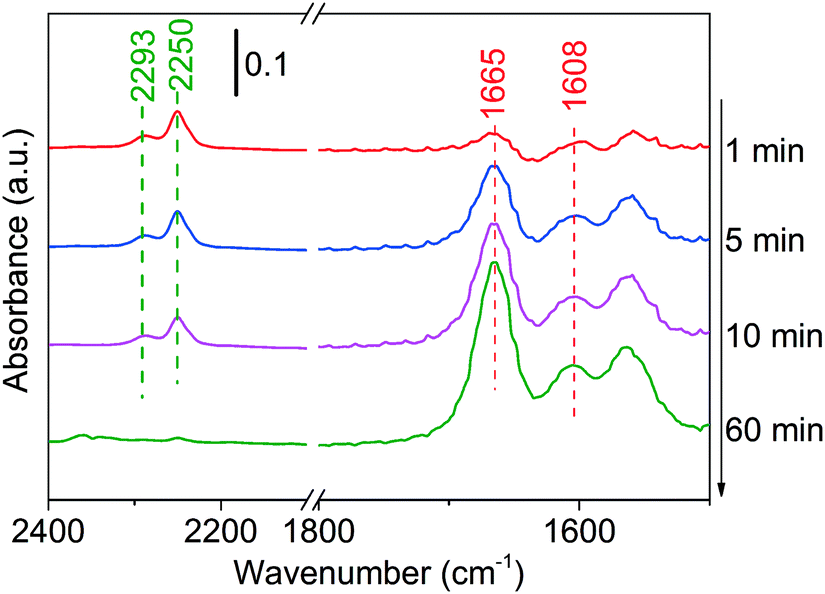 | ||
| Fig. 5 DRIFTS of adsorbed butanenitrile on Ni/Al2O3-DP surface as a function of time under N2 flow at 100 °C. At t = 0 min, butanenitrile was injected. | ||
3.5 Substrate scope and catalyst stability
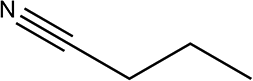
The Ni/Al2O3-DP catalyst is able to effectively convert a series of alcohols to nitriles. Straight-chain aliphatic alcohols gave nitriles as the only detectable products, regardless of carbon chain length (Table 5, entry 1–4). Cyclo-aliphatic alcohols, such as cyclohexanemethanol and cyclopentanemethanol (entry 5 and 6), were also easily converted into corresponding nitriles with high yield (86% and 95%) and excellent selectivity (93% and 99%). Benzyl alcohol and substituted benzyl alcohol were effectively converted to aromatic nitriles (entries 7–8), while 3-phenyl-1-propanol (entry 9) afforded moderate yield (68%). 2-Phenylethanol and other primary alcohols with heterocyclic rings, such as 3-pyridinemethanol, tetrahydrofurfural alcohol, and furfuryl alcohol (Table S3, entry 1–4†) were tested but lower yields were obtained. 4-Nitrobenzyl alcohol was transformed to 4-aminobenzonitrile, instead of 4-nitrobenzonitrile (Table S3, entry 5†), since the nitro group was hydrogenated on Ni catalyst.|
|
||||
|---|---|---|---|---|
| Entry | Substrate | Product | Temperature (°C) | Yield/selectivity (%) |
| Reaction conditions: 1 μL min−1 substrate, 200 mg Ni/Al2O3-DP, 20 mL min−1 N2, 8 mL min−1 NH3. a 0.5 μL min−1 substrate. GHSV = 1.12 × 104 h−1. | ||||
| 1 |
|
|
230 | 86/89 |
| 2a |

|
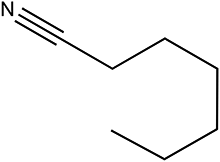
|
210 | 86/86 |
| 3a |
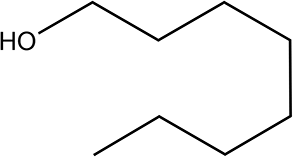
|
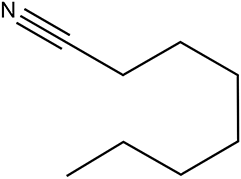
|
210 | 70/82 |
| 4a |
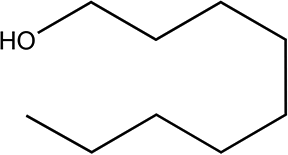
|
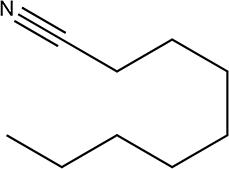
|
210 | 79/79 |
| 5a |
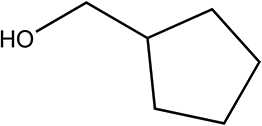
|
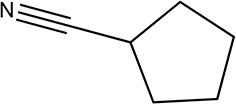
|
210 | 86/93 |
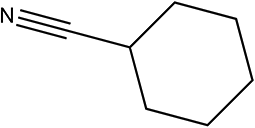
| 6a |
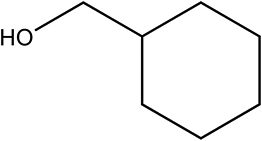
|
|
210 | 95/99 |
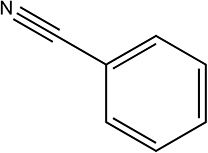
| 7a |
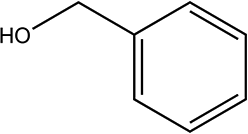
|
|
230 | 84/94 |
| 8a |
|
|
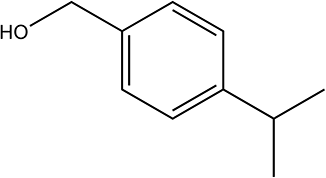
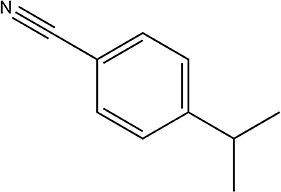
250 | 83/88 |
| 9 |
|
|
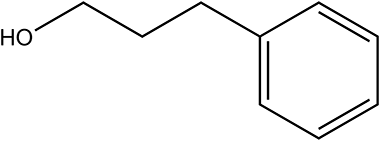
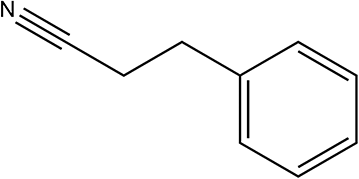
230 | 68/90 |
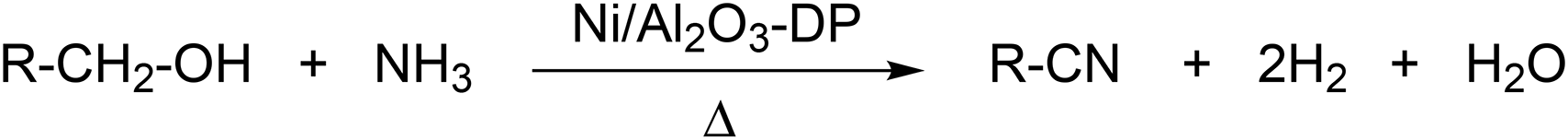
The catalytic stability test over a period of 48 hours was conducted (Fig. 6(a)), with initial yield of hexanenitrile at around 70%. The Ni/Al2O3-DP catalyst kept 90% of its original activity after 48 hours, and satisfactory carbon balance was maintained throughout the stability test. Indeed, TG analysis (Fig. 6(b)) over spent catalyst suggested 13 wt% carbon deposition, which corresponds to only about 1 mol% of carbon in the starting material accumulatively injected in 48 hours. ATR-IR (Fig. S13†) was further employed to analyse fresh and spent Ni/Al2O3-DP catalyst. The major bands for the spent catalyst were ascribed to the surface-bound carboxyl species (1554 cm−1 and 1458 cm−1), which, at least partially, accounted for the 1 mol% of carbon loss.
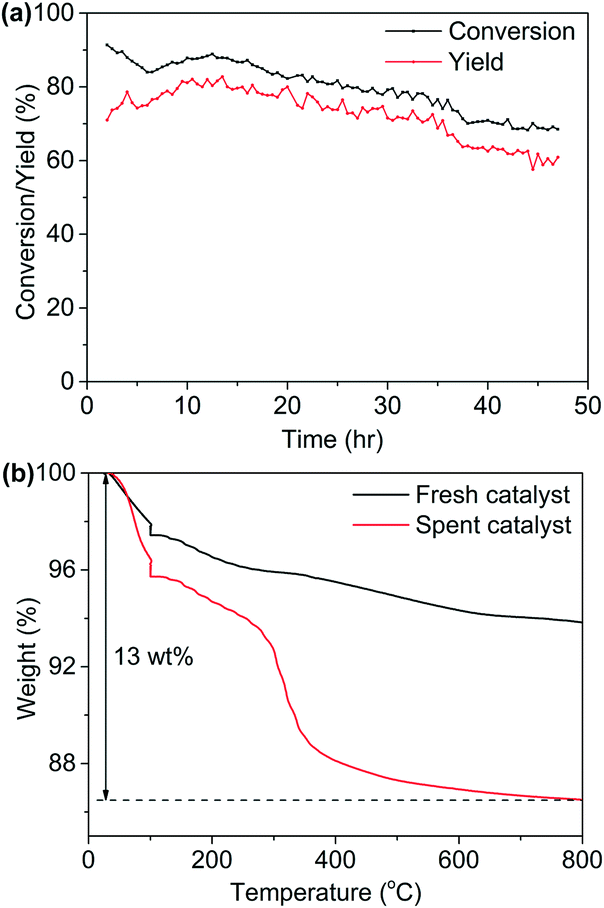 | ||
| Fig. 6 (a) Stability test for Ni/Al2O3-PD catalyst.1-Hexanol at 1 μL min−1 with 8 mL min−1 NH3 and 20 mL min−1 N2 flowed through 200 mg catalyst at 230 °C. GHSV = 1.12 × 104 h−1. (b) TG analysis for Ni/Al2O3-DP catalyst before and after reaction. | ||
Conclusions
We report in this study the development of a highly efficient catalytic system to promote nitrile production from alcohols and ammonia gas at low temperature in the absence of any oxidant. A simple, conventional Ni on alumina catalyst prepared by deposition–precipitation method is identified to be most active. The reaction progresses mainly via a tandem dehydrogenation–imination–dehydrogenation pathway, in which metal catalyses dehydrogenation reactions whereas the support promotes imine formation. Ni is superior to all noble metal catalysts tested, presumably because the dehydrogenation activity of Ni is not inhibited by NH3, while alumina is the most effective support promoting the reaction between aldehyde and ammonia to form imine. The right combination of metal centre and a slight acidic support is the key to excellent catalyst performance. A series of alcohols have been transformed into nitriles in high yields over the system, and the catalyst is stable up to 48 hours under continuous flow conditions at 230 °C. The wide substrate scope, the excellent product yield, the affordability of Ni as catalyst, and the long-term stability of the catalytic system show promise of current system for nitrile synthesis on a large scale.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National University of Singapore and Ministry of Education, Singapore (R-279-000-462-112 and R-279-000-464-133) and the funding from the “GSK-EDB Fund for Green and Sustainable Manufacturing”. Computation time was provided by the SuperComputer System, Institute for Chemical Research, Kyoto University. We thank Prof. Weiping Deng and Prof. Ye Wang from Xiamen University for providing the facility to conduct the pyridine adsorption-FTIR experiments.References
- J. Kim, H. J. Kim and S. Chang, Angew. Chem., Int. Ed., 2012, 51, 11948–11959 CrossRef CAS PubMed.
- M. J. Hülsey, H. Yang and N. Yan, ACS Sustainable Chem. Eng., 2018, 6, 5694–5707 CrossRef.
- C. Galli, Chem. Rev., 1988, 88, 765–792 CrossRef CAS.
- J. Lindley, Tetrahedron, 1984, 40, 1433–1456 CrossRef CAS.
- P. Anbarasan, T. Schareina and M. Beller, Chem. Soc. Rev., 2011, 40, 5049–5067 RSC.
- H.-J. Cristau, A. Ouali, J.-F. Spindler and M. Taillefer, Chem. – Eur. J., 2005, 11, 2483–2492 CrossRef CAS PubMed.
- J. Zanon, A. Klapars and S. L. Buchwald, J. Am. Chem. Soc., 2003, 125, 2890–2891 CrossRef CAS PubMed.
- T. Schareina, A. Zapf, W. Mägerlein, N. Müller and M. Beller, Tetrahedron Lett., 2007, 48, 1087–1090 CrossRef CAS.
- F. G. Buono, R. Chidambaram, R. H. Mueller and R. E. Waltermire, Org. Lett., 2008, 10, 5325–5328 CrossRef CAS PubMed.
- M. Sundermeier, S. Mutyala, A. Zapf, A. Spannenberg and M. Beller, J. Organomet. Chem., 2003, 684, 50–55 CrossRef CAS.
- A. J. Watson and J. M. Williams, Science, 2010, 329, 635–636 CrossRef CAS PubMed.
- S. Furukawa, R. Suzuki and T. Komatsu, ACS Catal., 2016, 6, 5946–5953 CrossRef CAS.
- S. Shi, M. Liu, L. Zhao, M. Wang, C. Chen, J. Gao and J. Xu, Chem. – Asian J., 2017, 12, 2404–2409 CrossRef CAS PubMed.
- W. Deng, Y. Wang, S. Zhang, K. M. Gupta, M. J. Hülsey, H. Asakura, L. Liu, Y. Han, E. M. Karp, G. T. Beckham, P. J. Dyson, J. Jiang, T. Tanaka, Y. Wang and N. Yan, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 5093–5098 CrossRef CAS PubMed.
- A. Martin and V. N. Kalevaru, ChemCatChem, 2010, 2, 1504–1522 CrossRef CAS.
- S. Iida and H. Togo, Tetrahedron, 2007, 63, 8274–8281 CrossRef CAS.
- N. Mori and H. Togo, Synlett, 2005, 9, 1456–1458 Search PubMed.
- S. lida and H. Togo, Synlett, 2007, 38, 407–410 Search PubMed.
- S. Yamazaki and Y. Yamazaki, Chem. Lett., 1990, 19, 571–574 CrossRef.
- G. D. McAllister, C. D. Wilfred and R. J. K. Taylor, Synlett, 2002, 1291–1292 CAS.
- F. E. Chen, Y. Y. Li, M. Xu and H. Q. Jia, Synthesis, 2002, 1804–1806 CAS.
- L. M. Dornan, Q. Cao, J. C. Flanagan, J. J. Crawford, M. J. Cook and M. J. Muldoon, Chem. Commun., 2013, 49, 6030–6032 RSC.
- W. Yin, C. Wang and Y. Huang, Org. Lett., 2013, 15, 1850–1853 CrossRef CAS PubMed.
- D. K. T. Yadav and B. M. Bhanage, Eur. J. Org. Chem., 2013, 2013, 5106–5110 CrossRef CAS.
- J.-B. Xie, J.-J. Bao, H.-X. Li, D.-W. Tan, H.-Y. Li and J.-P. Lang, RSC Adv., 2014, 4, 54007–54017 RSC.
- T. Oishi, K. Yamaguchi and N. Mizuno, Angew. Chem., Int. Ed., 2009, 48, 6286–6288 CrossRef CAS PubMed.
- T. Ishida, H. Watanabe, T. Takei, A. Hamasaki, M. Tokunaga and M. Haruta, Appl. Catal., A, 2012, 425-426, 85–90 CrossRef CAS.
- R. V. Jagadeesh, H. Junge and M. Beller, Nat. Commun., 2014, 5, 4123 CrossRef CAS PubMed.
- S. Shang, L. Wang, W. Dai, B. Chen, Y. Lv and S. Gao, Catal. Sci. Technol., 2016, 6, 5746–5753 RSC.
- C. Hamill, H. Driss, A. Goguet, R. Burch, L. Petrov, M. Daous and D. Rooney, Appl. Catal., A, 2015, 506, 261–267 CrossRef CAS.
- Y. Hu, S. Jin, Z. Zhang, L. Zhang, J. Deng and H. Zhang, Catal. Commun., 2014, 54, 45–49 CrossRef CAS.
- Y. Zhang, X. Zhao, H. Zhang, X. Yan and J. Zhao, Appl. Catal., A, 2016, 522, 45–53 CrossRef CAS.
- D. Zhang, Y. Zhang, Y. Wen, K. Hou and J. Zhao, Chem. Eng. Res. Des., 2011, 89, 2147–2152 CrossRef CAS.
- A. N. Bashkirov, N. S. Zakirov, E. A. Abdurakhmanov, G. A. Kliger, K. Muradov and Y. S. Snagovskii, Neftekhimiya, 1987, 27, 818–821 CAS.
- Y. Zhang, W. Xu and J. Zhao, RSC Adv., 2012, 2, 6590–6598 RSC.
- Y. Zhang, T. Wei, Y. Pian and J. Zhao, Appl. Catal., A, 2013, 467, 154–162 CrossRef CAS.
- C. Wang, G. Garbarino, L. F. Allard, F. Wilson, G. Busca and M. Flytzani-Stephanopoulos, ACS Catal., 2016, 6, 210–218 CrossRef CAS.
- B. M. Nagaraja, A. H. Padmasri, P. Seetharamulu, K. Hari Prasad Reddy, B. David Raju and K. S. Rama Rao, J. Mol. Catal. A: Chem., 2007, 278, 29–37 CrossRef CAS.
- C. S. Yi and D. W. Lee, Organometallics, 2009, 28, 947–949 CrossRef CAS PubMed.
- S. De, J. Zhang, R. Luque and N. Yan, Energy Environ. Sci., 2016, 9, 3314–3347 RSC.
- K.-i. Shimizu, K. Kon, W. Onodera, H. Yamazaki and J. N. Kondo, ACS Catal., 2013, 3, 112–117 CrossRef CAS.
- K.-i. Shimizu, N. Imaiida, K. Kon, S. M. A. Hakim Siddiki and A. Satsuma, ACS Catal., 2013, 3, 998–1005 CrossRef CAS.
- A. Tomer, F. Wyrwalski, C. Przybylski, J.-F. Paul, E. Monflier, M. Pera-Titus and A. Ponchel, J. Catal., 2017, 356, 111–124 CrossRef CAS.
- A. Tomer, Z. Yan, A. Ponchel and M. Pera-Titus, J. Catal., 2017, 356, 133–146 CrossRef CAS.
- A. Y. K. Leung, K. Hellgardt and K. K. M. Hii, ACS Sustainable Chem. Eng., 2018, 6, 5479–5484 CrossRef CAS.
- T. E. Bell, J. M. González-Carballo, R. P. Tooze and L. Torrente-Murciano, J. Mater. Chem. A, 2015, 3, 6196–6201 RSC.
- J. He, C. Zhao and J. A. Lercher, J. Am. Chem. Soc., 2012, 134, 20768–20775 CrossRef CAS PubMed.
- K.-i. Shimizu, S. Kanno, K. Kon, S. M. A. Hakim Siddiki, H. Tanaka and Y. Sakata, Catal. Today, 2014, 232, 134–138 CrossRef CAS.
- Y. Bang, S. J. Han, J. Yoo, J. H. Choi, J. K. Lee, J. H. Song, J. Lee and I. K. Song, Appl. Catal., B, 2014, 148-149, 269–280 CrossRef CAS.
- T. E. Bell, J. M. González-Carballo, R. P. Tooze and L. Torrente-Murciano, RSC Adv., 2017, 7, 22369–22377 RSC.
- C. Kim, C. Kim, K. Lee and H. Lee, Chem. Commun., 2014, 50, 6353–6356 RSC.
- T. S. Phan, A. R. Sane, B. Rêgo de Vasconcelos, A. Nzihou, P. Sharrock, D. Grouset and D. Pham Minh, Appl. Catal., A, 2018, 224, 310–321 CrossRef CAS.
- S. Song, Y. Wang and N. Yan, Mol. Catal., 2018, 454, 87–93 CrossRef.
- M. G. Edwards, R. F. R. Jazzar, B. M. Paine, D. J. Shermer, M. K. Whittlesey, J. M. J. Williams and D. D. Edney, Chem. Commun., 2004, 90–91, 10.1039/b312162c.
- C. Gunanathan and D. Milstein, Science, 2013, 341, 1229712 CrossRef PubMed.
- J. W. Han, J. S. Park, M. S. Choi and H. Lee, Appl. Catal., B, 2017, 203, 625–632 CrossRef CAS.
- J. Bing, C. Hu and L. Zhang, Appl. Catal., B, 2017, 202, 118–126 CrossRef CAS.
- T. A. Zepeda, B. Pawelec, R. Obeso-Estrella, J. N. Díaz de León, S. Fuentes, G. Alonso-Núñez and J. L. G. Fierro, Appl. Catal., B, 2016, 180, 569–579 CrossRef CAS.
- K.-i. Shimizu, K. Kon, K. Shimura and S. S. M. A. Hakim, J. Catal., 2013, 300, 242–249 CrossRef CAS.
- Z. Ren, Z. Wu, W. Song, W. Xiao, Y. Guo, J. Ding, S. L. Suib and P.-X. Gao, Appl. Catal., B, 2016, 180, 150–160 CrossRef CAS.
- X. Wang and U. S. Ozkan, J. Catal., 2004, 227, 492–501 CrossRef CAS.
- C. K. S. Choong, L. Huang, Z. Zhong, J. Lin, L. Hong and L. Chen, Appl. Catal., A, 2011, 407, 155–162 CrossRef CAS.
- H. Knözinger and P. Ratnasamy, Catal. Rev.: Sci. Eng., 1978, 17, 31–70 CrossRef.
- L. Cheng and X. P. Ye, Catal. Lett., 2009, 130, 100–107 CrossRef CAS.
- S. Zhang, J. Shan, L. Nie, L. Nguyen, Z. Wu and F. Tao, Surf. Sci., 2016, 648, 156–162 CrossRef CAS.
- M. A. Centeno, I. Carrizosa and J. A. Odriozola, J. Alloys Compd., 2001, 323–324, 597–600 CrossRef CAS.
- M. C. Kung and H. H. Kung, Catal. Rev.: Sci. Eng., 1985, 27, 425–460 CrossRef.
- J. J. Rafalko, J. Polym. Sci., Part B: Polym. Phys., 1984, 22, 1211–1222 CAS.
- B. Coq, D. Tichit and S. Ribet, J. Catal., 2000, 189, 117–128 CrossRef CAS.
- R. Sokoll, H. Hobert and I. Schmuck, J. Catal., 1990, 121, 153–164 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8cy01799a |
| This journal is © The Royal Society of Chemistry 2019 |