
Comparison of TiO2 and g-C3N4 2D/2D nanocomposites from three synthesis protocols for visible-light induced hydrogen evolution†
Ruyi
Zhong
ab,
Zisheng
Zhang
a,
Shuqi
Luo
a,
Z. Conrad
Zhang
 b,
Limin
Huang
*a and
Meng
Gu
*c
b,
Limin
Huang
*a and
Meng
Gu
*c
aDepartment of Chemistry, Southern University of Science and Technology, Shenzhen, 518055, China. E-mail: huanglm@sustc.edu.cn
bDalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian 116023, China
cDepartment of Materials Science and Engineering, Southern University of Science and Technology, Shenzhen, 518055, China. E-mail: gum@sustc.edu.cn
First published on 17th October 2018
Abstract
Knowledge of the interfacial structure of nanocomposite materials is a prerequisite for rational design of nanostructured photocatalysts. Herein, TiO2 and g-C3N4 2D/2D nanocomposites were fabricated from three distinct synthetic protocols (i.e., co-calcination, solvothermal treatment and charge-induced aggregation), showing different degrees of enhancement (1.4–6.1 fold) in the visible-light induced photocatalytic hydrogen evolution reaction compared to the simple physical mixture. We propose that the interfacial Ti–O–N covalent bonding promotes the charge carrier transfer and separation more effectively than the electrostatic interaction, thus accelerating the photocatalytic H2 production. Meanwhile, the exposed surface area of TiO2 in the composite needs to be enlarged for deposition of the co-catalyst. This research sheds light on the rational design of hybrid nanocomposites based on earth-abundant elements for photocatalysis.
Introduction
Hydrogen is a promising clean energy carrier, as an environmentally friendly alternative to conventional fossil fuels, owing to its high combustion energy and zero emission.1–3 Photocatalytic water splitting using solar energy by semiconductor photocatalysts has received intensive research interest in recent decades.4,5 Fujishima and Honda originally proposed non-toxic TiO2 as a stable benchmark photocatalyst under UV irradiation.6 However, the large bandgap of TiO2 (ca. 3.2 eV) restricts its utilization of the whole solar spectrum, as UV light accounts for only a proportion of 4% and visible light (400–800 nm) is dominant (50%). Doping TiO2 with various elements or self-doping with Ti3+ could extend its optical absorption to the visible-light range, which, however, usually suffers from rapid photogenerated electron–hole recombination and reduced stability of the doped materials.7,8Recently, g-C3N4 has been identified as a promising organic semiconductor for photocatalytic applications, including photoreduction of H2O to H2.5,9–14 Bulk g-C3N4 prepared from facile pyrolysis of nitrogen-rich precursors has a suitable band structure with a low band gap of ca. 2.7 eV to harvest visible light, but its photocatalytic performance is limited by its low surface area and fast recombination rate of photogenerated charge carriers.12–17 Exfoliation of g-C3N4 to 2D nanosheets by various chemical and physical approaches could increase the surface area and reduce the charge recombination, but the problem of re-stacking remains.9,10,12,13
Constructing heterojunction structures of TiO2 and g-C3N4 has been proven as a cost-effective way to avoid the above drawbacks of each component and realize a synergic effect in promoting the efficient generation and separation of charge carriers, thus boosting the photocatalytic activity.12,18,19 The heterostructure interfaces would offer a premise for such synergism during photocatalytic reactions by forming different types of band diagrams.12,18,19 Therefore many efforts have been made to fabricate TiO2 with g-C3N4 to form a composite with abundant heterostructure interfaces. Most of the fabrication processes of heterostructures are based on simultaneous formation of heterojunctions and one (or both) of the components.20–31 For instance, solvothermal synthesis of nanostructured TiO2 has been performed in the presence of pre-synthesized bulk g-C3N4,20 thermally exfoliated g-C3N4,22,23 oxidized g-C3N4,21,24 or protonated g-C3N4,25 forming particles attached onto sheets20–23 or nanosheet to nanosheet assemblies,24,25 outperforming bare TiO2 or g-C3N4 in photocatalytic reactions. The precursors for g-C3N4 can be mixed with the pre-formed TiO2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 28,29,31 or its precursors26,27,30 to undergo calcination26–30 or refluxing,31 forming core–shell29,31 or particle attached to sheet26–28 structures with improved photocatalytic activity. As for the as-formed binary components, co-calcination has been frequently applied, resulting in particle on nanosheet g-C3N4/TiO2 structures.32–34 In addition, charge-induced aggregation has been proposed for constructing intimate heterojunctions of 0D/2D Nb2O5/g-C3N4,15 2D/2D g-C3N4/graphene oxide,36 2D/2D graphene/SnNb2O6,37etc. Among the various morphologies, the 2D/2D heterojunction has unique merits compared with the 0D/1D, 1D/1D, 0D/2D and 1D/2D interfaces, owing to the increased contact region and the intimate interface regardless of the lattice mismatch, as well as the large lateral size with high surface area, to promote charge transfer and electron–hole separation, thereby improving the photocatalytic performance.19,24,25,36,37 Meanwhile, the formation of 2D/2D heterostructures is also beneficial for the catalyst stability against photocorrosion and agglomeration.19,24,25,36,37 However, relatively scarce studies have been reported concerning fabrication of 2D/2D heterostructures of TiO2 and g-C3N4,24,25 let alone comparison of different construction approaches and clarification of the interfacial interaction modes.
28,29,31 or its precursors26,27,30 to undergo calcination26–30 or refluxing,31 forming core–shell29,31 or particle attached to sheet26–28 structures with improved photocatalytic activity. As for the as-formed binary components, co-calcination has been frequently applied, resulting in particle on nanosheet g-C3N4/TiO2 structures.32–34 In addition, charge-induced aggregation has been proposed for constructing intimate heterojunctions of 0D/2D Nb2O5/g-C3N4,15 2D/2D g-C3N4/graphene oxide,36 2D/2D graphene/SnNb2O6,37etc. Among the various morphologies, the 2D/2D heterojunction has unique merits compared with the 0D/1D, 1D/1D, 0D/2D and 1D/2D interfaces, owing to the increased contact region and the intimate interface regardless of the lattice mismatch, as well as the large lateral size with high surface area, to promote charge transfer and electron–hole separation, thereby improving the photocatalytic performance.19,24,25,36,37 Meanwhile, the formation of 2D/2D heterostructures is also beneficial for the catalyst stability against photocorrosion and agglomeration.19,24,25,36,37 However, relatively scarce studies have been reported concerning fabrication of 2D/2D heterostructures of TiO2 and g-C3N4,24,25 let alone comparison of different construction approaches and clarification of the interfacial interaction modes.
Moreover, a third component has been frequently involved to form a ternary composite,38–42 for example metallic Ag was incorporated into g-C3N4/TiO2 to facilitate interfacial electron transfer,38 Cu into MoO3/g-C3N4 to boost visible light absorption for the surface plasmon resonance effect,39 Al2O3 into g-C3N4/ZnO to remedy the lattice mismatch,40etc. Nevertheless, ternary nanocomposites bring in larger complexity than binary nanocomposites, due to their more intricate interfaces.
Herein, we investigated differently fabricated TiO2 and g-C3N4 2D/2D nanocomposites for efficient and stable photocatalytic H2 evolution under visible-light irradiation. The preparation methods, in situ solvothermal treatment, co-calcination and charge-induced aggregation, were adopted with the aim of finely tuning the interfacial properties for the facilitation of electron–hole separation and electron transfer. By analyzing the photocatalytic performance and the physicochemical properties of each type of composite, formation of interfacial covalent Ti–O–N bonds is more favorable than electrostatic interactions for promoting electron transfer and suppressing charge recombination. Meanwhile, large surface area of the composite needs to be ensured for abundant deposition of the co-catalyst.
Experimental
Materials
A poly(ethylene oxide)-block-poly(propylene oxide)-block-poly(ethylene oxide) triblock copolymer (Pluronic P123, EO20PO70PEO20, MW = 5800) was purchased from Sigma-Aldrich (USA). Titanium isopropoxide (TTIP), hydrochloric acid, ethylene glycol, melamine, ethanol, sodium borohydride, H2PtCl6 and triethanolamine (TEOA) were purchased from Aladdin Industrial Corporation (Shanghai, China). All reagents were used as-received without further purification.Synthesis of raw TiO2 and g-C3N4
The ultrathin TiO2 nanosheets were prepared using a solvothermal self-assembly method.24,43 Namely, 200 mg of raw TiO2 was ground thoroughly with 200 mg of NaBH4 at room temperature. The mixture was transferred to a porcelain boat and heated in a N2 flow at 220 °C for 20 min with a ramp rate of 10 °C min−1. The product was collected, washed with H2O to remove excess NaBH4 and dried at 60 °C overnight.The modification of g-C3N4 was performed by hydrothermal treatment45 or acid treatment46 to generate O–CN and H–CN, respectively. Briefly, for O–CN, 200 mg of g-C3N4 was dispersed in 100 mL H2O by ultrasonication for 30 min and transferred to a 200 mL Teflon-lined autoclave for heating at 180 °C for 4 h. The solid sample was collected by centrifugation, washed with H2O and dried at 60 °C overnight. For H–CN, 500 mg of g-C3N4 was mixed with 12.5 mL of concentrated HCl (37.5 wt%) at room temperature under stirring for 1 h. The solid sample was collected by centrifugation, washed with H2O to remove excess HCl and dried at 60 °C overnight.
Fabrication of three TiO2 and g-C3N4 nanocomposites
As a control, 100 mg of raw TiO2 was ground manually with 100 mg of O–CN, and the resultant sample was denoted as “O–CN TiO2 mixed”.
Characterization
Zeta potentials were obtained by dynamic light-scattering analysis using a Zetasizer Nano ZS90 analyzer. 20 mg of each sample was dispersed in 50 mL of H2O by ultrasonication for the zeta potential analysis. Thermogravimetric (TGA) measurements were performed in a Mettler-Toledo TGA instrument. About 10 mg of each sample was placed in an alumina crucible and heated to 800 °C at a heating rate of 10 °C min−1 in an air flow. X-ray diffraction (XRD) patterns were acquired on a Rigaku Smartlab-9 kW X-ray diffractometer in the 2θ range from 5 to 80° and at a scan speed of 5° min−1. Fourier transform infrared spectra (FTIR) were recorded on a Bruker IFS 66 spectrometer with 128 scans at a resolution of 2 cm−1. The samples were finely ground with KBr for the transparent pellets. The specific surface area and pore structure of each sample were determined by nitrogen sorption analysis using a Micromeritics ASAP 2020. Scanning electron microscopy (SEM) images were obtained on a Tescan MIRA3 scanning electron microscope. Transmission electron microscopy (TEM) images were acquired on a JEM-2100 electron microscope operating at 200 kV. The samples for TEM were prepared by depositing an ethanolic suspension of each sample onto holey carbon-coated copper grids and drying. The electron microscopy data (EDS, HAADF STEM) were acquired using a Cs aberration-corrected FEI Titan Themis G2 at 200 kV equipped with an ultra-high brightness gun and super-X EDS detector. X-ray photoelectron spectroscopy (XPS) measurement was carried out with a Thermo SCIENTIFIC ESCALAB 250Xi instrument using monochromatized Al Kα as the excitation source. The C 1s signal at 284.8 eV was used to calibrate the binding energy (BE) and a Shirley-type background was applied for each spectrum. UV-vis diffuse reflectance spectra (UV-vis DRS) of the samples were recorded using a Lambda 950 UV/vis/NIR spectrometer (Perkin Elmer) equipped with a diffuse reflectance attachment of a Spectralon-coated integrating sphere against a BaSO4 reference. Photoluminescence (PL) spectra were collected on a fluorescence spectrometer (Horiba PTI QuantaMaster 400) at room temperature with a laser excitation wavelength of 330 nm.Photocatalytic testing
The photocatalytic hydrogen evolution reactions were performed using an online photocatalytic hydrogen generation system (CEL-SPH2N, AuLight, Beijing). A 300 W Xe lamp (CEL-HX300) equipped with a UV cut-off filter (UVIRCUT400, AuLight, Beijing, λ > 400 nm) was used as the visible light source, which was fan-cooled during the experiment. To maintain the ambient temperature of the reaction system under irradiation, a liquid trap system with water circulation was used. In a typical test, 50 mg of photocatalyst was dispersed in 50 mL of aqueous solution containing 20 vol% TEOA as a sacrificial reagent. 3 wt% Pt co-catalyst was photo-deposited uniformly onto the surface of the photocatalyst using H2PtCl6 as a precursor. Before irradiation, the suspension was degassed thoroughly by evacuation to remove dissolved gases. After an adequate photo-deposition period, the gaseous product was analyzed every hour. H2 evolution was quantitatively determined by online gas chromatography (GC-7890, thermal conductivity detector, molecular sieves 5 Å, N2 carrier, Yiyou, Shanghai).Results and discussion
Structural characterization of catalysts
Scheme 1 illustrates the three composite samples (O–CN/TiO2, CN/TiO2-cal and H–CN/H–TiO2) and their precursors (TiO2, TiO2-cal, H–TiO2, CN, O–CN and H–CN) investigated in this article. The solvothermal in situ growth fabrication approach was detailed in our previous work.24 Ultrathin TiO2 nanosheets are formed in situ by P123 template-assisted self-assembly and are attached to the edges of O–CN where oxygenated groups exist via Ti–O–N covalent linkages.24 The as-formed Ti–O–N bonds under solvothermal treatment have been demonstrated to facilitate the efficient charge separation at interfaces and enhance the photocatalytic H2 evolution activity. However, this composite still has residual P123 which may interfere with the charge and mass transfer in the photocatalytic process.25 Besides, other interaction modes of the interface of TiO2 and g-C3N4 for instance electrostatic Coulombic force and the influence of the exfoliation degree of g-C3N4 remain to be explored. Therefore, two distinct and versatile approaches, i.e. co-calcination and charge-induced self-assembly, were also applied, to further investigate the contribution of the architecture of the interface to the photocatalytic hydrogen evolution reaction. During co-calcination, the thermal exfoliation of g-C3N4,22,33,34,47 the removal of P123 organic residues25,48 and the increased crystallinity25,48 are expected, which could be beneficial for the formation of close 2D/2D interfacial contact. During surface charge-induced aggregation, the oppositely-charged nanosheets could result in improved interaction between TiO2 and g-C3N4 and thus, the charge transfer across the heterojunction to promote the photocatalytic activity.15,36,37 | ||
| Scheme 1 Schematic illustration of the three synthesis approaches for forming three composite samples (O–CN/TiO2, CN/TiO2-cal and H–CN/H–TiO2). | ||
The TGA/DTG curves of the three composite samples (O–CN/TiO2, CN/TiO2-cal and H–CN/H–TiO2), the physically mixed sample (O–CN TiO2 mixed) and the precursors (TiO2, H–TiO2, CN, O–CN and H–CN) are shown in Fig. S1.† For the nanocomposites, aside from the H2O desorption peak below 200 °C, the sharp DTG peak at about 300 °C is assigned to the P123 residues,24,25,48 and the peak starting from about 450 °C is due to the burn-off of g-C3N4 species. The weight losses at different temperature stages are summarized in Table 1. The similar weight percentages of g-C3N4 species for the three nanocomposites and the simple physical mixture rationalize the comparison of the physicochemical properties among these materials. The amount ratio of g-C3N4 to TiO2 is roughly 1:1 in all the composites in this work; the 1:1 ratio has been found to be the optimized value for the construction of heterojunctions in the O–CN/TiO2 sample (g-C3N4 to TiO2 ratios of 1:1, 1:2 and 2:1 were investigated) without compromising the large surface area.24 It is also noteworthy that the content of P123 residues decreases to zero, following the order O–CN/TiO2, H–CN/H–TiO2 and CN/TiO2-cal.
| Entry | Catalyst | TiO2a (wt%) | g-C3N4 speciesb (wt%) | P123 residuesc (wt%) | S BET (m2 g−1) | V tot (cm3 g−1) | V meso (cm3 g−1) | Average pore sizeg (nm) |
|---|---|---|---|---|---|---|---|---|
| a Weight percentage of TiO2 was determined from the plateau in the high temperature range of TGA curves for samples containing TiO2. b Weight percentage of residual P123 was determined from the weight loss around 300 °C of the TGA curves for samples containing TiO2. c Weight percentage of g-C3N4 species was determined from the main weight loss above 450 °C of the TGA curves for samples containing g-C3N4. d Surface area (SBET) was calculated from the N2 sorption isotherms by the Brunauer–Emmett–Teller (BET) method. e Total pore volume (Vtot) was calculated from the saturation plateau at high relative pressures. f Mesopore volume (Vmeso) was calculated by subtracting the micropore volume from the Vtot using the t-plot method. g Pore size (Dpore) was calculated from the adsorption branch of the isotherms by the Barrett–Joyner–Halenda (BJH) method. | ||||||||
| 1 | TiO2 | 59 | — | 13 | 330 | 0.41 | 0.36 | 5.0 |
| 2 | H–TiO2 | 68 | — | 10 | n.d. | n.d. | n.d. | n.d. |
| 3 | CN | 0 | 99 | — | 12 | 0.12 | 0.12 | 43.5 |
| 4 | O–CN | 0 | 98 | — | 55 | 0.20 | 0.20 | 14.7 |
| 5 | H–CN | 0 | 90 | — | 19 | 0.12 | 0.12 | 25.8 |
| 6 | CN/TiO2-cal | 57 | 45 | 0 | 73 | 0.30 | 0.30 | 16.3 |
| 7 | O–CN/TiO2 | 37 | 47 | 8 | 263 | 0.57 | 0.53 | 8.7 |
| 8 | H–CN/H–TiO2 | 37 | 50 | 5 | 163 | 0.17 | 0.13 | 4.3 |
| 9 | O–CN TiO2 mixed | 35 | 48 | 8 | n.d. | n.d. | n.d. | n.d. |
Table 1 also lists the textural properties of the composites and the precursors. The N2 sorption isotherms and pore size distributions are plotted in Fig. S2.† All the materials present type IV isotherms with H4 type hysteresis loops, which are caused by the stacking of nanosheets forming slit-like pores. The raw TiO2 shows a large surface area of 330 m2 g−1, while pristine CN shows an SBET of only 12 m2 g−1. The average pore size of CN is much larger than that of TiO2. These results indicate that the solvothermally synthesized TiO2 is highly exfoliated and of relatively small sheet size, while CN is largely bulky. The O–CN and H–CN increase the surface area to 55 and 19 m2 g−1, respectively, proving the delamination effect during these treatments.45,46 After formation of heterojunctions with TiO2 in the three distinct ways, the surface areas increase tremendously to 73, 263 and 163 m2 g−1 for CN/TiO2-cal, O–CN/TiO2 and H–CN/H–TiO2, respectively. The surface area of the composite is mainly contributed by the TiO2 component. The high surface area of O–CN/TiO2 implies the successful formation of ultrathin TiO2 nanosheets under solvothermal conditions in the presence of O–CN. The high surface area would supply more photocatalytically reactive centers to speed up the hydrogen evolution reaction. Besides, CN/TiO2-cal possesses merely mesopores (Vmeso = 0.30 cm3 g−1), whereas H–CN/H–TiO2 shows a Vmicro of 0.04 cm3 g−1 out of the Vtot of 0.17 cm3 g−1. These results suggest the different origins of the slit-like pores. It is likely that the mesopores in CN/TiO2-cal are from the vacancy after the removal of P123 residues and the stacking of the nanosheets. As for H–CN/H–TiO2, compact stacking driven by electrostatic Coulombic force can be envisaged, resulting in relatively high microporosity. Moreover, it has been found that very small pores around 2.14 nm in porous g-C3N4 from kaolinite-templated synthesis still provided reactive surface area for the photocatalytic hydrogen evolution with Pt as the co-catalyst.49 Therefore, there may be no diffusion limitation in micropores and the surface area could serve as an evaluation criterion for the amount of photocatalytically reactive centers.
The XRD patterns of the TiO2 and g-C3N4 composites and precursors are shown in Fig. 1. The characteristic peaks corresponding to the (101), (004), (200), (105), (211) and (204) planes of anatase are identified with the XRD database. These diffraction peaks are obvious in TiO2-cal and CN/TiO2-cal, whereas the raw TiO2 sheets, O–CN/TiO2 and H–CN/H–TiO2 are largely amorphous in the TiO2 phase with a minor rutile phase as well. Calcination at 450 °C improves the crystallization degree of TiO2 nanosheets and transforms them to a pure anatase phase, consistent with a reported observation.48 In the co-calcined CN/TiO2-cal, TiO2 nanosheets also grow into the anatase phase in the presence of CN. For the g-C3N4 precursors and the composite samples, the peaks corresponding to the (100) and (200) planes of g-C3N4 are observed, attributed to the in-plane repeated tri-s-triazine units and stacking of the conjugated N-containing aromatic ring, respectively.41 The crystallinity of O–CN is more or less the same as that of CN, while H–CN is less ordered than CN, in terms of the peak intensity of CN(200). The CN(200) for the three composites remains at the same position compared to that for the g-C3N4 precursors and the peak widths are in the objective order of crystallinity of the g-C3N4 part, indicating that the presence of TiO2 did not influence the crystal structure of g-C3N4. The XRD pattern of the composite sample can be treated as superposition of those of individual TiO2 and g-C3N4 precursors.
 | ||
| Fig. 1 XRD patterns of the three composite samples (O–CN/TiO2, CN/TiO2-cal and H–CN/H–TiO2) and the precursors (TiO2, TiO2-cal, H–TiO2, CN, O–CN and H–CN). The A, R and CN represent anatase, rutile and g-C3N4, respectively. | ||
The FTIR spectra are presented in Fig. 2. For the pristine TiO2 nanosheets, the absorption peak around 1635 cm−1 is attributed to O–H bending vibrations.38 The two small peaks at 2935 and 2868 cm−1 are attributed to C–H stretching vibrations of the residual P123 species. These organics also show the C–O stretching vibration and C–H bending vibration signals in the range of 1200–600 cm−1. The hydrogenated TiO2 shows lower intensity for P123 and these absorption bands disappear for the calcined TiO2, in line with the TGA results. For the g-C3N4 precursors (CN, O–CN and H–CN), the characteristic absorption peaks have similar shapes at the same positions. The strong bands in the range of 1650–1200 cm−1 are assigned to the typical stretching modes of the tri-s-triazine skeleton ring and the peak at about 800 cm−1 to the breathing mode of the triazine units.45,50 The FTIR spectra of the three composites embody both the characteristic absorption bands of the individual TiO2 and g-C3N4 precursors, indicating the coupling of TiO2 with g-C3N4 for each composite.
 | ||
| Fig. 2 FTIR spectra of the three composite samples (O–CN/TiO2, CN/TiO2-cal and H–CN/H–TiO2) and the precursors (TiO2, TiO2-cal, H–TiO2, CN, O–CN and H–CN). | ||
The morphology and microstructure of the three nanocomposites and the precursors were studied by TEM, as shown in Fig. 3. The CN, O–CN and H–CN appear to have relatively large and amorphous laminar morphologies, as reported elsewhere.24,36,46,50 The protonated H–CN and the mildly-oxygenated O–CN show more exfoliated and less integrated sheets than the pristine CN.24,36,46 The raw TiO2 shows poorly crystallized nanosheets with a thickness of ca. 1 nm and lengths of ca. 80 nm, consistent with a previous report.43 The calcination at 450 °C preserves the lamellar structure of the TiO2 sheets; the clear lattice fringe in the magnified region demonstrates the high degree of crystallization. Hydrogenated H–TiO2 shows the crystallinity between raw TiO2 and TiO2-cal, also with the presence of the anatase phase. Notably, the TiO2 nanosheets in H–TiO2 are more corrugated, perhaps due to the mutual electrostatic repulsive force between the negatively charged sites in H–TiO2.36 The TEM images of the three composites do not clearly show the interface of TiO2 and g-C3N4, but the facts that the lattice fringes belong to anatase TiO2 and the size of g-C3N4 sheets is relatively large help to distinguish the TiO2 from g-C3N4 phases. The SEM and low-magnification TEM images in Fig. S3† both reflect that TiO2 nanosheets were attached onto large O–CN nanosheets in the O–CN/TiO2 nanocomposite. The crystallinity of O–CN/TiO2, H–CN/H–TiO2 and CN/TiO2-cal roughly follows the order of the corresponding pure TiO2, in line with the XRD results. All the three nanocomposites show the intertwined TiO2 and g-C3N4 regions under HRTEM, proving the close linkages and the compacted interfaces for the three distinct fabrication ways.
 | ||
| Fig. 3 TEM images of the three composite samples (O–CN/TiO2, CN/TiO2-cal and H–CN/H–TiO2) and the precursors (TiO2, TiO2-cal, H–TiO2, CN, O–CN and H–CN). | ||
High-angle annular dark-field scanning TEM (HAADF-STEM) and energy-dispersive X-ray spectroscopy (EDS) mapping are employed to ascertain the heterojunction structure and composition in the three nanocomposites, as shown in Fig. 4. From the distributions of the four elements C, N, O and Ti and their combined STEM image, O–CN/TiO2 clearly shows many small ultrathin TiO2 nanosheets bonded to the edges of large O–CN sheets, as observed before.24 TiO2 nanosheets are relatively individually standing on the O–CN sheets, due to the P123 templating effect during solvothermal in situ growth synthesis. The interfacial Ti–O–N covalent bonds in O–CN/TiO2 have been demonstrated by EDS point analysis.24 In the co-calcined nanocomposite, though the TiO2 phase is also in intimate contact with CN sheets, the self-aggregation of TiO2 nanosheets is quite severe, which is attributed to the removal of intercalating P123 residues. In the case of H–CN/H–TiO2, the distributions of C, N, O and Ti are almost the same throughout the whole selected area. The 2D features of H–TiO2 and H–CN are well-preserved, and the charge-directed heteroaggregation upon ultrasonication results in effective formation of heterojunctions. The positively charged H–CN (with a zeta potential equal to 5.5 mV in H2O) is likely to stack alternately with the negatively charged H–TiO2 (with a zeta potential equal to −39.2 mV in H2O) into a layered 2D/2D heterostructure.
 | ||
| Fig. 4 HADDF STEM images and EDS elemental mapping images of C, N, O and Ti of the three nanocomposites (O–CN/TiO2, CN/TiO2-cal and H–CN/H–TiO2). | ||
XPS was utilized to investigate the oxidation state and surface chemical compositions of the three TiO2 and g-C3N4 nanocomposites, and bare TiO2 and CNs, as shown in Fig. 5. The C–H and C–C signals of C 1s from adventitious carbon were set to 284.8 eV for calibration. The O–CN/TiO2, H–CN/H–TiO2 and raw TiO2 nanosheets all exhibit Ti 2p3/2 and Ti 2p1/2 with binding energies at 458.8 and 464.5 eV, respectively, ascribed to Ti4+ species in the nanosheets with the presence of residual P123. The presence of Ti3+ after NaBH4 reduction hardly causes the alteration in the Ti 2p XPS spectra.44 The peak positions are lower than those of the reference anatase (Ti 2p3/2 = 459.4 eV, Ti 2p1/2 = 465.3 eV) or rutile (Ti 2p3/2 = 459.3 eV, Ti 2p1/2 = 465.3 eV) phases.43 The CN/TiO2-cal shows a small component peak at 458.8 eV for Ti 2p3/2, while the main component peak is at 460.1 eV, showing a shift of 1.3 eV to the higher binding energy, indicating the formation of bulky TiO2 species in the calcined sample.44,48
 | ||
| Fig. 5 Ti 2p, N 1s, O 1s and C 1s XPS spectra of the three nanocomposites (O–CN/TiO2, CN/TiO2-cal and H–CN/H–TiO2) and the precursors (TiO2, CN and O–CN). | ||
The N 1s XPS spectrum shows four peaks at ca. 398.4, 399.0, 399.8 and 401.1 eV in bare CN, corresponding to N atoms in C–N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C, N–(C)3, N–O and N–H, respectively.24,29 After hydrothermal treatment, the N–O component increases in the N 1s peak, attributed to the introduction of oxygen. The acid treatment would not change the positions of the component peaks but would lead to the increase of the N–H content.46 After hybridization of g-C3N4 with TiO2, the peak for N–O shifts toward the high binding energy region to different degrees in the three nanocomposites, indicative of the bonding state of the heterojunction. O–CN/TiO2 shows a shift of around 0.4 eV, indicating the formation of Ti–O–N covalent bonds, due to the partial substitution of Ti–O–N with H–O–N and the resultant reduced electron density for the relatively electron-deficient Ti atoms. CN/TiO2 shows an even larger shift, ca. 0.5 eV, owing to the formation of Ti–O–N covalent bonds and the removal of P123 during the calcination procedure. In contrast, H–CN/H–TiO2 shows only a shift of about 0.1 eV, in good accordance with the subtle change in the N 1s peak positions of 3CN:1Nb heterostructure prepared from the charge-induced aggregation of Nb2O5 and g-C3N4.15
C, N–(C)3, N–O and N–H, respectively.24,29 After hydrothermal treatment, the N–O component increases in the N 1s peak, attributed to the introduction of oxygen. The acid treatment would not change the positions of the component peaks but would lead to the increase of the N–H content.46 After hybridization of g-C3N4 with TiO2, the peak for N–O shifts toward the high binding energy region to different degrees in the three nanocomposites, indicative of the bonding state of the heterojunction. O–CN/TiO2 shows a shift of around 0.4 eV, indicating the formation of Ti–O–N covalent bonds, due to the partial substitution of Ti–O–N with H–O–N and the resultant reduced electron density for the relatively electron-deficient Ti atoms. CN/TiO2 shows an even larger shift, ca. 0.5 eV, owing to the formation of Ti–O–N covalent bonds and the removal of P123 during the calcination procedure. In contrast, H–CN/H–TiO2 shows only a shift of about 0.1 eV, in good accordance with the subtle change in the N 1s peak positions of 3CN:1Nb heterostructure prepared from the charge-induced aggregation of Nb2O5 and g-C3N4.15
The C 1s spectrum of CN is mainly composed of two peaks at 284.8 and 288.2 eV; the latter peak corresponds to N–CN in the triazine rings of g-C3N4.15,29 The small peak at ca. 286.0 eV is assigned to C–(N)3 species.29 The C 1s spectrum of TiO2 reflects the presence of P123 residues, which are slightly oxidized after the solvothermal synthesis. The nanocomposites of TiO2 and g-C3N4 show the peak at 288.2 eV featuring the N–CN of g-C3N4 and the peak at about 286.0 eV, combining the C–(N)3 groups of g-C3N4 and the C–O groups in P123 residues from the TiO2 part.
The O 1s peak in CN and O–CN is located at ca. 532.7 eV, arising from the C–O or N–O groups in g-C3N4. O–CN shows a higher amount of O than CN. As for the TiO2 nanosheets, O 1s peaks at 530.6 eV and 532.2 eV are identified, attributed to C–O from P123 residues and Ti–O, respectively. O–CN/TiO2 and H–CN/H–TiO2 show similarly two peaks for O atoms. However, CN/TiO2-cal exhibits two peaks for Ti–O with a distance of 1.4 eV and an area ratio of 4.7 (close to 4.9, the area ratio of the two peaks in Ti 2p). The blue shift of the main Ti–O peak is also likely due to the removal of P123 and the increased crystallinity. Meanwhile, the peak for O–C and O–N in CN/TiO2-cal shifts around 0.4 eV to high binding energy relative to CN, possibly resulting from the formation of Ti–O–N covalent bonds as a consequence of the relatively small electronegativity of Ti compared with H. O–CN/TiO2 should have such shifts as well, though these are overlapped by the O 1s signals from residual P123.
Photospectral analysis and photocatalytic hydrogen evolution performance of catalysts
Fig. 6 shows the UV-vis diffuse reflectance spectra (UV-vis DRS) of the different TiO2 and g-C3N4 nanocomposites and their precursors. The raw TiO2 and TiO2-cal have ultraviolet light absorption almost below 400 nm, while the hydrogenated H–TiO2 shows extended weak absorption in the range of 400–600 nm, characteristic of reported black TiO2 nanomaterials.7 All g-C3N4 samples have much greater absorbance in the visible light range of 400–600 nm. The post-treatment by mild oxidation in H2O does not influence much the absorption, whereas the treatment by acid slightly blue-shifts the absorption.45,46 After combining TiO2 with g-C3N4 in three ways, the absorption in both the visible and ultraviolet light regions is significantly enhanced. Compared to the individual precursors, the nanocomposites show the both characteristic light responses of TiO2 and g-C3N4. The spectra of the three nanocomposites are close to each other, not a simple superposition of each precursor, which implies the synergistic effect between g-C3N4 and TiO2 to enhance the photoabsorption performance due to the construction of heterojunctions. | ||
| Fig. 6 UV-vis DRS and the corresponding plots of (αhν)2 or (αhν)1/2versus photon energy (hν) of the three composite samples (O–CN/TiO2, CN/TiO2-cal and H–CN/H–TiO2) and the precursors (TiO2, TiO2-cal, H–TiO2, CN, O–CN and H–CN). The bandgap energies are indicated in the figure. | ||
The band gap energy (Eg) was calculated according to the plots of transformed Kubelka-Munk (KM) function versus the light energy for the samples, as shown in Fig. 6. The function is (αhv)2 = A(hv − Eg) or (αhv)1/2 = A(hv − Eg) for direct or indirect transition semiconductors, respectively, where A is a constant, hν is the photon energy, h is Planck's constant, ν is the frequency of vibration, and α is the absorption coefficient.22,38 The estimated band gap energy of TiO2-cal (2.91 eV) is smaller than that of raw TiO2 nanosheets (3.16 eV), due to the increased crystallinity.48 The hydrogenation of TiO2 further narrowed the Eg to 2.72 eV, due to the increase of oxygen vacancies and Ti3+ concentration.44 The band gap energies of CN, O–CN and H–CN are 2.73, 2.76 and 2.85, respectively, consistent with the reported values.45,46 The slight alterations are ascribed to the introduction of electron-withdrawing O groups and the quantum confinement effect, respectively.45,46 The band gap energies for the nanocomposites O–CN/TiO2, CN/TiO2-cal and H–CN/H–TiO2 are 2.44, 2.52 and 2.54, respectively, which are lower than those of the precursors of TiO2 or g-C3N4. These results indicate that the photo-excitation occurs between the valence band (VB) of g-C3N4 and the conduction band (CB) of the TiO2 part in these nanocomposites with efficient 2D/2D interfacial contact, ensuring the high light absorption efficiency and therefore the improved photocatalytic performance for H2 evolution.
The photocatalytic activity of the three TiO2 and g-C3N4 nanocomposites, the physically mixed sample and the precursors was assessed by photocatalytic hydrogen evolution under visible light irradiation (λ > 400 nm) with 20 vol% TEOA as the sacrificial agent and 3 wt% Pt as the co-catalyst, as shown in Fig. 7. The raw TiO2 nanosheets show no photocatalytic activity due to the zero photo-absorption below 400 nm. The hydrogenated H–TiO2 also shows a negligible H2 evolution rate, perhaps due to the weak visible-light absorption. For g-C3N4 precursors, the time evolution curve for H2 production of O–CN almost overlaps with CN, while the activity of H–CN is about 1.5 times that of CN in terms of the average H2 evolution rate in the time span of 4 h. The reason could lie in the subtle modification of the band structure in O–CN; the influence on the band structure after the introduction of oxygen into g-C3N4 remains controversial in the literature.45,51,52 The enhancement in the catalytic activity after acid treatment could be explained by the increased favorable defects, which expose more activity sites and decrease recombination sites.46 The simple physical mixture of TiO2 and g-C3N4 would result in exactly half the H2 evolution activity compared to the pure g-C3N4, as observed with the O–CN TiO2 mixed sample and O–CN. Weak van der Waals interaction occurs in the “heterojunction” of the physical mixture, but does not accelerate the photocatalytic process.
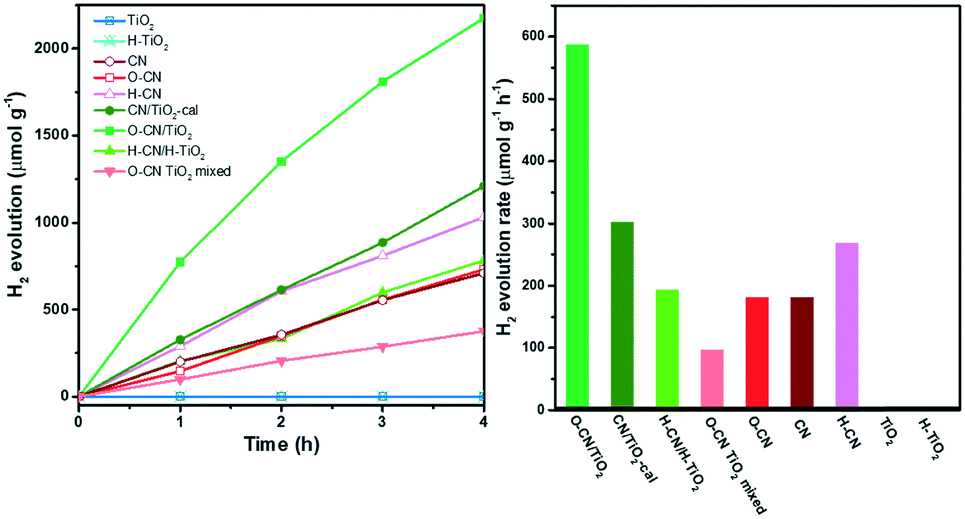 | ||
| Fig. 7 H2 evolution of the three composite samples (O–CN/TiO2, CN/TiO2-cal and H–CN/H–TiO2), the physically mixed sample (O–CN TiO2 mixed) and the precursors (TiO2, H–TiO2, CN, O–CN and H–CN) under visible light irradiation (left). The corresponding H2 evolution rates (right). | ||
The hybridization with TiO2 forming the three nanocomposites, however, significantly enhances the photocatalytic activity for H2 evolution, in comparison with the physical mixture of the corresponding TiO2 and g-C3N4 precursors, indicating the synergic interaction between the two components across the interface. Among the three nanocomposites, O–CN/TiO2 shows the highest H2 evolution rate, reaching a kinetic rate of 587 μmol g1 h−1, which is about 6.1 times that of the physical mixture. The photocatalytic activity stability of the O–CN/TiO2 nanocomposite was evaluated by extending the photo-irradiation time to 14 h. As shown in Fig. S4,† the average rate of H2 formation is 566 μmol g−1 h−1, close to the average rate during the initial 4 h, indicating the maintenance of excellent photocatalytic activity for H2 evolution on O–CN/TiO2. The increase factors for the photocatalytic activity of CN/TiO2-cal and H–CN/H–TiO2 are 3.4 and 1.4 times, respectively, relative to the physical mixture. Though the surface area of CN/TiO2-cal is the lowest among the three composites, the remarkable enhancement highlights the importance of forming covalent Ti–O–N bonds at the interface of TiO2 and g-C3N4 for the rapid charge carrier transfer and separation. The charge-induced aggregated H–CN/H–TiO2 sample, with a medium surface area and close 2D/2D face-to-face contact, does not avoid electron–hole recombination through electrostatic interaction as efficiently as through the covalent chemical bonds.
The PL spectra of the nanocomposite samples and the precursors are shown in Fig. 8, revealing the recombination of charge carriers mainly in the g-C3N4 part.29,34 The highest PL emission intensity is observed with the pristine CN, indicating the most severe recombination of electron–hole pairs, as a result of n–π* electronic transitions involving lone pairs of N atoms.20,23 The peak position is located at ca. 460 nm (i.e., 2.7 eV), in conformity with the band gap energy derived from its UV-vis DRS spectrum.35 The PL intensities decrease after the pretreatment of CN, consistent with previously reported results.45,46 The formation of the three nanocomposites with TiO2 further decreases the PL intensity, which is caused by the efficient separation of photogenerated electron–hole pairs between TiO2 and g-C3N4.20,34,53 The degree of PL quenching is in accordance with the photocatalytic hydrogen evolution results. The interfacial chemical bonds, i.e., Ti–O–N, in O–CN/TiO2 and CN/TiO2-cal most conspicuously facilitate the photogenerated charge separation and transfer, thereby effectively reducing the PL intensity and increasing the lifetime of photogenerated electrons and holes for the most prominently accelerated photocatalytic reactions. The recombination of photogenerated charge carriers is hardly inhibited in the O–CN TiO2 mixed sample and slightly retarded in H–CN/H–TiO2, reflecting the dependence of PL intensity on the interaction modes between TiO2 and g-C3N4, which also determines the photocatalytic performance.
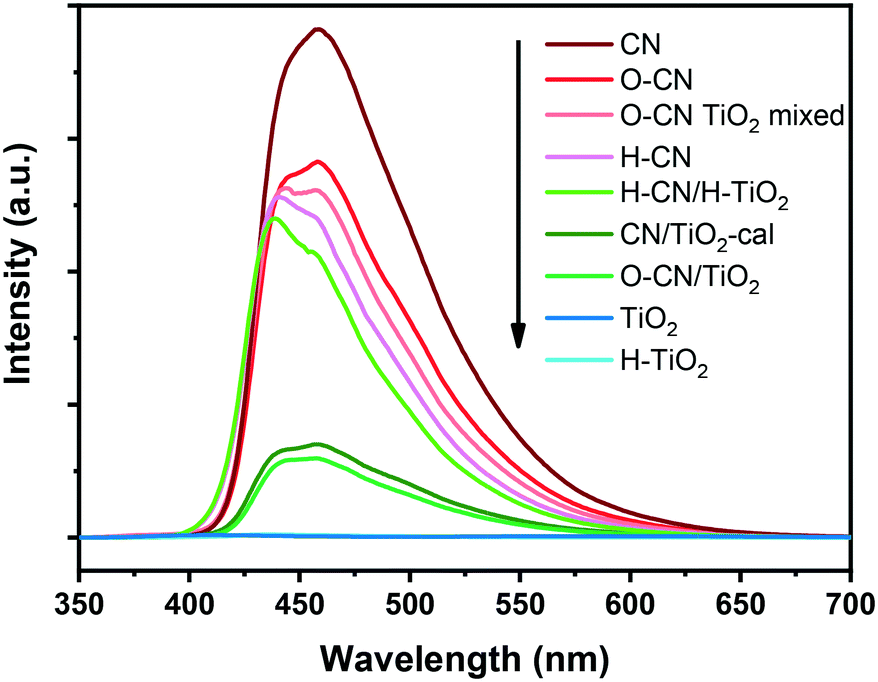 | ||
| Fig. 8 PL spectra of the three composite samples (O–CN/TiO2, CN/TiO2-cal and H–CN/H–TiO2), the physically mixed sample (O–CN TiO2 mixed) and the precursors (TiO2, H–TiO2, CN, O–CN and H–CN) under 330 nm excitation. | ||
The establishment of an intimate interfacial contact between TiO2 nanosheets and g-C3N4 is crucial to achieving high photocatalytic activity for H2 evolution. Scheme 2 shows a tentative photocatalytic H2 evolution mechanism catalyzed by TiO2 and g-C3N4 nanocomposites with deposited Pt under visible light irradiation. TiO2 nanosheets have the VB and CB at about 2.9 and −0.3 eV (vs. NHE), respectively.30,34 The calcination or hydrogenation by NaBH4 do not turn TiO2 into a strong visible-light absorber. Calcination largely reduces the surface area though the crystallinity increases and P123 organic residues are removed. The VB and CB of CN are located at about 1.5 and −1.2 eV, respectively.16,30,46 The mild oxidation or acid treatment results in slight modification of the band structure.46,52 These treatments and calcination50 indeed exfoliate CN, but still contribute a minor surface area to the nanocomposites with TiO2.
 | ||
| Scheme 2 The energy diagram of the TiO2 and g-C3N4 composites for photocatalytic H2 evolution under visible light irradiation. | ||
In the presence of the three TiO2 and g-C3N4 nanocomposites, at first, electrons in the VB of g-C3N4 are excited to its CB under visible-light irradiation, forming the electron–hole pairs. The electrons in the CB of g-C3N4 rapidly transfer to the CB of TiO2via the interfacial contact through either the covalent Ti–O–N bonds (in O–CN/TiO2 and CN/TiO2-cal) or the electrostatic Coulombic interaction (in H–CN/H–TiO2), realizing the efficient separation of photo-induced electrons and holes. The holes flowing from the VB of TiO2 (2.9 eV) to the VB of g-C3N4 (1.5 eV) are captured by TEOA. Simultaneously, the electrons flowing from the CB of g-C3N4 (−1.2 eV) to the CB of TiO2 (−0.3 eV) transfer to Pt for reduction of protons to H2. The Pt co-catalyst easily sinks the accumulated electrons, avoiding the excessive accumulation and recombination of charge carriers.12,19,49 Pt mainly deposits on the surface of TiO2 due to its large surface area from its ultrathin 2D structure. Therefore, the facilitated electron-transfer across the g-C3N4 and TiO2 interface and the large surface area of the TiO2 phase are the two most important factors for improving the photocatalytic efficiency of H2 evolution.
Conclusions
In summary, three distinct and typical approaches were applied to fabricate TiO2 and g-C3N4 2D/2D nanocomposites, i.e., solvothermal treatment, co-calcination and surface charge-induced heteroaggregation. The resultant three heterostructures exhibit higher photocatalytic activities towards the hydrogen evolution reaction under visible light irradiation, in comparison with the physical mixture of the corresponding g-C3N4 and TiO2 nanosheets, with an enhancement factor of 1.4–6.1. According to the analyses of the physicochemical properties, the formation of covalent Ti–O–N is evidenced to more efficiently facilitate the migration and separation of photo-induced charge carriers compared to electrostatic interactions, thus being more beneficial for H2 evolution. In addition, the maintenance of the large surface area outweighs the surface-cleanliness and crystallinity of the TiO2 component in the composites in terms of the photocatalytic H2 evolution activity. The well-designed comparative experiments and detailed characterization in this work may pave the way for rational design and synthesis of other composite materials with well-defined heterostructures for versatile applications.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the Southern University of Science and Technology Start Fund through Shenzhen Peacock Talent Program, the Basic Research Fund of Shenzhen (JCYJ20150507170334573), the Technical Research Fund of Shenzhen (JSGG20160427105120572), the National Natural Science Foundation of China (No. 21802065), and Guangdong Innovative and Entrepreneurial Research Team Program (No. 2016ZT06N532). This work was also supported by the Pico Center at SUSTech that receives support from the Presidential Fund and Development and Reform Commission of Shenzhen Municipality.References
- C. Acar, I. Dincer and G. F. Naterer, Int. J. Energy Res., 2016, 40, 1449–1473 CrossRef CAS.
- J. A. Turner, Science, 2004, 305, 972–974 CrossRef CAS PubMed.
- P. Lianos, Appl. Catal., B, 2017, 210, 235–254 CrossRef CAS.
- J. Schneider, M. Matsuoka, M. Takeuchi, J. Zhang, Y. Horiuchi, M. Anpo and D. W. Bahnemann, Chem. Rev., 2014, 114, 9919–9986 CrossRef CAS PubMed.
- S. Dong, J. Feng, M. Fan, Y. Pi, L. Hu, X. Han, M. Liu, J. Sun and J. Sun, RSC Adv., 2015, 5, 14610–14630 RSC.
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CrossRef CAS PubMed.
- S. G. Ullattil, S. B. Narendranath, S. C. Pillai and P. Periyat, Chem. Eng. J., 2018, 343, 708–736 CrossRef CAS.
- S. Chu, Y. Wang, Y. Guo, J. Feng, C. Wang, W. Luo, X. Fan and Z. Zou, ACS Catal., 2013, 3, 912–919 CrossRef CAS.
- W. Ong, L. Tan, Y. H. Ng, S. Yong and S. Chai, Chem. Rev., 2016, 116, 7159–7329 CrossRef CAS PubMed.
- J. Zhang, Y. Chen and X. Wang, Energy Environ. Sci., 2015, 8, 3092–3108 RSC.
- S. Martha, S. Mansingh, K. M. Parida and A. Thirumurugan, Mater. Chem. Front., 2017, 1, 1641–1653 RSC.
- S. Patnaik, S. Martha and K. M. Parida, RSC Adv., 2016, 6, 46929–46951 RSC.
- S. Patnaik, S. Martha, S. Acharya and K. M. Parida, Inorg. Chem. Front., 2016, 3, 336–347 RSC.
- S. Martha, A. Nashim and K. M. Parida, J. Mater. Chem. A, 2013, 1, 7816–7824 RSC.
- G. T. S. T. Da Silva, K. T. G. Carvalho, O. F. Lopes and C. Ribeiro, Appl. Catal., B, 2017, 216, 70–79 CrossRef CAS.
- Q. Zhang, S. Yuan, B. Xu, Y. Xu, K. Cao, Z. Jin, C. Qiu, M. Zhang, C. Su and T. Ohno, Catal. Today, 2018, 315, 184–193 CrossRef CAS.
- Y. Hong, Z. Fang, B. Yin, B. Luo, Y. Zhao, W. Shi and C. Li, Int. J. Hydrogen Energy, 2017, 42, 6738–6745 CrossRef CAS.
- Y. Wang, Q. Wang, X. Zhan, F. Wang, M. Safdar and J. He, Nanoscale, 2013, 5, 8326–8339 RSC.
- T. Su, Q. Shao, Z. Qin, Z. Guo and Z. Wu, ACS Catal., 2018, 8, 2253–2276 CrossRef CAS.
- Z. Lu, L. Zeng, W. Song, Z. Qin, D. Zeng and C. Xie, Appl. Catal., B, 2017, 202, 489–499 CrossRef CAS.
- K. Li, Z. Huang, X. Zeng, B. Huang, S. Gao and J. Lu, ACS Appl. Mater. Interfaces, 2017, 9, 11577–11586 CrossRef CAS PubMed.
- F. Chang, J. Zhang, Y. Xie, J. Chen, C. Li, J. Wang, J. Luo, B. Deng and X. Hu, Appl. Surf. Sci., 2014, 311, 574–581 CrossRef CAS.
- J. Ma, X. Tan, F. Jiang and T. Yu, Catal. Sci. Technol., 2017, 7, 3275–3282 RSC.
- R. Zhong, Z. Zhang, H. Yi, L. Zeng, C. Tang, L. Huang and M. Gu, Appl. Catal., B, 2018, 237, 1130–1138 CrossRef CAS.
- W. Gu, F. Lu, C. Wang, S. Kuga, L. Wu, Y. Huang and M. Wu, ACS Appl. Mater. Interfaces, 2017, 9, 28674–28684 CrossRef CAS PubMed.
- K. Li, S. Gao, Q. Wang, H. Xu, Z. Wang, B. Huang, Y. Dai and J. Lu, ACS Appl. Mater. Interfaces, 2015, 7, 9023–9030 CrossRef CAS PubMed.
- S. Zhou, Y. Liu, J. Li, Y. Wang, G. Jiang, Z. Zhao, D. Wang, A. Duan, J. Liu and Y. Wei, Appl. Catal., B, 2014, 158–159, 20–29 CrossRef CAS.
- N. Boonprakob, N. Wetchakun, S. Phanichphant, D. Waxler, P. Sherrell, A. Nattestad, J. Chen and B. Inceesungvorn, J. Colloid Interface Sci., 2014, 417, 402–409 CrossRef CAS PubMed.
- W. Wang, J. Fang, S. Shao, M. Lai and C. Lu, Appl. Catal., B, 2017, 217, 57–64 CrossRef CAS.
- S. Pany and K. M. Parida, Phys. Chem. Chem. Phys., 2015, 17, 8070–8077 RSC.
- Y. Wang, W. Yang, X. Chen, J. Wang and Y. Zhu, Appl. Catal., B, 2018, 220, 337–347 CrossRef CAS.
- J. Li, M. Zhang, Q. Li and J. Yang, Appl. Surf. Sci., 2017, 391, 184–193 CrossRef CAS.
- J. Li, M. Zhang, X. Li, Q. Li and J. Yang, Appl. Catal., B, 2017, 212, 106–114 CrossRef CAS.
- M. Reli, P. Huo, M. Šihor, N. Ambrožová, I. Troppová, L. Matějová, J. Lang, L. Svoboda, P. Kuśtrowski, M. Ritz, P. Praus and K. Kočí, J. Phys. Chem. A, 2016, 120, 8564–8573 CrossRef CAS PubMed.
- S. Patnaik, S. Martha, G. Madras and K. M. Parida, Phys. Chem. Chem. Phys., 2016, 18, 28502–28514 RSC.
- W. Ong, L. Tan, S. Chai, S. Yong and A. R. Mohamed, Nano Energy, 2015, 13, 757–770 CrossRef CAS.
- L. Yuan, M. Yang and Y. Xu, Nanoscale, 2014, 6, 6335–6345 RSC.
- Y. Chen, W. Huang, D. He, Y. Situ and H. Huang, ACS Appl. Mater. Interfaces, 2014, 6, 14405–14414 CrossRef CAS PubMed.
- S. Patnaik, G. Swain and K. M. Parida, Nanoscale, 2018, 10, 5950–5964 RSC.
- S. Liu, F. Li, Y. Li, Y. Hao, X. Wang, B. Li and R. Liu, Appl. Catal., B, 2017, 212, 115–128 CrossRef CAS.
- G. Jiang, K. Geng, Y. Wu, Y. Han and X. Shen, Appl. Catal., B, 2018, 366–375 CrossRef CAS.
- S. Patnaik, D. P. Sahoo, L. Mohapatra, S. Martha and K. M. Parida, Energy Technol., 2017, 5, 1687–1701 CrossRef CAS.
- Z. Sun, T. Liao, Y. Dou, S. M. Hwang, M. Park, L. Jiang, J. H. Kim and S. X. Dou, Nat. Commun., 2014, 5, 3813–3821 CrossRef CAS PubMed.
- B. Yan, P. Zhou, Q. Xu, X. Zhou, D. Xu and J. Zhu, RSC Adv., 2016, 6, 6133–6137 RSC.
- L. Ming, H. Yue, L. Xu and F. Chen, J. Mater. Chem. A, 2014, 2, 19145–19149 RSC.
- C. Dong, Z. Ma, R. Qie, X. Guo, C. Li, R. Wang, Y. Shi, B. Dai and X. Jia, Appl. Catal., B, 2017, 217, 629–636 CrossRef CAS.
- J. Xu, Y. Chen, Y. Hong, H. Zheng, D. Ma, B. Xue and Y. Li, Appl. Catal., A, 2018, 549, 31–39 CrossRef CAS.
- J. Yang, Y. Jiang, L. Li, E. Muhire and M. Gao, Nanoscale, 2016, 8, 8170–8177 RSC.
- Z. Zhang, L. Lu, Z. Lv, Y. Chen, H. Jin, S. Hou, L. Qiu, L. Duan, J. Liu and K. Dai, Appl. Catal., B, 2018, 232, 384–390 CrossRef CAS.
- S. C. Yan, Z. S. Li and Z. G. Zou, Langmuir, 2009, 25, 10397–10401 CrossRef CAS PubMed.
- Y. Huang, Y. Wang, Y. Bi, J. Jin, M. F. Ehsan, M. Fu and T. He, RSC Adv., 2015, 5, 33254–33261 RSC.
- C. Wang, H. Fan, X. Ren, J. Ma, J. Fang and W. Wang, ChemSusChem, 2018, 11, 700–708 CrossRef CAS PubMed.
- S. Nayak, L. Mohapatra and K. Parida, J. Mater. Chem. A, 2015, 3, 18622–18635 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: The results of TGA and N2 sorption measurements, SEM and low-magnification TEM images, and stability test results. See DOI: 10.1039/c8cy00965a |
| This journal is © The Royal Society of Chemistry 2019 |