
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhotocatalytic hydrogen peroxide production with an external quantum yield of almost 500%†
Yaozong
Yan
a,
Shin-ichi
Naya
 *b,
Hisashi
Sugime
ac,
Hiroaki
Tada
*d and
Tetsuro
Soejima
*ac
*b,
Hisashi
Sugime
ac,
Hiroaki
Tada
*d and
Tetsuro
Soejima
*ac
aGraduate School of Science and Engineering, Kindai University, 3-4-1, Kowakae, Higashi-Osaka, Osaka 577-8502, Japan. E-mail: soejima@apch.kindai.ac.jp
bEnvironmental Research Laboratory, Kindai University, 3-4-1, Kowakae, Higashi-Osaka, Osaka 577-8502, Japan. E-mail: shinichi.naya@itp.kindai.ac.jp
cDepartment of Applied Chemistry, Faculty of Science and Engineering, Kindai University, 3-4-1, Kowakae, Higashi-Osaka, Osaka 577-8502, Japan
dInstitutes of Innovation for Future Society, Nagoya University, Furo-cho, Chikusa-ku, Nagoya, 464-8603, Aichi, Japan
First published on 2nd May 2025
Abstract
From the perspective of energy and environmental issues, the development of green methods for H2O2 production is demanded as an alternative to the conventional energy-intensive anthraquinone method. Among the candidates, photocatalytic H2O2 production has recently attracted much interest. To withstand the use under harsh conditions, it is desirable that the photocatalyst be composed of robust inorganic materials. However, the external quantum yield (ϕex) of H2O2 production by inorganic photocatalysts remains below 20% in most studies. Here we demonstrate that a nanohybrid photocatalyst consisting of antimony-doped SnO2 and ZnO can produce H2O2 with a ϕex of ∼500% from O2-saturated ethanol aqueous solution under UV-light irradiation. In the photochemical reactions, the quantum yield is usually far below unity (<100%) due to the rapid recombination of photogenerated charge carriers. Breaking through this limit will pave the way for innovative photochemical reactions with the photocatalytic H2O2 synthesis brought closer to practical application.
Introduction
Hydrogen peroxide (H2O2) is of paramount importance because it is not only a versatile clean oxidant1 but also a promising fuel for fuel cells and photo-fuel cells.2,3 At present, most H2O2 is industrially produced by the anthraquinone auto-oxidation method involving multi-steps with large amounts of energy, hydrogen gas, and organic solvent consumed. In view of safety, low carbonization, and cost reduction, the development of green and sustainable on-site processes is imperative to produce the required amount of H2O2 at the required location. To this end, research on various green processes for H2O2 production is actively being conducted.1 Among them, photocatalytic H2O2 synthesis via two electron-oxygen reduction reaction (2e−-ORR, eqn (1)) utilizing sunlight as the driving force has recently attracted much attention.4–7 In this case, it should be noted that the standard electrode potential (E0vs. standard hydrogen electrode, SHE) for the reduction of H2O2 to H2O (eqn (2)) is much more positive than that for 2e−-ORR, which means that H2O2 once produced easily undergoes further reduction to H2O under the photocatalytic reaction conditions.| O2 + 2H+ + 2e− → H2O2 E0 = 0.695 V | (1) |
| H2O2 + 2H+ + 2e− → 2H2O E0 = 1.763 V | (2) |
| H2O2 → H2O + 1/2O2 ΔrG0298K = −116.78 kJ mol−1 | (3) |
Also, the decomposition of H2O2via disproportionation with a large negative Gibbs energy of reaction (ΔrG0) can spontaneously occur in the presence of a catalyst (eqn (3)). The intensive photocatalytic activity for H2O2 production is commonly evaluated using the external quantum yield (ϕex) or apparent quantum yield defined by eqn (4).
| ϕex(λ) = (NH2O2/Nphoton) × 2 × 100 | (4) |
Additionally, the concentration (C) and mole number (N) of the obtained H2O2 are important extensive indicators of the photocatalyst performance. The photocatalysts so far developed for H2O2 synthesis can be broadly classified into organic polymers8–10 and semiconducting metal oxides (MOs).11 Recent progress in the research on the H2O2 synthesis from O2 using polymer-based photocatalysts is remarkable. For example, photocatalytic performances of ϕex = 7.6% (λ = 420 nm), C = 5.4 mM and N = 0.27 mmol at photoirradiation time (tp) = 5 h were achieved in H2O2 production from O2 and H2O by using a resorcinol-formaldehyde resin photocatalyst.12 Also, an antimony-dispersed g-C3N4 photocatalyst was reported to yield H2O2 from O2 and H2O with ϕex = 17.6% (λ = 420 nm), C = 4.71 mM and N = 0.24 mmol at tp = 5 h.13 Further, a photoelectrochemical (PEC) cell with a polyterthiophene photoanode was developed to produce 110 mM H2O2 at tp = 11 h with the faradaic efficiency of ∼100% under illumination of simulated sunlight (one sun).14 On the other hand, from the viewpoint of stability, robust MO-based photocatalysts are more favorable since the organic polymers can be degraded by reactive oxygen species generated during H2O2 production.15 Recently, active research has dramatically improved the activity of MO-based photocatalysts for H2O2 production. For example, visible light-irradiation of Mo-doped BiVO4 decorated with CoOx and Pd nanoparticles (NPs) generated H2O2 from O2 and H2O with a ϕex of 5.8% (λ = 420 nm), and the C and N reached 1.2 mM and 0.017 mmol at tp = 1 h (saturated C (Csat) ≈ 1.6 mM and saturated N (Nsat) = 0.022 mmol at tp →∞), respectively, owing to the catalytic activities of CoOx for oxygen evolution reaction and Pd NPs for 2e−-ORR.16 The addition of appropriate electron and proton donors can further enhance the photocatalytic performances. In the presence of ethanol, Au NP-loaded TiO2 (Au/TiO2) particles immobilized on a porous hydrophobic substrate provided photocatalytic performances of ϕex = 17% (λ = 367 nm), C = 26.5 mM and N = 0.040 mmol at tp = 16 h (Csat = 28.1 mM and Nsat = 0.042 mmol), which results from an increased supply of O2 to the reaction sites.17 Also, photocatalytic performances of ϕex = 7.2% (λ = 365 nm), C = 3.0 mM and N = 0.30 mmol at tp = 1 h were reported using a TiO2/MoSx-Au NP photocatalyst, in which MoSx tunes the electronic state of Au NPs to facilitate the O2 adsorption.18 Further, very high values of ϕex = 55% (λ = 450 nm), C = 38 mM and N = 0.19 mmol at tp = 3 h (Csat = 51 mM and Nsat = 0.26 mmol) were recorded in the WO3-photocatalyzed H2O2 synthesis by adding 2-propanol mainly due to its excellent electron and proton donating ability.19 However, in all MO-photocatalyzed H2O2 production reported so far, the rate of H2O2 production significantly decreases with reaction time, and the amount produced saturates within a few hours due to further reduction (eqn (2)) and/or decomposition of H2O2 (eqn (3)). Therefore, to further enhance the C and N, it is essential to use MOs with low affinity for H2O2 as the photocatalyst materials. While most MOs are catalytically active for H2O2 decomposition, we have found that SnO2, Sb-doped SnO2 (ATO), and ZnO are inactive (Fig. S1†), and that ATO possesses high catalytic activity for 2e−-ORR.20
This study shows that a self-assembled nanohybrid photocatalyst consisting of ATO clusters and ZnO (ATO-CL/ZnO) produces H2O2 from an O2-saturated ethanol aqueous solution with an unprecedented ϕex value of ∼500%, and the C and N increase with increasing light exposure time to reach 162 mM and 8.0 mmol, respectively, at tp = 24 h (eqn (5)). The basic action mechanism of the ATO-CL/ZnO photocatalyst is discussed based on electrochemical (EC) and PEC data.
| O2 + C2H5OH → H2O2 + CH3CHO ΔrG0 = −73.69 kJ mol−1 | (5) |
The combination of the reaction in eqn (5) with the natural photosynthesis can form a carbon neutral cycle with H2O2 as the key substance because ethanol can be produced from biomasses.21
Experimental
Materials
Zinc oxide powder (crystallite size = 41.5 nm, specific surface area = 9.5 m2 g−1) was purchased from Sigma-Aldrich Co. ATO powder (Sb-doped amount = 11.6 mol%, crystallite size = 4.3 nm, specific surface area = 58.5 m2 g−1, SN-100P) was kindly gifted from Ishihara Sangyo Co. TiO2 powder (anatase, mean particles size = 150 nm, specific surface area = 8.1 m2 g−1, A-100) was purchased from Ishihara Sangyo Co. Fluorine-doped tin(IV) oxide film-coated glass (FTO, 7 Ω per square, TEC7) and Nafion film (Nafion 117, thickness = 0.18 mm, Sigma-Aldrich Co.) were used for PEC measurements. The following reagents were used as received: 2,9-dimethyl-1,10-phenanthrolinehemihydrate (C14H12N2·0.5H2O, FUJIFILM Wako Pure Chemical Co.), ethanol (C2H5OH > 99.5%, FUJIFILM Wako Pure Chemical Co.), ethanol-d6 (C2D5OD > 99%, FUJIFILM Wako Pure Chemical Co.), copper(II) sulfate pentahydrate(CuSO4·5H2O > 99.5%, FUJIFILM Wako Pure Chemical Co.), hydrogen tetrachloroaurate(III) tetrahydrate(HAuCl4·4H2O > 99%, FUJIFILM Wako Pure Chemical Co.), silver nitrate (AgNO3 > 99.8%, FUJIFILM Wako Pure Chemical Co.), cobalt(II) nitrate hexahydrate (Co(NO3)2·6H2O > 98%, FUJIFILM Wako Pure Chemical Co.), and sodium hydroxide (NaOH > 97%, FUJIFILM Wako Pure Chemical Co.), phosphate buffer solution (pH 6.9, Kishida Chemical Co.), and sodium perchlorate (NaClO4 > 96.0%, Kanto Chemical Co.). All aqueous solutions were prepared with distilled water.Characterization
The specific surface area was determined by the Brunauer–Emmett–Teller (BET) method. Adsorption–desorption isotherms of N2 were measured at 77 K using a micromeritics automatic surface area and porosimetry analyzer (TriStar 3000, Shimadzu). Before the measurements, the samples were degassed in a vacuum at 423 K for 1 h. X-ray diffraction (XRD) patterns were measured using a Rigaku MiniFlexII using Cu Kα radiation. Scanning electron microscopic (SEM) observation was carried out with Hitachi SU8230 at an applied voltage of 20–25 kV. Transmission electron microscopy (TEM) and high-resolution TEM (HR-TEM) measurements were performed using a JEOL JEM-2100F at an applied voltage of 200 kV. The diffuse reflectance spectra were measured in an ultraviolet-visible-near infrared (UV-vis-NIR) region by means of a Hitachi UH4150 UV-vis spectrophotometer equipped with an integrating sphere. To avoid the saturation, samples (30 mg) were diluted with BaSO4 (270 mg). The diffuse reflectance spectra were measured using BaSO4 as a reference to be transformed to the absorption spectra using the Kubelka–Munk function F(R∞) = (1 − R∞)2/2R∞, where R∞ is the reflectivity for thick enough sample. The photoluminescence (PL) spectra were collected by means of a JASCO FP-6000 spectrofluorometer with varying excitation wavelengths at 77 K. The particle size distribution of ATO, ZnO, and the mixture, and their ζ-potential in a 4% ethanol solution were measured using a Zetasizer Nano ZS (Malvern Instruments).Photocatalytic H2O2 production
ZnO (50 mg) and ATO (100 mg) were dispersed into 4 vol% or 90 vol% aqueous EtOH solution (50 mL) by using ultrasonic irradiation, unless otherwise stated. This suspension was placed in the inner cell (100 mL) of a double-jacketed reaction vessel, and the temperature of the reaction solution was maintained at 298 K by circulating constant temperature water in the outer cell during light irradiation. During the reaction, O2 gas bubbling was continued with a flow rate of 200 sccm. When the EtOH concentration (CEtOH) is 4 vol%, the rate of vaporization was negligibly small (∼0.04 mL h−1) under the conditions. However, the evaporation rate reached 0.6 mL h−1 at CEtOH = 90 vol%, so the reaction suspension was continuously replenished with 90 vol% EtOH aqueous solution at the same rate using a syringe pump (Pump 11, Harvard Apparatus). Experiments on isotope effects were performed at a tenth scale. After exposure to light for a given period of time, the particles were removed from the dispersion using a membrane filter. The H2O2 concentration in the resulting solution was determined by a spectrophotometric method with Cu(II) ion and 2,9-dimethyl-1,10-phenanthroline.22 We confirmed that this method allows accurate quantification of H2O2 even in the presence of EtOH and oxidation products (CH3CHO and CH3COOH).To determine the ϕex, monochromatic light was irradiated by using LED with various emission wavelength (CCS inc.). ZnO (10 mg) and ATO (10 mg) were dispersed in a 4 vol% aqueous EtOH solution (10 mL) by ultrasonic irradiation. This suspension was placed in the inner cell (100 mL) of a double-jacketed reaction vessel, and the temperature of the reaction solution was maintained at 298 K by circulating constant temperature water in the outer cell during light irradiation. During the reaction, O2 gas bubbling was continued with a flow rate of 200 sccm. After irradiation for 15 min, the particles were removed from the suspension using a membrane filter, and the H2O2 concentration was quantified by the spectrophotometric method with Cu(II) ion and 2,9-dimethyl-1,10-phenanthroline.22 The number of incident photons (Nphoton) was calculated from the relation of Nphoton = I/(hc/λ), where I (W cm−2) is the light intensity, h is Planck constant, c is the seed of light, λ is the wavelength of incident light. From the values of NH2O2 and Nphoton, the ϕex was calculated by eqn (4) as a function of I.
Photodeposition of Ag and CoOx
In the Ag photodeposition, ATO-CL/ZnO particles (30 mg) were ultrasonically dispersed in 4 vol% aqueous EtOH solution (10 mL) with AgNO3 (1 mM) for 10 s in a glass test tube. The resulting suspension was degassed by bubbling Ar with a stainless-steel syringe needle for 30 min with stirring. The test tube was capped with a rubber lid and set inside a double-jacketed glass reaction vessel. The suspension was irradiated by two ultraviolet-light emitting diodes (UV-LED) (λ = 365 nm, I = 120 mW cm−2, HLV-24UV365-4WNRBTNJ, CCS Inc.) at 298 K for 2 h. The resulting particles were collected and washed with water by centrifugal separation.In the CoOx photodeposition, ATO-CL/ZnO particles (30 mg) were ultrasonically dispersed in 10 mL of aqueous Co(NO3)2 solution (1 mM) for 10 s in a glass test tube. The suspension was bubbled with O2 gas using a stainless-steel syringe needle for 30 min with stirring. The test tube was capped with a rubber lid and set inside a double-jacketed glass reaction vessel. After the suspension was irradiated under the same irradiation conditions as the Ag photodeposition, the resulting particles were collected and washed with water by centrifugal separation.
Electrochemical measurements
EC measurements were performed using two-component three-electrode cell consisting of ATO-CL/ZnO nanorod array (NRA) (working electrode), Ag/AgCl (reference electrode)|0.1 M NaClO4 aqueous solution containing 4 vol% EtOH|Nafion|0.1 M NaClO4 aqueous solution containing 4 vol% EtOH|Pt film (counter electrode) in the dark. The active area of working electrode is 1 cm2 (1 cm × 1 cm). By the reported procedure, ZnO NRA was formed on FTO.23 ATO (2 mg) was dispersed into H2O (0.4 mL), and the resulting suspension was dropped onto ZnO NRA. The sample was dried at 353 K to obtain ATO-CL/ZnO NRA electrode. After bubbling by argon or O2 gas for 30 min, the linear sweep voltammetry curves were measured by means of a galvanostat/potentiostat (HZ-7000, Hokuto Denko) with scan rate = 20 mV s−1 in the dark. The faradaic efficiency (η) was calculated at irradiation time (t) = 30 min by eqn (6).| η = 2N(t)/(∫I(t)dt/F) | (6) |
Photoelectrochemical H2O2 production
Two-component three-electrode cells were fabricated with a structure of ZnO NRA (working electrode), Ag/AgCl (reference electrode)|0.1 M NaClO4 aqueous solution containing 4 vol% EtOH (80 mL)|Nafion|0.1 M NaClO4 aqueous solution containing 4 vol% EtOH (80 mL)|ATO/FTO (counter electrode). ATO (2 mg) was dispersed into H2O (0.4 mL), and the suspension was dropped onto FTO. The active area of working electrode is 1 cm2 (1 cm × 1 cm). The sample was dried at 353 K to obtain ATO/FTO electrode. After O2 bubbling each component for 30 min, the working electrode was illuminated by LED lamp (λex = 365 ± 20 nm, intensity = 120 mW cm−2) at 298 K. The photocurrent was measured by means of a galvanostat/potentiostat (HZ-7000, Hokuto Denko) at dark rest potential. The H2O2 concentration was quantified by the spectrophotometric method with Cu(II) ion and 2,9-dimethyl-1,10-phenanthroline.22 The faradaic efficiency (η) was also calculated by eqn (6).Results
Preparation and characterization of ATO-CL/ZnO photocatalysts
Commercial particles of ATO (Sb-doped amount = 11.6 mol%, SN-100P, Ishihara Sangyo) and ZnO (Sigma-Aldrich) were used as received. The specific surface area of ATO and ZnO particles were determined to be 58.5 m2 g−1 and 9.5 m2 g−1, respectively, by the BET method. In the XRD pattern of ATO particles, diffraction peaks are observed at 2θ = 26.66°, 33.94°, 37.98°, and 51.80° assignable to the diffraction from the (110), (101), (200), and (211) planes of tetragonal SnO2 (Fig. 1a). Also, the XRD pattern of ZnO has strong diffraction peaks at 2θ = 31.74°, 34.40°, and 36.24° due to the diffraction from the (10![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 0), (0002), and (101) planes of hexagonal ZnO. The crystallite sizes of ATO and ZnO were determined to be 4.3 nm and 41.5 nm, respectively, using Scherrer's equation from the ATO (110) and ZnO (100) diffraction line widths, respectively. TEM images show that the ZnO sample consists of faceted NPs involving short nanorods with a large variation in size (Fig. S2†), while the ATO sample forms clusters of aggregated particles smaller than 10 nm (Fig. S3†). After the ATO (100 mg) and ZnO NPs (50 mg) were dispersed in 4 vol% ethanol aqueous solution (50 mL) by ultrasonic irradiation for 1 min and stirred for 60 min, the solids were recovered by centrifugation to be washed and dried. The XRD pattern of the obtained sample is a superposition of those for ATO and ZnO. The TEM image of the hybrid sample shows that the ZnO surface is partly modified with ATO clusters (ATO-CL/ZnO, Fig. 1b) as more clearly shown by the TEM-energy dispersive X-ray spectroscopy (EDS) elemental mapping (Fig. 1c).
0), (0002), and (101) planes of hexagonal ZnO. The crystallite sizes of ATO and ZnO were determined to be 4.3 nm and 41.5 nm, respectively, using Scherrer's equation from the ATO (110) and ZnO (100) diffraction line widths, respectively. TEM images show that the ZnO sample consists of faceted NPs involving short nanorods with a large variation in size (Fig. S2†), while the ATO sample forms clusters of aggregated particles smaller than 10 nm (Fig. S3†). After the ATO (100 mg) and ZnO NPs (50 mg) were dispersed in 4 vol% ethanol aqueous solution (50 mL) by ultrasonic irradiation for 1 min and stirred for 60 min, the solids were recovered by centrifugation to be washed and dried. The XRD pattern of the obtained sample is a superposition of those for ATO and ZnO. The TEM image of the hybrid sample shows that the ZnO surface is partly modified with ATO clusters (ATO-CL/ZnO, Fig. 1b) as more clearly shown by the TEM-energy dispersive X-ray spectroscopy (EDS) elemental mapping (Fig. 1c).
 | ||
| Fig. 1 (a) XRD patterns of ATO, ZnO, and ATO-CL/ZnO. (b) TEM image of ATO-CL/ZnO. (c) EDS elemental mapping of ATO-CL/ZnO for the TEM image in (b). (d) Size distributions of ATO and ZnO NP suspensions, and their mixture in 4 vol% ethanol aq. (pH 6.4) measured by the DLS method. (e) ζ-Potentials of ATO and ZnO NP suspensions, and their mixture in 4% ethanol aq. (pH 6.4). HR-TEM images of ATO-CL/ZnO (f) and ht-ATO-CL/ZnO (g). In each image, the interface is indicated by a dotted rectangle. | ||
The size distribution of ATO (100 mg) and ZnO (50 mg) particles dispersed in 4 vol% ethanol aqueous solution (50 mL) before and after mixing was measured by dynamic light scattering (DLS) (Fig. 1d). In each suspension, ATO and ZnO particles exist as aggregates with the mean size of 188 nm and 335 nm, respectively. On mixing both the suspensions with excess ATO, the peak size of ZnO shifts to 447 nm, while a peak of non-coupled ATO particles is observed at 159 nm. Evidently, self-assembling of ATO and ZnO particles occurs only by mixing both the suspensions. ζ-Potentials were measured in 4 vol% ethanol aqueous solution (pH 6.4) by the electrophoretic method (Fig. 1e). The ζ-potentials of ATO and ZnO particles are −24.8 mV and +20.8 mV, respectively. ATO-CL/ZnO has a value of −18.6 mV, which is intermediate between the values of ATO and ZnO. Clearly, the hybridization of the ATO and ZnO particles is caused by the electrostatic attraction between them in the near-neutral aqueous solution. The hybrid sample prepared by mixing ATO (x/mg) and ZnO (y/mg) in 4 vol% ethanol aqueous solution (50 mL) is designated as ATO-CL(x)/ZnO(y) below.
In the HR-TEM image of ATO-CL(100)/ZnO(50) (Fig. 1f), the interplanar spacings of fringes observed in the small crystals of 0.26 nm and 0.31 nm are close to the values of the SnO2(101) and (001) planes, respectively. Also, the d-spacing of 0.24 nm in the large particle is in agreement with the value of ZnO (101). As indicated by the yellow arrows, some voids are observed near the interface between ATO and ZnO particles. ATO-CL(100)/ZnO(50) was further heated at 673 K for 1 h in air (ht-ATO-CL(100)/ZnO(50)). TEM-EDS mapping of ht-ATO-CL(100)/ZnO(50) shows that the ZnO particle surfaces are effectively modified with ATO clusters (Fig. S4†), similar to that of ATO-CL(100)/ZnO(50) before calcination (Fig. 1b and c). Among the many particles observed, all ZnO particles were coupled to ATO clusters without independent ZnO particles. Importantly, the HR-TEM image of ht-CL(100)/ZnO(50) shows that the voids observed in the unheated sample almost disappear, and some of the ATO NPs in the cluster are tightly bonded to the ZnO surface (Fig. 1g). Obviously, the heat treatment makes the ZnO and ATO particles more firmly bonded at the interface and increases the contact area.
Diffuse reflectance UV-vis-NIR absorption spectra of ZnO, ATO, and ATO-CL(100)/ZnO(50) were measured. In the Kubelka–Munk-transformed absorption spectra of ZnO and ATO (Fig. S5†), the absorption due to the interband transition rises at light wavelength (λ) < 380 nm and λ < ∼340 nm, respectively. ATO has also strong absorption by free electrons in the NIR region,23 and the absorption tail extending from 340 nm to ∼400 nm indicates the existence of mid-gap levels due to defects. From the Tauc plots for ZnO and ATO (Fig. S6†), the allowed and inhibited direct band gaps were determined to be 3.27 eV for ZnO and 3.53 eV for ATO, respectively, which are close to the reported values of 3.26 eV![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 24 and 3.60 eV.25 The absorption spectrum of ATO-CL(100)/ZnO(50) consists of the superposition of the ATO and ZnO spectra.
24 and 3.60 eV.25 The absorption spectrum of ATO-CL(100)/ZnO(50) consists of the superposition of the ATO and ZnO spectra.
Photocatalytic H2O2 production
The photocatalytic activities of ATO, ZnO, and ATO-CL(100)/ZnO(50) for H2O2 generation from aerated 4% ethanol solution (pH 6.4, 50 mL) were evaluated at 298 K under UV-light irradiation (λ = 365 nm) by comparing that of the typical Au/TiO2 photocatalyst (Fig. 2a and S7†).21 During the reaction, O2 gas bubbling was continued with a flow rate of 200 standard cubic centimetre per min (sccm), where the change in H2O2 concentration accompanying the evaporation of the solvent was negligible. Au/TiO2 initially shows high photocatalytic activity for H2O2 production, which significantly decreases over photoirradiation time. As a result, the H2O2 concentration reaches an almost constant value of 18.1 ± 0.7 mM at tp ≥ 12 h. While ATO itself is inactive, ZnO produces H2O2 with an almost constant rate of 1.2 mM h−1, and the H2O2 concentration at tp = 24 h of 28.8 mM exceeds the value of 18.8 mM in the Au/TiO2 system. Strikingly, in the ATO-CL/ZnO system, the concentration of H2O2 continues to increase to reach 105 mM at tp = 24 h. Also, gas chromatography analysis confirmed that acetaldehyde is produced in the gas phase as an oxidation product of ethanol. The photocatalytic activity of ATO-CL/ZnO strongly depends on the mixing ratio of ATO and ZnO with a maximum at x/y = 2 (Fig. S8†). | ||
| Fig. 2 (a) Time courses for H2O2 generation from 4 vol% ethanol aqueous solutions (pH 6.4, 50 mL) in the ATO (100), ZnO(50), ATO-CL(100)/ZnO(50) systems, and for comparison, Au/TiO2 system under UV-light irradiation (λ = 365 nm) using two LED lamps (each light intensity = 120 mW cm−2). (b) Ethanol concentration effect on the photocatalytic activities of ht-ATO-CL(100)/ZnO(50) under the same irradiation conditions as (a). (c) Action spectrum of external quantum yield for the photocatalytic H2O2 generation in the ATO-CL(10)/ZnO(10) system (pH 6.4, 50 mL). (d) Plots of external quantum yield vs. light intensity. The data in the O2-saturated and air-saturated solutions are solid triangles and open triangle, respectively. | ||
In the ATO-CL/ZnO system, the amount of H2O2 generated was small without ethanol in the reaction solution (9.1 μM), and then, the photocatalytic activity was assessed by increasing the ethanol concentration from 4 vol% to 90 vol% (Fig. 2b). In this case, the evaporation rate reached 0.6 mL h−1, so the reaction suspension was continuously replenished with 90 vol% ethanol aqueous solution at the same rate using a syringe pump. In the ATO-CL/ZnO system, the photocatalytic activity significantly decreases (Fig. S9†). On the contrary, the photocatalytic activity of ht-CL(100)/ZnO(50) drastically increases with the H2O2 concentration to reach 162 mM at tp = 24 h. These results indicate that at least partial tight bonding between ATO and ZnO is essential for ATO-CL/ZnO to exhibit high photocatalytic activity (Fig. 1f and g). In high-concentration ethanol aqueous solutions, heat treatment of a mixture of ATO and ZnO particles at 673 K is effective for this purpose, but above 773 K, the photocatalytic activity of ZnO decreases (Fig. S10†), probably due to the reduction in surface area. To check the stability of ht-CL(100)/ZnO(50), the XRD patterns of the samples collected after 24 h of reaction were measured. The XRD patterns show almost no change before and after the reaction (Fig. S11†).
Catalytic and photocatalytic activities of ATO-CL(100)/ZnO(50) and Au/TiO2 for H2O2 decomposition were studied in dark and under UV-light irradiation, respectively (Fig. S12†). In the Au/TiO2 system, the concentration of H2O2 decreases in dark due to the adsorption and the catalytic decomposition of H2O2, which is considerably accelerated by photoirradiation. On the other hand, in the ATO-CL/ZnO system, the H2O2 concentration remains almost constant in dark and even under photoirradiation.
The photocatalytic activity of ATO(100)/ZnO(50) was evaluated using LEDs with varying emission wavelengths as the light source otherwise under the same conditions (Fig. 2c). The action spectrum of ϕex rises at λ = 365 nm, well tracing the absorption spectrum of ZnO, and thus, this photocatalytic reaction is mainly driven by the band gap excitation of ZnO. In this case, the optical absorption of ZnO is hardly blocked by ATO CLs since ATO is nearly transparent to the UV region where the ZnO has strong absorption (Fig. S5†). Further, the light intensity-dependence of ϕex was examined at λ = 365 nm (Fig. 2d). The ϕex increases with lowering light intensity, and surprisingly, reaches ∼500% at an incident photon flux of 1.6 × 1014 cm−2 s−1. When the reaction was carried out in air-saturated solution, the ϕex of 109% lowers to 13% at an incident photon flux of 1.6 × 1016 cm−2 s−1.
Discussion
To gain insight into the origin for the extremely high photocatalytic activity of ATO-CL/ZnO, the redox sites were firstly studied using the labeling and visualizing technique by photodeposition.26 To specify the reduction sites, Ag NPs were photodeposited on ht-ATO-CL/ZnO from a degassed AgNO3 aqueous solution containing 4% ethanol under UV irradiation (λ = 365 nm). TEM-EDS elemental mapping for the resulting sample shows that Ag NPs are deposited on the ATO CL and close to the interface with ZnO (Fig. 3a). Also, to specify the oxidation sites, CoOx photodeposition on ht-ATO-CL/ZnO was carried out from an O2-saturated Co(NO3)2 aqueous solution under the same irradiation conditions.27,28 In contrast to the Ag photodeposition, CoOx species are selectively formed on the surface of ZnO (Fig. 3b). These results indicate that in the ATO-CL/ZnO system, ATO CL and ZnO act as the reduction sites and oxidation sites, respectively, through effective charge separation.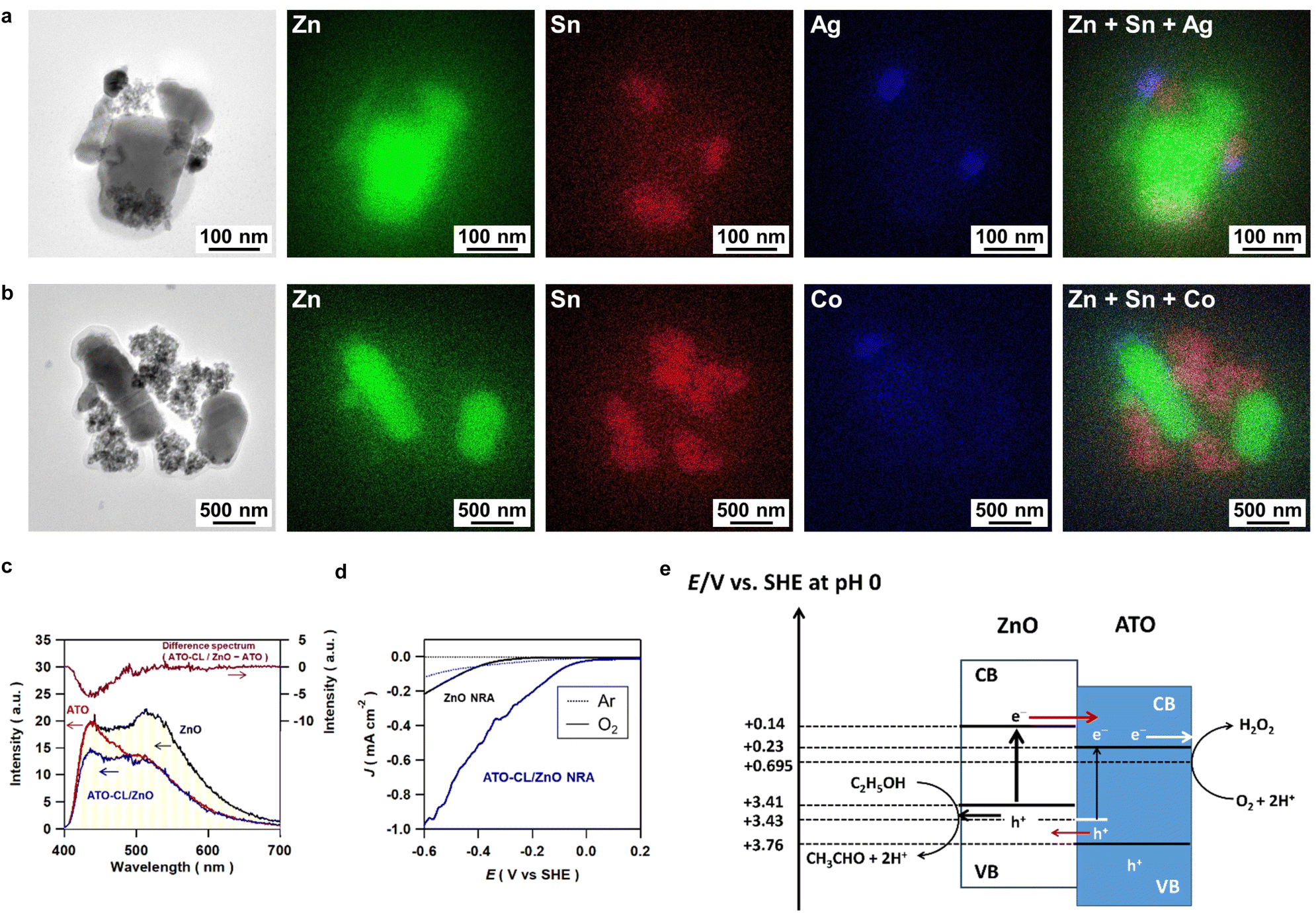 | ||
| Fig. 3 (a) TEM-EDS elemental mapping of Ag-photodeposited ht-ATO-CL(20)/ZnO(10). (b) TEM-EDS elemental mapping of CoOx-photodeposited ht-ATO-CL(20)/ZnO(10). (c) PL spectra of ATO, ZnO, and ATO-CL(100)/ZnO(50) NPs on excitation by light with wavelength of 365 nm at 77 K. (d) Dark current (J)-potential (E) curves of ZnO NRA and ATO-CL/ZnO NRA electrodes in argon gas (dotted lines) and oxygen gas (solid lines)-bubbled 0.1 M NaClO4 electrolyte solution containing 4 vol% ethanol. (e) A reaction mechanism proposed for the present ATO-CL/ZnO-photocatalyzed H2O2 production. | ||
Next, photoluminescence (PL) spectra were measured under excitation by light with λ = 365 nm at 77 K (Fig. 3c). Under these conditions, the interband transition of ZnO and the excitation of the electrons from the mid-gap states of ATO to the conduction band (CB) are possible. In the spectrum of ZnO, broad emission bands are present around 429 nm (E1) and 525 nm (E2), which are assignable to the near band-to-band emission and the defect-level-related emission, respectively. ATO has emission at 429 nm (E3) with a shoulder around 500 nm due to the defect-level-related emission. As shown by the difference spectrum (ATO-CL/ZnO–ATO), the E2 and E1 signals of ZnO disappear with the coupling with ATO, which can be attributed to the interfacial electron transfer from the CB of ZnO to ATO in a manner similar to the ATO/TiO2 system.29 On the contrary, the E3 signal of ATO weakens, suggesting simultaneous occurrence of the hole transfer from the deep defect levels of ATO to the valence band (VB) of ZnO.
Further, ZnO nanorod array was formed on fluorine-doped tin oxide electrode (ZnO NRA/FTO) for EC measurements (Fig. S13†).30 The dark current (J)-potential (E) curves were measured using a three-electrode EC cell with the structure of ZnO NRA/FTO or ATO-CL/ZnO NRA/FTO (working electrode)|0.1 M NaClO4 aq. containing 4 vol% ethanol (electrolyte solution, 80 mL)|Ag/AgCl (reference electrode)|Pt (auxiliary electrode) (Fig. 3d). In a degassed electrolyte solution, only a small current flows at −0.6 ≤ E ≤ 0 V in each electrode system. On the other hand, in the O2-saturated electrolyte solution, a cathodic current due to ORR rises at E = −0.3 V in the ZnO NRA/FTO electrode system. The current is further increased significantly in the ATO-CL/ZnO NRA/FTO electrode system with the current onset potential shifted to ∼0 V.
Based on the results above, the action mechanism of ATO-CL/ZnO is discussed. The energy diagram was constructed using the flatband potentials and band gaps of ZnO (0.14 V, 3.27 eV)31 and ATO (0.229 V, 3.53 eV)32 (Fig. 3e). When ATO and ZnO NPs were dispersed in near neutral 4 vol% ethanol aqueous solution, the surfaces of ATO and ZnO NPs are strongly negatively and positively charged, respectively. As a result, they are coupled in situ through the electrostatic interaction to yield ATO-CL/ZnO nanohybrids. Upon UV-light irradiation (λ = 365 nm) of ATO-CL/ZnO, the electrons in the VB of ZnO are promoted to the CB with simultaneous excitation of the deep defect levels in ATO to the CB (eqn (7)). As indicated by the Ag and CoOx photodeposition and the PL spectra, the CB electrons in ZnO (E ≈ 0.14 V) are transferred to ATO with the lower-lying CB minimum (E ≈ 0.48 V), and at the same time, the holes generated in the deep defect levels in ATO (E ≈ 3.43 V) can be moved to the VB of ZnO (E ≈ 3.41 V) (eqn (8)). Consequently, effective charge separation can be achieved by the interfacial electron and hole transfer in the opposite direction. The electrons in the CB of ATO efficiently reduce O2 to H2O2 (E0(O2/H2O2) = 0.695 V) with the assistance by its electrocatalysis for 2e−-ORR (eqn (9)),20 while the VB holes in ZnO oxidizes ethanol (eqn (10)).33,34 In this case, even if part of the ATO cluster is in contact with ZnO, the large-surface-area ATO cluster facilitates 2e−-ORR because of the delocalization of the injected electrons throughout the cluster due to the interparticle electron transfer.35 Further, the amount of H2O2 increases almost in proportion to irradiation time owing to the low catalytic and photocatalytic activity of ATO-CL/ZnO for H2O2 decomposition. In 90 vol% ethanol aqueous solution, the significant coupling effect can only be obtained after heat treatment of ATO-CL/ZnO because of the decrease in the amounts of the surface charges, which points out the importance of the intimate junction between ATO and ZnO for the charge separation, and further the photocatalytic activity.36
| ATO/ZnO + hν → ATO(e−⋯h+)/ZnO(e−⋯h+) | (7) |
| ATO(e−⋯h+)/ZnO(e−⋯h+) → ATO(e−)/ZnO(h+) | (8) |
| ATO(e−)/ZnO(h+) + 1/2O2 + H+ → ATO/ZnO(h+) + 1/2H2O2 | (9) |
| ATO/ZnO(h+) + CH3CH2OH → ATO/ZnO + CH3ĊHOH + H+ | (10) |
Consequently, the remarkable photocatalytic performances of ATO-CL/ZnO and ht-ATO-CL/ZnO for H2O2 production can stem from the following features: (1) effective charge separation via the interfacial electron and hole transfer through the intimate junction between the components, (2) high electric conductivity and electrocatalytic activity of ATO for 2e−-ORR, (3) low catalytic and photocatalytic activities of ATO and ZnO for H2O2 decomposition. However, these features alone cannot explain the origin of the ϕex above 100%.
Then, to explicate the reactions on the reduction sites (ATO) and oxidation sites (ZnO) of the ATO-CL/ZnO photocatalyst, a two-compartment three-electrode PEC cell with the structure ZnO NRA/FTO (working electrode), Ag/AgCl (reference electrode)|0.1 M NaClO4 aq. containing 4 vol% ethanol (80 mL)|Nafion|O2-saturated 0.1 M NaClO4 aq. (80 mL)|ATO/FTO (auxiliary electrode) was fabricated as a model for the particulate system (Fig. 4a). Light irradiation of the ZnO NRA/FTO electrode (λ = 365 nm) for 30 min yielded 10 μM H2O2 in the cathode compartment and 30 μM H2O2 in the anode compartment for which the faradaic efficiencies were calculated to be 81% and 243%, respectively. Obviously, H2O2 is produced in the ATO-CL/ZnO-photocatalyzed reaction via not only 2e−-ORR on the ATO electrode (Fig. 4a, right) but also ethanol oxidation on the ZnO photoelectrode (Fig. 4b, left). Even if ethanol is oxidized on the ZnO photoelectrode with a current doubling effect,37 the faradaic efficiency should remain at an upper limit of 200%.
 | ||
| Fig. 4 (a) Photoelectrochemical cell with the structure ZnO NRA/FTO (working electrode), Ag/AgCl (reference electrode)|0.1 M NaClO4 aq. containing 4 vol% ethanol|Nafion|O2-saturated 0.1 M NaClO4 aq.|ATO/FTO (auxiliary electrode). For simplicity, the Ag/AgCl reference electrode is omitted in this figure. (b) Additive effect of various alcohols on the ATO-CL(100)/ZnO(50)-photocatalyzed H2O2 production under UV-light irradiation (λ = 365 nm, light intensity = 1 mW cm−2, photon flux = 1.6 × 1016 cm−2 s−1). (c) Time courses for ATO-CL(100)/ZnO(50)-photocatalyzed H2O2 production in the 4 vol% C2H5OH and C2D5OD aqueous solutions at 288 K. | ||
To explain the faradaic efficiency above 200% in the photoanodic reaction, we propose the following chain reaction mechanism (Fig. 4a, left). When CH3CH2OH is oxidized by the holes in the VB of ZnO, CH3ĊHOH radical and H+ are generated (eqn (10)). CH3ĊHOH radical reacts with O2 to yield CH3CH(OH)OȮ radical (eqn (11)). Extraction of H atom of CH3CH2OH by CH3CH(OH)OȮ radical affords CH3CH(OH)OOH with CH3ĊHOH radical reproduced (eqn (12)). CH3CH(OH)OOH is decomposed to H2O2 and CH3CHO (eqn (13)).
| CH3ĊHOH + O2 → CH3CH(OH)OȮ | (11) |
| CH3CH(OH)OȮ + CH3CH2OH → CH3CH(OH)OOH + CH3ĊHOH | (12) |
| CH3CH(OH)OOH → H2O2 + CH3CHO | (13) |
It has recently been observed in a TiO2 photoelectrode system that a large photocurrent due to the current doubling of C2H5OH in a degassed electrolyte solution is halved by in the presence of O2.38 This result indicates that the current doubling of C2H5OH is inhibited due to the rapid reaction of CH3ĊHOH with O2 in the solution (eqn (11)). For the reaction to occur according to this reaction scheme, the alcohol must have at least one H atom on the α-carbon. To confirm this, the effect of adding various alcohols on the ATO-CL/ZnO-photocatalyzed H2O2 production was investigated. As shown by the data (Fig. 4b), the external quantum efficiency decreases in the following order: primary alcohols (ethanol, methanol) > secondary alcohol (2-propanol) > tertiary alcohol (tert-butanol). In fact, the addition of butanol bearing no H atom at the α-position hardly accelerates the reaction.
4-Nitrophenol was reported to be a scavenger of ethyl radial (CH3ĊH2).39 Then, the effect of adding 4-nitrophenol on the photocatalytic activity of ATO-CL/ZnO was examined. The amount of H2O2 produced after 15 min UV-light irradiation decreases with increasing concentration of 4-nitrophenol (Fig. S14†). Further, since the propagation process in the chain reaction involves the C–H bond cleavage, the kinetic isotope effect was investigated by measuring the rates of reaction in the C2H5OH and C2D5OD systems at 288 K. In both the systems, the reaction apparently follows the zero-order rate law at tp ≤ 2 h (Fig. 4c), and the rate of H2O2 generation in the C2D5OD system (0.52 mM h−1) decreases as compared to the value in the C2H5OH system (0.69 mM h−1). All these results support the chain reaction mechanism, which explains the faradaic efficiency of PEC H2O2 production at the ZnO NRA photoanode exceeding 200% and the external quantum yield of ATO-CL/ZnO-photocatalyzed reaction reaching approximately 500%.
In the TiO2 photocatalytic reaction, the degradation of alcohols and aldehydes is known to proceed in the presence of O2via the autooxidation involving radical chain reactions.40 Also, the research group of Hashimoto and Fujishima previously studied PEC oxidation of ethanol on a Pd patterned film-coated TiO2 photoelectrode in the presence of O2. The authors demonstrated that H2O2 is produced via a similar radical chain reaction (eqn (10)–(13)) on the oxidation site (or TiO2 surface) but not on the reduction site (or Pd surface).41 However, in the TiO2-photocatalyzed ORR in aerated ethanol aqueous solution, the concentration of H2O2 produced saturates only at ∼0.35 mM.42 This is because H2O2 is chemisorbed onto TiO2 and the resulting surface complex readily undergoes reductive photocatalytic decomposition.43
Conclusions
In the photocatalytic H2O2 production, increasing the quantum yield and production amount is a major challenge, while it is also important from the viewpoint of artificial photosynthesis to synthesize H2O2 from O2 and H2O under visible light irradiation. The key findings in this study are summarized as follows (Scheme 1). Only mixing of ZnO and ATO NPs in near-neutral solution spontaneously forms a nanohybrid consisting of ATO clusters and ZnO due to the electrostatic interaction between them (ATO-CL/ZnO). Noble metal-free ATO-CL/ZnO possesses the features of effective separation of reduction sites (ATO) and oxidation sites (ZnO) through the interfacial charge transfer between the components, high electric conductivity and electrocatalytic activity of ATO for 2e−-ORR, and very low affinity of ATO and ZnO for H2O2. Consequently, UV-light irradiation (λ = 365 nm) of ATO-CL/ZnO in O2-saturated ethanol solution yields 162 mM H2O2 (8.0 mmol) at tp = 24 h. Further, an unparalleled high external quantum yield of ∼500% can be achieved through 2e−-ORR by the CB electrons over ATO and the VB hole-induced radical chain reaction over ZnO. Finally, it should be emphasized that the present photocatalytic and PEC systems yield H2O2via both the reduction and oxidation processes, which is a great advantage over water splitting. Rational design of the entire reaction system including the photocatalyst materials can break through the quantum yield limit of unity (100%) to open up the practical on-site H2O2 production. | ||
| Scheme 1 Schematic description of the action mechanism of ATO-CL/ZnO photocatalysis for the production of H2O2 from ethanol aqueous solution. | ||
Data availability
The authors confirm that the data supporting the findings of this study are available within the article and its ESI.†Author contributions
Y. Y. and H. S. conducted catalyst synthesis, characterization, photocatalytic experiments, and S. N. carried out EC and PEC experiments, and T. S. and H. T. supervised the work and data analysis.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by JSPS KAKENHI Grant-in-Aid for Scientific Research (C) no. 21K05236 and 23K04545, the Futaba Foundation, Nippon Sheet Glass Foundation for Materials Science and Engineering, Kato Foundation for Promotion of Science, and Izumi Science and Technology Foundation.Notes and references
- J. M. Campos-Martin, G. Blanco-Brieva and J. L. G. Fierro, Hydrogen peroxide synthesis: an outlook beyond the anthraquinone process, Angew. Chem., Int. Ed., 2006, 45, 6962–6984 CrossRef CAS.
- K. Mase, M. Yoneda, Y. Yamada and S. Fukuzumi, Seawater usable for production and consumption of hydrogen peroxide as a solar fuel, Nat. Commun., 2016, 7, 11470 CrossRef CAS.
- H. Tada, Hydrogen peroxide photo-fuel cells, Dalton Trans., 2023, 52, 14733–14741 RSC.
- H. Hou, X. Zeng and X. Zhang, Production of hydrogen peroxide by photocatalytic processes, Angew. Chem., Int. Ed., 2020, 59, 17356–17376 CrossRef CAS PubMed.
- N. Karamoschos and D. Tasis, Photocatalytic evolution of hydrogen peroxide: A minireview, Energies, 2022, 15, 6202 CrossRef CAS.
- S. Lee, H.-S. Bae and W. Choi, Selective control and characteristics of water oxidation and dioxygen reduction in environmental photo(electro)catalytic systems, Acc. Chem. Res., 2023, 56, 867–877 CrossRef CAS PubMed.
- S. Mansingh, K. K. Das, N. Priyadarshini, D. P. Sahoo, D. Prusty, J. Sahu, U. A. Mohanty and K. Parida, Minireview elaborating S-scheme charge dynamic photocatalysts: Journey from Z to S, mechanism of charge flow, characterization proof, and H2O2 evolution, Energy Fuels, 2023, 37, 9873–9894 CrossRef CAS.
- Y. Kofuji, Y. Isobe, Y. Shiraishi, H. Sakamoto, S. Tanaka, S. Ichikawa and T. Hirai, Carbon nitride−aromatic diimide−graphene nanohybrids: Metal-free photocatalysts for solar-to-hydrogen peroxide energy conversion with 0.2% efficiency, J. Am. Chem. Soc., 2016, 138, 10019–10025 CrossRef CAS.
- L. Shi, L. Yang, W. Zhou, Y. Liu, L. Yin, X. Hai, H. Song and J. Ye, Photoassisted construction of holey defective g-C3N4 photocatalysts for efficient visible-light-driven H2O2 production, Small, 2018, 4, 1703142 CrossRef.
- Q. Tian, L. Jing, S. Ye, J. Liu, R. Chen, C. A. H. Price, F. Fan and J. Liu, Nanospacial charge modulation of monodispersed polymeric microsphere photocatalysts for exceptional hydrogen peroxide production, Small, 2021, 17, 2103224 CrossRef CAS.
- L. Wang, J. Zhang, Y. Zhang, H. Yu, Y. Qu and J. Yu, Inorganic metal-oxide photocatalyst for H2O2 production, Small, 2022, 18, 2104561 CrossRef CAS PubMed.
- Y. Shiraishi, T. Takii, T. Hagi, S. Mori, Y. Kofuji, Y. Kitagawa, S. Tanaka, S. Ichikawa and T. Hirai, Resorcinol-formaldehyde resins as metal-free semiconductor photocatalysts for solar-to-hydrogen peroxide energy conversion, Nat. Mater., 2019, 18, 985–993 CrossRef CAS PubMed.
- Z. Teng, Q. Zhang, H. Yang, K. Kato, W. Yang, Y.-R. Lu, S. Liu, C. Wang, A. Yamakata, C. Su, B. Liu and T. Ohno, Atomically dispersed antimony on carbon nitride for the artificial photosynthesis of hydrogen peroxide, Nat. Catal., 2021, 4, 374–384 CrossRef CAS.
- W. Fan, B. Zhang, X. Wang, W. Ma, D. Li, Z. Wang, M. Dupuis, J. Shi, S. Liao and C. Li, Efficient hydrogen peroxide synthesis by metal-free polyterthiophene via photoelectrocatalytic dioxygen reduction, Energy Environ. Sci., 2020, 13, 238–245 RSC.
- J. D. Xiao, Q. Han, Y. Xie, J. Yang, Q. Su, Y. Chen and H. Cao, Is C3N4 chemically stable toward reactive oxygen species in sunlight-driven water treatment?, Environ. Sci. Technol., 2017, 51, 13380–13387 CrossRef CAS PubMed.
- T. Liu, Z. Pan, J. J. M. Vequizo, K. Kato, B. Wu, A. Yamakata, K. Katayama, B. Chen, C. Chu and K. Domen, Overall photosynthesis of H2O2 by an inorganic semiconductor, Nat. Commun., 2022, 13, 1034 CrossRef CAS.
- Z. Liu, X. Sheng, D. Wang and X. Feng, Generation utilizing photocatalytic oxygen reduction at a triphase interface, iScience, 2019, 17, 67–73 CrossRef CAS PubMed.
- X. Zhang, D. Gao, B. Zhu, B. Cheng, J. Yu and H. Yu, Enhancing photocatalytic H2O2 production with Au co-catalysts through electronic structure modification, Nat. Commun., 2024, 15, 3212 CrossRef CAS PubMed.
- H. Kominami, K. Kamitani and A. Tanaka, Rapid production of 38 mM H2O2 in an alcoholic suspension of a WO3 photocatalyst under visible light, Chem. Commun., 2024, 60, 7017–7020 RSC.
- H. Tada and S. Naya, Antimony-doped tin oxide catalysts for green and sustainable chemistry, J. Phys. Chem. C, 2022, 126, 13539–13547 CrossRef CAS.
- H. Tada, M. Teranishi and S. Naya, Hydrogen peroxide production by inorganic photocatalysts consisting of gold nanoparticle and metal oxide towards oxygen cycle chemistry, J. Phys. Chem. C, 2023, 127, 5199–5209 CrossRef CAS.
- K. Kosaka, H. Yamada, S. Matsui, S. Echigo and K. Shishida, Comparison among the methods for hydrogen peroxide measurements to evaluate advanced oxidation processes: application of a spectrophotometric method using copper(II) ion and 2,9-dimethyl-1,10-phenanthroline, Environ. Sci. Technol., 1998, 32, 3821–3824 CrossRef CAS.
- J. M. Xu, L. Li, S. Wang, H. L. Ding, Y. X. Zhang and G. H. Li, Influence of Sb doping on the structural and optical properties of tin oxide nanocrystals, CrystEngComm, 2013, 15, 3296–3300 RSC.
- S. Liang, X. Gou, J. Cui, Y. Luo, H. Qu, T. Zhang, Z. Yang, Q. Yang and S. Sun, Novel cone-like ZnO mesocrystals with co-exposed (10-11) and (000-1) facets and enhanced photocatalytic activity, Inorg. Chem. Front., 2018, 5, 2257–2267 RSC.
- H. Suzuki, J. Yamauchi, S. Naya, H. Sugime and H. Tada, Noble metal-free inorganic photocatalyst consisting of antimony-doped tin oxide nanorod and titanium oxide for two-electron oxygen reduction reaction, ChemPhysChem, 2022, 23, e202200029 Search PubMed.
- H. Tada, A. Hattori, Y. Tokihisa, K. Imai, N. Tohge and S. Ito, A patterned−TiO2/SnO2 bilayer type photocatalyst, J. Phys. Chem. B, 2000, 104, 4585–4587 CrossRef CAS.
- T. Takata, J. Jiang, Y. Sakata, M. Nakabayashi, N. Shibata, V. Nandal, K. Seki, T. Hisatomi and K. Domen, Photocatalytic water splitting with a quantum efficiency of almost unity, Nature, 2020, 581, 411–414 CrossRef CAS.
- M. Okazaki, Y. Suganami, N. Hirayama, H. Nakata, T. Oshikiri, T. Yokoi, H. Misawa and K. Maeda, Site-selective deposition of a cobalt cocatalyst onto a plasmonic Au/TiO2 photoanode for improved water oxidation, ACS Appl. Energy Mater., 2020, 3, 5142–5146 CrossRef CAS.
- Y. Park, H. Kim, R. C. Pawar, S. Kang and C. S. Lee, Photocatalytic evaluation of ATO/TiO2 heterojunction films fabricated by a nanoparticle deposition system, Mater. Chem. Phys., 2018, 203, 118–124 CrossRef CAS.
- L. E. Greene, M. Law, J. Goldberger, F. Kim, J. C. Johnson, Y. Zhang, R. J. Saykally and P. Yang, Low-temperature wafer-scale production of ZnO nanowire arrays, Angew. Chem., Int. Ed., 2003, 42, 3031–3034 CrossRef CAS.
- J. Bandara and H. C. Weerasinghe, Enhancement of photovoltage of dye-sensitized solid-state solar cells by introducing high-band-gap oxide layers, Sol. Energy Mater. Sol. Cells, 2005, 88, 341–350 CrossRef CAS.
- G. Boschloo and D. Fitzmaurice, Spectroelectrochemistry of highly doped nanostructured tin dioxide electrodes, J. Phys. Chem. B, 1999, 103, 3093–3098 CrossRef CAS.
- O. Akhavan, Photocatalytic reduction of graphene oxides hybridized by ZnO nanoparticles in ethanol, Carbon, 2011, 49, 11–18 CrossRef CAS.
- R. Ghalta, A. Chauhan and R. Srivastava, Selective hydrogenation of cinnamaldehyde, furanic, and aromatic aldehydes over a Z-Scheme heterojunction photocatalyst constituted of Pd NPs supported on g-C3N4/ZnO nanocomposite, ACS Appl. Nano Mater., 2024, 7, 1462–1476 CrossRef CAS.
- K. Tvrdy, P. A. Frantsuzov and P. V. Kamat, Photoinduced electron transfer from semiconductor quantum dots to metal oxide nanoparticles, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 29–34 Search PubMed.
- H. Tada, S. Naya and M. Fujishima, Nanohybrid crystals with heteroepitaxial junctions for solar-to-chemical transformations, J. Phys. Chem. C, 2020, 124, 25657–25666 Search PubMed.
- W. P. Gomes, T. Freund and S. R. Morrison, Chemical reactions involving holes at the zinc oxide single crystal anode, J. Electrochem. Soc., 1968, 115, 818–823 Search PubMed.
- H. Tada, T. Masuda, S. Naya, H. Sugime, S. Tanaka and M. Fujishima, Efficient photon-to-current and hydrogen conversion by decomposition of formic acid on TiO2 photoanode, J. Phys. Chem. C, 2024, 128, 9016–9023 Search PubMed.
- J.-J. Yao, N.-Y. Gao, C. Li, L. Li and B. Xu, Mechanism and kinetics of parathion degradation under ultrasonic irradiation, J. Hazard. Mater., 2010, 175, 138–145 Search PubMed.
- J. Schwitzgebel, J. G. Ekerdt, H. Gerischer and A. Heller, Role of the Oxygen Molecule and of the Photogenerated Electron in TiO2-Photocatalyzed Air Oxidation Reactions, J. Phys. Chem., 1995, 99(15), 5633–5638 CrossRef CAS.
- K. Ikeda, H. Sakai, R. Baba, K. Hashimoto and A. Fujishima, Photocatalytic reactions involving radical chain reactions using microelectrodes, J. Phys. Chem. B, 1997, 101, 2617–2620 CrossRef CAS.
- M. Teranishi, S. Naya and H. Tada, In situ liquid phase synthesis of hydrogen peroxide from molecular oxygen using gold nanoparticle-loaded titanium(IV) dioxide photocatalyst, J. Am. Chem. Soc., 2010, 132, 7850–7851 CrossRef CAS.
- X. Li, C. Chen and J. Zhao, Mechanism of photodecomposition of H2O2 on TiO2 surfaces under visible light irradiation, Langmuir, 2001, 17, 4118–4122 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5sc01447f |
| This journal is © The Royal Society of Chemistry 2025 |