DOI:
10.1039/D5SC01121C
(Edge Article)
Chem. Sci., 2025,
16, 9135-9142
Building metal-induced inherent chirality: chiral stability/phosphorescence enhancement via a ring expansion strategy of Pt(II) complexes†
Received
12th February 2025
, Accepted 25th April 2025
First published on 28th April 2025
Abstract
Chirality is indeed a fundamental aspect of nature, particularly in chemistry and biology. Herein, we have demonstrated a straightforward strategy to create a novel type of metal-induced inherent chirality by expanding coordination rings. Typically, tetracoordinated d8 Pt(II) complexes with two five-membered coordination rings are 2D square-planar, resulting in the absence of chirality. However, if one of the coordination rings is expanded to six or seven members, the molecular structures of Pt(II) complexes would transition from a 2D plane to a 3D boat-like configuration. This not only facilitates the construction of inherently chiral scaffolds but also addresses the problem of emission aggregation-caused quenching. Furthermore, the strategy of ring expansion can enhance the chiral stability and phosphorescence quantum yield (up to 81.0%). Thus, the present work introduces a new structural motif with promising potential for application as chiroptical and luminescent materials.
Introduction
Chirality, referring to molecules that cannot fully overlap with their mirror images, is a fundamental property present in nature,1 and thus the development of novel chiral molecules plays a pivotal role in biochemistry, asymmetric synthesis,2 supramolecular chemistry,3 chiral optics,4 and so on. In general, chirality of organic molecules can be classified into four categories: point, axial, helical, and planar.5 The term “inherently chiral” was first suggested by Böhmer for chiral calixarenes, which do not strictly fall into definitions of other types of chirality.6 Later, a more general definition was formulated in 2004 by the group of Mandolini and Schiaffino.7 It states that inherent chirality derives from the “introduction of a curvature in an ideal planar structure that is devoid of symmetry axes in its bidimensional representation”.8 Since then, the concept of inherent chirality has been increasingly used to denote chirality arising from the rigid structures in both medium-sized rings (no less than a seven-membered ring)9 and large-sized rings (such as rotaxanes, catenanes, fullerenes, cavitands, and capsular assemblies).10 However, these pure organic molecules usually exhibit fluorescence.9a,11
Cyclometalated Pt(II) complexes have been extensively investigated in the past two decades owing to their rich phosphorescence properties and wide applications in phosphorescent organic light emitting diodes,12 chemosensors,13 bio-imaging,14 circularly polarized luminescence (CPL),5a,d,15 and so on. Typically, tetracoordinated d8 Pt(II) complexes with two five-membered coordination rings are 2D square-planar,16 resulting in the lack of chirality. Moreover, these square-planar molecules are far more likely to encounter the problem of emission “aggregation-caused quenching” (ACQ), because they readily form dimers through not only intermolecular π–π stacking interactions but also PtII–PtII interactions.14 Therefore, the chirality of Pt(II) complexes is primarily introduced by chiral ligands.5d,15,17 Although there are limited examples of chiral Pt(II) complexes with achiral ligands,18 their chiroptical properties, especially CPL, have seldom been reported before, mainly due to not only the weak chirality but also low phosphorescence quantum yields (Φ). Herein, we have demonstrated a straightforward strategy to build a novel type of metal-induced inherent chirality by expanding coordination rings (Fig. 1). When using N1^O, N2^O, N^P, N^PS, and N^PO bidentate ligands, one of the coordination rings expands into six or seven members. The molecular structures of Pt(II) complexes would change from 2D plane into 3D boat-like configuration, which not only facilitates the construction of inherently chiral scaffolds but also resolves the ACQ problem. Moreover, the strategy of ring expansion can improve the chiral stability and phosphorescence quantum yield (up to 81.0%). Furthermore, the resultant enantiomers of complexes can be separated by chiral high-performance liquid chromatography (HPLC), and their absolute configuration is confirmed by X-ray diffraction, circular dichroism, CPL, and density functional theory calculation.
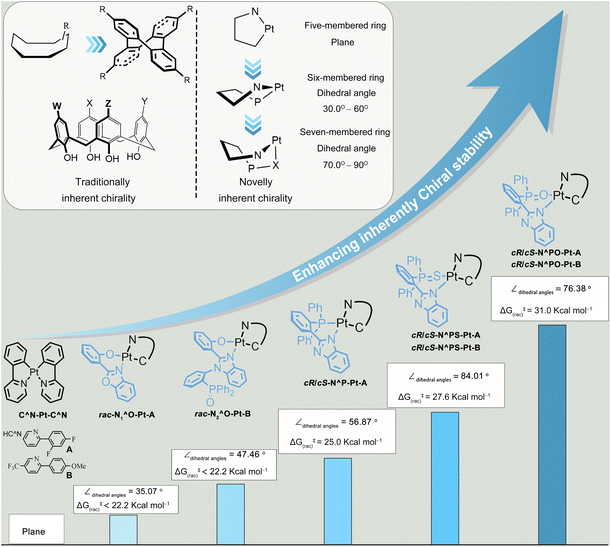 |
| | Fig. 1 The designed scheme and chemical structures of inherently chiral Pt(II) complexes (∠dihedral angles: angle between the phenyl plane and the PtC^N coordination plane; ΔG(rac‡): racemization energy barriers of inherently chiral Pt(II) complexes). | |
Results and discussion
Synthesis and characterization
The synthetic methods for the ligands and Pt(II) complexes are detailed in the ESI.† Initially, we utilized N1^O, N2^O, and N^P ligands to synthesize the Pt(II) complexes with six-membered coordination rings. N^PS and N^PO ligands were used to prepare the Pt(II) complexes with seven-membered coordination rings. Single crystals of rac-N1^O-Pt-A (CCDC: 2378928), rac-N2^O-Pt-B (CCDC: 2378929), rac-N^P-Pt-A (CCDC: 2378930), rac-N^PS-Pt-A (CCDC: 2378935), rac-N^PO-Pt-A (CCDC: 2378936), rac-N^PS-Pt-B (CCDC: 2378939), and rac-N^PO-Pt-B (CCDC: 2386027) were obtained through slow diffusion and evaporation of a hexane/CH2Cl2 solution.
First, chiral HPLC was used to analyze and separate enantiomers of the Pt(II) complexes. For rac-N1^O-Pt-A and rac-N2^O-Pt-B (Fig. S1†), there exists only one chromatographic peak in the HPLC trace. The possible reason is that the steric hindrance of N1^O and N2^O is too small to stabilize chirality in solution. With strong steric hindrance from two phenyl groups in triphenyl phosphine, rac-N^P-Pt-A, rac-N^PS-Pt-A, rac-N^PO-Pt-A, rac-N^PS-Pt-B, and rac-N^PO-Pt-B have two separated chromatographic peaks featuring a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio of the area (Fig. 2). The first peak (black line) refers to the optically pure cR-N^P-Pt-A, cR-N^PS-Pt-A, cR-N^PO-Pt-A, cR-N^PS-Pt-B, and cR-N^PO-Pt-B, while the second one (red line) corresponds to the cS-N^P-Pt-A, cS-N^PS-Pt-A, cS-N^PO-Pt-A, cS-N^PS-Pt-B, and cS-N^PO-Pt-B (see the later discussion). Enantiopure cR and cS-Pt(II) complexes (>98% ee) were collected by chiral HPLC, albeit the recovered yield being inferior to 30%.
:1 ratio of the area (Fig. 2). The first peak (black line) refers to the optically pure cR-N^P-Pt-A, cR-N^PS-Pt-A, cR-N^PO-Pt-A, cR-N^PS-Pt-B, and cR-N^PO-Pt-B, while the second one (red line) corresponds to the cS-N^P-Pt-A, cS-N^PS-Pt-A, cS-N^PO-Pt-A, cS-N^PS-Pt-B, and cS-N^PO-Pt-B (see the later discussion). Enantiopure cR and cS-Pt(II) complexes (>98% ee) were collected by chiral HPLC, albeit the recovered yield being inferior to 30%.
 |
| | Fig. 2 Top: chiral HPLC traces of rac-N^P-Pt-A, rac-N^PS-Pt-A, rac-N^PO-Pt-A, rac-N^PS-Pt-B, and rac-N^PO-Pt-B. Bottom: chiral HPLC traces of enantiopure (cR) (black line) and (cS) (red line) Pt(II) complexes. | |
Photophysical properties and calculations
Table 1 presents the UV/visible absorption and emission data of Pt(II) complexes at room temperature. As illustrated in Fig. 3, rac-N1^O-Pt-A, rac-N^P-Pt-A, rac-N^PO-Pt-A, and rac-N^PS-Pt-A with a common HC^N ligand of 2-(4′,6′-difluorophenyl)pyridine (A) exhibit low-energy absorption bands at λabs = 402, 380, 375, and 358 nm, respectively, demonstrating a gradual blue shift. This trend contrasts with the trend of corresponding dihedral angles, which are 35.07°, 56.87°, 76.38°, and 84.01° for rac-N1^O-Pt-A, rac-N^P-Pt-A, rac-N^PO-Pt-A, and rac-N^PS-Pt-A, respectively. A bigger dihedral angle would lead to a smaller degree of conjugation and a blue-shifted absorption band consequently (see the later discussion). rac-N^PO-Pt-A and rac-N^PO-Pt-B with the same N^PO but different C^N ligands display low-energy absorption bands at λabs = 375 and 419 nm, respectively. All the above results indicate that not only N1^O, N2^O, N^P, N^PS, and N^PO but also HC^N ligands have an obvious impact on absorption spectra.
Table 1 Room-temperature photophysical properties of binuclear Pt(II) complexes
| Compounds |
Medium |
λ
abs/nm (ε/dm3 mol−1 cm−3) |
λ
em/nm |
Φ
PL
|
τ
P/μs |
|
rac-N^P-Pt-A
|
CH2Cl2 |
316 (1.84 × 104), 380 (2.99 × 103) |
— |
— |
— |
| Powder |
— |
468/496/535 |
0.45% |
1.11 |
|
rac-N^PS-Pt-A
|
CH2Cl2 |
267 (1.68 × 104), 318 (9.01 × 103), 358 (4.25 × 103) |
— |
— |
— |
| Powder |
— |
466/500/535 |
6.61% |
0.97 |
|
rac-N^PO-Pt-A
|
CH2Cl2 |
247 (2.47 × 104), 317 (1.02 × 104), 375 (1.60 × 103) |
— |
— |
— |
| Powder |
— |
473/502/544 |
29.4% |
0.94 |
|
rac-N^PS-Pt-B
|
CH2Cl2 |
296 (1.22 × 104), 410 (1.43 × 103) |
— |
— |
— |
| Powder |
— |
506/538 |
3.39% |
8.51 |
|
rac-N^PO-Pt-B
|
CH2Cl2 |
311 (1.48 × 104), 419 (1.89 × 103) |
— |
— |
— |
| Powder |
— |
521/549 |
81.0% |
9.27 |
 |
| | Fig. 3 Experimental absorption spectra of Pt(II) complexes in CH2Cl2 solution (5.0 × 10−5 mol dm−3). | |
Density functional theory (DFT) and time-dependent DFT (TD-DFT) calculations were conducted using the Gaussian 09 program package to investigate the mechanisms of excited states and transitions. In dilute CH2Cl2 solution, both the absorption spectral bands and molar absorption coefficients (ε) of Pt(II) complexes are well-replicated by theoretical computations (Fig. 3 and S2–S4†). The low-energy absorption bands of rac-N^PS-Pt-A, rac-N^PO-Pt-A, and rac-N^P-Pt-A are 358 nm, 375 nm, and 380 nm, respectively, which are accurately reproduced by TD-DFT calculations (calculated λabs = 362 nm, 365 nm, and 388 nm for cS-N^PS-Pt-A, cS-N^PO-Pt-A, and cS-N^P-Pt-A, respectively). These low-energy absorption bands are primarily attributed to the transition from the highest occupied molecular orbital (HOMO) to the lowest unoccupied molecular orbital (LUMO) with contributions exceeding 80%, and the corresponding oscillator strength (fosc) values are 0.0075, 0.0288, and 0.0389 for cS-N^PS-Pt-A, cS-N^PO-Pt-A, and cS-N^P-Pt-A, respectively. The energy level diagram and frontier molecular orbitals indicate that the electron cloud of the HOMO is predominantly contributed by the electron-rich Pt(II) ions and the 1H-benzo[d]imidazole segments of the N^P, N^PS, and N^PO ligands, while the LUMO's electron cloud is mainly derived from the electron-deficient HC^N ligands (Fig. 4). Consequently, the low-energy absorption bands are primarily attributed to metal-to-ligand charge transfer (MLCT) from Pt(II) ions to C^N ligands, as well as ligand-to-ligand charge transfer (LLCT) from 1H-benzo[d]imidazole to HC^N ligands. Not only N^P, N^PS, and N^PO ligands but also HC^N ligands have obvious contributions to low-energy absorption bands, which is in accord with UV/visible absorption data (Table 1).
 |
| | Fig. 4 Energy level diagram and frontier molecular orbitals of cS-N^P-Pt-A, cS-N^PS-Pt-A, and cS-N^PO-Pt-A (in CH2Cl2). | |
All the Pt(II) complexes are weakly emissive or non-emissive in a dilute degassed CH2Cl2 solution. In comparison with solution samples, their powders show significant aggregation-induced emission (AIE)19 with promising quantum yields (3.76%, 4.40%, 0.45%, 6.61%, 29.4%, 3.39%, and 81.0% for rac-N1^O-Pt-A, rac-N2^O-Pt-B, rac-N^P-Pt-A, rac-N^PS-Pt-A, rac-N^PO-Pt-A, rac-N^PS-Pt-B, and rac-N^PO-Pt-B, respectively) (Fig. 5, S5, Tables 1 and S1†). Based on their relatively long emission decay lifetimes in the microsecond range (μs, Fig. S6, S7,† and Table 1) and significant Stokes shifts (ranging from 140 nm to 170 nm) between the absorption and emission bands, the emission from Pt(II) complexes can be classified as phosphorescence, originating from triplet excited states. As illustrated in Fig S8,† the frontier molecular orbitals in the triplet excited states of Pt(II) complexes (in CH2Cl2 solution) are analogous to those in singlet excited states, suggesting that their phosphorescence predominantly arises from 3MLCT and 3LLCT.
 |
| | Fig. 5 PL spectra of rac-N^P-Pt-A, rac-N^PS-Pt-A, rac-N^PO-Pt-A, rac-N^PS-Pt-B, and rac-N^PO-Pt-B in the powder and doped film. Photographs of rac-N^PO-Pt-A, and rac-N^PO-Pt-B in the powder under a 365 nm UV lamp. | |
X-ray single-crystal structures
The molecular structure and packing in X-ray single crystals of rac-N^PS-Pt-A and rac-N^PO-Pt-A are illustrated in Fig. 6. Unlike the traditional inherently chiral molecules, according to the definition of calixarenes, curvature is denoted as ‘c’, with clockwise as ‘cR’ and counterclockwise as ‘cS’.12,13 The core skeletons of these Pt(II) complexes are chiral (Fig. 1), and such metal-induced chirality can also be defined as inherent chirality for simplicity. An ideal observer standing on the cabin (the concave side of the surface) will see a seven-membered ring (Pt, O, P, C, C, C, and N atoms) at the side of the boat. The clockwise or counterclockwise array of the three highest priority atoms is designated as (cR) or (cS), respectively, as shown in Fig. 6. As anticipated, there are a pair of (cR)/(cS) enantiomers in their single crystals [cR-N^PO-Pt-A and cS-N^PO-Pt-A; cR-N^PS-Pt-A and cS-N^PS-Pt-A], corroborating the viability of our design strategy. Additionally, the enantiomers of rac-N1^O-Pt-A, rac-N2^O-Pt-B, rac-N^PS-Pt-B, and rac-N^PO-Pt-B (Fig. S9–S11†) are also discernible within the single-crystal structures.
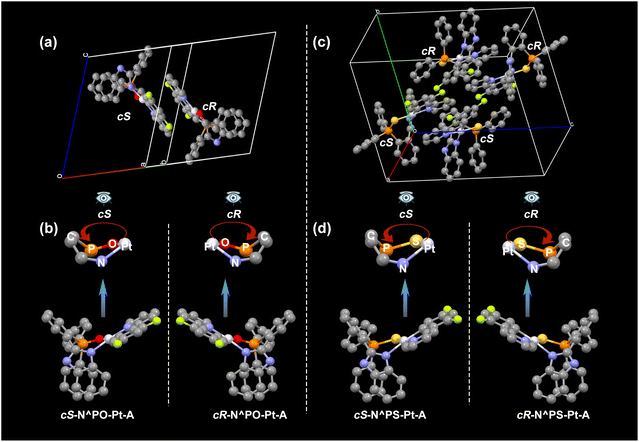 |
| | Fig. 6 X-ray single-crystal structures of rac-N^PO-Pt-A: (a) one crystal cell and (b) cR-N^PO-Pt-A and cS-N^PO-Pt-A molecular structures and the definition of (cR)/(cS) chirality (some atoms are omitted); X-ray single-crystal structures of rac-N^PS-Pt-A: (c) one crystal cell and (d) cR-N^PS-Pt-A and cS-N^PS-Pt-A molecular structures and the definition of (cR)/(cS) chirality (some atoms are omitted). | |
The photoluminescence properties of Pt(II) complexes are significantly influenced by their molecular structures and arrangements. There are no freely rotating groups for rac-N1^O-Pt-A, and rac-N2^O-Pt-B; however, their solutions are weakly emissive. This suggests the molecular backbone vibration to promote the non-radiative decay process.11,20 The other Pt(II) complexes contain not only molecular backbone vibration but also C–P-bond-linked phenyl groups, which are capable of free motion and consequently offer a possible pathway for the non-radiative annihilation of their excited states in solution. On the other hand, there are many strong intermolecular interactions, such as F–H (2.627–2.933 Å), C–H (2.665–2.886 Å), N–H (2.551–2.862 Å), O–H (2.410–2.713 Å), and H–H (2.350–2.769 Å) interactions (Fig. S9–S12†) in the single crystals of rac-N^P-Pt-A, rac-N^PS-Pt-A, rac-N^PO-Pt-A, rac-N^PS-Pt-B, and rac-N^PO-Pt-B. These strong intermolecular interactions can inhibit the rotation of C–P single bonds. The metal–metal interactions can significantly alter the photophysical properties of the complexes, such as red-shifted spectra and changes in the nature of the charge transfer transitions.21 Moreover, these resultant Pt(II) complexes adopt a 3D boat-like structure, which hinders intermolecular face-to-face π–π stacking interaction and intermolecular PtII–PtII interactions (5.871–8.033 Å, Fig. S10–12†) between adjacent complexes. Under the above synergistic effects, these Pt(II) complexes exhibit a substantial AIE phenomenon19 (0.45%, 6.61%, 29.4%, 3.39%, and 81.0% for rac-N^P-Pt-A, rac-N^PS-Pt-A, rac-N^PO-Pt-A, rac-N^PS-Pt-B, and rac-N^PO-Pt-B, respectively).
In theory, the emission quantum yield of a square-planar Pt(II) complex is strongly dependent on the planeness of coordination. The lesser the torsion is, the higher the quantum yield. For all the AIE-active Pt(II) complexes, the Pt atoms and HC^N ligands are nearly coplanar. However, their emission quantum yields are quite different. The torsion angle of C1–C2–Pt–P, C1–C2–Pt–S, C1–N1–Pt–O, C1–C2–Pt–S, and C1–N1–Pt–O is 16.99°, 1.68°, 1.01°, 3.87°, and 3.01° for cS-N^P-Pt-A, cS-N^PS-Pt-A, cS-N^PO-Pt-A, (cS)N^PS-Pt-B, and cS-N^PO-Pt-B, respectively. Additionally, the torsion angle of C3–N1–Pt–N2, C3–N1–Pt–N2, C2–C3–Pt–N2, C3–N1–Pt–N2, and C2–C3–Pt–N2 is 15.54°, 9.66°, 7.53°, 9.60°, and 4.42° for (cS)N^P-Pt-A, cS-N^PS-Pt-A, cS-N^PO-Pt-A, cS-N^PS-Pt-B, and cS-N^PO-Pt-B, respectively. cS-N^PO-Pt-A, and cS-N^PO-Pt-B have a less distorted square-planar geometry, resulting in their high emission quantum yields (Fig. 7).
 |
| | Fig. 7 The relationship between emission quantum yields and torsion angles of coordinated atoms among the representative products: cS-N^P-Pt-A, cS-N^PS-Pt-A, cS-N^PO-Pt-A, cS-N^PS-Pt-B, and cS-N^PO-Pt-B. | |
Racemization experiment
The chirality of these Pt(II) complexes originated from their 3D boat-type structures. If the cocked phenyl group in N1^O, N2^O, N^P, N^PS, and N^PO ligands flips down into the coplane with the coordination system, racemization would occur. Therefore, the chiral stability is determined by the dihedral angle between the phenyl group and the coordination system. The dihedral angles of these Pt(II) complexes exhibit a positive correlation with both the size of the ring and the peripheral substituent groups. It is reported that enantiomers with racemization energy barriers more than 22.2 kcal mol−1 could be separable at room temperature.22 The chirality of rac-N1^O-Pt-A and rac-N2^O-Pt-B is not stable in solution, because they don't have a large steric group of triphenyl phosphine. The chirality of other Pt(II) complexes with a triphenyl phosphine group is stable and separable. The dihedral angle of cS-N1^O-Pt-A, cS-N2^O-Pt-B, cS-N^P-Pt-A, cS-N^PO-Pt-A, and cS-N^PS-Pt-A is measured to be 35.07°, 47.46°, 56.87°, 76.38°, and 84.01°, respectively (Fig. 8a). Following Curran's methodology,23 the fitting of cS-N^P-Pt-A, cS-N^PO-Pt-A, and cS-N^PS-Pt-A is to calculate the flipping energy barrier. The energy barriers calculated by fitting are shown in Fig. 8b, which are 25.0 kcal mol−1 (half life, t1/2 = 1.50 days at 25 °C), 27.6 kcal mol−1 (t1/2 = 0.31 years at 25 °C), and 31.0 kcal mol−1 (t1/2 = 94 years at 25 °C), respectively (Tables S1–S3†). The sulfur atom in the N^PS ligands is undoubtedly larger than the oxygen atom in the N^PO ligands. Therefore, the increased repulsive force from the sulfur atom results in a more distorted seven-membered ring in cS-N^PS-Pt-A compared to cS-N^PO-Pt-A, which is evidenced by the larger dihedral angle of cS-N^PS-Pt-A (84.01°) than that of cS-N^PO-Pt-A (76.38°).
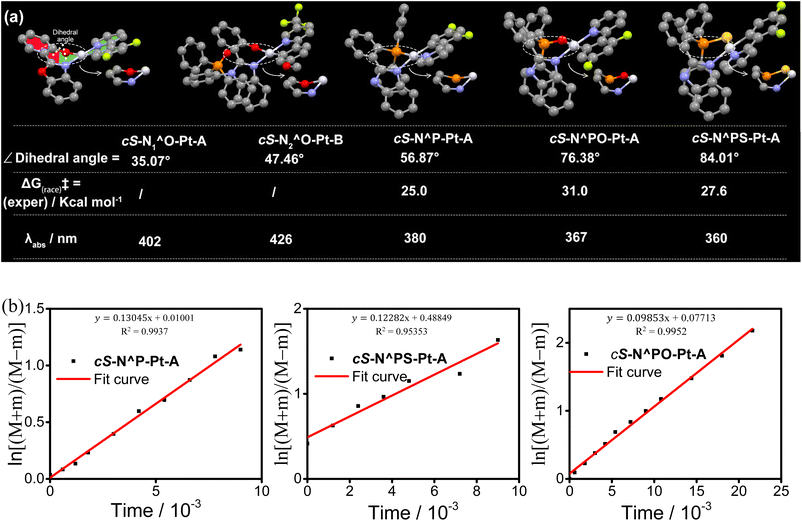 |
| | Fig. 8 (a) X-ray single-crystal structures, dihedral angles, and experimental interconversion energy barriers of cS-N1^O-Pt-A, cS-N2^O-Pt-B, cS-N^P-Pt-A, cS-N^PO-Pt-A, and cS-N^PS-Pt-A; (b) investigation of rotation barriers of cS-N^P-Pt-A, cS-N^PO-Pt-A, and (cS)N^PS-Pt-A. | |
Chiroptical properties
The chiroptical properties of Pt(II) complexes were examined using circular dichroism (CD) and CPL spectroscopy (Fig. 9). The CD spectra of the enantiomers in dilute CH2Cl2 solution (3.0 × 10−5 mol dm−3) exhibit perfect mirror images. The calculated CD spectra closely correspond to the experimental spectra, further confirming the absolute stereochemistry. Analyzing the calculations, the CD peaks in the range of 350–450 nm can be attributed to the MLCT and LLCT. The chirality of square-planar d8 Pt(II) complexes is predominantly introduced through the use of chiral ligands, and there are only limited examples of chiral Pt(II) complexes derived from achiral ligands. However, the |glum| of these chiral Pt(II) complexes typically range around 10−3, and they often exhibit low photoluminescence quantum yields (Φ). The CPL spectra of the resulting Pt(II) complexes in the solid state are shown in Fig. 9 and closely correspond to their photoluminescence (PL) spectra. The |gPL| values at the emission maxima (λem) are approximately 1.0–2.0 × 10−3, which are comparable to those observed in typical small organic molecules.24 Nevertheless, both the luminescence dissymmetry factors and quantum yields require further enhancement to meet practical application standards. Recent studies have shown that chiral Pt(II) complexes can achieve markedly enhanced glum values via the construction of chiral supramolecular architectures through intermolecular interactions,12g,25 highlighting a compelling strategy for advancing next-generation CPL-active materials.
 |
| | Fig. 9 (a) CD spectra of Pt(II) complexes in dilute CH2Cl2 solution (3.0 × 10−5 mol dm−3); (b) experimental and calculated CD spectra of cR/cS-N^P-Pt-A complexes in dilute CH2Cl2 solution; (c) CPL spectra of Pt(II) complexes in powder. | |
Conclusions
In this study, we developed a straightforward and efficient strategy to construct a novel type of (cR)/(cS) inherent chirality through metal coordination. These Pt(II) complexes, featuring a boat-type conformation, face challenges related to the absence of metal-induced chirality and the risk of ACQ for square-planar complexes. They exhibit intense blue-green and green phosphorescence with high quantum yields and moderate asymmetry factors. These findings provide valuable insights into the design of metal-doped chiral materials and their potential applications.
Data availability
The data supporting this article have been included as part of the ESI.† Deposition numbers 2378928, 2378929, 2378930, 2378935, 2378936, 2378939, and 2386027 contain the supplementary crystallographic data for this paper. The data can be obtained free of charge via the joint Cambridge Crystallographic Data Centre (CCDC).
Author contributions
Y. S. Shi, K. Tang, and J. C. Zhang conducted all of the reactions and data collection. J. T. Song conducted DFT calculations and experimental mechanistic studies on some aspects of characterization of the compounds, wrote the manuscript. Y. Q. Zhou contributed to crystal testing. P. Hu, B.-Q. Wang, and K.-Q. Zhao supervised and directed the project. H. F. Xiang conceptualized and directed the project, revised the manuscript. All the authors contributed to scientific discussions.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors gratefully acknowledge financial support from the National Natural Science Foundation of China (No. 22405184 and 21871192), Sinopec Seed Program Project (No. 30000000-25-ZC0607-0398) and the Natural Science Foundation of Sichuan Province, China (No. 2025ZNSFSC0118). We would like to thank Prof. Dr Peng Wu and Dr Yanying Wang of Analytical & Testing Center, Sichuan University for their assistance in transient fluorescence experiments. We would also like to thank Prof. Dr Zong-Xiang Xu of Department of Chemistry, Southern University of Science and Technology for X-ray diffraction work and single-crystal analysis.
Notes and references
- K. P. Meurer and F. Vogtle, Top. Curr. Chem., 1985, 127, 1–76 CrossRef CAS.
-
(a) P. Kočovský, Š. Vyskočil and M. Smrčina, Chem. Rev., 2003, 103, 3213–3246 CrossRef;
(b) D. Zhang, A. Martinez and J.-P. Dutasta, Chem. Rev., 2017, 117, 4900–4942 CrossRef CAS.
-
(a) M. Liu, L. Zhang and T. Wang, Chem. Rev., 2015, 115, 7304–7397 CrossRef CAS PubMed;
(b) L.-J. Chen, H.-B. Yang and M. Shionoya, Chem. Soc. Rev., 2017, 46, 2555–2576 RSC.
- L. Zhang, H.-X. Wang, S. Li and M. Liu, Chem. Soc. Rev., 2020, 49, 9095–9120 RSC.
-
(a) Z.-L. Gong, T.-X. Dan, J.-C. Chen, Z.-Q. Li, J. Yao and Y.-W. Zhong, Angew. Chem. Int. Ed., 2024, 63, e202402882 (
Angew. Chem.
, 2024
, 136
, e202402882
) CrossRef CAS;
(b) N. Hellou, M. Srebro-Hooper, L. Favereau, F. Zinna, E. Caytan, L. Toupet, V. Dorcet, M. Jean, N. Vanthuyne, J. A. G. Williams, L. Di Bari, J. Autschbach and J. Crassous, Angew. Chem. Int. Ed., 2017, 56, 8236–8239 (
Angew. Chem.
, 2017
, 129
, 8348–8351
) CrossRef CAS PubMed;
(c) G. Mazzeo, S. Ghidinelli, R. Ruzziconi, M. Grandi, S. Abbate and G. Longhi, ChemPhotoChem, 2022, 6, e202100222 CrossRef CAS;
(d) M. Ikeshita, N. Hara, Y. Imai and T. Naota, Inorg. Chem., 2023, 62, 13964–13976 CrossRef CAS.
- V. Böhmer, D. Kraft and M. Tabatabai, J. Inclusion Phenom. Mol. Recognit. Chem., 1994, 19, 17–39 CrossRef.
- A. Dalla Cort, L. Mandolini, C. Pasquini and L. Schiaffino, New J. Chem., 2004, 28, 1198–1199 RSC.
- A. Szumna, Chem. Soc. Rev., 2010, 39, 4274–4285 RSC.
-
(a) Y. Liu, Z. Ma, Z. Wang and W. Jiang, J. Am. Chem. Soc., 2022, 144, 11397–11404 CrossRef CAS PubMed;
(b) J.-H. Li, X.-K. Li, J. Feng, W. Yao, H. Zhang, C.-J. Lu and R.-R. Liu, Angew. Chem. Int. Ed., 2024, 63, e202319289 (
Angew. Chem.
, 2024
, 136
, e202319289
) CrossRef CAS PubMed.
-
(a) S. Tong, J.-T. Li, D.-D. Liang, Y.-E. Zhang, Q.-Y. Feng, X. Zhang, J. Zhu and M.-X. Wang, J. Am. Chem. Soc., 2020, 142, 14432–14436 CrossRef CAS PubMed;
(b) W. H. Powell, F. Cozzi, G. P. Moss, C. Thilgen, R. J.-R. Hwu and A. Yerin, Pure Appl. Chem., 2002, 74, 629–695 CrossRef CAS;
(c) E. M. G. Jamieson, F. Modicom and S. M. Goldup, Chem. Soc. Rev., 2018, 47, 5266–5311 RSC;
(d) C. A. Schalley, W. Reckien, S. Peyerimhoff, B. Baytekin and F. Vögtle, Chem.–Eur. J., 2004, 10, 4777–4789 CrossRef CAS PubMed.
-
(a) Z. Zhao, X. Zheng, L. Du, Y. Xiong, W. He, X. Gao, C. Li, Y. Liu, B. Xu, J. Zhang, F. Song, Y. Yu, X. Zhao, Y. Cai, X. He, R. T. K. Kwok, J. W. Y. Lam, X. Huang, D. Lee Phillips, H. Wang and B. Z. Tang, Nat. Commun., 2019, 10, 2952 CrossRef PubMed;
(b) J.-W. Han, X.-S. Peng and H. N. C. Wong, Natl. Sci. Rev., 2017, 4, 892–916 CrossRef CAS.
-
(a) M. A. Baldo, D. F. O'Brien, Y. You, A. Shoustikov, S. Sibley, M. E. Thompson and S. R. Forrest, Nature, 1998, 395, 151–154 CrossRef CAS;
(b) E. V. Puttock, M. T. Walden and J. A. G. Williams, Coord. Chem. Rev., 2018, 367, 127–162 CrossRef CAS;
(c) C.-W. Chan, L.-K. Cheng and C.-M. Che, Coord. Chem. Rev., 1994, 132, 87–97 CrossRef CAS;
(d) X. Wu, D.-G. Chen, D. Liu, S.-H. Liu, S.-W. Shen, C.-I. Wu, G. Xie, J. Zhou, Z.-X. Huang, C.-Y. Huang, S.-J. Su, W. Zhu and P.-T. Chou, J. Am. Chem. Soc., 2020, 142, 7469–7479 CrossRef CAS;
(e) H. Zhang, W. Wang, C. Liu, Z. Peng, C. Du and B. Zhang, J. Mater. Chem. C, 2021, 9, 9627–9636 RSC;
(f) M. A. Soto, V. Carta, R. J. Andrews, M. T. Chaudhry and M. J. MacLachlan, Angew. Chem. Int. Ed., 2020, 59, 10348–10352 (
Angew. Chem.
, 2020
, 132
, 10434–10438
) CrossRef CAS PubMed;
(g) Y. Wang, N. Li, L. Chu, Z. Hao, J. Chen, J. Huang, J. Yan, H. Bian, P. Duan, J. Liu and Y. Fang, Angew. Chem. Int. Ed., 2024, 63, e202403898 (
Angew. Chem.
, 2024
, 136
, e202403898
) CrossRef CAS;
(h) H. Leopold, M. Tenne, A. Tronnier, S. Metz, I. Münster, G. Wagenblast and T. Strassner, Angew. Chem. Int. Ed., 2016, 55, 15779–15782 (
Angew. Chem.
, 2016
, 128
, 16011–16014
) CrossRef CAS.
- K. M.-C. Wong and V. W.-W. Yam, Coord. Chem. Rev., 2007, 251, 2477–2488 CrossRef CAS.
- H. Xiang, J. Cheng, X. Ma, X. Zhou and J. J. Chruma, Chem. Soc. Rev., 2013, 42, 6128–6185 RSC.
-
(a) B. Li, Y. Li, M. H.-Y. Chan and V. W.-W. Yam, J. Am. Chem. Soc., 2021, 143, 21676–21684 CrossRef CAS PubMed;
(b) J. Gong and X. Zhang, Coord. Chem. Rev., 2022, 453, 214329 CrossRef CAS;
(c) G. Park, D. Y. Jeong, S. Y. Yu, J. J. Park, J. H. Kim, H. Yang and Y. You, Angew. Chem. Int. Ed., 2023, 62, e202309762 (
Angew. Chem.
, 2023
, 135
, e202309762
) Search PubMed.
- L. Chassot and A. Von Zelewsky, Helv. Chim. Acta, 1983, 66, 2443–2444 CrossRef CAS.
- J. R. Brandt, X. Wang, Y. Yang, A. J. Campbell and M. J. Fuchter, J. Am. Chem. Soc., 2016, 138, 9743–9746 Search PubMed.
-
(a) T. R. Schulte, J. J. Holstein, L. Krause, R. Michel, D. Stalke, E. Sakuda, K. Umakoshi, G. Longhi, S. Abbate and G. H. Clever, J. Am. Chem. Soc., 2017, 139, 6863–6866 CrossRef CAS PubMed;
(b) G. Hirata, Y. Kobayashi, R. Sato, Y. Shigeta, N. Yasuda and H. Maeda, Chem.–Eur. J., 2019, 25, 8797–8804 CrossRef CAS PubMed;
(c) J. Song, H. Xiao, L. Fang, L. Qu, X. Zhou, Z.-X. Xu, C. Yang and H. Xiang, J. Am. Chem. Soc., 2022, 144, 2233–2244 Search PubMed;
(d) J. Song, H. Xiao, B. Zhang, L. Qu, X. Zhou, P. Hu, Z.-X. Xu and H. Xiang, Angew. Chem. Int. Ed., 2023, 62, e202302011 (
Angew. Chem.
, 2023
, 135
, e202302011
) Search PubMed.
- S.-Y. Yang, Y. Chen, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, Chem. Soc. Rev., 2024, 53, 5366–5393 RSC.
- Y. Tu, Z. Zhao, J. W. Y. Lam and B. Z. Tang, Natl. Sci. Rev., 2021, 8, nwaa260 CrossRef PubMed.
-
(a) J. L.-L. Tsai, T. Zou, J. Liu, T. Chen, A. O.-Y. Chan, C. Yang, C.-N. Lok and C.-M. Che, Chem. Sci., 2015, 6, 3823–3830 RSC;
(b) C. Y.-S. Chung and V. W.-W. Yam, Chem. Sci., 2013, 4, 377–387 Search PubMed.
- J. S. J. Tan and R. S. Paton, Chem. Sci., 2019, 10, 2285–2289 RSC.
- D. B. Guthrie and D. P. Curran, Org. Lett., 2009, 11, 249–251 Search PubMed.
- E. M. Sánchez-Carnerero, A. R. Agarrabeitia, F. Moreno, B. L. Maroto, G. Muller, M. J. Ortiz and S. de la Moya, Chem.–Eur. J., 2015, 21, 13488–13500 CrossRef PubMed.
- X. Zhu, Z. Wang, Y. Jia, F. Yang, Y. Zhang, S. Zhao and X. He, J. Mater. Chem. C, 2023, 11, 11671–11680 RSC.
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Yuqiao
Zhou
a,
Yuqiao
Zhou








