
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRh-catalyzed mechanochemical transfer hydrogenation for the synthesis of periphery-hydrogenated polycyclic aromatic compounds†
Yoshifumi
Toyama
 a,
Takumu
Nakamura
b,
Yushin
Horikawa
a,
Yuta
Morinaka
b,
Yohei
Ono
b,
Akiko
Yagi
ac,
Kenichiro
Itami
*cd and
Hideto
Ito
*a
a,
Takumu
Nakamura
b,
Yushin
Horikawa
a,
Yuta
Morinaka
b,
Yohei
Ono
b,
Akiko
Yagi
ac,
Kenichiro
Itami
*cd and
Hideto
Ito
*a
aGraduate School of Science, Nagoya University, Chikusa, Nagoya 464-8602, Japan
bTokyo Research Center, Advanced Materials Research Laboratory, Advanced Integration Research Center, Research Division, Tosoh Corporation, 2743-1 Hayakawa, Ayase, Kanagawa 252-1123, Japan
cInstitute of Transformative Bio-Molecules (WPI-ITbM), Nagoya University, Chikusa, Nagoya 464-8602, Japan
dMolecule Creation Laboratory, Cluster for Pioneering Research, RIKEN, Wako, Saitama 351-0198, Japan
First published on 17th April 2025
Abstract
Hydrogenated nanographene has attracted attention as a new class of nanocarbon material owing to its potential applications in various research fields. However, the synthesis of periphery-hydrogenated nanographenes or polycyclic aromatic hydrocarbons (PAHs) is a significant challenge because of the harsh conditions and poor solubility of the starting materials. Conventional solution-state conditions require high-pressure hydrogen gas and lengthy reaction times. In this study, we developed a novel approach utilizing rhodium-catalyzed mechanochemical transfer hydrogenation, which enables hydrogenation without using hydrogen gas. Various hydrogenated PAHs were rapidly obtained using a simple protocol under ambient atmosphere and air, with one PAH showcasing intriguing properties such as aggregation-induced emission. Thus, the demonstrated mechanochemical hydrogenation method is expected to contribute to the rapid and efficient synthesis of a novel class of sp2/sp3-carbon-conjugated hydrocarbons.
Introduction
Nanographene and polycyclic aromatic hydrocarbons (PAHs) are regarded as promising materials owing to their unique electronic, optical, and magnetic properties.1 These materials are also attractive for applications in fields such as materials science and organic electronics, mainly because of their extended conjugated carbon structures, which allow for fine-tuning of their properties through molecular design. Recently, a notable increase in interest has been observed surrounding a novel class of nanocarbon material, namely, hydrogenated nanographenes/PAHs.2–4 Some of these molecules, such as hydrogenated anthracene, corannulene, and coronene, have been proposed to exist as interstellar materials.5 Of particular interest are periphery-hydrogenated PAHs, which possess high solubility and enhanced emission properties compared to those of mother PAHs. Moreover, these PAHs are anticipated to exhibit augmented electron–phonon interactions and universal characteristics such as a negative electron affinity associated with rigid sp3-carbon-based nanocarbon materials.3,4 Despite their prospective utilities, the accessibility of periphery-hydrogenated PAHs has been considerably constrained, primarily due to the difficulty of their synthesis.The synthesis of periphery-hydrogenated PAHs has been achieved by employing transition metal catalysts such as Pd, Pt, Ni, Co, and Rh under high-pressure hydrogen gas atmospheres.6 In the 1970s, hydrogenated PAHs such as hydrogenated pyrene, perylene, and benzo[a]anthracene were synthesized as a mixture of isomers under harsh conditions with high-pressure hydrogen gas at high temperature for many hours (Fig. 1A).7–9 In 2004 and 2019, Müllen and Narita reported the synthesis of peralkylated coronene from hexabenzo[bc,ef,hi,kl,no,qr]coronene and peralkylated circumbiphenyl from ovalene using Pd/C and H2 (65–150 bar) at 65–120 °C (Fig. 1B).3,4 As demonstrated previously, the hydrogenation of larger PAHs still necessitates harsh conditions. The presence of bulky moieties, such as adamantane, of the aromatic core requires harsh conditions for hydrogenation albeit small PAH structures.10 Another difficulty in the hydrogenation of PAHs is that PAHs are typically poorly soluble in conventional organic solvents due to strong intermolecular π–π and C–H/π interactions.11 As a result, only few studies have been reported on the hydrogenation of PAHs, and the optimal hydrogenation techniques have still been under development.12
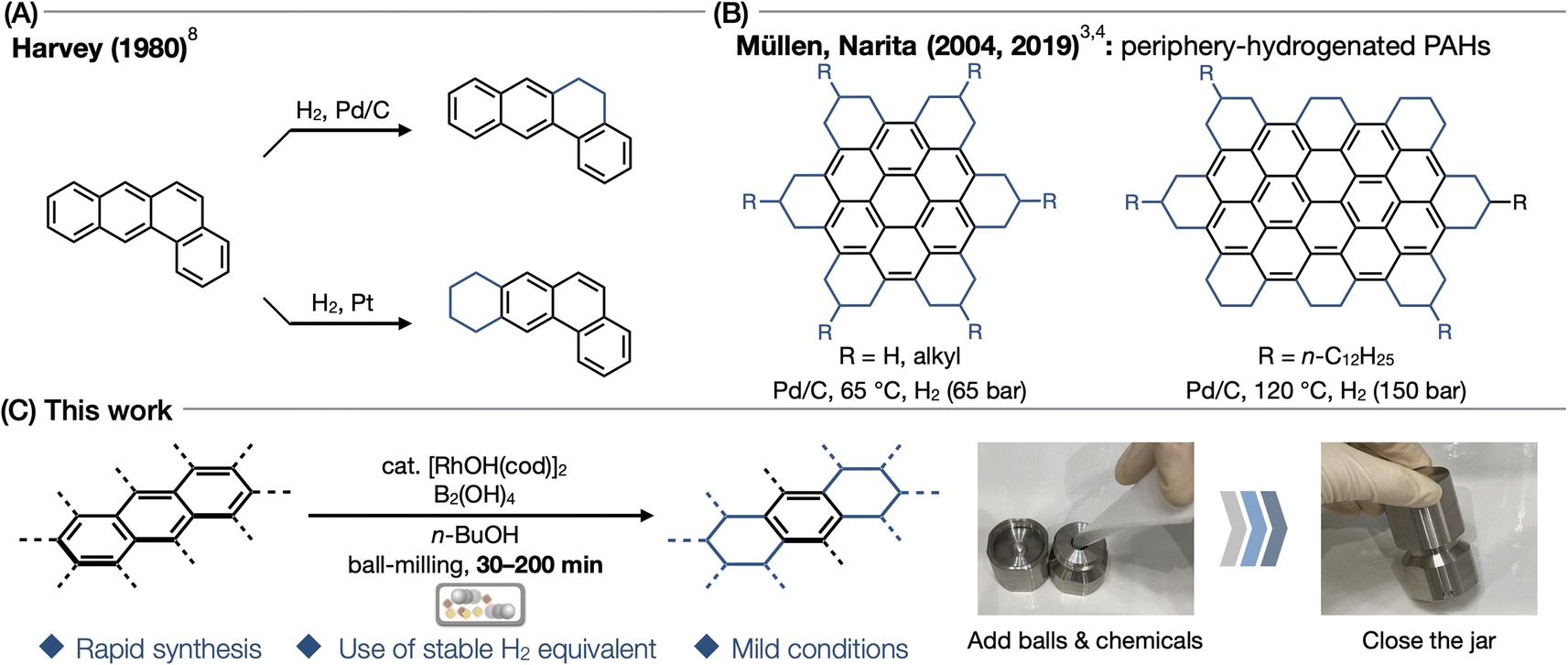 | ||
| Fig. 1 (A) Hydrogenation of PAHs with transition metal catalysts. (B) Previous reports of periphery-hydrogenated nanographenes. (C) This work. | ||
Recently, catalytic transfer hydrogenation methods for simple arenes without the use of H2 gas were reported by Glorius13 and Dou,14 (Fig. 1B). A key common feature of both reactions is the use of alcohol with ammonia borane (NH3BH3) or tetrahydroxydiboron B2(OH)4/EtOH as a source of hydrogen. During our recent research on the synthesis and transformation of nanocarbons by solution-state and/or mechanochemical methods,15–17 we envisaged that this hydrogen-gas-free hydrogenation system would be beneficial for the mechanochemical hydrogenation of poorly soluble PAHs. Another benefit is that the general characteristics of mechanochemical reactions, such as the use of minimal or no solvent and rapid reaction under mild reaction conditions, are appropriate for achieving this challenging hydrogenation. Sajiki also demonstrated the utility of mechanochemical (planetary ball-milling) hydrogenation of simple arenes;18 however, its applicability to larger PAHs has not been demonstrated. Furthermore, while related hydrogenations of ketone, alkene, alkyne, and nitro compound have been reported,19–23 to the best of our knowledge, no study has been conducted on the mechanochemical catalytic transfer hydrogenation of PAHs.
Herein, we present the mechanochemical transfer hydrogenation of various PAHs in the presence of a Rh catalyst with diboronic acid and alcohol as a hydrogen source (Fig. 1C). Direct mixing of the Rh catalyst, diboronic acid, arenes, and n-butanol using a ball-milling machine in air enabled the hydrogenation of arenes within short duration. Depending on the reaction conditions and substrates, diverse unprecedented hydrogenation reactions occur to afford per- and/or periphery-hydrogenated PAHs.
Results and discussion
First, we optimized the reaction conditions for mechanochemical arene hydrogenation using phenylcyclohexane (1) (0.20 mmol, 1.0 equiv.) as a model substrate (Table 1). Following a previous study on catalytic transfer hydrogenation by Dou with slight modifications,14 B2(OH)4 (8.0 equiv.) and n-BuOH (0.20 mL, 11 equiv.) was selected as an additive for the hydrogen source, with 5 mol% [RhOH(cod)]2 (cod = 1,5-cyclooctadiene) as the catalyst (Table 1). In the optimal reactions, all reagents without n-BuOH, a stainless-steel ball (diameter of 7 mm), and n-BuOH were added in the given order to a 1.5 mL stainless-steel jar in air. The jar was then closed and placed in a mixer mill (Retsch MM400). After 60 min of ball milling at 30 Hz with direct heating using a heat-gun15,24 (heat-gun preset temperature: 100 °C), the jar was opened, and the content was diluted with CHCl3 and passed through a short pad of silica gel to remove the catalyst. 1H NMR analysis and isolation resulted in the full conversion of 1 and the almost stoichiometric formation of cyclohexylcyclohexane (2) (99% NMR yield and 93% isolated yield). In contrast, the use of NH3BH3, [RhCl(cod)]2, and 2,2,2-trifluoroethanol, whose conditions were proven to be effective by Glorius for the in-solution hydrogenation of benzene, naphthalene, pyridine, and indole derivatives,13 dramatically diminished the yield of the desired product 2 (entry 2, 8%). Although the reaction was not complete within 1 min (entry 3, 21%), completion was observed even after 10 min reaction (entry 4, 99%). The reaction was then conducted at room temperature (20–25 °C) to afford 2 in 57% yield in 60 min along with the recovery of unreacted 1 (entry 5). Subsequently, various Rh sources such as [RhCl(cod)2], Rh(BF4)(cod)2, and RhCl(CAAC)(cod)25 were examined instead of using [RhOH(cod)]2 (entries 6–8), and the yield of each product did not exceed the yield of the reaction with [RhOH(cod)2]. Unexpectedly, RhCl(CAAC)(cod),25 which is known as an effective hydrogenation catalyst in the solution-state reaction, did not display any catalytic activity in the present mechanochemical system. When using ethanol with heating at 100 °C, the yield of 2 was reduced to 71% (entry 9). The volatile ethanol vapor was observed to leak from the reaction jar owing to heat-gun heating; thus, liquid-assisted grinding26 did not work sufficiently. Other alcohol additives, i-PrOH and 2,2,2-trifluoroethanol, were also ineffective (entries 10 and 11). Interestingly, the reaction proceeded even when using solid alcohol reagents, D-(+)-glucose and β-cyclodextrin to afford 2 in 29% and 24% NMR yields, respectively (entries 12 and 13). As a result, the use of n-BuOH—a liquid primary alcohol with a high-boiling point (approximately 118 °C)—was considered optimal for the present mechanochemical hydrogenation reaction.|
|
||
|---|---|---|
| Entry | Deviation from the standard conditions | Yield of 2b |
| a Reactions were conducted in a 1.5 mL stainless-steel jar using a stainless-steel ball with a diameter of 7 mm. b 1H NMR yield using dibromomethane as an internal standard. c Isolated yield. Heat-gun preset temperature (100 °C) was indicated as the reaction temperature. | ||
| 1 | None | 99% (93%)c |
| 2 | NH3BH3(1.5 equiv.), [RhCl(cod)]2 (10 mol%), and 2,2,2-trifluoroethanol (0.20 mL) were used instead of B2(OH)4, [RhOH(cod)]2, and n-BuOH | 8% |
| 3 | Ball-milling for 1 min | 21% |
| 4 | Ball-milling for 10 min | 99% |
| 5 | Without heating (room temperature, 20–25 °C) | 57% |
| 6 | [RhCl(cod)]2 instead of [RhOH(cod)]2 | 57% |
| 7 | 10 mol% of Rh(BF4)(cod)2 instead of [RhOH(cod)]2 | 14% |
| 8 | 10 mol% of RhCl(CAAC)(cod)24 instead of [RhOH(cod)]2 | 9.5% |
| 9 | EtOH instead of n-BuOH | 71% |
| 10 | i-PrOH instead of n-BuOH | 35% |
| 11 | 2,2,2-Trifluoroethanol instead of n-BuOH | Trace |
| 12 | D-(+)-Glucose (2.2 equiv.) instead of n-BuOH | 29% |
| 13 | β-Cyclodextrin (0.5 equiv.) instead of n-BuOH | 24% |

We then conducted the mechanochemical hydrogenation of naphthalene (3) to compare its activity with that of conventional solution-state hydrogenation. The reaction under Dou's conditions14 using B2(OH)4/EtOH for solution-state transfer hydrogenation at 50 °C for 30 min resulted in the production of tetralin (4a) in 45% NMR yield (Fig. 2A). In this reaction, naphthalene was recovered in 45% NMR yield. Next, a room-temperature mechanochemical reaction with a reduced amount of EtOH for 10 min resulted in a similar result in terms of yield, although the reaction seemed to be faster than the solution-state reaction. However, a distinct difference was observed when the reaction time was extended to 30 min for mechanochemical hydrogenation; the fully hydrogenated product cis-decalin (4b) was isolated in 65% yield, whereas trans-decalin (4c) was not formed. These control experiments suggest that mechanochemical hydrogenation has advantages over conventional solution-based reactions in terms of the catalytic activity, reaction time, and temperature. In Dou's solution-state hydrogenation using [RhOH(cod)]2/B2(OH)4/EtOH, the formation of heterogeneous Rh nanoparticles is expected to be one of the active species.14 In our mechanochemical reaction, we also expected that the similar heterogeneous Rh nanoparticle-catalyzed hydrogenations occur because of preferential formation of cis-decalin (4b), which is a typical kinetic product in the heterogeneous hydrogenation of 2 catalyzed by metal nanoparticles such as Ru and Rh.27,28 Considering that the conventional high-pressure, high-temperature, long-time hydrogenation of arenes eventually yields the thermodynamically most stable trans-products under reversible hydrogenation–dehydrogenation equilibrium, our mechanochemical transfer hydrogenation at 100 °C for 10–60 min seems to be a mild and kinetically controlled reaction. In addition, we hypothesized that the further acceleration effect in our mechanochemical system can be derived from an increase in the frequency of contact between the catalyst and the substrate because of its highly concentrated conditions.
 | ||
| Fig. 2 Comparison of mechanochemical and solution-state conditions in the hydrogenation of (A) naphthalene and (B) adamantane-annulated biphenyl. Ball milling reactions were conducted in a 1.5 mL stainless-steel jar using a stainless-steel ball (diameter of 7 mm). a1H NMR yield using dibromomethane as an internal standard. bIsolated yield. | ||
Then, we applied this methodology to an arene containing a bulky substituent, adamantane-annulated arene.29 In 2024, we reported the synthesis of all-sp3 carbon-containing diamondoids via hydrogenation of adamantane-annulated biphenyl (5).10 We conducted catalytic hydrogenation of 5 with Rh–Pt/(DMPSi–Al2O3)30,31 and Sc(OTf)3 under 10 atom hydrogen gas at 125 °C for 72 h, affording two structural isomers of diamondoids, 6a (as a thermodynamically favored product) and 6b (as a kinetically favored product). Motivated by the harsh reaction conditions of the previous hydrogenation reaction and the higher catalytic activity of the proposed mechanochemical hydrogenation reaction, we examined the mechanochemical hydrogenation of 5. As a result, fully and partially hydrogenated products 6b and 6c were obtained in 17 and 74% yields, respectively. The structure of the newly isolated 6c was determined by X-ray crystallography (see ESI†). Notably, a difference in stereoselectivity was observed between the solution- and solid-state conditions. We assumed that the present mechanochemical hydrogenation preferentially afforded 6b because the reaction proceeded in a shorter time at a lower temperature than that under solution-state conditions. Consequently, greater synthetic advantages in terms of faster reactions under milder conditions were observed in the present mechanochemical reaction.
We explored the substrate scope of various PAHs with a highly efficient and easy-to-handle mechanochemical hydrogenation (Fig. 3). The use of anthracene (7a), 9-anthraceneboronic acid bis(pinacol) ester (7b) and 9-anthracenecarboxylic acid (7c) resulted in the production of octahydroanthracene derivatives (8a–8c) in yields of 69%, 64% and 46%, respectively. In contrast, the reaction of methyl-9-anthracenecarboxylate (7d) afforded tetrahydroanthracene 8d and dihydroanthracene 8d′ (see ESI†) as a mixture (40%, 8d/8d′ = 89![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :11). The use of 9-fluoroanthracene (7e) also afforded tetrahydroanthracene 8e in 30% yield. The other polyaromatic compounds containing bromo, chloro, amino, formyl, hydroxy and ether groups, were found to be ineffective in this mechanochemical reaction in terms of low conversion of starting materials, poor functional group tolerability and formation of inseparable multiple isomers (see Fig. S2 in the ESI† for details). Then, we used π-extended acene such as tetracene (7f) and pentacene (7g) resulted in the formation of octahydrotetracene (8f) in 65% yield and hexadecahydropentacene (8g) as a single stereoisomer in 18% yield. Hydrogenation of 9,10-diphenylanthracene (7h) generated a separable mixture of terphenyl derivative 8h (41%) and 1,4-diphenylnaphthalene derivative 8h′ (49%). In the reaction using 5,6,11,12-tetraphenyltetracene (rubrene, 7i), both the sterically less hindered acene termini were hydrogenated to give 1,4,5,8-tetraphenylnaphthalene 8i in 51% yield. In addition, a larger scale synthesis of 8i was performed using a 10 mL stainless-steel jar and two stainless-steel balls (both with diameters of 10 mm) with 0.50 mmol (266.7 mg) 7i, affording 8i in a moderate yield of 36% (97.1 mg) after the reaction at same temperature for the same reaction time. The generation of the product in lower yield in the large-scale reaction can be attributed to the valence of the reaction scale, container surface area, and number of ball impacts; nevertheless, the yield and efficiency can be improved by further examination of the reaction parameters.32 We then demonstrated the hydrogenation of larger PAHs, which typically show a low solubility and thus, a low reactivity for conventional hydrogenation. Using triphenylene (7j), dibenzo[g,p]chrysene (7k), and tribenzo[b,e,k]fluoranthene (7l) resulted in the formation of periphery-hydrogenated polyaromatic compounds 8j, 8k, and 8l in 82%, 10%, and 53% yields, respectively. During the hydrogenation of 7k, various partially hydrogenated products were formed, including 8k, which made it difficult to isolate 8k in high yield (see ESI† for details). The reactions with perylene (7m) and naphtho[8,1,2-bcd]perylene (7n)15 also proceeded well to afford periphery-hydrogenated perylene derivatives 8m (71%, 3:1 cis/trans mixture), 8m′ (12%), and 8n (61%). The use of highly fused polyaromatic compounds such as coronene (7o), corannulene (7p), and dibenzo[fg,ij]-naphtho[1,2,3,4-rst]pentaphene (7q) afforded the periphery-hydrogenated PAHs 8o, 8p, and 8q in 19%, 9%, and 11% yields, respectively. The reason for the low yields of these compounds is similar to that of 8k: the formation of various partially hydrogenated products and isomers made separation difficult using silica gel column chromatography and size exclusion chromatography. Although the relative stereochemistry of 8p could not be determined, it was obtained as a single stereoisomer. Single crystals of 8i and 8o were successfully obtained by recrystallization from CHCl3/pentane, and their structures were elucidated by X-ray diffraction analysis. Unlike flat rubrene (5,6,11,12-tetraphenyltetracene),338i possesses a highly twisted structure in the central naphthalene unit because of its congested peri-substituents and cyclohexane rings. Interestingly, only a few examples of synthesizing 1,4,5,8-tetraphenylnaphthalene are reported regardless of its simple structure.34 In addition, highly twisted naphthalene structure with 26° of twisted angle (Φ) was observed, whose value is even larger than that of flat rubrene33 (Φ = 0°), considerable to those of π-extended rubrenes35 (Φ = 20° in tetraphenylnaphthalene core), and smaller than that of octaphenyltetrabenzohexacene36 (a twistacene) (average twisted angle of central tetracene core: Φaverage = 62°) (see ESI† for details). In the crystallographic analysis of 8o, the configuration of the four protons on the tertiary sp3-carbons in 8o was found to be syn-form, suggesting that the present multi-hydrogenation appears to be cis-hydrogenation, as in a typical heterogeneous hydrogenation using metal nanoparticles such as Pd/C. This is in accordance with the mechanistic expectation of Dou.14 that Rh nanoparticles are the active species in the catalytic system with [RhOH(cod)]2/B2(OH)4/alcohol.
:11). The use of 9-fluoroanthracene (7e) also afforded tetrahydroanthracene 8e in 30% yield. The other polyaromatic compounds containing bromo, chloro, amino, formyl, hydroxy and ether groups, were found to be ineffective in this mechanochemical reaction in terms of low conversion of starting materials, poor functional group tolerability and formation of inseparable multiple isomers (see Fig. S2 in the ESI† for details). Then, we used π-extended acene such as tetracene (7f) and pentacene (7g) resulted in the formation of octahydrotetracene (8f) in 65% yield and hexadecahydropentacene (8g) as a single stereoisomer in 18% yield. Hydrogenation of 9,10-diphenylanthracene (7h) generated a separable mixture of terphenyl derivative 8h (41%) and 1,4-diphenylnaphthalene derivative 8h′ (49%). In the reaction using 5,6,11,12-tetraphenyltetracene (rubrene, 7i), both the sterically less hindered acene termini were hydrogenated to give 1,4,5,8-tetraphenylnaphthalene 8i in 51% yield. In addition, a larger scale synthesis of 8i was performed using a 10 mL stainless-steel jar and two stainless-steel balls (both with diameters of 10 mm) with 0.50 mmol (266.7 mg) 7i, affording 8i in a moderate yield of 36% (97.1 mg) after the reaction at same temperature for the same reaction time. The generation of the product in lower yield in the large-scale reaction can be attributed to the valence of the reaction scale, container surface area, and number of ball impacts; nevertheless, the yield and efficiency can be improved by further examination of the reaction parameters.32 We then demonstrated the hydrogenation of larger PAHs, which typically show a low solubility and thus, a low reactivity for conventional hydrogenation. Using triphenylene (7j), dibenzo[g,p]chrysene (7k), and tribenzo[b,e,k]fluoranthene (7l) resulted in the formation of periphery-hydrogenated polyaromatic compounds 8j, 8k, and 8l in 82%, 10%, and 53% yields, respectively. During the hydrogenation of 7k, various partially hydrogenated products were formed, including 8k, which made it difficult to isolate 8k in high yield (see ESI† for details). The reactions with perylene (7m) and naphtho[8,1,2-bcd]perylene (7n)15 also proceeded well to afford periphery-hydrogenated perylene derivatives 8m (71%, 3:1 cis/trans mixture), 8m′ (12%), and 8n (61%). The use of highly fused polyaromatic compounds such as coronene (7o), corannulene (7p), and dibenzo[fg,ij]-naphtho[1,2,3,4-rst]pentaphene (7q) afforded the periphery-hydrogenated PAHs 8o, 8p, and 8q in 19%, 9%, and 11% yields, respectively. The reason for the low yields of these compounds is similar to that of 8k: the formation of various partially hydrogenated products and isomers made separation difficult using silica gel column chromatography and size exclusion chromatography. Although the relative stereochemistry of 8p could not be determined, it was obtained as a single stereoisomer. Single crystals of 8i and 8o were successfully obtained by recrystallization from CHCl3/pentane, and their structures were elucidated by X-ray diffraction analysis. Unlike flat rubrene (5,6,11,12-tetraphenyltetracene),338i possesses a highly twisted structure in the central naphthalene unit because of its congested peri-substituents and cyclohexane rings. Interestingly, only a few examples of synthesizing 1,4,5,8-tetraphenylnaphthalene are reported regardless of its simple structure.34 In addition, highly twisted naphthalene structure with 26° of twisted angle (Φ) was observed, whose value is even larger than that of flat rubrene33 (Φ = 0°), considerable to those of π-extended rubrenes35 (Φ = 20° in tetraphenylnaphthalene core), and smaller than that of octaphenyltetrabenzohexacene36 (a twistacene) (average twisted angle of central tetracene core: Φaverage = 62°) (see ESI† for details). In the crystallographic analysis of 8o, the configuration of the four protons on the tertiary sp3-carbons in 8o was found to be syn-form, suggesting that the present multi-hydrogenation appears to be cis-hydrogenation, as in a typical heterogeneous hydrogenation using metal nanoparticles such as Pd/C. This is in accordance with the mechanistic expectation of Dou.14 that Rh nanoparticles are the active species in the catalytic system with [RhOH(cod)]2/B2(OH)4/alcohol.
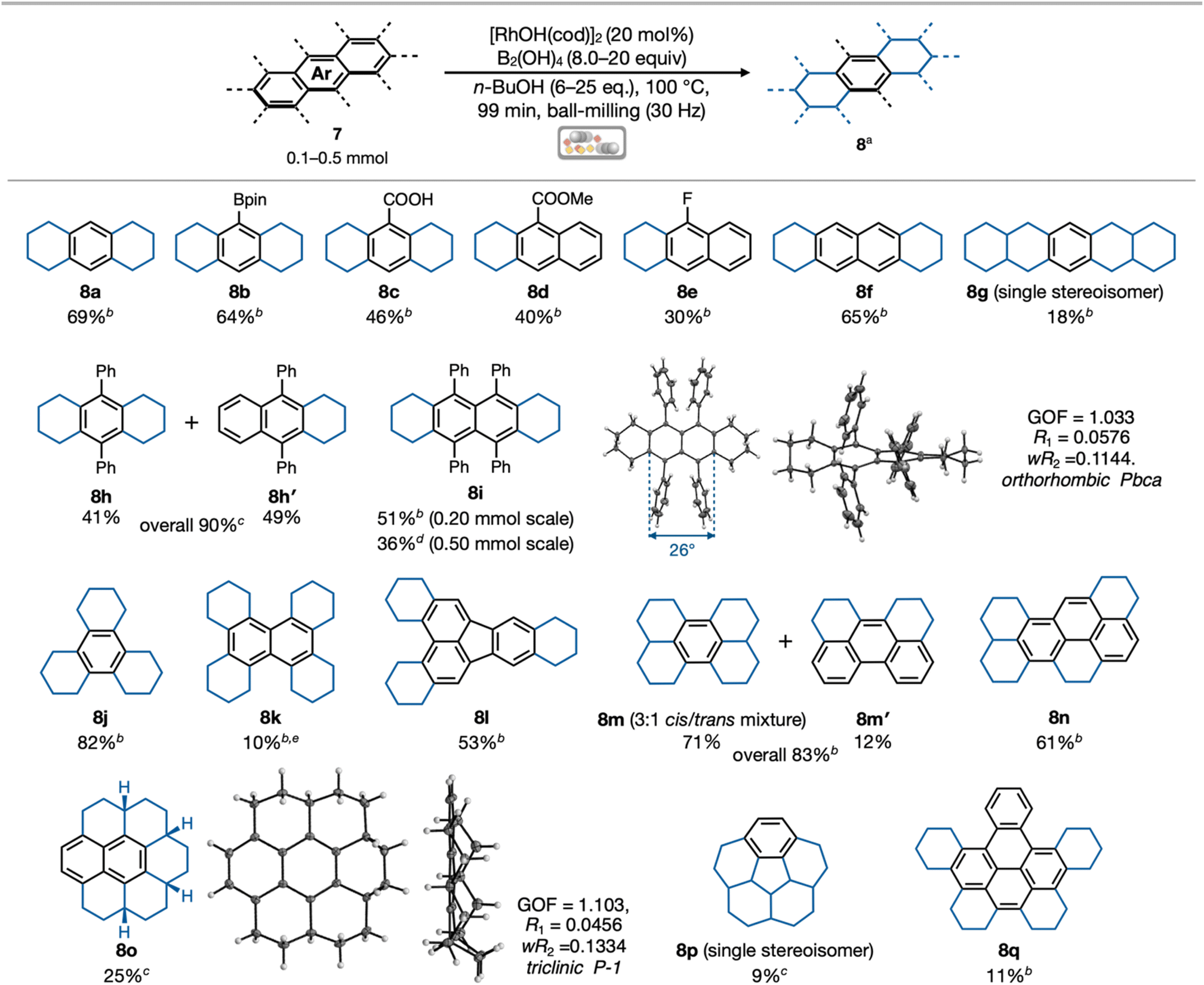 | ||
| Fig. 3 Substrate scope in the mechanochemical transfer hydrogenation of PAHs and the ORTEP drawing of 8i/8o. aIsolated yield. bReactions were conducted in a 1.5 mL stainless-steel jar using a stainless-steel ball with a diameter of 7 mm. cReactions were conducted in a 5.0 mL stainless-steel jar using a stainless-steel ball with a diameter of 10 mm. dReactions were conducted in a 10 mL stainless-steel jar using two stainless-steel balls (both with a diameters of 10 mm) for 198 min. eReaction time of ball milling was 60 min. | ||
Because the obtained 8i is a rare example of a 1,4,5,8-tetraphenylnaphthalene derivative and a lower homolog of rubrene and twistacene with a highly twisted structure, its electronic and photophysical properties are of interest. In the UV-vis absorption measurements of a solution of 8i in THF (c = 1.53 × 10−5 M), two absorption maxima were observed at λabs = 260 and 322 nm; the absorption band was extended up to 400 nm (Fig. 4A). In addition, 8i exhibited fluorescence upon excitation with 340 nm light, and broad emission band was observed along with an emission maximum at λem = 407. The absorption maxima were slightly red-shifted and emission maxima was slightly blue-shifted, compared with pristine 1,4,5,8-tetraphenylnaphthalene32 (λabs = 248 and 334 nm, λem = 437 in cyclohexane). In the depiction of frontier molecular orbitals (MOs) calculated by the B3LYP/6-31G+(d,p) level of theory, HOMO and LUMO were mainly localized on naphthalene moiety, while the orbitals were slightly spread on phenyl rings and σ-bonds on benzylic C–H. In addition, terphenyl-like MO distributions appeared in other low-lying HOMO − 1 and HOMO − 2 and high-lying LUMO + 1 and LUMO + 2 (see ESI†). The photophysical properties of 8i were further analyzed using time-dependent density functional theory (TD-DFT) calculations at the B3LYP/6-31+G(d,p) level of theory. The observed absorption maximum at λabs = 322 nm can be attributed to the allowed HOMO → LUMO transition (λ = 352 nm, oscillator strength (f) = 0.2104), whereas the second transition corresponding to HOMO → LUMO + 1 and HOMO − 1 → LUMO (λ = 323 nm, f = 0.0021) and other transitions appear forbidden (see ESI† for other transitions).
 | ||
| Fig. 4 (A) UV-vis absorption and emission spectra of 8i (c = 1.53 × 10−5 M) in THF. Excitation light was 340 nm. (B) Possible transitions calculated by TD-DFT at the B3LYP/6-31G+(d,p) level of theory and depiction of frontier MOs. | ||
Next, we investigated the solubility-derived properties of 8i. This is typical of polyaromatic compounds, and planar PAHs generally show lower solubility in common organic solvents. A comparison of the solubilities of the mother- and periphery-hydrogenated aromatics is of interest. Indeed, rubrene shows low solubility (<0.2 mg mL−1) in its saturated solution of hexane (see ESI†). Octahydrorubrene (8i) exhibited a higher solubility (0.6 mg mL−1 in hexane) than rubrene (see ESI†). Due to the slightly increased solubility and highly congested tetraphenyl moiety of 8i, 8i showed aggregation-induced emission (AIE)37 in a THF/H2O-mixed solvent system (Fig. 5) in the solutions of seven different ratios of THF/H2O (100:0, 70:30, 40:60, 35:65, 25:75, 10:90, and 1:99). As illustrated in Fig. 5, the emission intensity at 407 nm gradually decreased as the THF/H2O ratio shifted from 100:0 to 1:99, and new red-shifted emission peaks at 450 and 480 nm appeared at a ratio of 35:65. This is considered an AIE. The emission intensities became maximum values with enhanced fluorescent quantum yield (ΦF = 72%) at the 10:90 ratio, whereas the intensities and ΦF declined at the 1:99 possibly because the solvent composition became almost exclusively polar water solvents. While these AIE properties are similar to those of typical AIE molecules such as polyphenylarenes and tetraarylethenes,37 the AIE properties of 8i are unprecedented as 1,4,5,8-tetraphenylnaphthalene derivatives.
 | ||
| Fig. 5 (A) AIE emission of 8i (c = 1.93 × 10−6 M) in THF/H2O. Fluorescence spectra were recorded upon excitation at 340 nm. (B) Fluorescence photographs of solutions of 8i in THF/water mixtures with different water contents. fw is defined as fraction of water over total volumes of THF and H2O. | ||
Conclusions
In summary, we developed a mechanochemical transfer hydrogenation of simple arenes and various PAHs as a novel synthetic method for periphery-hydrogenated PAHs. Using [RhOH(cod)]2 as the catalyst and B2(OH)4/n-BuOH as the hydrogen donor under mechanochemical conditions, rapid and highly effective hydrogenation occurred, even under ambient pressure and atmosphere, affording sp3-carbon-rich nanocarbons in a few hours. Furthermore, one of the hydrogenated products, the octahydrorubrene derivative (8i), exhibited characteristic photophysical properties such as AIE. We expect that the proposed mechanochemical hydrogenation method will contribute to the rapid and efficient synthesis of a novel class of sp2/sp3-carbon-conjugated hydrocarbons. Further studies on reactive species in the current catalytic system and application to larger nanographene is currently undergoing in our laboratory.Data availability
Experimental and characterization data, including crystallographic data [6c (CCDC no. 2425787), 8i (CCDC no. 2389852), and 8o (CCDC no. 2389851)], photophysical measurements, and NMR spectra, as well as computational investigations. The data supporting this article have been included as part of the ESI.†Author contributions
Y. T. synthesized, analyzed, and characterized all compounds and performed theoretical calculations with partial support from T. N., Y. M., and Y. O. Y. H. synthesized 6c and elucidated its structure by X-ray crystallographic analysis. H. I. designed the project and target compounds, supervised the experiments, and conducted data analyses. H. I. and K. I. directed the project. The draft manuscript was written by Y. T. and H. I., and all the authors finalized the manuscript through proofreading. All authors approved the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was supported by the Sumitomo Foundation (2300884 to H. I.), the Foundation of Public Interest of Tatematsu (22B025 to H. I.), the Kondo Memorial Foundation (2022-03, to H. I.), the NAGAI Foundation of Science and Technology (to H. I.), and JST-CREST (JPMJCR19R1 to A. Y.). Y. T. thanks the Interdisciplinary Frontier Next-Generation Researcher Program of the Tokai Higher Education and Research System for the fellowships. We thank Takato Mori and Daiki Imoto for their assistance with the X-ray crystallographic analyses. The computations were performed at the Research Center for Computational Science, Okazaki, Japan (Project No. 23-IMS-C061 and 24-IMS-C059).Notes and references
- (a) S. Allard, M. Forster, B. Souharce, H. Thiem and U. Scherf, Angew. Chem., Int. Ed., 2008, 47, 4070 CrossRef CAS; (b) C. Wang, H. Dong, W. Hu, Y. Liu and D. Zhu, Chem. Rev., 2012, 112, 2208 CrossRef CAS PubMed; (c) Y. Segawa, H. Ito and K. Itami, Nat. Rev. Mater., 2016, 1, 15002 CrossRef CAS; (d) A. Narita, X. Wang, X. Fengb and K. Müllen, Chem. Soc. Rev., 2015, 44, 6616 RSC.
- (a) R. M. Ferullo, C. E. Zubieta and P. G. Belelli, Phys. Chem. Chem. Phys., 2019, 21, 12012 RSC; (b) J. Son, S. Lee, S. J. Kim, B. C. Park, H. Lee, S. Kim, J. H. Kim, B. H. Hong and J. Hong, Nat. Commun., 2016, 7, 13261 CrossRef CAS PubMed; (c) S. Cazaux, L. Boschman, N. Rougeau, G. Reitsma, R. Hoekstra, D. Teillet-Billy, S. Morisset, M. Spaans and T. Schlathölter, Sci. Rep., 2016, 6, 19835 CrossRef.
- M. D. Watson, M. G. Debije, J. M. Warman and K. Müllen, J. Am. Chem. Soc., 2004, 126, 766 CrossRef CAS PubMed.
- X. Yao, X. Y. Wang, C. Simpson, G. M. Paternò, M. Guizzardi, M. Wagner, G. Cerullo, F. Scotognella, M. D. Watson, A. Narita and K. Müllen, J. Am. Chem. Soc., 2019, 141, 4230 CrossRef CAS.
- (a) M. Leccese, R. Jaganathan, L. Slumstrup, J. D. Thrower, L. Hornekær and R. Martinazzo, Mon. Not. R. Astron. Soc., 2023, 519, 5567 CrossRef CAS; (b) H. Andrews, A. Candian and A. G. G. M. Tielens, Astron. Astrophys., 2016, 595, A23 CrossRef; (c) J. D. Thrower, E. E. Friis, A. L. Skov, B. Jørgensena and L. Hornekær, Phys. Chem. Chem. Phys., 2014, 16, 3381 RSC.
- (a) R. H. Baker and R. D. Schuetz, J. Am. Chem. Soc., 1947, 69, 1250 CrossRef CAS; (b) W. Kitching, H. A. Olszowy, G. M. Drew and W. Adcock, J. Org. Chem., 1982, 47, 5153 CrossRef CAS; (c) R. J. Bonilla, B. R. James and P. G. Jessop, Chem. Commun., 2000, 941 RSC; (d) B. Han, M. Zhang, H. Jiao, H. Ma, J. Wang and Y. Zhang, RSC Adv., 2021, 11, 39934 RSC; (e) A. A. Philippov, A. M. Chibiryaev and O. N. Martyanov, Catal. Today, 2021, 379, 15 Search PubMed.
- (a) L. Rampazzo and A. Zeppa, J. Electroanal. Chem., 1979, 105, 221 CrossRef CAS; (b) S. Friedman, S. Meltina, A. Svedi and I. Wender, J. Org. Chem., 1959, 24, 1287 CrossRef CAS.
- (a) P. P. Fu and R. G. Harvey, Tetrahedron Lett., 1977, 5, 415 CrossRef; (b) P. P. Fu, H. M. Lee and R. G. Harvey, J. Org. Chem., 1980, 45, 2797 CrossRef CAS.
- W. Lijinsky, G. Advani, L. Keefer, H. Y. Ramahi and L. Stach, J. Chem. Eng. Data, 1972, 17, 100 CrossRef CAS.
- Y. Toyama, T. Yoshihara, H. Shudo, H. Ito, K. Itami and A. Yagi, Chem. Lett., 2024, 53, upad037 CrossRef CAS.
- (a) S. Origuchi, M. Kishimoto, M. Yoshizawa and S. Yoshimoto, Angew. Chem., Int. Ed., 2018, 57, 15481 CrossRef CAS PubMed; (b) M. Müller, V. S. Iyer, C. Kübel, V. Enkelmann and K. Müllen, Angew. Chem., Int. Ed. Engl., 1997, 36, 1607 CrossRef.
- G. Principi, F. Agresti, A. Maddalena and S. L. Russo, Energy, 2009, 34, 2087 CrossRef CAS.
- C. Gelis, A. Heusler, Z. Nairoukh and F. Glorius, Chem.–Eur. J., 2020, 26, 14090 CrossRef CAS PubMed.
- Y. Wang, Z. Chang, Y. Hu, X. Lin and X. Dou, Org. Lett., 2021, 23, 1910 CrossRef CAS PubMed.
- (a) Y. Toyama, A. Yagi, K. Itami and H. Ito, Chemrxiv, 2024 Aug. 30th, preprint, DOI:10.26434/chemrxiv-2024-ts4vf; (b) K. Fujishiro, Y. Morinaka, Y. Ono, T. Tanaka, L. T. Scott, H. Ito and K. Itami, J. Am. Chem. Soc., 2023, 145, 8163 CrossRef CAS PubMed.
- (a) R. Takahashi, A. Hu, P. Gao, Y. Gao, Y. Pang, T. Seo, J. Jiang, S. Maeda, H. Takaya, K. Kubota and H. Ito, Nat. Commun., 2021, 12, 6691 CrossRef CAS PubMed; (b) P. Gao, J. Jiang, S. Maeda, K. Kubota and H. Ito, Angew. Chem., Int. Ed., 2022, 134, e202207118 CrossRef; (c) R. Takahashi, P. Gao, K. Kubota and H. Ito, Chem. Sci., 2023, 14, 499 RSC; (d) K. Kubota, N. Shizukuishi, S. Kubo and H. Ito, Chem. Lett., 2024, 53, upae056 CrossRef CAS; (e) K. Kubota, K. Kondo, T. Seo, M. Jin and H. Ito, RSC Adv., 2023, 13, 28652 RSC.
- (a) S. L. James, C. J. Adams, C. Bolm, D. Braga, P. Collier, T. Friščić, F. Grepioni, K. D. M. Harris, G. Hyett, W. Jones, A. Krebs, J. Mack, L. Maini, A. G. Orpen, I. P. Parkin, W. C. Shearouse, J. W. Steedk and D. C. Waddelli, Chem. Soc. Rev., 2012, 41, 413 Search PubMed; (b) G. W. Wang, Chem. Soc. Rev., 2013, 42, 7668 RSC; (c) J. L. Do and T. Friščić, ACS Cent. Sci., 2017, 3, 13 Search PubMed; (d) K. Kubota and H. Ito, Trends Chem., 2020, 2, 1066 CrossRef CAS.
- (a) Y. Sawama, N. Yasukawa, K. Ban, R. Goto, M. Niikawa, Y. Monguchi, M. Itoh and H. Sajiki, Org. Lett., 2018, 20, 2892 CrossRef CAS PubMed; (b) Y. Sawama, K. Ban, K. A. Suyama, H. Nakatam, M. Mori, T. Yamada, T. Kawajiri, N. Yasukawa, K. Park, Y. Monguchi, Y. Takagi, M. Yoshimura and H. Sajiki, ACS Omega, 2019, 4, 11522 CrossRef CAS PubMed.
- (a) V. J. Kolcsár and G. Szőllősi, ChemCatChem, 2022, 14, e202101501 Search PubMed; (b) V. J. Kolcsár and G. Szőllősi, Mol. Catal., 2022, 520, 112162 Search PubMed.
- A. Y. Li, A. Segalla, C. J. Li and A. Moores, ACS Sustainable Chem. Eng., 2017, 5, 11752 Search PubMed.
- T. Portada, D. Margetić and V. Štrukil, Molecules, 2018, 23, 3163 CrossRef PubMed.
- M. Carta, A. L. Sanna, A. Porcheddu, S. Garroni and F. Delogu, Sci. Rep., 2023, 13, 2470 Search PubMed.
- B. G. Fiss, A. J. Richard and T. Friščić, Can. J. Chem., 2021, 99, 93 Search PubMed.
- (a) T. Seo, N. Toyoshima, K. Kubota and H. Ito, J. Am. Chem. Soc., 2021, 143, 6165 CrossRef CAS PubMed; (b) Y. Gao, C. Feng, T. Seo, K. Kubota and H. Ito, Chem. Sci., 2022, 13, 430 RSC.
- (a) B. L. Tran, J. L. Fulton, J. C. Linehan, J. A. Lercher and R. M. Bullock, ACS Catal., 2018, 8, 8441 CrossRef CAS; (b) M. P. Wiesenfeldt, T. Knecht, C. Schlepphorst and F. Glorius, Angew. Chem., Int. Ed., 2018, 57, 8297 CrossRef CAS PubMed.
- (a) T. Seo, T. Ishiyama, K. Kubota and H. Ito, Chem. Sci., 2019, 10, 8202 RSC; (b) Z. J. Jiang, Z. H. Li, J. B. Yu and W. K. Su, J. Org. Chem., 2016, 81, 10049 CrossRef CAS PubMed; (c) J. L. Howard, Y. Sagatov, L. Repusseau, C. Schotten and D. L. Browne, Green Chem., 2017, 19, 2798 Search PubMed; (d) P. Ying, J. Yu and W. Su, Adv. Synth. Catal., 2021, 363, 1246 Search PubMed.
- E. Bresó-Femenia, B. Chaudret and S. Castillón, Catal. Sci. Technol., 2015, 5, 2741 Search PubMed.
- C. Hubert, E. G. Bilé, A. Denicourt-Nowicki and A. Roucoux, Green Chem., 2011, 13, 1766 Search PubMed.
- T. Yoshihara, H. Shudo, A. Yagi and K. Itami, J. Am. Chem. Soc., 2023, 145, 11754 CrossRef CAS PubMed.
- H. Miyamura, A. Suzuki, T. Yasukawa and S. Kobayashi, J. Am. Chem. Soc., 2018, 140, 11325 CrossRef CAS PubMed.
- H. Miyamura and S. Kobayashi, Angew. Chem., Int. Ed., 2022, 61, e202201203 CrossRef CAS.
- (a) G. Báti, S. Laxmi and M. C. Stuparu, ChemSusChem, 2023, 16, e202301087 CrossRef; (b) D. M. Baier, T. Rensch, D. Dobreva, C. Spula, S. Fanenstich, M. Rappen, K. Bergheim, S. Grätz and L. Borchardt, Chem.: Methods, 2023, 3, e202200058 CAS; (c) A. Stolle, R. Schmidta and K. Jacoba, Faraday Discuss., 2014, 170, 267 RSC.
- O. D. Jurchescu, A. Meetsma and T. T. Palstra, Acta Crystallogr., 2006, B62, 330 CAS.
- E. D. Bergmann, S. Blumberg, P. Bracha and S. Epstein, Tetrahedron, 1964, 20, 195 CrossRef CAS.
- W. Matsuoka, K. P. Kawahara, H. Ito, D. Sarlah and K. Itami, J. Am. Chem. Soc., 2023, 145, 658 CrossRef CAS PubMed.
- R. G. Clevenger, B. Kumar, E. M. Menuey and K. V. Kilway, Chem.–Eur. J., 2018, 24, 3113 CrossRef CAS PubMed.
- (a) Y. Hong, J. W. Y. Lam and B. Z. Tang, Chem. Soc. Rev., 2011, 40, 5361 RSC; (b) J. Mei, Y. Hong, J. W. Y. Lam, A. Qin, Y. Tang and B. Z. Tang, Adv. Mater., 2014, 26, 5429 CrossRef CAS PubMed; (c) J. Mei, N. L. C. Leung, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, Chem. Rev., 2015, 115, 11718 CrossRef CAS PubMed; (d) X. Cai and B. Liu, Angew. Chem., Int. Ed., 2020, 59, 9868 CrossRef CAS PubMed; (e) Z. Zhao H Zhang, J. W. Y. Lam and B. Z. Tang, Angew. Chem., Int. Ed., 2020, 59, 9888 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Syntheses, NMR, UV-vis absorption, emission, DFT calculations, and crystallographic table. CCDC 2389851, 2389852 and 2425787. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5sc01489a |
| This journal is © The Royal Society of Chemistry 2025 |