
Efficient synthesis of P-chiral biaryl phosphonates by stereoselective intramolecular cyclization†
Guangqing
Xu
,
Minghong
Li
,
Shouliang
Wang
and
Wenjun
Tang
*
State Key Laboratory of Bio-Organic and Natural Products Chemistry, Shanghai Institute of Organic Chemistry, 345 Ling Ling Rd, Shanghai 200032, P. R. China. E-mail: tangwenjun@sioc.ac.cn
First published on 3rd August 2015
Abstract
A series of P-chiral biaryl phosphonates were efficiently synthesized from diaryl 2-bromo arylphosphonates in high yields (up to 92%) and good enantioselectivities (up to 88% ee) through a palladium-catalyzed asymmetric cyclization with a novel P-chiral biaryl monophosphorus ligand. The P-chiral biaryl phosphonate can be rapidly transformed to both antipodes of a P-chiral dialkyl biaryl monophosphorus structure. The method provides a convenient access to various P-chiral biaryl monophosphines.
Since Knowles first introduced P-chiral phosphines CAMP and DIPAMP for rhodium-catalyzed asymmetric hydrogenation almost half a century ago,1P-chiral phosphorus ligands have played significant roles in the rapid development of the asymmetric catalysis area.2 Efficient construction of P-chiral phosphorus compounds has become a hot subject of research.3 Various efficient methods were developed including chemical resolutions,4 asymmetric synthesis by using chiral auxiliaries or reagents,5 and recently catalytic asymmetric methods.6 Because of the increasing applications of P-chiral biaryl monophosphorus ligands in organic synthesis,7 we propose to develop a general and efficient synthetic method for P-chiral biaryl monophosphorus ligands from a P-chiral biaryl phosphonate A through two consecutive stereospecific substitutions at the phosphorus center (Fig. 1). The challenge is whether the P-chiral biaryl phosphonate A can be efficiently synthesized from the readily accessible ortho-bromo arylphosphonate B through an enantioselective palladium-catalyzed desymmetric intramolecular cyclization.8 Herein we disclose our study on this asymmetric cyclization and its transformations toward P-chiral biaryl monophosphorus ligands.
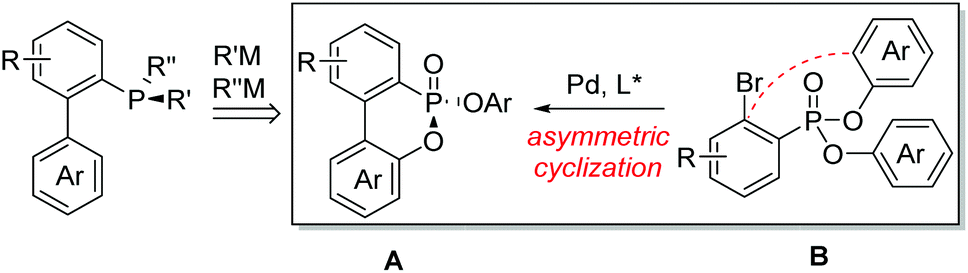 | ||
| Fig. 1 A new strategy for the synthesis of P-chiral biaryl monophosphorus ligands. | ||
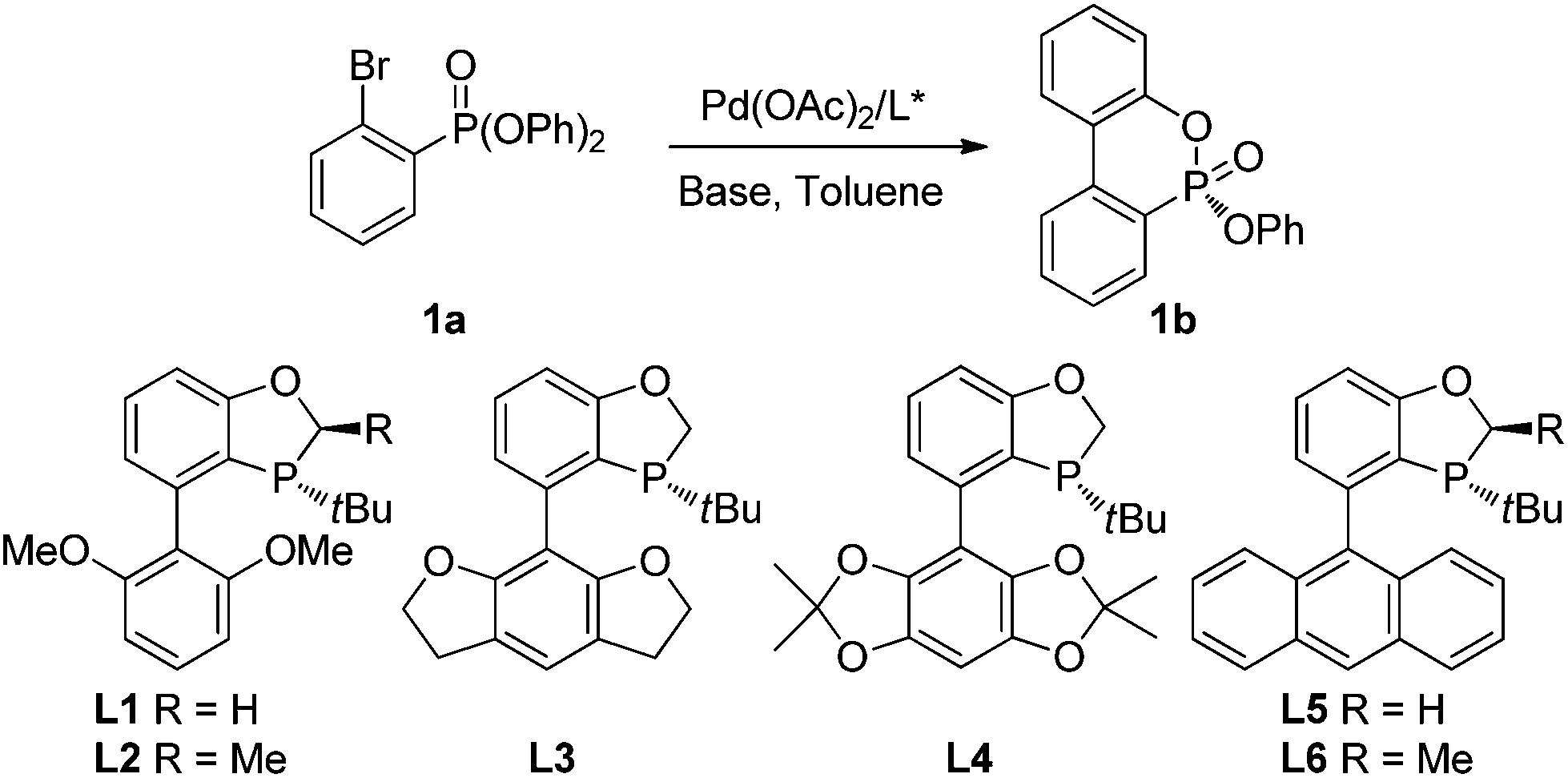
We chose diphenyl(2-bromophenyl)phosphonate (1a) as the substrate for study. As shown in Table 1, the palladium-catalyzed asymmetric cyclization of 1a proceeded smoothly at 80 °C in toluene with KOAc as the base to afford the cyclization product 1b in excellent yields in the presence of a P-chiral monophosphorus ligand. Among the several P-chiral biaryl monophosphorus ligands employed (entries 1–5),9 the newly developed ligand L3 with a tetrahydrobenzodifuran moiety provided an excellent yield (93%) and a good enantioselectivity (77% ee) with potassium acetate as the base. Apparently, the substituents on the low aryl ring of the P-chiral biaryl ligands exert significant influence on the enantioselectivity. Moderate ees were achieved with acyclic or cyclic alkoxy moieties such as methoxy substituents, furans and dioxolanes (entries 1, 3 and 4). In contrast, AntPhos (L5) proved to be ineffective (entry 5). Ligand L2 with a methyl group at the 2 position of the oxophosphole ring also provided a diminished ee (entry 2). When L3 was employed for further optimization, a dramatic base effect was observed. A more hindered base KOPiv afforded an inferior yield and ee value (entry 6). Meanwhile, 1-AdCOOK could provide comparable enantioselectivity to KOAc but with lower yield (entry 7). When PhCOOK was employed as a base, a higher ee value (88%) was achieved, albeit with a low yield (34%, entry 8). The low yield could be largely due to its relatively weak basicity. We thus employed PhCH2COOK as the base. Although the cyclization yield was comparable to that with KOAc, its enantioselectivity was slightly inferior (entry 9). With Ph2CHCOOK as the base, we obtained a similar yield to that with KOAc, but with a slightly better ee value (entry 10). When the reaction temperature was reduced to 70 °C, the ee value of 1b was improved to 82% (entry 11). Change of the solvent to cyclohexane, 1,4-dioxane, THF, and 1,2-dichloroethane (DCE) did not enhance the enantioselectivity (entries 12–15). When the mole ratio of Pd/L3 increased from 1/1.2 to 1/2 (4 mol% Pd), a better ee value (88%) was achieved along with an acceptable yield (entry 16). Other bases were also tested, but no further improvement of the ee value was achieved.10
|
|
||||||
|---|---|---|---|---|---|---|
| Entriesa | L* | Base | Solvent | T (°C) | Yieldb (%) | % eec |
| a Unless otherwise specified, the reactions were performed at the designated reaction temperature in organic solvent (1 mL) with aryl bromide (0.2 mmol) under nitrogen for 24 h in the presence of Pd(OAc)2 (5 mol%), L* (6 mol%), and base (0.3 mmol), the absolute configuration of 1b was assigned by analogy according to the X-ray crystal structure of 2f. b Isolated yield. c ee values were determined by chiral HPLC on a chiralcel AD-H column. d Pd(OAc)2 (4 mol%), L3 (8 mol%). | ||||||
| 1 | L1 | KOAc | Toluene | 80 | 91 | 71 |
| 2 | L2 | KOAc | Toluene | 80 | 91 | 16 |
| 3 | L3 | KOAc | Toluene | 80 | 93 | 77 |
| 4 | L4 | KOAc | Toluene | 80 | 93 | 66 |
| 5 | L5 | KOAc | Toluene | 80 | 81 | 1 |
| 6 | L3 | KOPiv | Toluene | 80 | 70 | 70 |
| 7 | L3 | 1-AdCOOK | Toluene | 80 | 76 | 77 |
| 8 | L3 | PhCOOK | Toluene | 80 | 34 | 83 |
| 9 | L3 | PhCH2COOK | Toluene | 80 | 94 | 75 |
| 10 | L3 | Ph2CHCOOK | Toluene | 80 | 93 | 78 |
| 11 | L3 | Ph2CHCOOK | Toluene | 70 | 70 | 82 |
| 12 | L3 | Ph2CHCOOK | CyHex | 70 | 88 | 76 |
| 13 | L3 | Ph2CHCOOK | Dioxane | 70 | 26 | 37 |
| 14 | L3 | Ph2CHCOOK | THF | 70 | 19 | 74 |
| 15 | L3 | Ph2CHCOOK | DCE | 70 | 97 | 74 |
| 16d | L3 | Ph2CHCOOK | Toluene | 70 | 83 | 88 |
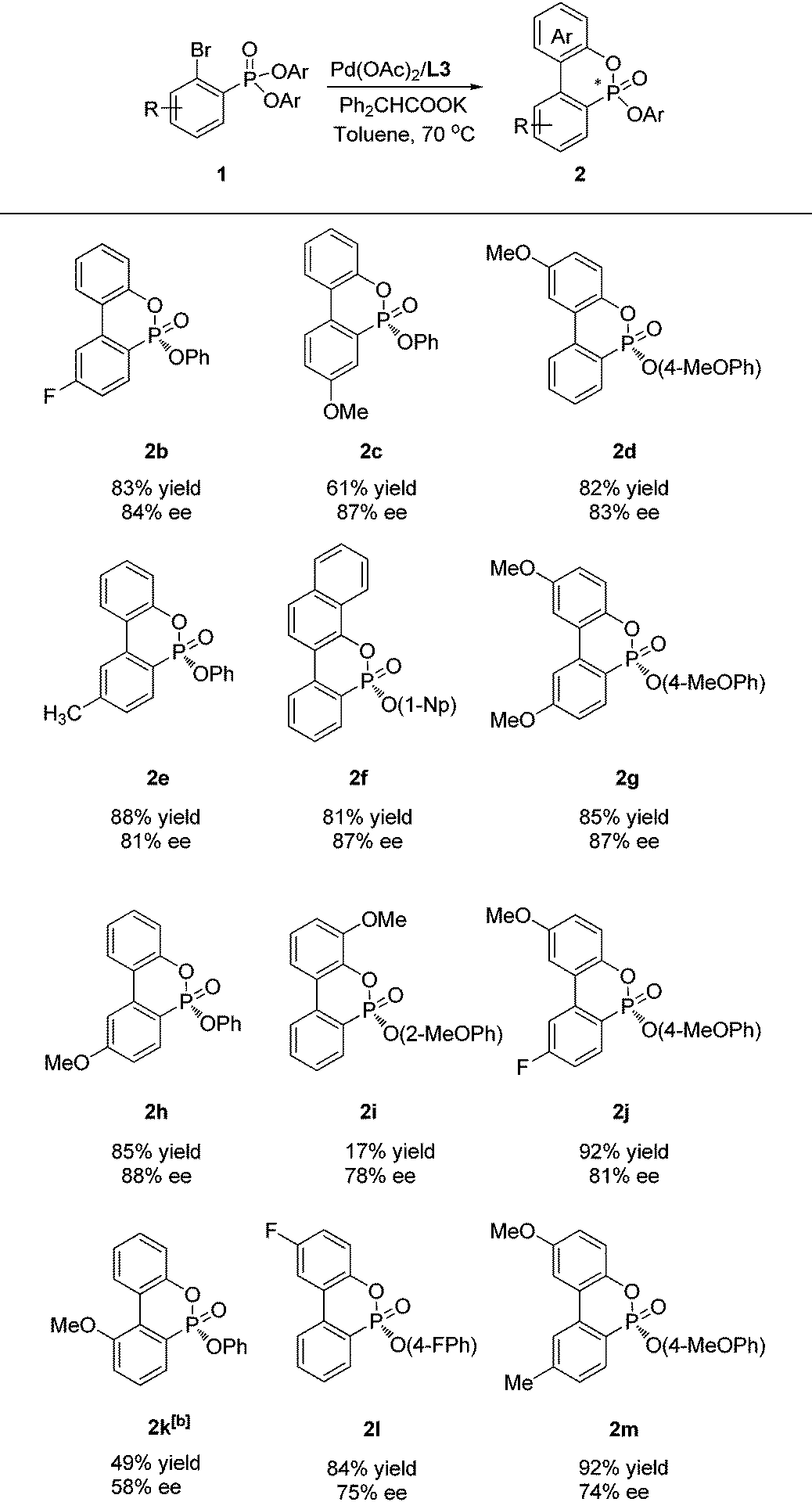
We then investigated the substrate scope of this asymmetric cyclization under optimized conditions (Table 2). Thus, a series of substituted diphenyl ortho-bromo phenylphosphonates (1b, e, h, c) were successfully cyclized to provide the corresponding P-chiral phosphonates in high yields and good enantioselectivities with L3 as the ligand. Substituents such as methyl, methoxy, and fluoro groups at the meta- or para-position were well tolerated. A substrate with a methoxy substituent adjacent to the bromine atom 1k provided the corresponding cyclization product 2k in only 27% ee and 52% yield. However, an improved ee (58%) value was achieved when L6 was employed as the ligand. In addition, various di(substituted aryl)ortho-bromo phenylphosphonates were also applicable to provide the corresponding cyclization products (2d, 2f–g, 2j, 2l–2m) in good yields and enantioselectivity. Di(ortho-methoxyphenyl)ortho-bromo phenylphosphonate (1i) also provided a decent ee value (78%) albeit with a low yield of 2i. The absolute configuration of 2f was determined as R by X-ray crystallographic analysis.11
| a Unless otherwise specified, the reactions were performed in toluene (1 mL) at 70 °C under nitrogen for 24 h with aryl bromide (0.2 mmol), Pd(OAc)2 (4 mol%), L3 (8 mol%), and Ph2CHCOOK (0.3 mmol); isolated yields; ee values were determined by chiral HPLC. The absolute configuration of 2f was determined by X-ray crystallography, others were assigned by analogy. b L6 as a ligand. |
|---|
|
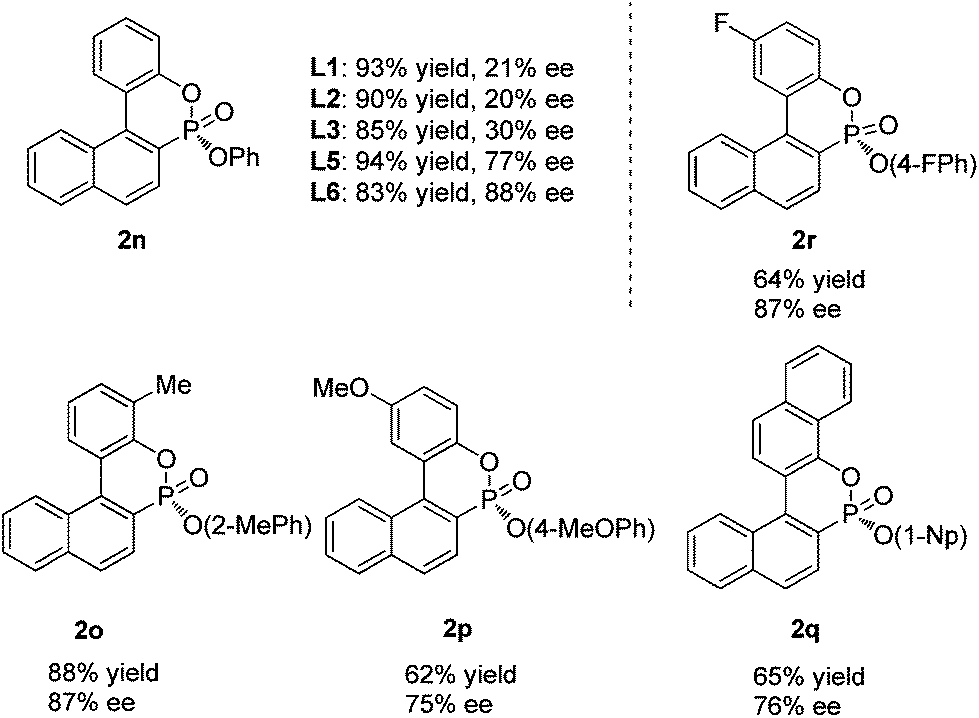
Interestingly, when diphenyl (1-bromo-2-naphthyl)phosphonate (1n) was employed for cyclization under similar reaction conditions, the cyclization product 2n was formed in only 30% ee and in 85% yield. In order to obtain a better enantioselectivity, we further screened the P-chiral biaryl monophosphorus ligands in our laboratory. As can be seen in Table 3, ligands L1–3 all provided very poor enantioselectivities. To our surprise, AntPhos (L5) formed the cyclization product in 77% ee. L6 with a methyl substituent on the oxophosphole ring deriving from L5 afforded the cyclization product in 88% ee and 83% yield. It was thus chosen as the ligand for this series of substrates. By using these conditions, various di(substitutedaryl) (1-bromo-2-naphthyl)phosphonates (1o–r) were also subjected to the cyclization and the corresponding cyclization products (2o–r) were formed in good yields and high enantioselectivities. The di(para-methoxy)phosphonate substrate 1p and di(1-naphthyl)phosphonate substrate 1q afforded the corresponding products 2p and 2q in slightly lower ee values, respectively.
| a Unless otherwise specified, the reactions were performed for 24 h under nitrogen at 70 °C in toluene (1 mL) with naphthyl bromide (0.2 mmol), Pd(OAc)2 (5 mol%), L6 (6 mol%), and KOAc (0.3 mmol); isolated yields; ee values were determined by chiral HPLC; the absolute configurations were assigned by analogy. |
|---|
|
The P-chiral phosphonates 2a–q can be envisioned as useful precursors for a variety of P-chiral biaryl phosphorus ligands. Because both aryloxy substituents of the phosphonate can be displaced stereospecifically by different alkyl lithium or Grignard reagents sequentially, both antipodes of a P-chiral biaryl structure could be prepared from a single P-chiral phosphonate product. In order to demonstrate this utility (Scheme 1), the P-chiral biaryl phosphonate 2a was treated first with isopropyllithium in the presence of Et2AlCl to form isopropyl substituted product 3 without erosion of enantioselectivity. Subsequent treatment of 3 with methyllithium stereospecifically provided P-chiral dialkyl biarylphosphine oxide 4.12 Alternatively, treatment of 2a (80% ee) with methyllithium and isopropyllithium sequentially provided ent-4 in an unoptimized yield with light erosion of the ee value (73% ee). Stereospecific reduction of 4 and ent-4 with a reported procedure13 could provide both antipodes of a P-chiral dialkyl biaryl phosphine, respectively.
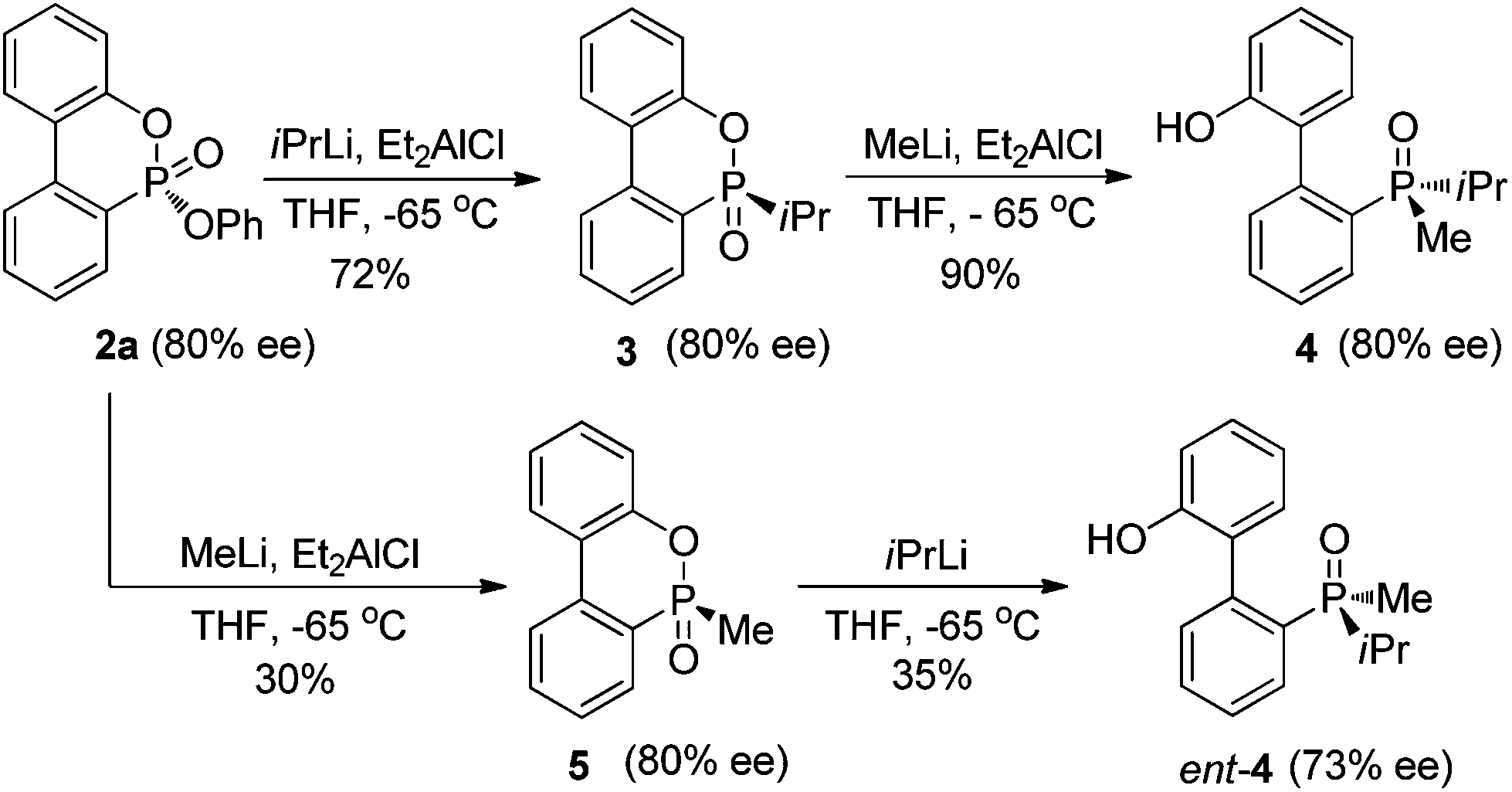 | ||
| Scheme 1 Stereospecific transformation of P-chiral phosphonate 2a to P-chiral biaryl phosphine oxides 4 and ent-4. | ||
In summary, we have developed an efficient Pd-catalyzed desymmetric intramolecular cyclization of diaryl ortho-bromo aryl phosphonates that have led to a series of P-chiral biaryl phosphonates in high yields (up to 92%) and good enantioselectivities (up to 88% ee) under very mild conditions. The P-chiral biaryl phosphonates have been demonstrated as excellent precursors to both antipodes of P-chiral dialkyl biaryl monophosphines. This method has provided convenient access to various P-chiral biaryl monophosphine ligands, which should have increasing applications in the area of asymmetric catalysis.
Acknowledgements
We are grateful to the NSFC (21432007, 21272254), STCSM (13J1410900), the “Thousand Plan” Youth program.Notes and references
- (a) W. S. Knowles and M. J. Sabacky, Chem. Commun., 1968, 1445 RSC; (b) W. S. Knowles, M. J. Sabacky, B. D. Vineyard and D. J. Weinkauff, J. Am. Chem. Soc., 1975, 97, 2567 CrossRef CAS.
- (a) P. C. J. Kamer and P. W. N. M. Van Leeuween, Phosphorus(III) Ligands in homogeneous Catalysis: Design and Synthesis, Wiley & Sons, West Sussex, 2012 CrossRef; (b) W. Tang and X. Zhang, Chem. Rev., 2003, 103, 3029 CrossRef CAS PubMed; (c) P-Stereogenic Ligands in Enantioselective Catalysis, ed. A. Grabulosa, RSC, Cambridge, 2011 Search PubMed.
- For reviews on the synthesis of P-chiral phosphines, see: (a) K. M. Pietrusiewicz and M. Zablocka, Chem. Rev., 1994, 94, 1375 CrossRef CAS; (b) A. Grabulosa, J. Granell and G. Muller, Coord. Chem. Rev., 2007, 251, 25 CrossRef CAS; (c) J. S. Harvey and V. Gouverneur, Chem. Commun., 2010, 46, 7477 RSC; (d) O. I. Kolodiazhnyi, Tetrahedron: Asymmetry, 2012, 23, 1 CrossRef CAS.
- For selective examples, see: (a) K. Tani, L. D. Brown, J. Ahmed, J. A. Ibers, M. Yokota, A. Nakamura and S. Otsuka, J. Am. Chem. Soc., 1977, 99, 7876 CrossRef CAS; (b) N. K. Roberta and S. B. Wild, J. Am. Chem. Soc., 1979, 101, 6254 CrossRef; (c) T. Imamoto, K. V. L. Crépy and K. Katagiri, Tetrahedron: Asymmetry, 2004, 15, 2213 CrossRef CAS; (d) D. Liu and X. Zhang, Eur. J. Org. Chem., 2005, 646 CrossRef CAS.
- For selective examples, see: (a) O. Korpiun and K. Mislow, J. Am. Chem. Soc., 1967, 89, 4784 CrossRef CAS; (b) D. Gatineau, L. Giordano and G. Buono, J. Am. Chem. Soc., 2011, 133, 10728 CrossRef CAS PubMed; (c) O. Berger and J.-L. Montchamp, Angew. Chem., Int. Ed., 2013, 52, 11377 CrossRef CAS PubMed; (d) S. Jugé, M. Stephan, J. A. Laffitte and J. P. Genet, Tetrahedron Lett., 1990, 31, 6357 CrossRef; (e) Z. S. Han, N. Goyal, M. A. Herbage, J. D. Sieber, B. Qu, Y. Xu, Z. Li, J. T. Reeves, J.-N. Desrosiers, S. Ma, N. Grinberg, H. Lee, H. P. R. Mangunuru, Y. Zhang, D. Krishnamurthy, B. Z. Lu, J. J. Song, G. Wang and C. H. Senanayake, J. Am. Chem. Soc., 2013, 135, 2474 CrossRef CAS PubMed.
- For selective examples, see: (a) J. R. Moncarz, N. F. Laritcheva and D. S. Glueck, J. Am. Chem. Soc., 2002, 124, 13356 CrossRef CAS PubMed; (b) V. S. Chan, I. C. Stewart, R. G. Bergman and F. D. Toste, J. Am. Chem. Soc., 2006, 128, 2786 CrossRef CAS PubMed; (c) C. Scriban and D. S. Glueck, J. Am. Chem. Soc., 2006, 128, 2788 CrossRef CAS PubMed; (d) N. F. Blank, J. R. Moncarz, T. J. Brunker, C. Scriban, B. J. Anderson, O. Amir, D. S. Glueck, L. N. Zakharov, J. A. Golen, C. D. Incarvito and A. L. Rheingold, J. Am. Chem. Soc., 2007, 129, 6847 CrossRef CAS PubMed; (e) V. S. Chan, R. G. Bergman and F. D. Toste, J. Am. Chem. Soc., 2007, 129, 15122 CrossRef CAS PubMed; (f) C. Scriban, D. S. Glueck, J. A. Golen and A. L. Rheingold, Organometallics, 2007, 26, 1788 CrossRef CAS; (g) B. J. Anderson, M. A. Guino-o, D. S. Glueck, J. A. Golen, A. G. DiPasquale, L. M. Liable-Sands and A. L. Rheingold, Org. Lett., 2008, 10, 4425 CrossRef CAS PubMed; (h) V. S. Chan, M. Chiu, R. G. Bergman and F. D. Toste, J. Am. Chem. Soc., 2009, 131, 6021 CrossRef CAS PubMed; (i) T. W. Chapp, D. S. Glueck, J. A. Golen, C. E. Moore and A. L. Rheingold, Organometallics, 2010, 29, 378 CrossRef CAS; (j) C. Li, W.-X. Li, S. Xu and W.-L. Duan, Chin. J. Org. Chem., 2013, 33, 799 CrossRef CAS; (k) Y. Huang, Y. Li, P.-H. Leung and T. Hayashi, J. Am. Chem. Soc., 2014, 136, 4865 CrossRef CAS PubMed; (l) C. Li, B.-L. Bian, S. Xu and W.-L. Duan, Org. Chem. Front., 2014, 1, 541 RSC; (m) Z.-J. Du, J. Guan, G.-J. Wu, P. Xu, L.-X. Gao and F.-S. Han, J. Am. Chem. Soc., 2015, 137, 632 CrossRef CAS PubMed.
- (a) J. Yin and S. L. Buchwald, J. Am. Chem. Soc., 2000, 122, 12051 CrossRef CAS; (b) X. Shen, G. O. Jones, D. A. Watson, B. Bhayana and S. L. Buchwald, J. Am. Chem. Soc., 2010, 132, 11278 CrossRef CAS PubMed; (c) W. Tang, N. D. Patel, G. Xu, X. Xu, J. Savoie, S. Ma, M.-H. Hao, S. Keshipeddy, A. G. Capacci, X. Wei, Y. Zhang, J. J. Gao, W. Li, S. Rodriguez, B. Z. Lu, N. K. Yee and C. H. Senanayake, Org. Lett., 2012, 14, 2258 CrossRef CAS PubMed; (d) K. Li, N. Hu, R. Luo, W. Yuan and W. Tang, J. Org. Chem., 2013, 78, 6350 CrossRef CAS PubMed; (e) G. Xu, W. Fu, G. Liu, C. H. Senanayake and W. Tang, J. Am. Chem. Soc., 2014, 136, 570 CrossRef CAS PubMed; (f) K. Du, P. Guo, Y. Chen, Z. Cao, Z. Wang and W. Tang, Angew. Chem., Int. Ed., 2015, 54, 3033 CrossRef CAS PubMed.
- During preparation of the manuscript, two examples of palladium-catalyzed enantioselective C–H arylation for the synthesis of P-stereogenic phosphinic amides were reported: (a) Z.-Q. Lin, W.-Z. Wang, S.-B. Yan and W.-L. Duan, Angew. Chem., Int. Ed., 2015, 54, 6265 CrossRef CAS PubMed; (b) L. Liu, A.-A. Zhang, Y. Wang, F. Zhang, Z. Zuo, W.-X. Zhao, C.-L. Feng and W. Ma, Org. Lett., 2015, 17, 2046 CrossRef CAS PubMed.
- For other applications of ligands L1–2 and L5–6 in catalysis, see ref. 7c–f and: (a) W. Tang, A. G. Capacci, X. Wei, W. Li, A. White, N. D. Patel, J. Savoie, J. J. Gao, S. Rodriguez, B. Qu, N. Haddad, B. Z. Lu, D. Krishnamurthy, N. K. Yee and C. H. Senanayake, Angew. Chem., Int. Ed., 2010, 49, 5879 CrossRef CAS PubMed; (b) W. Tang, S. Keshipeddy, Y. Zhang, X. Wei, J. Savoie, N. D. Patel, N. K. Yee and C. H. Senanayake, Org. Lett., 2011, 13, 1366 CrossRef CAS PubMed; (c) Q. Zhao, C. Li, C. H. Senanayake and W. Tang, Chem. – Eur. J., 2013, 19, 2261 CrossRef CAS PubMed; (d) C. Li, G. Xiao, Q. Zhao, H. Liu, T. Wang and W. Tang, Org. Chem. Front., 2014, 1, 225 RSC; (e) G. Xu, Q. Zhao and W. Tang, Chin. J. Org. Chem., 2014, 34, 1919 CrossRef CAS.
- Ph3CCOOK, Ph2CHCOOCs, and potassium 2-(naphthalen-1-yl)acetate were also tested as bases and the highest ee value was 85%.
- CCDC 1062715 contains the supplementary crystallographic data for this paper.
- 1H NMR and 31P NMR spectra showed two atropisomers in a ratio of 2.2/1 at 25 °C.
- For examples of reduction of chiral phosphine oxides, see ref. 5c–e and the following literatures: (a) T. Imamoto, S.-i. Kikuchi, T. Miura and Y. Wada, Org. Lett., 2001, 3, 87 CrossRef CAS PubMed; (b) K. V. Rajendran and D. G. Gilheany, Chem. Commun., 2012, 48, 817 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed procedures of cross-coupling reactions, characterization data, and spectra. CCDC 1062715. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5qo00142k |
| This journal is © the Partner Organisations 2015 |