
Efficient synthesis of P-chiral biaryl phosphonates by stereoselective intramolecular cyclization†
Abstract
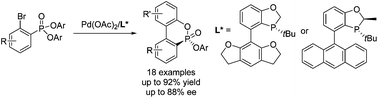
A series of P-chiral biaryl phosphonates were efficiently synthesized from diaryl 2-bromo arylphosphonates in high yields (up to 92%) and good enantioselectivities (up to 88% ee) through a palladium-catalyzed asymmetric cyclization with a novel P-chiral biaryl monophosphorus ligand. The P-chiral biaryl phosphonate can be rapidly transformed to both antipodes of a P-chiral dialkyl biaryl monophosphorus structure. The method provides a convenient access to various P-chiral biaryl monophosphines.
- This article is part of the themed collection: Celebrating 70 Years of Shanghai Institute of Organic Chemistry
 Please wait while we load your content...
Please wait while we load your content...

