
Base-promoted cascade reaction of isocyanides, selenium and amines: a practical approach to 2-aminobenzo[d][1,3]selenazines under metal-free conditions†
Yi
Fang
,
Shun-Yi
Wang
*,
Xiao-Bin
Shen
and
Shun-Jun
Ji
*
Key Laboratory of Organic Synthesis of Jiangsu Province, College of Chemistry, Chemical Engineering and Materials Science & Collaborative Innovation Center of Suzhou Nano Science and Technology, Soochow University, Suzhou, 215123, P. R. China. E-mail: shunyi@suda.edu.cn; shunjun@suda.edu.cn; Fax: +86-512-65880307; Tel: +86-512-65880307
First published on 28th July 2015
Abstract
A new practical approach for constructing 2-aminobenzo[d][1,3]selenazines by base mediated multicomponent reaction of isocyanides, selenium and amines under metal-free conditions is reported. A series of selenazine derivatives are observed in moderate to excellent yields.
Organoselenium compounds are full of diversity and specialized wide applications in organic synthesis.1 What's more, they are recognized as active reagents in medicinal and biological chemistry. Ebselen (Scheme 1, top), one of the representative organoselenium compounds, shows great biological and medicinal activities.2–5 The compounds containing an isoselenourea skeleton could be employed as precursors for the generation of selenolate anions under basic conditions.6 On the other hand, the isoselenourea derivatives also show encouraging effects on the antitumor and antibacterium activities (Scheme 1).7
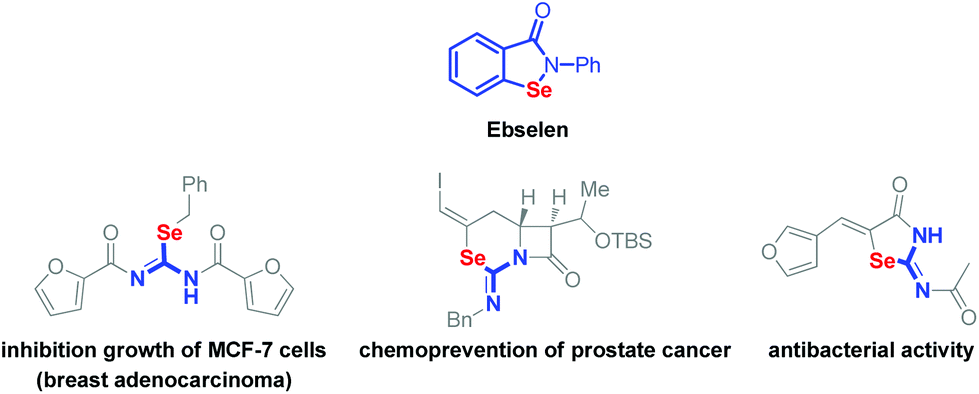 | ||
| Scheme 1 Biologically active compounds containing an isoselenourea skeleton. | ||
Traditional methods for the construction of isoselenoureas are the alkylation reactions of selenoureas with alkyl sulfates8 or alkyl halides.9 However, these methods have the drawbacks such as low yields and utilization of toxic reagents.10 Isocyanides are important building blocks in organic synthesis. In 1972, Sonoda developed a practical method to build up isoselenocyanates by the base-promoted reactions of isocyanides with elemental selenium.11 The reaction of isoselenocyanates with amines could provide selenoureas efficiently (Scheme 2).12
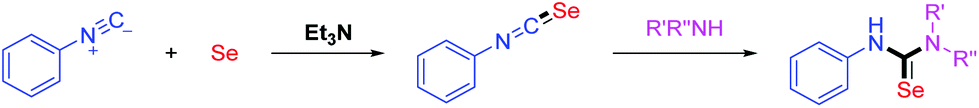 | ||
| Scheme 2 Convenient route to selenoureas. | ||
Multicomponent reactions (MCRs) involving isocyanides have shown great diversity and significance in modern organic chemistry.13 In recent years, ortho-functionalized aryl isocyanides have attracted great attention due to their various applications in organic synthesis.14 Herein, we report a new practical synthesis of 2-amino-benzo[d][1,3]-selenazines by multicomponent reaction of ortho-functionalized aryl isocyanides, elemental selenium and amines via isoselenocyanate formation and subsequently intramolecular Michael addition reactions under metal-free conditions (Scheme 3).
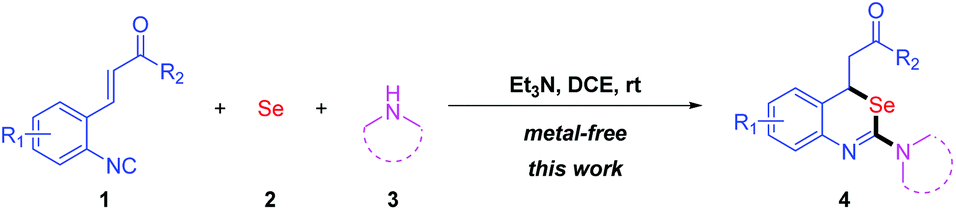 | ||
| Scheme 3 Synthesis of 2-aminobenzo[d][1,3]selenazines by a metal-free multicomponent reaction. | ||
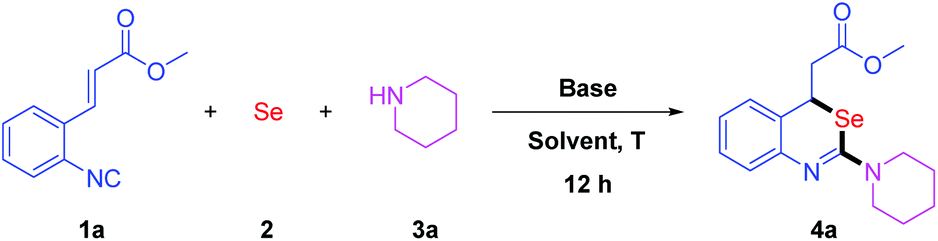
We initiated the studies by the reaction of methyl (E)-3-(2-isocyanophenyl)acrylate 1a with elemental selenium 2 and piperidine 3a, in the presence of DBU. The reaction was performed in THF for 12 h at ambient temperature. To our delight, the desired product 4a was obtained in 62% yield (Table 1, entry 1). Inspired by this result, we further optimized the reaction conditions by screening the solvents. Ether solvents, such as 1,4-dioxane, MTBE (methyl tert-butyl ether) and DME (dimethoxyethane), gave the products in moderate yields (Table 1, entries 2–4). Furthermore, DMF and DMSO decreased the yields to 43% and 21%, respectively (Table 1, entries 5 and 6). Other solvents such as toluene, acetone, EtOAc and MeCN failed to give better results (Table 1, entries 7–10). We fortunately discovered that the chlorine-containing solvents increased the yields dramatically (Table 1, entries 11–13). The desired product 4a was observed in 92% yield when DCE (1,2-dichloroethane) was used as the solvent. Next, we optimized the additive base. While DBU was replaced by DABCO, iPr2NEt, Cs2CO3 or LiOMe, lower yields were obtained (Table 1, entries 14 and 16–18). It was found that Et3N could increase the yield slightly (Table 1, entry 15). The employment of 1.5 equivalents of Et3N (Table 1, entry 19) turned out to be more efficient than 1.2 equivalents. When a catalytic amount of Et3N was used, we could only observe the product in low yield (Table 1, entry 20). We supposed that Et3N played an important role in the formation of isoselenocyanates. Therefore, the optimal conditions were 1a (0.3 mmol, 1.0 eq.), 2 (0.45 mmol, 1.5 eq.) and 3a (0.45 mmol, 1.5 eq.) in DCE (2 mL) in the presence of Et3N (0.45 mmol, 1.5 eq.) at room temperature for 12 h.
|
|
||||
|---|---|---|---|---|
| Entry | Base | Solvent | T | Yieldb (%) |
| a Reaction conditions: 1a (0.3 mmol), 2 (0.36 mmol), 3a (0.36 mmol), base (0.36 mmol), solvent (2 mL), rt, under air. b Yields were determined by LC analysis using biphenyl as an internal standard. c 2 (1.5 equiv.), 3a (1.5 equiv.) and Et3N (1.5 equiv.) were used. d Et3N (10 mol%) was used. | ||||
| 1 | DBU | THF | rt | 62 |
| 2 | DBU | Dioxane | rt | 57 |
| 3 | DBU | MTBE | rt | 35 |
| 4 | DBU | DME | rt | 51 |
| 5 | DBU | DMSO | rt | 21 |
| 6 | DBU | DMF | rt | 43 |
| 7 | DBU | Toluene | rt | 36 |
| 8 | DBU | Acetone | rt | 68 |
| 9 | DBU | EtOAc | rt | 57 |
| 10 | DBU | MeCN | rt | 65 |
| 11 | DBU | DCE | rt | 92 |
| 12 | DBU | DCM | rt | 83 |
| 13 | DBU | CHCl3 | rt | 85 |
| 14 | DABCO | DCE | rt | 83 |
| 15 | Et3N | DCE | rt | 93 |
| 16 | iPr2NEt | DCE | rt | 90 |
| 17 | Cs2CO3 | DCE | rt | 43 |
| 18 | LiOMe | DCE | rt | 69 |
| 19c | Et3N | DCE | rt | 98 |
| 20d | Et3N | DCE | rt | 15 |
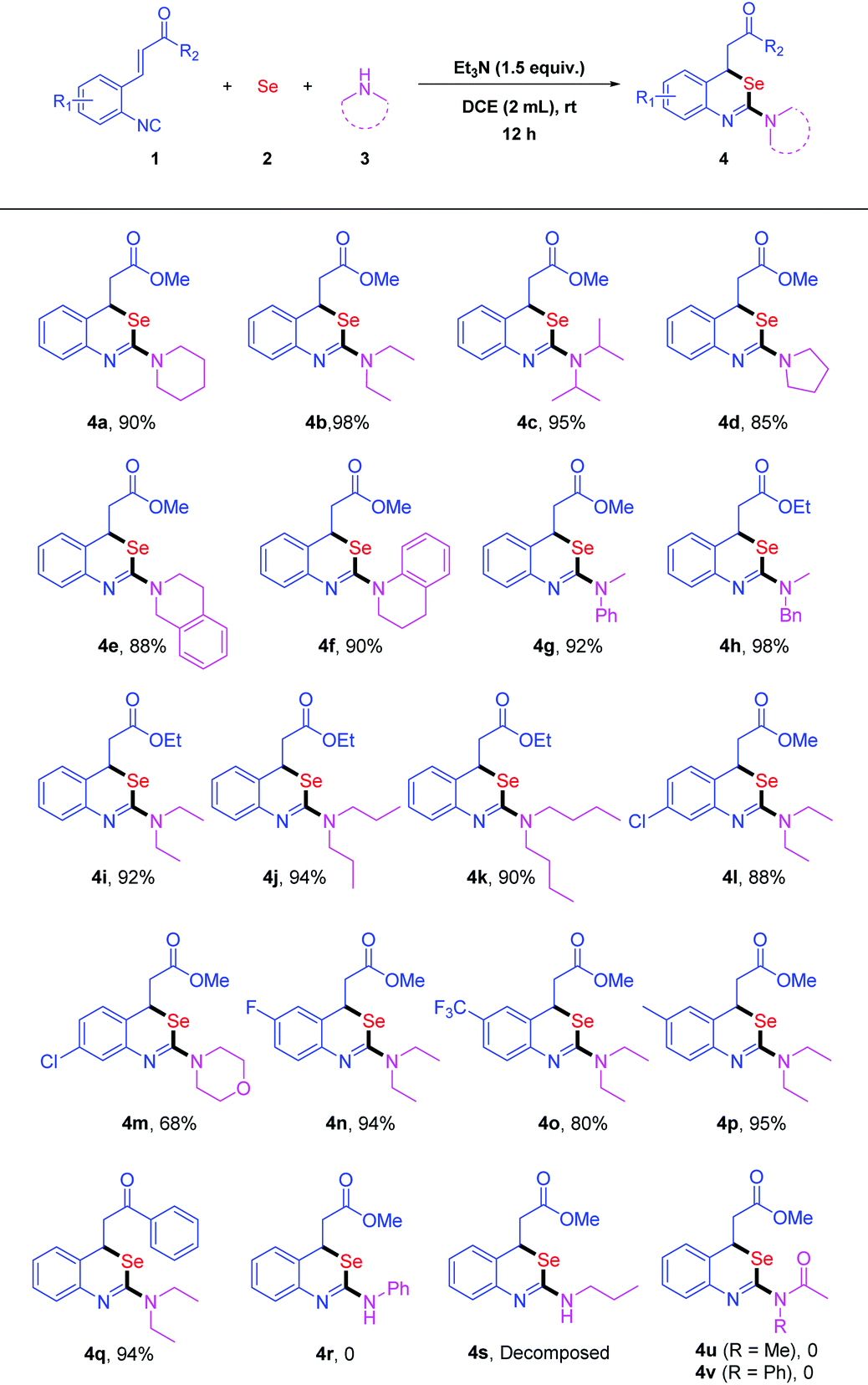
With the optimal reaction conditions in hand, we explored the substrate scope of amines (Table 2). Various secondary amines were successfully applied to the reactions of isocyanide 1a with selenium powder 2 under the optimized conditions. The desired products 4a–k could be isolated in 85% to 98% yields (Table 2). The reactions of diethylamine 3b and N-methyl-1-phenylmethanamine 3h with 1a and 2 afforded 4b and 4h both in 98% yields, respectively. The use of pyrrolidine 3d and 1,2,3,4-tetrahydroisoquinoline 3e resulted in 4d and 4e in 85% and 88% yields, respectively. These results indicated that the acyclic secondary amines were more reactive than the cyclic ones. Morpholine 3m with 1a and 2 could also lead to the desired product 4m in 68% yield. Next, we investigated a number of ortho-functionalized isocyanides. When 1i was subjected to the reaction, benzo[d][1,3]-selenazine derivative 4i was obtained in 92% yield (Table 2, 4i). What's more, the reactants, bearing both electron-withdrawing and electron-donating groups on the aromatic ring, all could be transformed to the corresponding benzo[d][1,3]selenazines 4l–p smoothly in good to excellent yields. The reaction of chalcone 1q could also undergo smoothly to give 4q in 94% yield. Unfortunately, the reaction of aniline failed to furnish the desired product 4r due to its weak nucleophilicity. When primary amine n-propylamine was applied to the reaction, the desired product 4s was not stable and decomposed to release the elemental selenium under air. Additionally, amides were used instead of amines to investigate the reactivity. We failed to obtain the product either 4u or 4v disappointedly. The negative results might be due to the weak nucleophilicity of amides as well.
To our delight, the optimal conditions could also be applied to the reaction of (S)-diphenyl(pyrrolidin-2-yl)methanol as the nucleophile and the desired products were isolated as diastereomers 4t and 4t′ in 66% and 25% yields, respectively (Scheme 4). The configurations of 4t and 4t′ are predicted by a computational study. Computational studies show that the 4t configuration is 3.3 kcal mol−1 lower in energy than the 4t′ configuration (for details see the ESI†). This result shows that the configuration of the Michael addition product was affected by the steric structure of the nucleophile.
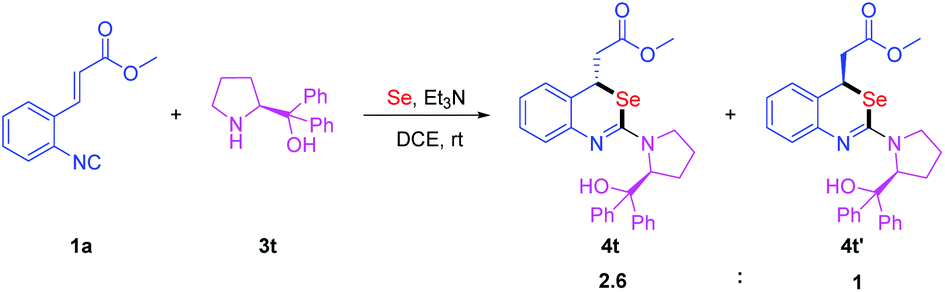 | ||
| Scheme 4 Investigation using (S)-diphenyl(pyrrolidin-2-yl)methanol. | ||
In view of the potential biological and medicinal activities of the 2-aminobenzo[d][1,3]selenazines, we explored the gram-scale reaction of 1a (5 mmol) with elemental selenium and diethylamine 3b under the standard reaction conditions. To our delight, the desired product 4b was observed in 90% yield (Scheme 5).
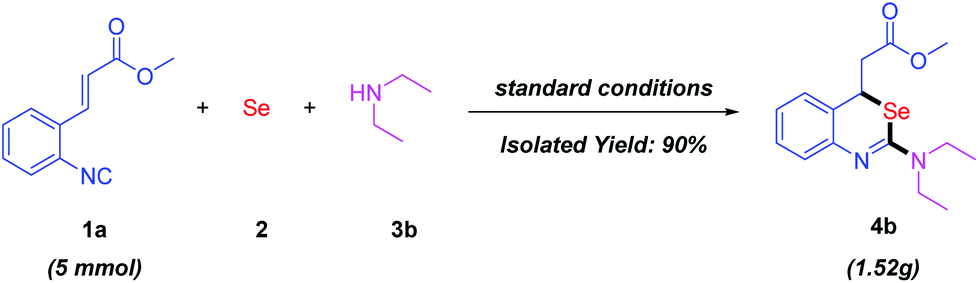 | ||
| Scheme 5 A gram-scale synthesis of 4b. | ||
Based on the above experimental results and literature reports,15 we proposed a plausible mechanism in Scheme 6. Initially, an isoselenocyanate A is generated in situ by the reaction of isocyanide 1 with elemental selenium in the presence of Et3N. Then, amine 3 reacts with isoselenocyanate A to give carbamimidoselenoate B through intermolecular nucleophilic attack. Following a subsequent Michael addition of B and protonation, 4 is formed.
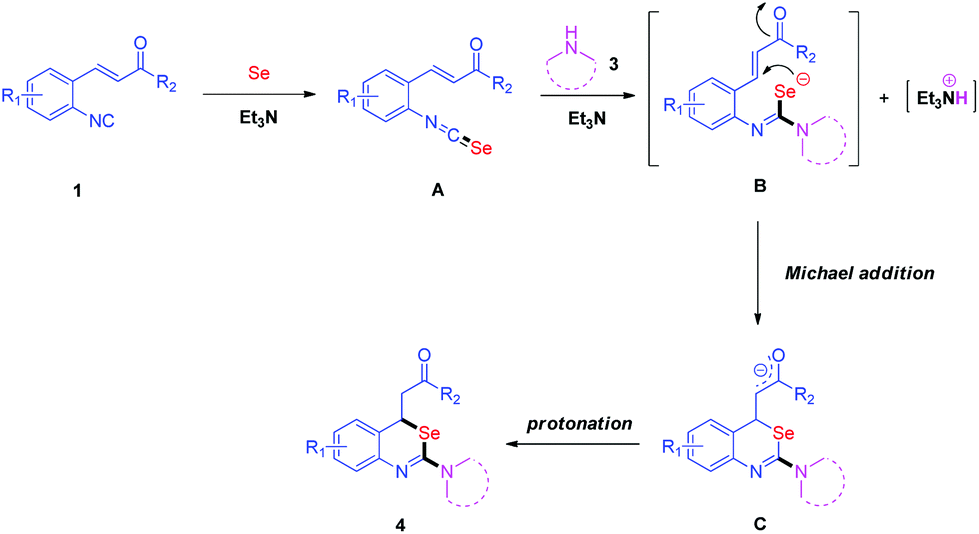 | ||
| Scheme 6 Plausible reaction mechanism. | ||
Conclusions
In summary, we have established a practical approach for the synthesis of 2-aminobenzo[d][1,3]selenazines via a base-promoted cascade reaction involving isocyanides, elemental selenium and amines under metal-free conditions. This approach provides a direct construction of selenazine derivatives with potential biological and medicinal activities under mild conditions. The investigations of extending our strategy to the synthesis of enatiopure selenazines are ongoing in our group.Acknowledgements
We gratefully thank Prof. X.-G. Bao (Soochow Univ.) for computational studies. We gratefully acknowledge the Natural Science Foundation of China (no. 21172162, 21372174), the Young National Natural Science Foundation of China (no. 21202111), the Ph.D. Programs Foundation of Ministry of Education of China (2013201130004), the Young Natural Science Foundation of Jiangsu Province (BK2012174), Key Laboratory of Organic Synthesis of Jiangsu Province (KJS1211), PAPD, and Soochow University for financial support.Notes and references
- For selected examples: (a) K. C. Nicolaou, N. Winssinger, R. Hughes, C. Smethurst and S. Y. Cho, Angew. Chem., Int. Ed., 2000, 39, 1084–1088 CrossRef CAS; (b) K. Okamoto, Y. Nishibayashi, S. Uemura and A. Toshimitsu, Angew. Chem., Int. Ed., 2005, 44, 3588–3591 CrossRef CAS PubMed; (c) S. S. Khokhar and T. Wirth, Angew. Chem., Int. Ed., 2004, 43, 631–633 CrossRef CAS PubMed; (d) S. A. Shahzad and T. Wirth, Angew. Chem., Int. Ed., 2009, 48, 2588–2591 CrossRef CAS PubMed; (e) D. M. Freudendahl, S. Santoro, S. A. Shahzad, C. Santi and T. Wirth, Angew. Chem., Int. Ed., 2009, 48, 8409–8411 CrossRef CAS PubMed; (f) J. Trenner, C. Depken, T. Weber and A. Breder, Angew. Chem., Int. Ed., 2013, 52, 8952–8956 CrossRef CAS PubMed.
- (a) G. Mugesh, W.-W. du Mont and H. Sies, Chem. Rev., 2001, 101, 2125 CrossRef CAS PubMed, and references therein. (b) C. W. Nogueira, G. Zeni and J. B. T. Rocha, Chem. Rev., 2004, 104, 6255 CrossRef CAS PubMed; (c) B. K. Sharma and G. Mugesh, J. Am. Chem. Soc., 2005, 127, 11477 CrossRef PubMed; (d) K. P. Bhabak and G. Mugesh, Chem. – Eur. J., 2007, 13, 4594 CrossRef CAS PubMed; (e) B. K. Sharma and G. Mugesh, Chem. – Eur. J., 2008, 14, 10603 CrossRef PubMed; (f) K. P. Bhabak and G. Mugesh, Chem. – Asian J., 2009, 4, 974 CrossRef CAS PubMed; (g) B. K. Sharma, D. Manna, M. Minoura and G. Mugesh, J. Am. Chem. Soc., 2010, 132, 5364 CrossRef PubMed; (h) K. P. Bhabak and G. Mugesh, Acc. Chem. Res., 2010, 43, 1408–1419 CrossRef CAS PubMed.
- (a) H. J. Reich and C. P. Jasperse, J. Am. Chem. Soc., 1987, 109, 5549 CrossRef CAS; (b) T. G. Back and B. P. Dyck, J. Am. Chem. Soc., 1997, 119, 2079 CrossRef CAS; (c) C. Jacob, G. I. Giles, N. M. Giles and H. Sies, Angew. Chem., Int. Ed., 2003, 42, 4742 CrossRef CAS PubMed.
- (a) R. S. Glass, F. Farooqui, M. Sabahi and K. W. Ehler, J. Org. Chem., 1989, 54, 1092 CrossRef CAS; (b) R. Zhao, H. Masayasu and A. Holmgren, Proc. Natl. Acad. Sci. U.S.A., 2002, 99, 8579 CrossRef CAS PubMed; (c) A. C. Terentis, M. Freewan, T. S. S. Plaza, M. J. Raftery, R. Stocker and S. R. Thomas, Biochemistry, 2010, 49, 591 CrossRef CAS PubMed.
- (a) A. Muller, E. Cadenas, P. Graf and H. Sies, Biochem. Pharmacol., 1984, 33, 3235 CrossRef CAS; (b) M. Parnham and H. Sies, Expert Opin. Invest. Drugs, 2000, 9, 607 CrossRef CAS PubMed.
- (a) S. Chu and H. G. Mautner, J. Org. Chem., 1962, 27, 2899 CrossRef CAS; (b) G. C. Chen, C. H. Banks, K. J. Irgolic and R. A. Zingaro, J. Chem. Soc., Perkin Trans. 1, 1980, 2287 RSC; (c) M. Burutus, Y. Mollier and M. Stavaux, Nouv. J. Chim., 1986, 10, 51 ( Chem. Abstr. , 1987 , 106 , 195792 ) Search PubMed; (d) C. K. Mirabelli, D. T. Hill, L. F. Faucette, F. L. McCabe, G. R. Girard, D. B. Bryan, B. M. Sutton, J. O. Bartus, S. T. Crooke and R. K. Johnson, J. Med. Chem., 1987, 30, 2181 CrossRef CAS; (e) K. Hideg, C. P. Sar, O. H. Hankovszky and G. Jerkovich, Synthesis, 1991, 616 CrossRef CAS; (f) K. Hideg, C. P. Sar, O. H. Hankovszky, T. Tamas and G. Jerkovich, Synthesis, 1993, 390 CrossRef CAS; (g) T. Kalai, M. Balog, J. Jeko and K. Hideg, Synthesis, 1999, 973 CrossRef CAS; (h) G. T. Manh, F. Purseigle, D. Dubreuil, J. P. Pradere, A. Guingant, R. Danion-Bougot, D. Danion and L. Toupet, J. Chem. Soc., Perkin Trans. 1, 1999, 2821 RSC; (i) B. D. Jhonston and B. M. Pinto, J. Org. Chem., 2000, 65, 4607 CrossRef PubMed; (j) H. E. Russel, R. W. A. Luke and M. Bradley, Tetrahedron Lett., 2000, 41, 5287 CrossRef; (k) T. Blum, J. Ermwrt and H. H. J. Coenen, J. Labelled Comp. Radiopharm., 2001, 44, 587 CrossRef CAS PubMed ; Chem. Abstr., 2001, 135, 344558.
- (a) B. Romanoa, M. Fonta, I. Encíob, J. A. Palopa and C. Sanmartín, Eur. J. Med. Chem., 2014, 83, 674–684 CrossRef PubMed; (b) R. Terazawaa, D. R. Garudb, N. Hamadaa, Y. Fujitaa, T. Itoha, Y. Nozawaa, K. Nakaned, T. Deguchid, M. Koketsue and M. Ito, Bioorg. Med. Chem., 2010, 18, 7001–7008 CrossRef PubMed; (c) I. B. Levshin, A. A. Tsurkan, É. A. Rudzit and G. N. Neshchadim, Pharm. Chem. J., 1981, 15, 242–245 CrossRef.
- M. L. Wolfrom and F. A. H. Rice, J. Am. Chem. Soc., 1947, 69, 1833 CrossRef CAS.
- (a) R. A. Zingaro, F. C. Bennett Jr. and G. W. Hammer, J. Org. Chem., 1953, 18, 292 CrossRef CAS; (b) J. S. Warner and T. F. Page Jr., J. Org. Chem., 1966, 31, 606–607 CrossRef CAS; (c) D. Lloyd and R. W. Miller, Tetrahedron, 1977, 33, 1379 CrossRef CAS; (d) T. Kálai, N. M. Bárácz, G. Jerkovich, O. H. Hankovszky and K. Hideg, Synthesis, 1995, 1278 CrossRef; (e) S. Chu and H. G. Mautner, J. Org. Chem., 1962, 27, 2899 CrossRef CAS; (f) C. K. Mirabelli, D. T. Hill, L. F. Faucette, F. L. McCabe, G. R. Girard, D. B. Bryan, B. M. Sutton, J. O. Bartus, S. T. Crooke and R. K. Johnson, J. Med. Chem., 1987, 30, 2181 CrossRef CAS; (g) K. Hideg, C. P. Sár, O. H. Hankovszky and G. Jerkovich, Synthesis, 1991, 616 CrossRef CAS; (h) T. Kálai, M. Balog, J. Jekö and K. Hideg, Synthesis, 1999, 973 CrossRef; (i) G. T. Manh, F. Purseigle, D. Dubreuil, J. P. Pradère, A. Guingant, R. Danion-Bougot, D. Danion and L. Toupet, J. Chem. Soc., Perkin Trans. 1, 1999, 2821 RSC.
- (a) Y. Zhou and M. K. Denk, Tetrahedron Lett., 2003, 44, 1295–1299 CrossRef CAS; (b) M. Koketsu, Synth. Commun., 2002, 32, 3075–3079 CrossRef CAS PubMed.
- N. Sonoda, G. Yamamoto and S. Tsutsumi, Bull. Chem. Soc. Jpn., 1972, 45, 2937 CrossRef CAS.
- (a) A. N. Proshin, I. V. Serkov, A. S. Lermontov, E. F. Shevtsova, M. E. Neganova and S. O. Bachurin, Russ. Chem. Bull., 2013, 1, 142–146 CrossRef; (b) G. L. Sommen, A. Linden and H. Heimgartner, Eur. J. Org. Chem., 2005, 3128–3137 CrossRef CAS PubMed; (c) D. R. Garud and M. Koketsu, Org. Lett., 2008, 10, 3319–3322 CrossRef CAS PubMed; (d) N. Tanahashia and M. Koketsu, Tetrahedron Lett., 2011, 52, 4650–4653 CrossRef PubMed; (e) M. Koketsu, T. Kiyokuni, T. Sakai, H. Ando and H. Ishihara, Chem. Lett., 2006, 35, 626–627 CrossRef CAS.
- For reviews, see (a) I. Ugi, Angew. Chem., 1962, 74, 9–22 ( Angew. Chem. Int. Ed. Engl. , 1962 , 1 , 8–21 ) CrossRef CAS PubMed; (b) A. Dömling and I. Ugi, Angew. Chem., 2000, 112, 3300–3344 ( Angew. Chem. Int. Ed. , 2000 , 39 , 3168–3210 ) CrossRef; (c) H. Bienayme, C. Hulme, G. Oddon and P. Schmitt, Chem. – Eur. J., 2000, 6, 3321–3329 CrossRef CAS; (d) J. Zhu, Eur. J. Org. Chem., 2003, 1133–1144 CrossRef CAS PubMed; (e) V. Nair, C. Rajesh, A. U. Vinod, S. Bindu, A. R. Sreekanth, J. S. Mathen and L. Balagopal, Acc. Chem. Res., 2003, 36, 899–907 CrossRef CAS PubMed; (f) A. Dömling, Chem. Rev., 2006, 106, 17–89 CrossRef PubMed; (g) L. El Kaim and L. Grimaud, Tetrahedron, 2009, 65, 2153–2171 CrossRef CAS PubMed; (h) E. Ruijter, R. Scheffelaar and R. V. A. Orru, Angew. Chem., Int. Ed., 2011, 50, 6234–6246 CrossRef CAS PubMed; (i) S. Sadjadi and M. M. Heravi, Tetrahedron, 2011, 67, 2707–2752 CrossRef CAS PubMed.
- For selected examples: (a) M. Tobisu, K. Koh, T. Furukawa and N. Chatani, Angew. Chem., Int. Ed., 2012, 51, 11363 CrossRef CAS PubMed; (b) B. Zhang, C. Muck-Lichtenfeld, C. G. Daniliuc and A. Studer, Angew. Chem., Int. Ed., 2013, 52, 10792 CrossRef CAS PubMed; (c) Q. Wang, X. Dong, T. Xiao and L. Zhou, Org. Lett., 2013, 15, 4846 CrossRef CAS PubMed; (d) H. Jiang, Y. Cheng, R. Wang, M. Zheng, Y. Zhang and S. Yu, Angew. Chem., Int. Ed., 2013, 52, 13289 CrossRef CAS PubMed; (e) Y.-M. Li, M. Sun, H.-L. Wang, Q.-P. Tian and S.-D. Yang, Angew. Chem., Int. Ed., 2013, 52, 3972 CrossRef CAS PubMed; (f) Y.-R. Chen and W.-L. Duan, J. Am. Chem. Soc., 2013, 135, 16754 CrossRef CAS PubMed; (g) M. Tobisu, H. Fujihara, K. Koh and N. Chatani, J. Org. Chem., 2010, 75, 4841–4847 CrossRef CAS PubMed; (h) Y. Han-yal, H. Tokuyama and T. Fukuyama, Angew. Chem., Int. Ed., 2011, 50, 4884–4887 CrossRef PubMed; (i) B. Zhang and A. Studer, Org. Lett., 2014, 16, 1216–1219 CrossRef CAS PubMed.
- (a) S. Fujiwara and N. Sonoda, J. Org. Chem., 2002, 67, 6275–6278 CrossRef CAS PubMed; (b) Y. Asanuma, S. Fujiwara, T. Shin-ike and N. Kambe, J. Org. Chem., 2004, 69, 4845–4848 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5qo00150a |
| This journal is © the Partner Organisations 2015 |