
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOne-step preparation of conjugated homopolymer sub-microspheres via a controllable supramolecular approach toward optoelectronic applications†
Mengna
Yu‡
a,
Bin
Liu‡
a,
Jinyi
Lin
*bc,
Lubing
Bai
b,
Haifeng
Ling
a,
Wensai
Zhu
b,
Linghai
Xie
 *a,
Mingdong
Yi
a,
Jiangpu
Wang
b and
Wei
Huang
*ab
*a,
Mingdong
Yi
a,
Jiangpu
Wang
b and
Wei
Huang
*ab
aCenter for Molecular Systems and Organic Devices (CMSOD), Key Laboratory for Organic Electronics and Information Displays, Institute of Advanced Materials (IAM), Jiangsu National Synergetic Innovation Center for Advanced Materials (SICAM), Nanjing University of Posts & Telecommunications, 9 Wenyuan Road, Nanjing 210023, China. E-mail: iamlhxie@njupt.edu.cn
bKey Laboratory of Flexible Electronics (KLOFE), Institute of Advanced Materials (IAM), Jiangsu National Synergetic Innovation Center for Advanced Materials (SICAM), Nanjing Tech University (NanjingTech), 30 South Puzhu Road, Nanjing 211816, China. E-mail: wei-huang@njtech.edu.cn; iamjylin@njtech.edu.cn
cDepartment of Physics and Centre for Plastic Electronics, The Blackett Laboratory, Imperial College London, Prince Consort Road, London SW7 2AZ, UK
First published on 6th March 2017
Abstract
Supramolecular poly{9-[4-(octyloxy)phenyl]fluoren-9-ol-2,7-diyl} (PPFOH)-based conjugated polymer sub-microspheres (CPSMs) with diameters ranging from 150 nm to several micrometres are constructed via “one step’ solvent evaporation induced interface self-assembly (SEIS). Their diameter and packing mode can be optimized by controlling the preparation parameters, which pave the way to construct colloidal crystal and light-emitting diodes.
In the last several decades, conjugated polymers (CPs) have been widely applied in optoelectronic devices, such as polymer light-emitting diodes (PLEDs), polymer solar cells (PSC), polymer lasers and sensors, because of their mechanical flexibility, and low-cost manufacturing as well as their versatile chemical design and synthesis.1–3 Compared to the p–n (also called donor–acceptor) molecular engineering strategy for constructing optoelectronic materials,4,5 the micro/nano-approach can act as an alternative with a higher degree of freedom. For example, it not only controls the energy transfer, charge separation and photophysical characteristics of CPs by manipulating molecular self-assembly behaviour, but also simplifies the fabrication processes of flexible optoelectronic devices.6,7 Among various CP-based multi-dimensional micro/nanostructures, conjugated polymer sub-microspheres (CPSMs) can be used as microscopic building blocks to fabricate organic optoelectronic devices by high-throughput eco-friendly solution processing,8,9 particularly the self-assembly of three dimensional (3D) photonic crystals.10,11 Other than the sub-microspheres prepared from conventional non-conjugated or doped polymers,12–14 CPSMs-based photonic crystals are expected to show enhanced electroluminescence (EL), no-threshold laser oscillation, and highly efficient photoelectric conversion,15–18 such as long-lived excitons, and light confinement.10,11,19,20 Thus, it is significant to develop one class of CPs system, as well as a facile method to construct pure CPSMs.
Up to date, the majority of reported polymer micro/nanoparticles was constructed by direct polymerization techniques, such as dispersion and miniemulsion polymerization,3,6,21 or post-polymerization method (multi-steps mini-emulsification and precipitation).10,11,22 In order to fabricate conjugated polymer micro/nanoparticle's active films or mesoscopic structure, it need multi-steps purification to remove stabilizer for mini-emulsification (diameters ranging from 100 nm to several micrometres) and/or slowly dry a suspension to distribute/pack particles on precleaned substrates for the precipitation method (sizes ranging from several nanometres to hundreds nanometres) attributed to the lower particles concentration in precursor suspension.22,23 What's more, the critical matter is that the synergistic effect of rigidity and hydrophobicity of the aromatic conjugated backbone structure makes it difficult to give isotropic spherical assemblies by post-polymerization, which is out of question for the non-conjugated polymers like polystyrene (PS) and poly(methyl methacrylate) (PMMA).24,25 As a result of high planarity, rigidity and hydrophobicity, CPs chains tend to pack together and self-assemble into anisotropic nano-aggregates by post-polymerization method, ultimately generate irregular zero-dimensional (0D) nanoparticles or dots,22,25 one-dimensional (1D) fibril,7 even two-dimensional (2D) layered structures,26 but structurally isotropic spheres are rarely reported.27,28 Recently, alternating copolymers with two different π-conjugated components were demonstrated to form well-shaped CPSMs quantitatively with diameters ranging from a few hundreds of nanometres to several micrometres, ascribed to large steric hindrance on their backbone that reduces planarity of the polymers and inhibits anisotropic packed growth.10 However, up to now, it is rarely post-polymerization method to construct well-defined, widely size-tunable spherical sub-microstructures prepared from π-conjugated homopolymer toward potential optoelectronic application, especial colloidal crystal.
Supramolecular functionalization imparts π-conjugated homopolymer with abundant self-assembly behaviours to construct multi-dimensional superstructures.29,30 Accordingly, supramolecular strategy not only endows CPs with self-assembly ability, but also reserves their optoelectronic properties when taking into consideration of the practical applications.31,32 Over fast precipitation and evaporation methods, supramolecular approach provides a flexible driving force to induce the formation of CPSMs, which can be attributed to the thermodynamically favourable lowest surface per volume, similar to amphiphilic homopolymers and block copolymers. Herein, we explore the preparation of spherical assemblies of supramolecular conjugated polymer (PPFOH),33 modified with the hydroxyl groups, which are generated by supramolecular interactions during the process of “one step” solvent evaporation induced interface self-assembly (SEIS) (Scheme 1). In our previous work, we constructed PPFOH-based small irregular nanoparticles with the size tuning from 200 nm to 40 nm via reprecipitation method.31 Here, amphiphilicity of PPFOH in the N,N-dimethylformamide (DMF, high boiling point of 153 °C) makes polymer chains self-assemble into isotropic CPSMs with the lowest surface energy during slow evaporation without daylight. PPFOH CPSMs tend to show well-shaped CPSMs quantitatively with diameters ranging from a few hundreds of nanometres to several micrometres, as well as a size-dependent emission property. Tentatively, preliminary explorations of PPFOH-based CPSMs toward potential application in photonic crystal and PLEDs are also described here.
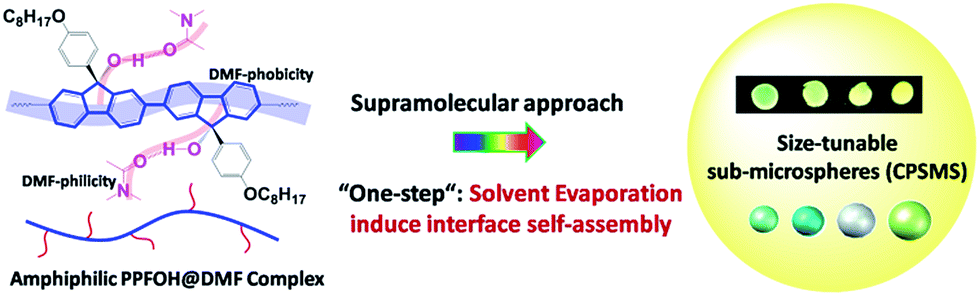 | ||
| Scheme 1 Illustrate model of PPFOH-based CPSMs via solvent evaporation induced interface self-assembly (SEIS) method. | ||
As demonstrated in our previous work, PPFO8 (without hydroxyl) exhibited extremely poor solubility in DMF for the large difference of solubility parameters between PPFO8 (∼9.5 (cal cm−3)1/2) and DMF (12.1 (cal cm−3)1/2), indicated DMF is an immiscible solvent for non-polar polyfluorene backbone structures (Scheme 1).33 On the contrary, PPFOH showed good solubility in DMF for the formation of hydrogen bond between hydroxyl and carbonyl of DMF. In order to further confirm this assumption, the 1H NMR spectra of TFOHC8 (trimer of 9-ol-fluorene) in CDCl3 (Fig. S1†) were investigated, where the TFOHC8 acted as a model of PPFOH to directly examine whether the hydrogen bonds formation or not between DMF and PPFOH. Firstly, when only TFOHC8 was dissolved in CDCl3, an apparent signal at δ (2.495 ppm) can be observed, which was attributed to the H atom of hydroxyl at 9 position of the fluorene unit. With the addition of a small amount of DMF, the aromatic signals downshifted a little, but the δ of H atom in hydroxyl upshifted from 2.495 to 4.276 ppm, which suggested the formation of a strong interaction between DMF (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) and hydroxyl (–OH). Meanwhile, the δ of H atom at b site of DMF shifted from 7.955 to 7.896 ppm, which further confirmed the formation of hydrogen bonds. On this basis, we propose such a model that hydroxyl groups prefer to interact with DMF molecules (DMF-philicity, similar to hydrophilicity) but DMF molecules are immiscible units with the backbones of polyfluorene (DMF-phobicity, hydrophobicity), which makes PPFOH@DMF complex show a slightly amphiphilic property in DMF solution (Scheme 1). Consequently, similar to the spherical self-assembly of the amphiphilic homopolymers and block copolymers,29 amphiphilic PPFOH@DMF complexes tend to self-assemble into spherical nanostructures by the control of external conditions.
O) and hydroxyl (–OH). Meanwhile, the δ of H atom at b site of DMF shifted from 7.955 to 7.896 ppm, which further confirmed the formation of hydrogen bonds. On this basis, we propose such a model that hydroxyl groups prefer to interact with DMF molecules (DMF-philicity, similar to hydrophilicity) but DMF molecules are immiscible units with the backbones of polyfluorene (DMF-phobicity, hydrophobicity), which makes PPFOH@DMF complex show a slightly amphiphilic property in DMF solution (Scheme 1). Consequently, similar to the spherical self-assembly of the amphiphilic homopolymers and block copolymers,29 amphiphilic PPFOH@DMF complexes tend to self-assemble into spherical nanostructures by the control of external conditions.
Subsequently, we attempted to prepare the regular nanostructures of PPFOH by making use of the amphiphilic property in DMF solution. Reasonably, CPSMs can be obtained from DMF solution of PPFOH on a solid substrate (such as silicon wafer, Fig. 1) via solvent evaporation processing. It is worth noting that conjugated polymers akin to PPFOH modified with hydroxyl groups also show excellent self-assembly behaviour to construct high quality CPSMs (Fig. S2†), which preliminary demonstrated the universality of amphiphilic CPs in terms of spherical self-assembly. Furthermore, the diameters of CPSMs increased gradually with the increasing initial concentrations of DMF solutions (Fig. 1e). As shown in Fig. 1a–d, the average diameters of CPSMs are approximately 150–200 nm for 1 mg mL−1, 400 nm for 10 mg mL−1, 700 nm for 30 mg mL−1, and 2000 nm for 50 mg mL−1, respectively. The increased sizes of CPSMs are likely derived from the high density of the PPFOH chains in the higher concentration. Meanwhile, PPFOH-based CPSMs can also be generated on other substrates, such as glass, quartz plate as well as Cu grid covered with carbon membrane (Fig. S3†), which demonstrated that “one step” SEIS method in terms of preparing CPSMs thin-film can be implemented on various kinds of substrates or electrodes. Moreover, it is easy to find that CPSMs show more uniform spherical shapes on the interface with the lowest surface energy, because of the effective molecular rearrangement during the DMF molecules evaporation process. In addition, PPFOH-based CPSMs were revealed to be solid by transmission electron microscope (TEM) analysis (Fig. S4†). As shown in Fig. S5,† a larger, stronger and continuous ring diffraction in GIXD pattern suggested the isotropic amorphous states in CPSMs, which also supported the assumption of isotropic property of CPSMs. In order to further demonstrate the validity of amphiphilic property of PPFOH, we tried dimethyl sulfoxide (DMSO) and dimethylacetamide (DMAc) solutions as well, where the hydrophilicity can be induced by the formation of hydrogen bonds between solvent and hydroxyl of PPFOH, and induced polymer chains self-assemble into CPSMs (Fig. S6†). By contrary, continuous PPFOH films instead of CPSMs were obtained in other solutions, such as chloroform, tetrahydrofuran and toluene solutions (Fig. S7†), which further indicated that amphiphilic property of PPFOH in DMF solution is the driving force for PPFOH chains self-assembling into CPSMs films.
 | ||
| Fig. 1 SEM images of the PPFOH-based CPSMs. PPFOH CPSMs obtained at different initial concentrations in DMF solution: (a) 1 mg mL−1, (b) 10 mg mL−1, (c) 30 mg mL−1, (d) 50 mg mL−1. (e) Diameter as a function of the concentrations. (f–i) SEM images of three-dimensional (3D) colloidal crystals of PPFOH CPSMs. (j) Schematic representation of PPFOH photonic crystal on silicon substrates fabricated by droplet-pinned solvent evaporation method. | ||
Not surprising, no CPSMs were formed for the corresponding PPFO8 solution at the same condition for the poor solubility in DMF. Actually, the humidity during the preparation process significantly affected the formation of CPSMs, which was ascribed to the excellent water absorption property and longer evaporation speed of DMF. Moderate humidity environment (higher than 20% RH and lower than 70%) is more beneficial for CPSMs generation than low humidity (<20% RH). Therefore, when placed in a drying oven, there were no CPSMs but film obtained (Fig. S8†). Besides, we also prepared PPFOH spin-coated film from DMF solution. As shown in the SEM images of the film (Fig. S9†), inhomogeneous and aggregated CPSMs but smaller sizes formed on the continuous film. In this regard, DMF evaporation rate is an important factor to dominate the formation of PPFOH-based CPSMs, that rapid evaporation is unfavorable for the formation of spheres. Here, we propose a possible formation process: first, PPFOH is dissolved in DMF solution due to the formation of hydrogen bonds between hydroxyl groups linked to the polymer backbones and carbonyl groups on the DMF molecule. With the DMF evaporation, DMF-hydrophobic backbone chains fold and then form spheres, which are surrounded by the hydrophilic hydrogen bonds outside effectively. The absorption of water molecules can also disrupt hydrogen-bonds between PPFOH and DMF, further promoted the formation of spheres. During solvent evaporation process, PPFOH eventually assembles into well-defined CPSMs. Our results suggested that supramolecular functionalization can afford semi-rigid conjugated polymer with amphiphilic property in a certain solvent and give rise to self-assemble into size-controllable CPSMs.
The close packing of PPFOH CPSMs can be a model to construct fluorescent colloidal crystals after fine manipulation (Fig. 1c and d). We tried to create three-dimensional (3D) colloidal crystals of CPSMs preliminarily. According to the formation mechanism of CPSMs, we further designed a simple droplet-pinned solvent evaporation method (Fig. 1j).34 The operating principle of this approach is illustrated as follows: during drying of a droplet of the polymer solution on a substrate, CPSMs nucleate near the contact line and grow gradually along the receding direction (toward the centre) of the droplet, ultimately closely stack together. Large-scale PPFOH CPSMs film (up to 100 μm) with regular arrangement was obtained by this method (Fig. 1f and g). PPFOH CPSMs in the large-scale film show the similar diameter and smooth surface with a size about 600 nm (Fig. 1h and i).11 Certainly, more attempts are needed to create colloidal crystals, such as precisely controlling of the size and deviation. Here, we provide a significant potential candidate for photonic crystals using supramolecular functionalization of π-conjugated polymers.
The optical properties of PPFOH CPSMs and solutions were investigated by the UV-vis absorption and photoluminescence (PL) measurements (Fig. 2). It is easy to distinguish that the fluorescent profiles of PPFOH CPSMs are composed of two emission regimes: the blue emission at 440–460 nm and the green-yellow emission at 540–560 nm, which is different from the pristine PFO nanoparticles prepared via re-precipitation with the absence of green-yellow band emission,35 but similar to the amphiphilic oligofluorene- or polyfluorene-based nanoparticles (also confirmed a slightly amphiphilic property of PPFOH@DMF complex in DMF solution).36–38 Interestingly, the low band emission of the larger CPSMs (>700 nm) red-shifted about 10 nm in comparison with those of smaller CPSMs (<700 nm). And the intensities of the broad and shoulder absorption peaks in the wavelength of 450–550 nm increased with the increasing CPSMs sizes (Fig. S10†). Two reasons may explain these properties: one is attributed to the size-dependent spectroscopic properties in closely stacked PPFOH CPSMs (lattice softening),39 the other is the stronger aggregation in the larger CPSMs. Time-resolved fluorescence spectroscopy measurements were carried out to determine the transition property of PPFOH CPSMs film, and the corresponding lifetimes were summarized in Table S1.† PPFOH in DMF solution exhibited single-exponential (1A*) decay at three detection wavelengths of 420, 440 and 480 nm, respectively, and their lifetimes were about 480 ps corresponding to three peaks of the polyfluorenes fluorescent spectra.40 But the energy transfer was significantly enhanced in the PPFOH CPSMs. It is clear to find that the high energy band emission (420–480 nm) disappeared or reduced sharply because of an increasing possibility of non-irradiative decay pathway for energy transfer from 450 nm (host, donor wavelength) to 560 nm (guest, acceptor wavelength). The lifetime analysis was implemented at 450 and 560 nm, which were considered as donor and acceptor wavelengths, respectively (Fig. S10 and Table S1†). The emission peak at ca. 560 nm exhibited double-exponential decay, and the lifetimes were about 2–4 ns and 6–9 ns, much longer than the blue emission at 450 nm. It can be deduced that the aggregate species (low energy emission), together with the pristine polyfluorene chains in CPSMs (high energy emission), formed a self-doping nano-system with energy transfer from host to guest. Meanwhile, we also believed that this green band (g-band) emission may be attributed to the aggregation excimer. Of course, our PPFOH also cannot completely avoid the oxidation in the preparation and purification processing, result in fluorenone-induced green emission. Also, our recent work indicated that the green emission may be ascribed to the random, distorted and folded fluorene conformation and/or therefore truncation of the conjugation length as the result of strong aggregation in CPSMs.41
 | ||
| Fig. 2 (a) Photoluminescence (PL) spectra of PPFOH CPSMs with different diameters. (b) Green-yellow index of PPFOH with the function of the CPSMs diameter. (c) Concentration dependent PL spectra of PPFOH@DMF solution, the inset showing the photograph of fluorescent dispersions of PPFOH in DMF under the UV lamp (365 nm) (left to right: 0.001 mg mL−1 to 15 mg mL−1). PL spectra of concentrated solutions are measured by using the front-face detection from a 1 mm cuvette to extremely minimize the effect of self-absorbance on PL spectra. (d) PL spectra of 1 mg mL−1 PPFOH/DMF solution taken at different drying time at 20 °C (without daylight), the inset showing the photographs of PPFOH/DMF solution dropped on the silicon substrate during the solvent evaporation process (top to down: 0 min to 125 min). All above were excited at 380 nm. | ||
The size-dependent PL spectra of CPSMs are shown in Fig. 2a. We clearly observed that the green-yellow index of CPSMs increased with the increasing of diameters (Fig. 2b). The possible reason is that the aggregate exciton is sensitive to the diameters of CPSMs owing to their comparable sizes, leading to a more effective energy transfer between single polymer chain (donor) and aggregates (acceptor). Noted that the stronger self-absorption in the large size CPSMs film can also explain the size-dependent emission. Besides, the size-dependent g-band emission is nearly linear proportional to the increased concentrations of PPFOH/DMF solutions (Fig. 2c), which implies that the g-band emission is closely associated with the intensity of molecular aggregates. Compared with our previous work, where PPFOH nanoparticle formed immediately (kinetic processing),31 PPFOH CPSMs prepared by SEIS method undergo an extended period of time (thermodynamic processing) that resulted in the possible formation of aligned and stretched polymer chains and further induced excimer emission in the CPSMs (Fig. 2d).22 The emission peaks in the wavelength range of 540–560 nm increased with the increasing of drying time during the evaporation process. Therefore, molecular aggregation of π-stacked fluorene segments in spheres is an alternative explanation for the origin of the g-band emission. Even if consideration of fluorenone-mechanism, more efficient intra- or interchain energy transfer can also explain the size-dependent emission.
We finally fabricated PPFOH spheres-based polymer nanostructure-based light-emitting devices (PNLEDs) to examine their semiconducting properties. Devices with the configuration of Au/p-SiC/PPFOH spheres/ITO were fabricated to investigate the performance of CPSMs.42 CPSMs films on the p-SiC (4H, Cree Inc.) can be easily prepared by drop-coating technique (Fig. 3a). Noted that larger PPFOH spheres with several hundred nanometers may stack together and further result into larger film thickness. In that case, no light emission could be observed from the PNLEDs. In general, PPFOH nanospheres with the diameters of 150 nm may tend to efficiently disperse on the p-SiC substrates. The average PPFOH–CPSMs film thickness in the PNLEDs is about 150 nm. Fig. 3b shows the current–voltage (I–V) curve of the PNLEDs and the schematic diagram of the proposed PNLEDs configuration. Hence, the I–V curve exhibited typical rectifying behaviour. The PNLEDs based on the CPSMs have turn-on voltage (current) of ∼25 V (0.60 mA). The EL spectra of PNLEDs at various forward biased voltages are shown in Fig. 3c and no light emission was observed from the PNLEDs under reverse bias. The photograph of the devices indicated the white light emission (Fig. 3c, inset). The EL spectra of PNLEDs can be fitted by two Gaussian curves with peak wavelengths at 440 and 540 nm, respectively. White light emission (CIE: 0.33, 0.35) can be observed by naked eyes from the surface of the PNLEDs in dark room (Fig. 3c and d). Notably, EL spectra showed a broad emission as compared with the PL spectra owing to the decreased green-yellow index. The EL intensity of the PNLEDs increased sharply when the applied voltages were larger than the turn-on voltage. In addition, no light emission under either optical or electrical excitation can be measured for the p-SiC sample without the PPFOH nanostructure layer in our experiments, hence the EL spectrum observed was due to the presence of PPFOH CPSMs.
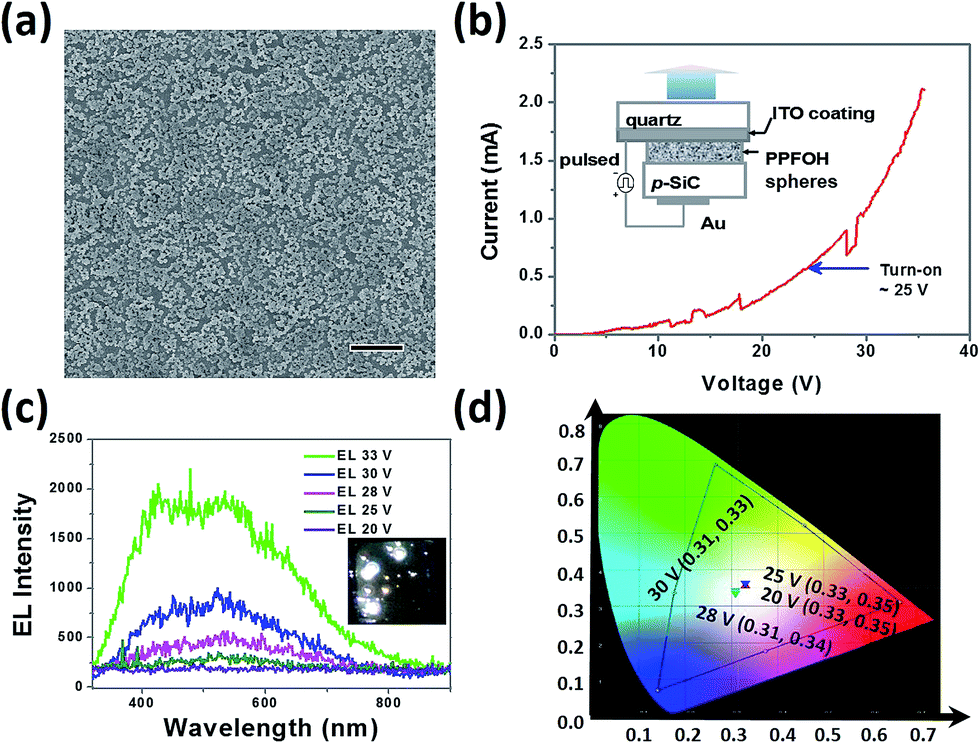 | ||
| Fig. 3 (a) SEM images of the PPFOH-based films on the p-SiC, scale bar is 5 μm. (b) Current–voltage (I–V) curve of the PNLEDs; the inset is the device configuration of the PNLEDs based on PPFOH spheres. (c) EL spectra at different voltages; the inset is the photograph of device with white light emission. (d) CIE coordinates of PNLEDs biased at various voltages. | ||
Conclusions
In summary, we proposed the supramolecular functionalization strategy to design versatile hairy-rod type π-conjugated polymer amphiphiles that act as the precursor of supramolecular self-assembly for CPSMs. PPFOH CPSMs with different sizes have been obtained by controlling the initial concentration via SEIS method, which is a new and convenient method to prepare CPSMs thin-film on substrates or electrodes. The fascinating size-dependent luminescent properties of PPFOH CPSMs provide a flexible method to tune their emission colours. White light emission with CIE coordinates of (0.33, 0.35) has been achieved by single-component polyfluorene spheres in the PNLEDs. The closely packing of CPSMs would facilitate the construction of colloidal crystals. Supramolecular π-conjugated polymers are a series of potential next-generation advanced materials for organic optoelectronics.Acknowledgements
We sincerely thank Professor Donal D. C. Bradley (University of Oxford) for valuable discussions. The project was supported by the National Key Basic Research Program of China (973) (2015CB932200), National Natural Science Foundation of China (21504041, 21502091, 21274064, 61136003), Doctoral Fund of Ministry of Education of China (20133223110007), Project funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions, PAPD (YX03001), Synergetic Innovation Center for Organic Electronics and Information Displays, China Postdoctoral Science Foundation (2015M580419), Jiangsu Planned Projects for Postdoctoral Research Funds (1501019B), the Six Peak Talents Foundation of Jiangsu Province (XCL-CXTD-009), Excellent Science and Technology Innovation Team of Jiangsu Higher Education Institutions (2013), Natural Science of the Education Committee of Jiangsu Province (15KJB430019). Dr Jinyi Lin gratefully acknowledges SICAM Fellowship, Oxford University, the Plastic Electronics Centre for Doctoral Training, UK EPSRC (EP/G037515/1) and Imperial College London.Notes and references
- J. Burroughes, D. Bradley, A. Brown, R. Marks, K. Mackay, R. Friend, P. Burns and A. Holmes, Nature, 1990, 347, 539–541 CrossRef CAS.
- M. Campoy-Quiles, T. Ferenczi, T. Agostinelli, P. G. Etchegoin, Y. Kim, T. D. Anthopoulos, P. N. Stavrinou, D. D. Bradley and J. Nelson, Nat. Mater., 2008, 7, 158–164 CrossRef CAS.
- L.-H. Xie, C.-R. Yin, W.-Y. Lai, Q.-L. Fan and W. Huang, Prog. Polym. Sci., 2012, 37, 1192–1264 CrossRef CAS.
- W.-L. Yu, H. Meng, J. Pei and W. Huang, J. Am. Chem. Soc., 1998, 120, 11808–11809 CrossRef CAS.
- W. Huang, H. Meng, W. L. Yu, J. Gao and A. J. Heeger, Adv. Mater., 1998, 10, 593–596 CrossRef CAS.
- L.-H. Xie, S.-H. Yang, J.-Y. Lin, M.-D. Yi and W. Huang, Philos. Trans. R. Soc., A, 2013, 371, 20120337 CrossRef.
- F. S. Kim, G. Ren and S. A. Jenekhe, Chem. Mater., 2010, 23, 682–732 CrossRef.
- E. Fisslthaler, S. Sax, U. Scherf, G. Mauthner, E. Moderegger, K. Landfester and E. J. List, Appl. Phys. Lett., 2008, 92, 183305 CrossRef.
- E. Fisslthaler, A. Blümel, K. Landfester, U. Scherf and E. J. List, Soft Matter, 2008, 4, 2448–2453 RSC.
- T. Adachi, L. Tong, J. Kuwabara, T. Kanbara, A. Saeki, S. Seki and Y. Yamamoto, J. Am. Chem. Soc., 2013, 135, 870–876 CrossRef CAS PubMed.
- A. J. Kuehne, M. C. Gather and J. Sprakel, Nat. Commun., 2012, 3, 1088 CrossRef.
- T. Kanai, T. Sawada, A. Toyotama and K. Kitamura, Adv. Funct. Mater., 2005, 15, 25–29 CrossRef CAS.
- A. Molinos-Gómez, M. Maymó, X. Vidal, D. Velasco, J. Martorell and F. López-Calahorra, Adv. Mater., 2007, 19, 3814–3818 CrossRef.
- S. Furumi, T. Kanai and T. Sawada, Adv. Mater., 2011, 23, 3815–3820 CAS.
- S. Satoh, H. Kajii, Y. Kawagishi, A. Fujii, M. Ozaki and K. Yoshino, Jpn. J. Appl. Phys., 1999, 38, L1475 CrossRef.
- R. Polson, A. Chipouline and Z. V. Vardeny, Adv. Mater., 2001, 13, 760–764 CrossRef CAS.
- M. Deutsch, Y. A. Vlasov and D. J. Norris, Adv. Mater., 2000, 12, 1176–1180 CrossRef CAS.
- H. Yabu, A. Tajima, T. Higuchi and M. Shimomura, Chem. Commun., 2008, 4588–4589 RSC.
- S. John and T. Quang, Phys. Rev. Lett., 1997, 78, 1888 CrossRef CAS.
- K. Yoshino, S. Lee, S. Tatsuhara, Y. Kawagishi, M. Ozaki and A. Zakhidov, Appl. Phys. Lett., 1998, 73, 3506–3508 CrossRef CAS.
- J. P. Rao and K. E. Geckeler, Prog. Polym. Sci., 2011, 36, 887–913 CrossRef CAS.
- J. Pecher and S. Mecking, Chem. Rev., 2010, 110, 6260–6279 CrossRef CAS.
- D. Tuncel and H. V. Demir, Nanoscale, 2010, 2, 484–494 RSC.
- P. M. Beaujuge and J. M. Fréchet, J. Am. Chem. Soc., 2011, 133, 20009–20029 CrossRef CAS.
- X. L. Chen and S. A. Jenekhe, Macromolecules, 2000, 33, 4610–4612 CrossRef CAS.
- H. Sirringhaus, P. Brown, R. Friend, M. M. Nielsen, K. Bechgaard, B. Langeveld-Voss, A. Spiering, R. A. Janssen, E. Meijer and P. Herwig, Nature, 1999, 401, 685–688 CrossRef CAS.
- X. Xiao, W. Bai, L. Cai and J. Lin, Chem. Lett., 2014, 43, 331–333 CrossRef CAS.
- X. Xiao, W. Bai and J. Lin, Polym. Bull., 2014, 71, 2103–2112 CrossRef CAS.
- F. J. Hoeben, P. Jonkheijm, E. Meijer and A. P. Schenning, Chem. Rev., 2005, 105, 1491–1546 CrossRef CAS PubMed.
- R. C. Evans, J. Mater. Chem. C, 2013, 1, 4190–4200 RSC.
- J. Y. Lin, J. Wong, L. H. Xie, X. C. Dong, H. Y. Yang and W. Huang, Macromol. Rapid Commun., 2014, 35, 895–900 CrossRef CAS.
- D. M. Bassani, L. Jonusauskaite, A. Lavie-Cambot, N. D. McClenaghan, J.-L. Pozzo, D. Ray and G. Vives, Coord. Chem. Rev., 2010, 254, 2429–2445 CrossRef CAS.
- J. Lin, Z. Yu, W. Zhu, G. Xing, Z. Lin, S. Yang, L. Xie, C. Niu and W. Huang, Polym. Chem., 2013, 4, 477–483 RSC.
- H. Li, B. C. Tee, J. J. Cha, Y. Cui, J. W. Chung, S. Y. Lee and Z. Bao, J. Am. Chem. Soc., 2012, 134, 2760–2765 CrossRef CAS.
- C. Wu and J. McNeill, Langmuir, 2008, 24, 5855–5861 CrossRef CAS.
- L. Zhu, J. Qin and C. Yang, J. Phys. Chem. B, 2010, 114, 14884–14889 CrossRef CAS.
- L. Zhu, C. Yang and J. Qin, Chem. Commun., 2008, 6303–6305 RSC.
- F. Wang, M.-Y. Han, K. Y. Mya, Y. Wang and Y.-H. Lai, J. Am. Chem. Soc., 2005, 127, 10350–10355 CrossRef CAS.
- N. Kurokawa, H. Yoshikawa, N. Hirota, K. Hyodo and H. Masuhara, ChemPhysChem, 2004, 5, 1609–1615 CrossRef CAS.
- J.-Y. Lin, W.-S. Zhu, F. Liu, L.-H. Xie, L. Zhang, R. Xia, G.-C. Xing and W. Huang, Macromolecules, 2014, 47, 1001–1007 CrossRef CAS.
- Y.-Y. Liu, J.-Y. Lin, Y.-F. Bo, L.-H. Xie, M.-D. Yi, X.-W. Zhang, H.-M. Zhang, T.-P. Loh and W. Huang, Org. Lett., 2015, 18, 172–175 CrossRef.
- J. Xiao, H. Yang, Z. Yin, J. Guo, F. Boey, H. Zhang and Q. Zhang, J. Mater. Chem., 2011, 21, 1423–1427 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra28859f |
| ‡ Mengna Yu and Bin Liu contributed equally. |
| This journal is © The Royal Society of Chemistry 2017 |