
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOne-step synthesis of Fe2O3 nano-rod modified reduced graphene oxide composites for effective Cr(VI) removal: removal capability and mechanism
Chaopei Kongab,
Miao Li *a,
Jiacheng Lia,
Xuejiao Maab,
Chuanping Fengb and
Xiang Liua
*a,
Jiacheng Lia,
Xuejiao Maab,
Chuanping Fengb and
Xiang Liua
aSchool of Environment, Tsinghua University, Beijing 100084, China. E-mail: miaoli@tsinghua.edu.cn
bSchool of Water Resources and Environment, China University of Geosciences (Beijing), Beijing 100083, China
First published on 2nd July 2019
Abstract
Reduced graphene oxide (rGO) supported Fe2O3 nanorod composites were prepared via a one-step hydrothermal method and further utilized for hexavalent chromium (Cr(VI)) removal from aqueous environments. The composite material exhibited an excellent removal efficiency for chromium (47.28 mg L−1), which was attributed to the electrostatic attraction and chemical reduction of chromium by the material. The removal mechanism was studied by SEM, BET, XPS, and FTIR. The results demonstrated that rGO was successfully modified by Fe2O3 nanorods (approximately 50 nm wide). Compared with graphene oxide (GO), the compound was much more easily separated from the solution after completing the removal. Furthermore, XPS characterization showed that Cr(VI) could also be reduced to low-toxicity Cr(III) by hydroxyl groups. In the variables test, it was found that the removal process was pH-dependent. The results of the designed experiments for exploring the adsorption kinetics, isotherms and thermodynamics indicated that the removal process obeyed a pseudo-second-order kinetics model, Langmuir isotherm model and that it was a spontaneous exothermal process. This study provides the possibility of hydrothermal synthesis of Fe2O3/rGO for use as an excellent material to remove Cr(VI) from aqueous environments.
1. Introduction
Groundwater is a precious clean water resource, and clean water resources are crucial to the development of a country. Heavy metal pollutants in groundwater have caused increasingly harmful health effects in organisms. This issue has attracted much attention in the past few decades. Hexavalent chromium is one of the most toxic heavy metal pollutants in groundwater.1 Chromium is a commonly used element in various industries, including leather tanning, electroplating, dying, ore processing, and metallurgy.2,3 There are two essential forms of chromium in the aqueous phase: Cr(III), a biologically essential element with low toxicity, and hexavalent chromium (Cr(VI)). Cr(VI) is highly toxic, mutagenic and carcinogenic and can diffuse through cell membranes, leading to health risks. The World Health Organization has established that the level of chromium in drinking water should be less than 50 ng mL−1.4 In China, the Cr(VI) concentration in drinking water should be less than 0.05 mg L−1 according to the National Drinking Water Standard (GB5749-2006).5 Therefore, efficient technology must be developed for Cr(VI) removal from drinking water.There are several available methods to remove Cr(VI) from aqueous solutions, such as physical (membrane filtration7 and ion exchange8), biological (microbial degradation10), and chemical (chemical precipitation,6 reduction9 and adsorption11). Among these techniques, chemical methods are attracting increasingly more attention because of their effectiveness, low cost, versatility and great potential for the removal of Cr(VI). In recent years, fabrication of nanomaterials with adsorption and reduction capabilities has become a research hotspot.
Graphene, a carbon allotrope, is composed of a single atomic layer of sp2-bonded carbon. Graphene possesses excellent properties, including a huge specific surface area and superior electrical conductivity.12 Thus, graphene has become a hotspot in materials science and technology since it was discovered in 2004. However, owing to the strong van der Waals forces and π–π stacking effects between the graphene sheets, graphene easily aggregates, which reduces its dispersibility in water and limits its applications. Meanwhile, among the graphene analogues, graphene oxide (GO), prepared by the Hummers' method, has abundant functional groups located on its basal planes and edges. These functional groups have been investigated as efficient adsorbents for the removal of heavy metal ions, such as Ni(II), Pb(II), U(VI), and Cr(VI).13,14 However, due to its small particle size and hydrophilicity, GO is difficult to separate from the aqueous phase through centrifugation and filtration methods after the removal process.15 Therefore, it is essential to modify GO to enhance its separability.
Iron oxide materials are widely regarded as an interesting material due to their innocuousness and chemical stability. These materials are applied in many fields, including catalysts, adsorbents, magnetic resonance imaging, gas sensors, and ion exchangers.16 Among these materials, Fe2O3 is abundant, low cost and environmentally friendly. It is used as an adsorbent for Cr(VI) removal owing to its effectiveness and selectiveness. Pure Fe2O3 tends to agglomerate in water, which is not favorable for the adsorption process. In addition, the adsorption capacity of pure Fe2O3 is limited. Ai et al. reported that the adsorption capacity of Fe@Fe2O3 core–shell nanowires was 10.06 mg g−1 (Cr(VI) concentration: 20 mg L−1; dose of adsorbent: 1.0 g L−1).17
Therefore, iron oxides have been combined with matrix materials to synthetize composites for the removal of heavy metal ions from aqueous environments recently. These synthesized composites possess advantages from both of the starting components. Wan et al. used a template synthesis method combined with chemical co-precipitation to prepare γ-Fe2O3@cellulose aerogels as the adsorbent for Cr(VI) removal.16 Baikousi et al. conducted synthesis and characterization of γ-Fe2O3/carbon hybrids that were utilized in Cr(VI) removal experiments.18
Herein, we have attempted to use a hydrothermal method to develop Fe2O3/rGO composites and investigate its removal ability for Cr(VI). This work aims to study the effects of preparation times at different temperatures on Cr(VI) removal performance of Fe2O3/rGO composites. Furthermore, we characterize the changes of the physical and chemical properties of unused and used Fe2O3/rGO to confirm the removal sites. The Cr(VI) reaction kinetics and adsorption isotherms are performed to demonstrate the efficiency of the new hybrids. Meanwhile, a possible mechanism schematic for the preparation of the composites and its Cr(VI) removal is proposed.
2. Experimental
2.1. Materials
Natural flake graphite powder (Sigma-Aldrich) was used to synthesize GO. Iron nitrate nonahydrate (Fe(NO3)·9H2O) was purchased from Sinopharm Chemical Reagent Company (Shanghai, China) to prepare composite. Besides, all other reagents used in this work were also purchased from Sinopharm Chemical Reagent Company (Shanghai, China) and were of analytical reagent grade.2.2. Synthesis of Fe2O3/rGO composites
GO was prepared from natural flake graphite powder using a modified Hummers' method.20,21 GO (0.12 g) was sonicated for 1 hour in 60 mL deionized water to obtain a homogeneous GO dispersion, to which 0.4 g Fe(NO3)·9H2O was added with continued sonicating for 10 minutes. Then pH of the suspension was adjusted to about 11.7 using 50% sodium hydroxide solution and sonication was continued for 10 minutes again. Afterwards, the obtained mixture was transferred into a 100 mL Teflon-lined stainless-steel autoclave and hydrothermally treated at different temperatures (120, 140, 160, and 180 °C) for some hours (2, 4, and 6 h). After cooling naturally to room temperature, the sample was washed twice with 500 mL deionized water, followed by freeze-drying. A schematic sketch of the synthesis process was shown in Fig. 8.2.3. Characterization
The morphology of as-prepared sample was observed by a field emission scanning electron microscope (SEM, Zeiss Merlin; JEM-6301). X-ray photoelectron spectra (XPS, ESCALAB250Xi, Thermo Fisher, USA) was carried out to analyze the elements and chemical states on the surface of the sample. The structure analysis was conducted using a Fourier transform infrared (FTIR) spectrometer (Nicolet 6700). N2 adsorption/desorption isotherms were performed on a Quantachrome SI-MP system and the surface area was calculated by the Brunauer–Emmett–Teller (BET) method. Before analysis, the sample was degassed at 160 °C.2.4. Chromium removal experiment
All Cr(VI) solutions (i.e., 12.66, 16.63, 20.28, 25.06, 28.96, 33.22, 37.65 mg L−1) were prepared by diluting the stock solution of 200 mg L−1 with deionized water. Uptake studies were carried out in batch mode using a thermostatic shaker with a speed of 140 rpm for 36 hours to achieve the equilibrium.The effect of pH, adjusted by HCl or NaOH solutions from 2 to 10, initial concentrations and temperatures on removal were investigated. In addition, samples were added into solutions in each experiment, where the pH of solutions was 2.24 without additional instruction. After equilibrium, the materials and solutions were separated through a 0.2 μm polyethersulfone (water system) filter, and solution concentrations were determined by UV-Visible spectrophotometer (HACH6000, USA) at a wavelength of 540 nm.
At last, the time-dependent capacity (qt (mg g−1)) and equilibrium removal capacity (qe (mg g−1)) of Cr(VI) were calculated as eqn (1) and (2):
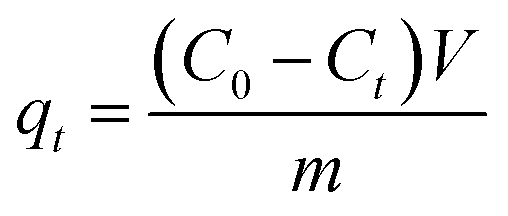 | (1) |
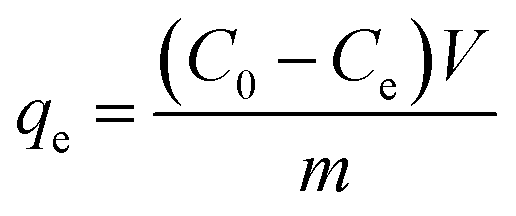 | (2) |
3. Results and discussion
3.1. Effects of preparation times at different temperatures on the removal performance
The removal efficiency and rate of materials prepared for different times at different temperatures are shown in Fig. 1. The Cr(VI) removal rate increased with the increase of synthesis time (Fig. 1(a) and (b)). It could be due to that time affected the formation of Fe2O3 at lower temperatures. And Fe2O3 contributed to improve the Cr(VI) removal. However, it can be seen in Fig. 1(c) and (d) that the Cr(VI) removal efficiency and rate reduced as the synthesis time increasing (160 and 180 °C). That resulted from the decrease of oxygen-containing functional groups on the material surface as the time increased. Furthermore, as shown in Fig. 1(e), the materials prepared at 160 °C for 2 h possessed the best removal efficiency and rate.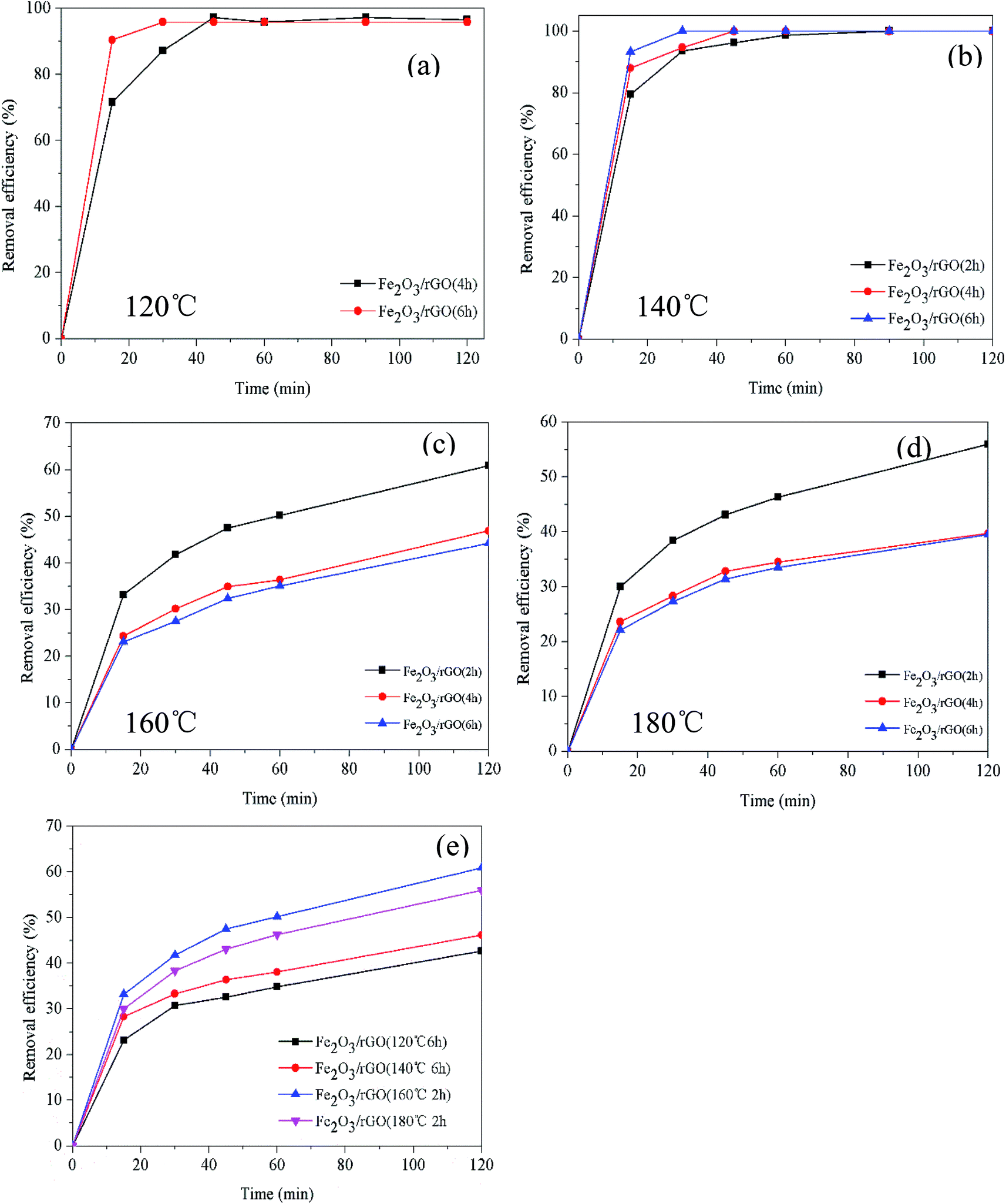 | ||
| Fig. 1 Effect of preparation time on Cr(VI) removal at different temperatures (C0 = 2.0 mg L−1; dosage = 0.5 g L−1; V = 100 mL; t = 2 h) (a) and (b); (C0 = 5.0 mg L−1; dosage = 0.2 g L−1; V = 100 mL; t = 2 h) (c) and (d); comparison of the best samples in each group (e) (C0 = 5.0 mg L−1; dosage = 0.2 g L−1; V = 100 mL; t = 2 h). | ||
3.2. Characterization and performance of removal mechanism
Scanning electron microscopy (SEM) images revealed the surface morphologies of GO, used Fe2O3/rGO and unused Fe2O3/rGO particles in Fig. 2. In Fig. 2(a), GO presented a sheet-like two-dimensional nanomaterial structure with many wrinkles on its surface, which is favorable for Fe2O3 particles anchored on the surface of the GO layers. From the low- and high-magnification SEM images of the Fe2O3/rGO material in Fig. 2(b) and (c), respectively, it can be seen that the samples displayed a flaky powder that was decorated with Fe2O3 nanorods. Fig. 2(d) showed that there was no obvious change between the used and unused materials.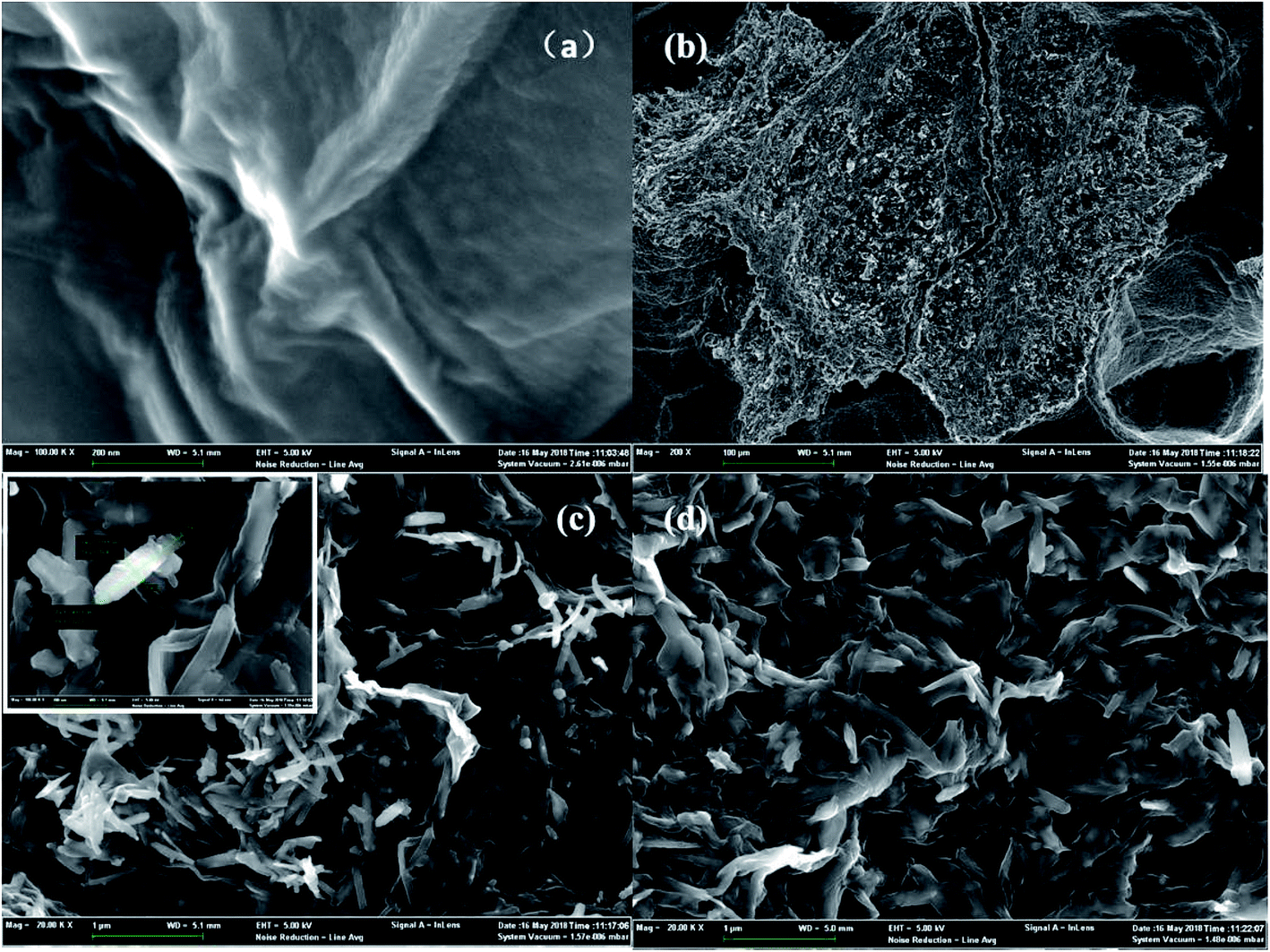 | ||
| Fig. 2 SEM of GO (a); low-magnifications SEM images of unused Fe2O3/rGO (b); high-magnifications SEM images of unused Fe2O3/rGO (c); used Fe2O3/rGO (d). | ||
To obtain the porous nature and Brunauer–Emmett–Teller (BET) specific surface area of the as-prepared samples, N2 adsorption and desorption isotherms were evaluated. As shown in Fig. 3(a), the adsorption curve did not show any adsorption limit at the relatively higher relative pressure (P/P0) region. Therefore, according to the International Union of Pure and Applied Chemistry classification, the sample displayed a Type IV isothermal model with a H3 hysteresis loop.22,23 This classification indicated that the samples were either flaky or layered and that mesopores existed within the materials. This result was confirmed in Fig. 3(b), where the pore size distribution for Fe2O3/rGO showed a bimodal-type pore size distribution with peak centers located at 3.81 (revealed in Fig. 3(c)) and 28.12 nm. Fig. 3(b) also showed that the Fe2O3/rGO materials had not only small-size mesopores but also large-size mesopores. Furthermore, the BET specific surface area, total pore volume, and average pore diameter of Fe2O3/rGO were 35.57 m2 g−1, 0.1128 cm3 g−1, and 3.812 nm, respectively.
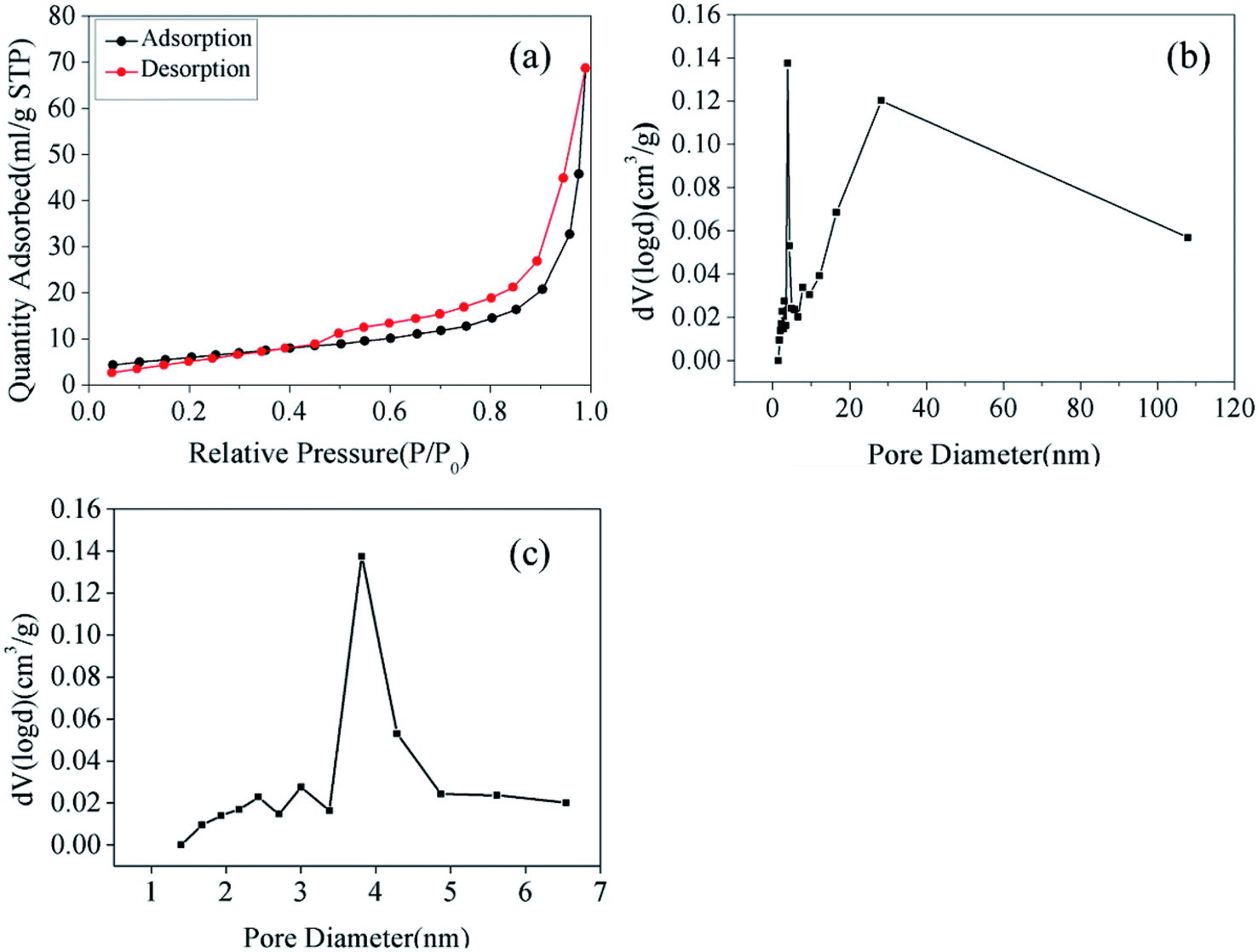 | ||
| Fig. 3 N2 adsorption/desorption isotherms (a); pore size distribution of Fe2O3/rGO (b); pore size distribution of Fe2O3/rGO ranging from 1 to 7 nm (c). | ||
The XRD pattern of Fe2O3/rGO composites is shown in Fig. 4. The prominent diffraction peaks at 2θ = 21.3°, 33.4°, 36.7°, 41.3°, 53.0°, 59.1°, 61.7° and 63.8° were assigned to the (012), (104), (110), (113), (024), (116), (214) and (300) planes of Fe2O3, indicating the formation of highly crystalline Fe2O3.24 However, no diffraction peaks of GO were observed in the Fig. 4, probably because the intercalation of Fe2O3 in composites changed the crystalline structures of GO during the synthesis.
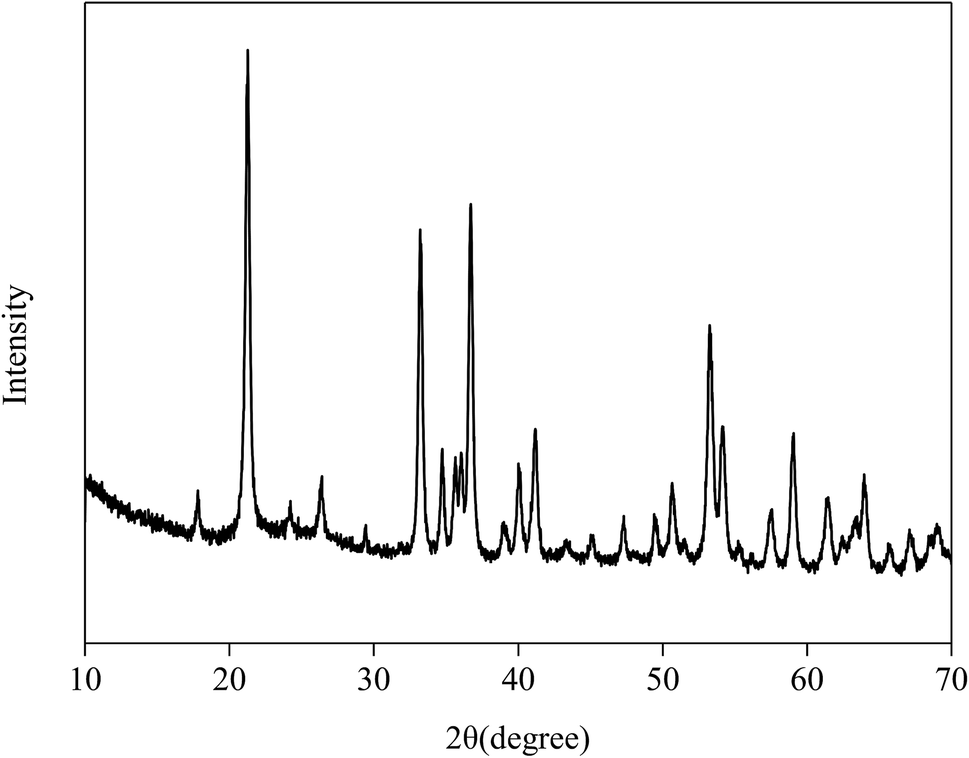 | ||
| Fig. 4 The XRD pattern of Fe2O3/rGO. | ||
Fig. 5(a) showed the TEM images of Fe2O3/rGO, showing that Fe2O3 was nanorod. The result was consistent with SEM. Fig. 5(b) was the electron diffraction (ED) pattern of a single Fe2O3 nanorod, revealing the single crystal nature of Fe2O3 nanoparticles. The diffraction data were determined to be taken from the [001] zone axis. The growth direction of Fe2O3 nanorod was along the [100] direction.25
 | ||
| Fig. 5 TEM images (a) and SAED patterns (b) of Fe2O3/rGO. | ||
X-ray photoelectron spectroscopy (XPS) analysis of the unused and used Fe2O3/rGO and GO was conducted to identify the oxidative states of the various elements. This analysis helped to investigate the interactions between the samples and Cr(VI). The survey scan results are shown in Fig. 6(a). These results showed that, compared with GO, the Fe 2p peaks of the unused Fe2O3/rGO appeared at the binding energies of 711.4 and 724.9 eV (Fig. 6(a) inset). This signal is the characteristic doublet Fe 2p peak of Fe2O3 with a splitting energy of 13.5 eV, suggesting that Fe2O3 was successfully anchored on GO.26 Moreover, a satellite peak located at 719.5 eV further confirmed the successful anchorage. After Cr(VI) removal, two new peaks centered at 577.5 and 587.2 eV, corresponding to Cr 2p3/2 and Cr 2p1/2, were observed in Fig. 6(b). These new peaks were consistent with Cr(III) and Cr(VI), which suggested that the adsorbed Cr(VI) was partially reduced to Cr(III). C 1s spectra of unused and used Fe2O3/rGO are shown in Fig. 6(c) and (d), respectively. It can been seen that the carbon functional groups of the unused and used Fe2O3/rGO can be decomposed into four component peaks, namely, C–C, C–H, C–OH, and C–OOH. Comparing the C 1s spectrum of used Fe2O3/rGO with that of the unused spectrum, the percentages of C–OH and C–OOH declined, suggesting that these functional groups were involved in the reaction. We obtained a similar conclusion from Fig. 6(e) and (f).
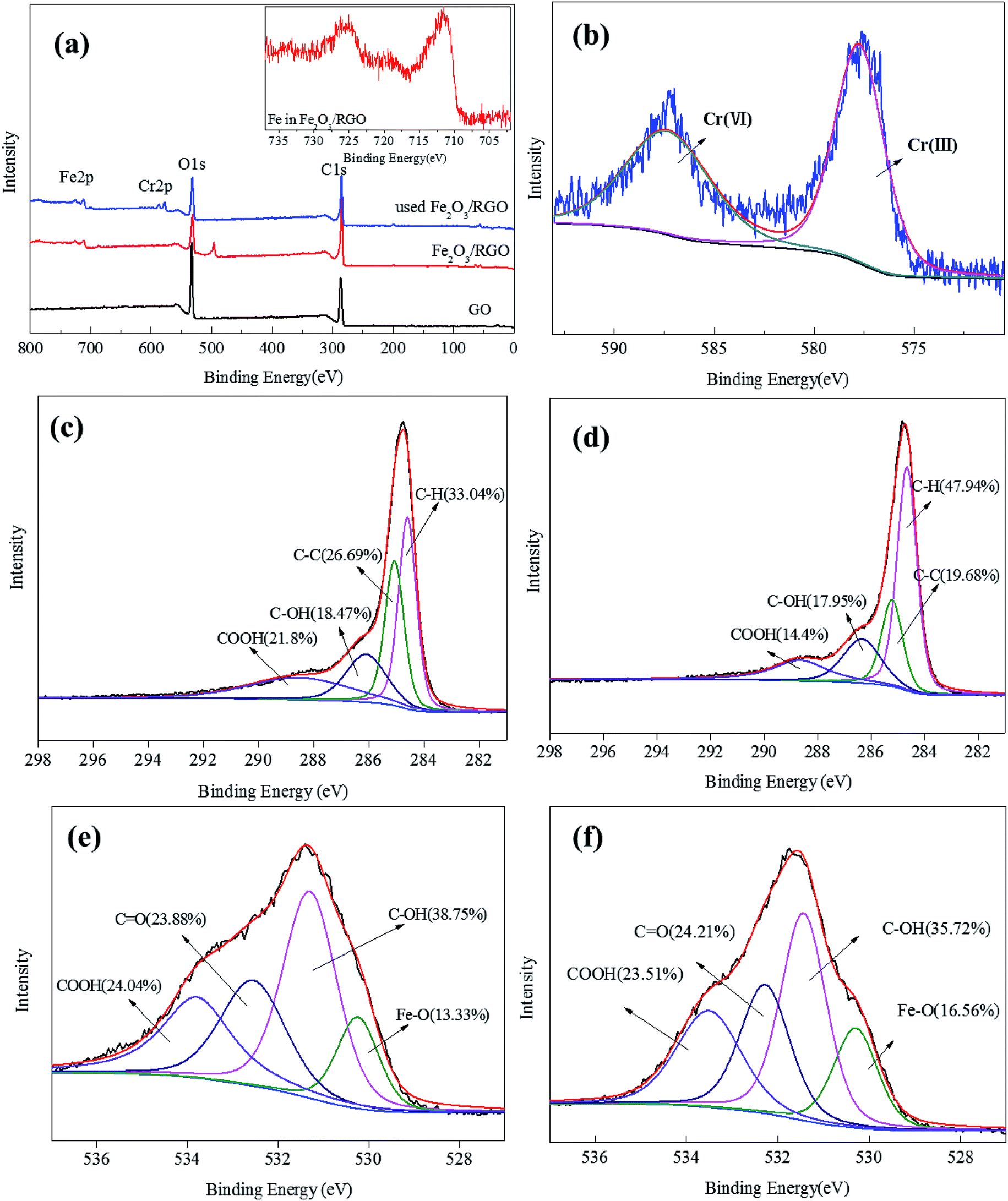 | ||
| Fig. 6 The spectra survey scans of GO, Fe2O3/rGO and used Fe2O3/rGO (a); Cr 2p spectra of used Fe2O3/rGO (b); C 1s spectra of unused Fe2O3/rGO (c); C 1s spectra of used Fe2O3/rGO (d); O 1s spectra of unused Fe2O3/rGO (e); O 1s spectra of used Fe2O3/rGO (f). | ||
As shown in Fig. 7, the Fourier-transform infrared (FTIR) spectrum of GO, unused Fe2O3/rGO and used Fe2O3/rGO were performed to investigate the transmutation of the functional groups. In the FTIR spectra of GO, the characteristic peaks at 1729 and 1067 cm−1 were ascribed to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and C–O stretching vibration of the carboxyl groups. A broad band at 3422 cm−1 corresponded to the hydroxyl group.26 The peaks at 1225 and 1372 cm−1 were attributed to epoxy C–O stretching vibrations and O–H deformation vibrations, respectively.27 The peak at 1619 cm−1 corresponded to CC.28 In comparison, the peaks of the oxygen-containing functional groups, such as COOH and OH, almost disappeared in the FTIR spectrum of Fe2O3/rGO. This observation suggested that GO was reduced through a hydrothermal synthesis process. Meanwhile, two new peaks at 883 and 783 cm−1 were ascribed to the Fe–OH bending vibration,19 which was favorable for heavy metal removal.29 The peak at 579 cm−1 was assigned to Fe–O. According to the reports, there should be new peaks corresponding to CrO and Cr–O after Cr(VI) removal based on FTIR analysis.30,31 However, there was no significant change in the FTIR spectra of used Fe2O3/rGO in this work, probably because the change was too weak to be observed.
O and C–O stretching vibration of the carboxyl groups. A broad band at 3422 cm−1 corresponded to the hydroxyl group.26 The peaks at 1225 and 1372 cm−1 were attributed to epoxy C–O stretching vibrations and O–H deformation vibrations, respectively.27 The peak at 1619 cm−1 corresponded to CC.28 In comparison, the peaks of the oxygen-containing functional groups, such as COOH and OH, almost disappeared in the FTIR spectrum of Fe2O3/rGO. This observation suggested that GO was reduced through a hydrothermal synthesis process. Meanwhile, two new peaks at 883 and 783 cm−1 were ascribed to the Fe–OH bending vibration,19 which was favorable for heavy metal removal.29 The peak at 579 cm−1 was assigned to Fe–O. According to the reports, there should be new peaks corresponding to CrO and Cr–O after Cr(VI) removal based on FTIR analysis.30,31 However, there was no significant change in the FTIR spectra of used Fe2O3/rGO in this work, probably because the change was too weak to be observed.
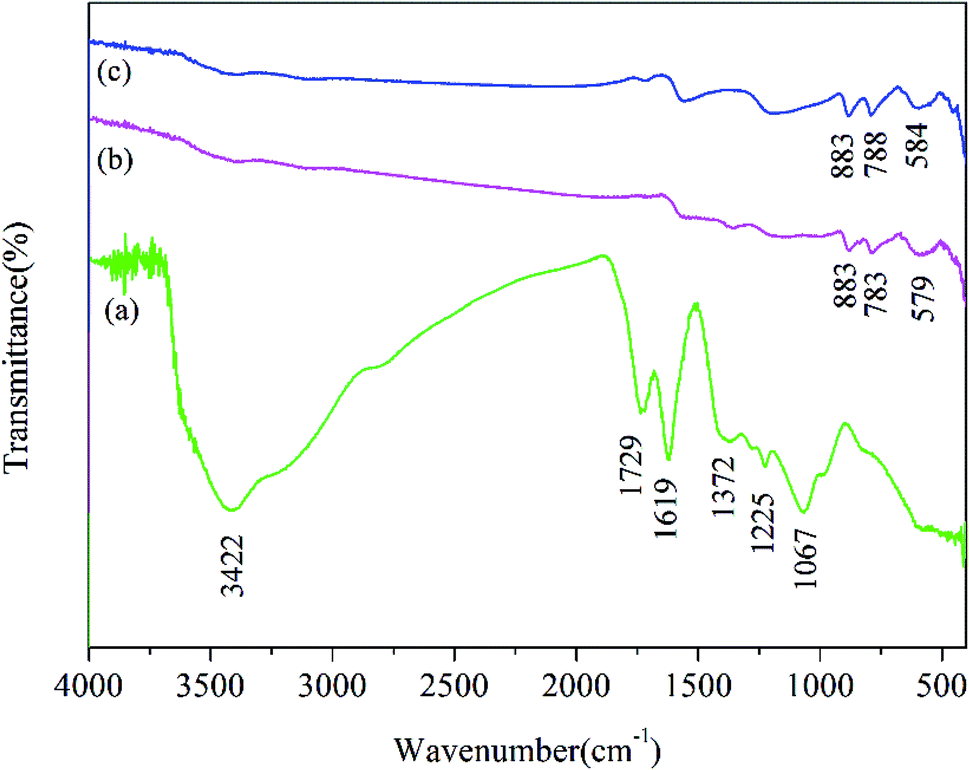 | ||
| Fig. 7 FTIR spectrum of GO (a), Fe2O3/rGO (b) and used Fe2O3/rGO (c). | ||
In view of the above results, we proposed the following removal mechanism. First, most of the Cr(VI) anion is bound to Fe2O3/rGO via electrostatic attraction with protonated hydroxyl and carboxyl groups, as shown in the following equations:32
| surface–OH2+ + HCrO4− ↔ surface–OH2CrO4− + H+ | (3) |
| surface–COOH2+ + HCrO4− ↔ surface–COOH2CrO4− + H+ | (4) |
Next, Cr(VI) is reduced to Cr(III) through electron donors, such as hydroxyl functional groups. The equation is as follows:33
| HCrO4− + 7H+ + 3e ↔ Cr3+ + 4H2O | (5) |
Then, one part of the reduced Cr(III) is released into the aqueous solution, and the other part is precipitated on the surface of Fe2O3/rGO. Moreover, we found that Fe2O3/rGO contained Fe–OH, which is beneficial for the removal of Cr(VI), as shown in the following eqn (6):34
| surface–FeOH + HCrO4− → surface–Fe–CrO4− + H2O | (6) |
The removal mechanism diagram is shown in Fig. 8.
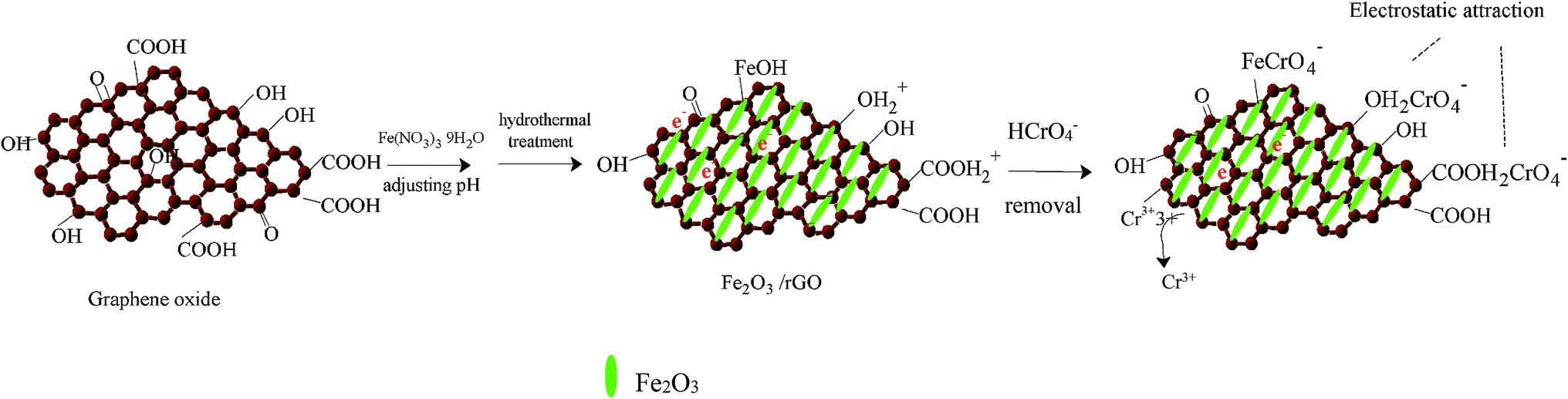 | ||
| Fig. 8 Schematic illustration for the preparation of Fe2O3/rGO and removal mechanism. | ||
3.3. Effect of influencing factors in the removal process
The pH of the solution is one of the most important variables affecting the removal process, because it does not only affect the material surface charge but also the metal ion speciation. The effect of pH on the removal of Cr(VI) is shown in Fig. 9(a). The removal capacity was highly dependent on the initial pH. The maximum removal capacity was 35.00 mg g−1, occurring at pH 2. The removal capacity decreased evidently with the initial solution pH increasing from 2 to 5, and the removal capacity decreased slightly as the solution pH increased from 6 to 10. This result meant that low pH was beneficial to the Cr(VI) removal process. At low pH (1–6), the dominant species of Cr(VI) is HCrO4−, while CrO42− is the dominant species at pH values above 6. Therefore, the presence of these ions had an effect on the removal capacity. What is more, the surface of the materials was highly protonated and the materials were positively charged at low pH values. This was favorable for the uptake of Cr(VI) anions and resulted in the high removal efficiency. At the same time, low pH values also favored the redox reaction in the removal process. Cr(VI) was partially reduced to Cr(III) by the reductive surface hydroxyl groups. Then, one part of the obtained Cr(III) was released into the aqueous solution at low pH, and the other part was precipitated onto the surface of the Fe2O3/rGO. Besides, the OH− ions would compete with the Cr(VI) anions with pH increasing.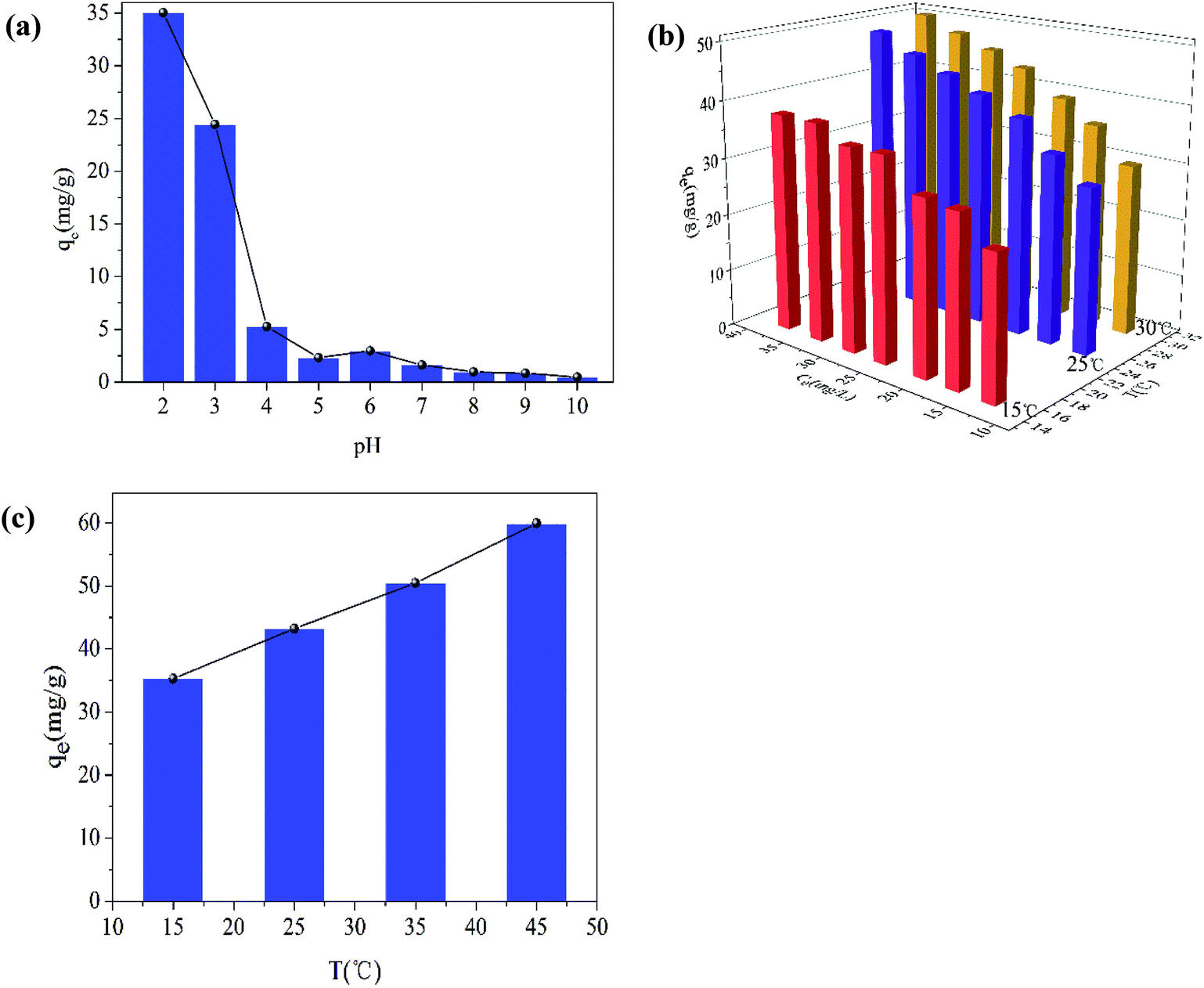 | ||
| Fig. 9 Effect of pH on Cr(VI) removal (C0 = 16.63 mg L−1; dose of Fe2O3/rGO = 0.4 g L−1; T = 25 ± 1 °C; t = 36 h; 140 rpm) (a); effect of the initial Cr(VI) concentration (b); effect of the temperatures (c) (dose of Fe2O3/rGO = 0.4 g L−1; pH = 2.2; t = 36 h; 140 rpm). | ||
The effect of the initial Cr(VI) concentration on the removal efficiency was studied at different concentrations (12.66, 16.63, 20.28, 25.06, 28.96, 33.22, and 37.65 mg L−1). In Fig. 9(b), it can been seen that the removal capacity was increased as the initial Cr(VI) concentration increased, implying that the removal capacity was related to the initial concentration. This effect could be the reason that a higher initial concentration resulted in a higher collision rate between the Cr(VI) ions and the active sites of Fe2O3/rGO. Thus, increasing the concentration gradient, a driving force contributing to overcoming the mass transfer resistance between the aqueous and solid phases, was favorable to the Cr(VI) removal.35 It facilitated the ion diffusion from liquid phase to solid phase.36
The influence of temperatures on Cr(VI) removal was another important factor. As shown in Fig. 9(c), as the temperatures increased from 15 to 45 °C, the removal capacity was also enhanced, meaning that higher temperatures were beneficial to Cr(VI) removal. It was an endothermic process. As the temperature increased, the strengthened irregular movement of Cr(VI) ions might lead to the higher collision rate between Fe2O3/rGO particles and Cr(VI) ions.37 In addition, higher temperatures also contribute to the expansion of pore size and the activation of the adsorbent surface, as reported by Namasivayam et al.38,39
3.4. Regeneration study
The regeneration experiment of Fe2O3/rGO at 5.0 mg L−1 Cr(VI) concentration was studied by repeating the removal process on spent materials. Three regeneration cycles are showed in Fig. 10. For the first removal process, the Cr(VI) removal capacity was 9.69 mg g−1. During the three regeneration cycles, the Cr(VI) removal capacities were 9.65, 9.44 and 9.00 mg g−1 respectively. The recyclability studies showed that the removal capacity of Fe2O3/rGO decreased by 7.7% after three cycles. Therefore, the materials can be repeatedly used in wastewater treatment.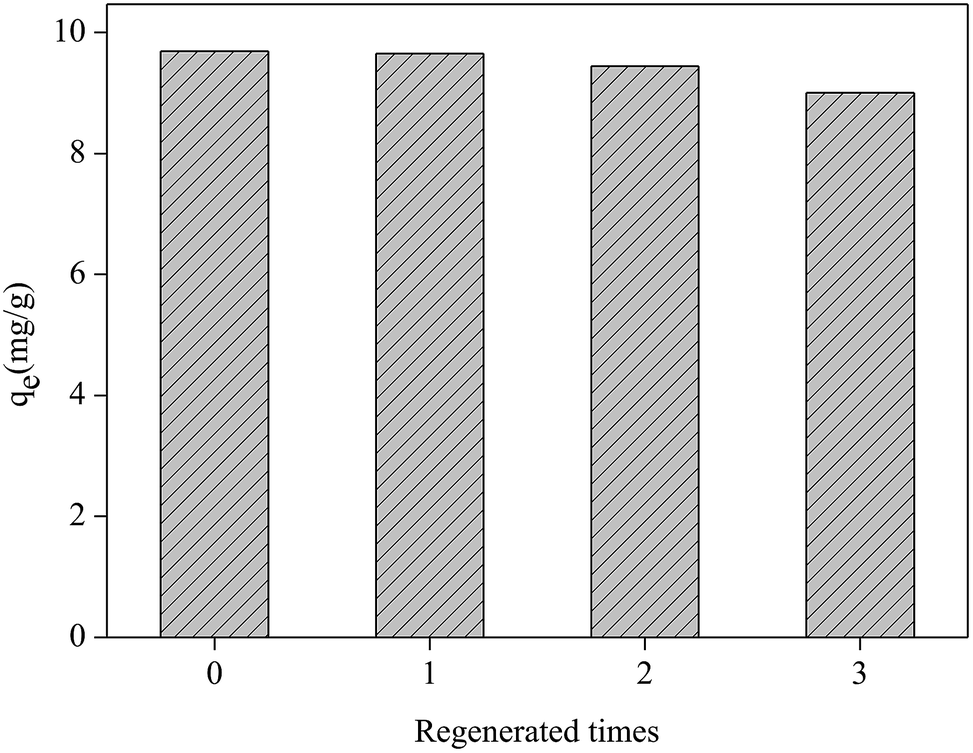 | ||
| Fig. 10 Regeneration test of the Fe2O3/rGO (dosage = 0.2 g L−1; C0 = 5.0 mg L−1; V = 50 mL; tremoval = 3 h; 0.1 mol L−1 NaOH, desorption agent; tdesorption = 3 h). | ||
3.5. Kinetics study
In this study, pseudo-first-order and pseudo-second-order kinetics models were used to analyse the experimental data to investigate the removal mechanisms and potential rate-controlling steps, which included mass transport and chemical reaction processes. The two models were expressed in the following equations:40,41
lg(qe − qt) = lg![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) qe − k1t qe − k1t
| (7) |
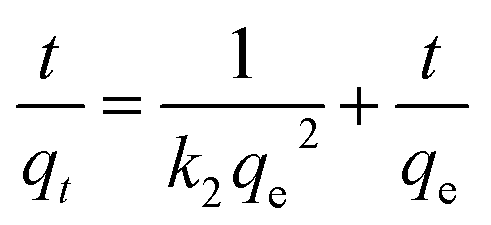 | (8) |
The kinetic parameters were given in Table 1, obtained from fitting the results of the Cr(VI) removal at different initial concentrations.
| Kinetic model | Parameter | Initial concentration (mg L−1) | ||
|---|---|---|---|---|
| 13.05 | 25.45 | 34.95 | ||
| Pseudo-first-order | qe(exp) (mg g−1) | 31.4375 | 40.0625 | 47.2813 |
| qe1 (mg g−1) | 17.6071 | 29.4001 | 33.8182 | |
| k1 (min−1) | 0.0024 | 0.0010 | 0.0010 | |
| R2 | 0.9819 | 0.9826 | 0.9602 | |
| Pseudo-second-order | qe2 (mg g−1) | 32.2581 | 41.6146 | 48.5437 |
| k2 (mg g−1 min−1) | 0.0004 | 0.0001 | 0.0001 | |
| R2 | 0.9975 | 0.9891 | 0.9881 | |
| Intra-particle diffusion | kid,1 (mg g−1 min−0.5) | 1.1135 | 1.4053 | 1.5935 |
| kid,2 (mg g−1 min−0.5) | 0.2640 | 0.5798 | 0.6363 | |
| C1 (mg g−1) | 6.9377 | 1.4887 | 1.9531 | |
| C2 (mg g−1) | 20.2416 | 14.5966 | 18.4125 | |
The values of the correlation coefficients of the pseudo-first-order and pseudo-second-order models were both high, but the correlation coefficients of the pseudo-second-order model were higher than those of the pseudo-first-order model. Additionally, the calculated qe values obtained from the pseudo-second-order model were closer to the experimental qe(exp). Therefore, the pseudo-second-order model was more suitable for describing the removal process, indicating that the chemical removal might be the rate-controlling step.42
The intraparticle diffusion model was applied to further investigate the rate-limiting step and diffusion mechanisms. The intraparticle diffusion model was expressed in the following equation:43
| qt = kidt0.5 + Ci | (9) |
3.6. Isotherm study
Adsorption isotherms were essential to analyse the surface properties and to evaluate the removal capacity of Fe2O3/rGO.44 Two isotherm models were applied to fitting the experimental data, namely Langmuir and Freundlich. The Langmuir model assumes that the adsorption process consists of monolayer adsorption on a homogeneous surface with binding sites possessing the same energy.45 Conversely, the Freundlich isotherm is based on multilayer adsorption that takes place on a inhomogeneous surface.46The linear forms of the Langmuir and Freundlich isotherm models were expressed in eqn (10) and (11), respectively:47
 | (10) |
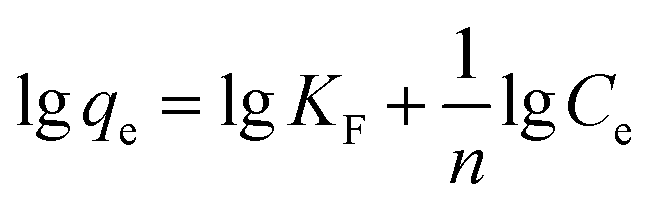 | (11) |
Furthermore, RL is an equilibrium parameter to describe the Langmuir isotherm characteristics. It can be given by eqn (12):48
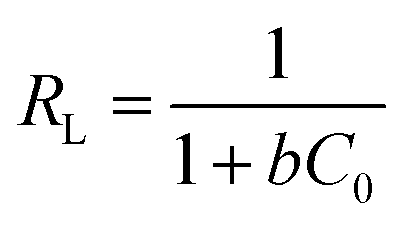 | (12) |
The fitting results were listed in Table 2. As shown in Table 2, all correlation coefficient values (R2 > 0.95) were high, indicating that the Langmuir and Freundlich isotherm models fitted well with the experimental data at different temperatures. However, the correlation coefficient values of the Langmuir model were higher than those of the Freundlich model, suggesting that the monolayer adsorption on a homogeneous surface was dominant process. The maximum adsorption capacities calculated from the Langmuir model were 41.67 and 51.81 at 288 and 298 K, respectively. In addition, the values of RL and 1/n were between 0 and 1, implying that it was a beneficial adsorption process.
| Temperature (K) | Langmuir constants | Freundlich constants | |||||
|---|---|---|---|---|---|---|---|
| qmax (mg g−1) | b (L mg−1) | R2 | RL | KF (mg g−1) | 1/n | R2 | |
| 288 | 41.667 | 0.481 | 0.995 | 0.053 | 20.930 | 0.201 | 0.951 |
| 298 | 51.814 | 0.544 | 0.991 | 0.046 | 27.736 | 0.197 | 0.982 |
3.7. Thermodynamic study
A thermodynamic study was conducted to observe the effect of temperature and to confirm the spontaneity of removal process. The thermodynamic parameters, such as the Gibbs free energy change (ΔG0), enthalpy change (ΔH0), and entropy change (ΔS0) were calculated using the following equations:|
ΔG0 = −RTln(qe/Ce)
| (13) |
| ΔG0 = ΔH0 − TΔS0 | (14) |
| Temperature (K) | ΔG0 (kJ mol−1) | ΔH0 (kJ mol−1 K−1) | ΔS0 (kJ mol−1 K−1) |
|---|---|---|---|
| 288 | −1.91 | 36.01 | 0.13 |
| 298 | −3.16 | ||
| 308 | −4.27 | ||
| 318 | −5.92 |
The values of ΔG0 was negative, indicating that the removal process was spontaneous and feasible. The positive value of ΔH0 showed the endothermic nature of the removal process. Additionally, the positive value of ΔS0 implied that the randomness increased at the solid–solution interface.
4. Conclusions
In summary, the material prepared at 160 °C for 2 h possessed the best removal efficiency and rate for Cr(VI) from aqueous solutions. They tended to showed a much better removal capacity at low pH value. This removal process was spontaneous and endothermic. Moreover, the removal process was not only ascribed to the electrostatic attraction between the protonated hydroxyl and carboxyl groups and Cr(VI), but also Cr(VI) reduction. Cr(VI) was mainly reduced by electrons, provided by the hydroxyl groups. Fe2O3 particles were highly dispersed due to the high surface area of rGO.The intraparticle diffusion was not the only rate-limiting step. This work developed a promising material for the removal of Cr(VI) from aqueous environments with easy recovery and high efficiency.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The research was supported by Key technology and project of Jinan water environment control (No. 201509002) and the Major Science and Technology Program for Water Pollution Control and Treatment (2017ZX07202002).References
- V. Gupta, V. Chandra, I. Tyagi and M. Verma, J. Colloid Interface Sci., 2016, 478, 54–62 CrossRef CAS PubMed.
- P. Xu, S. Huang, Y. Lv, Y. Chen, M. Liu and H. Fan, RSC Adv., 2018, 8, 5749–5759 RSC.
- L. Bentchikou, F. Mechelouf, F. Neggaz and A. Mellah, J. Turk. Chem. Soc., Sect. B, 2017, 1, 43–52 CAS.
- World Health Organization, Guidelines for drinking-water quality, World Health Organization, 4th edn, 2011 Search PubMed.
- Ministry of Health, Sanistary Standard for Drinking Water; UDC613.3/GB5749-2006, Ministry of Health, Beijing, 2006 Search PubMed.
- M. Matlock, S. Howerton and A. Atwood, Water Res., 2002, 36, 4757–4764 CrossRef CAS PubMed.
- S. Juang and C. Shiau, J. Membr. Sci., 2000, 165, 159–167 CrossRef.
- A. Dabrowski, Z. Hubicki, P. Podkościelny and E. Robens, Chemosphere, 2004, 56, 91–106 CrossRef CAS PubMed.
- R. Patterson, S. Fendorf and M. Fendorf, Environ. Sci. Technol., 1997, 31, 2039–2044 CrossRef CAS.
- S. Younan, Z. Sakita, R. Albuquerque, R. Keller and H. Bremer-Neto, J. Sci. Food Agric., 2016, 96, 3977–3982 CrossRef CAS PubMed.
- A. Demirbas, J. Hazard. Mater., 2008, 157, 220–229 CrossRef CAS PubMed.
- W. Wang, Z. Wang, J. Liu, Z. Zhang and L. Sun, Sci. Rep., 2017, 7, 43755 CrossRef PubMed.
- Y. Li, S. Zhu, Q. Liu, Z. Chen, J. Gu, C. Zhu, T. Lu, D. Zhang and J. Ma, Water Res., 2013, 47, 4188–4197 CrossRef CAS PubMed.
- M. Shaban, R. Abukhadra, M. Rabia, A. Elkader and A. El-Halim, Rendiconti Lincei, 2018, 29, 141–154 CrossRef.
- X. Deng, L. Lü, H. Li and F. Luo, J. Hazard. Mater., 2010, 183, 923–930 CrossRef CAS PubMed.
- C. Wan and J. Li, ACS Sustainable Chem. Eng., 2015, 3, 2142–2152 CrossRef CAS.
- Z. Ai, Y. Cheng, L. Zhang and J. Qiu, Environ. Sci. Technol., 2008, 42, 6955–6960 CrossRef CAS PubMed.
- M. Baikousi, B. Bourlinos, A. Douvalis, T. Bakas, F. Anagnostopoulos, J. Tuček, K. Safářová, R. Zboril and A. Karakassides, Langmuir, 2012, 28, 3918–3930 CrossRef CAS PubMed.
- H. Cong, X. Ren, P. Wang and S. Yu, ACS Nano, 2012, 6, 2693–2703 CrossRef CAS PubMed.
- W. Hummers and R. Offeman, J. Am. Chem. Soc., 1958, 80, 1339 CrossRef CAS.
- L. Xiao, D. Wu, S. Han, Y. Huang, S. Li, M. He, F. Zhang and X. Feng, ACS Appl. Mater. Interfaces, 2013, 5, 3764–3769 CrossRef CAS PubMed.
- G. Leofanti, M. Padovan, G. Tozzola and B. Venturelli, Catal. Today, 1998, 90, 207–219 CrossRef.
- C. Li, N. Chen, Y. Zhao, R. Li and C. Feng, Chemosphere, 2016, 163, 81–89 CrossRef CAS PubMed.
- Z. Song, W. Liu, W. Wei, C. Quan, N. Sun, Q. Zhou, G. Liu and X. Wen, J. Alloys Compd., 2016, 685, 355–363 CrossRef CAS.
- Y. Zou, J. Kan and Y. Wang, J. Phys. Chem. C, 2011, 115, 20747–20753 CrossRef CAS.
- Y. Liu, W. Jin, Y. Zhao, G. Zhang and W. Zhang, Appl. Catal., B, 2017, 206, 642–652 CrossRef CAS.
- S. Han, L. Hu, Z. Liang, S. Wageh, A. Al-Ghamdi, Y. Chen and X. Fang, Adv. Funct. Mater., 2014, 24, 5719–5727 CrossRef CAS.
- S. Li, X. Lu, Y. Xue, J. Lei, T. Zheng and C. Wang, PLoS One, 2012, 7, e43328 CrossRef CAS PubMed.
- A. Katsoyiannis and I. Zouboulis, Water Res., 2002, 36, 5141–5155 CrossRef PubMed.
- P. Wang and C. Lo, Water Res., 2009, 43, 3727–3734 CrossRef CAS PubMed.
- K. Kumar, S. Jiang and W. Tseng, J. Mater. Chem. A, 2015, 3, 7044–7057 RSC.
- C. Jung, J. Heo, J. Han, N. Her, S. Lee, J. Oh, J. Ryu and Y. Yoon, Sep. Purif. Technol., 2013, 106, 63–71 CrossRef CAS.
- L. Fan, C. Luo, M. Sun and H. Qiu, J. Mater. Chem., 2012, 22, 24577–24583 RSC.
- A. Katsoyiannis and I. Zouboulis, Water Res., 2002, 36, 5141–5155 CrossRef PubMed.
- M. Gheju, I. Balcu and G. Mosoarca, J. Hazard. Mater., 2016, 310, 270–277 CrossRef CAS PubMed.
- G. Sharma, M. Naushad, H. Al-Muhtaseb, A. Kumar, R. Khan, S. Kalia, Shweta, M. Bala and A. Sharma, Int. J. Biol. Macromol., 2017, 95, 484–493 CrossRef CAS PubMed.
- D. Wang, G. Zhang, L. Zhou, M. Wang, D. Cai and Z. Wu, Langmuir, 2017, 33, 7007–7014 CrossRef CAS PubMed.
- C. Namasivayam and T. Yamuna, Chemosphere, 1995, 30, 561–578 CrossRef CAS.
- J. WooáLee and S. BináKim, Nanoscale, 2011, 3, 3583–3585 RSC.
- S. Lagergren, K. Sven. Vetenskapsakad. Handl., 1898, 24, 1–39 Search PubMed.
- S. Ho and G. McKay, Process Biochem., 1999, 34, 451–465 CrossRef.
- L. Lv, N. Chen, C. Feng, J. Zhang and M. Li, RSC Adv., 2017, 7, 27992–28000 RSC.
- N. Ünlü and M. Ersoz, J. Hazard. Mater., 2006, 136, 272–280 CrossRef PubMed.
- H. Dehghani, M. Taher, K. Bajpai, B. Heibatie, I. Tyagi, M. Asif, S. Agarwal and V. Gupta, Chem. Eng. J., 2015, 279, 344–352 CrossRef.
- O. Altın, H. Özbelge and T. Doğu, J. Colloid Interface Sci., 1998, 198, 130–140 CrossRef.
- B. Albadarin, C. Mangwandi, H. Al-Muhtaseb, M. Walker, J. Allen and M. Ahmad, Chem. Eng. J., 2015, 179, 193–202 CrossRef.
- J. Lalley, C. Han, X. Li, D. Dionysiou and N. Nadagoudaet, Chem. Eng. J., 2016, 284, 1386–1396 CrossRef CAS.
- H. Dehghani, D. Sanaei, I. Ali and A. Bhatnagar, J. Mol. Liq., 2016, 215, 671–679 CrossRef.
| This journal is © The Royal Society of Chemistry 2019 |