
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
WO3–EDA hybrid nanoplates and nanowires: synthesis, characterization, formation mechanism and thermal decomposition†
Dávid Hunyadi *a,
Eszter Majzika,
Judit Mátyásia,
József Ballaa,
Attila Domjánb,
Ágnes Szegedic and
Imre Miklós Szilágyiad
*a,
Eszter Majzika,
Judit Mátyásia,
József Ballaa,
Attila Domjánb,
Ágnes Szegedic and
Imre Miklós Szilágyiad
aDepartment of Inorganic and Analytical Chemistry, Budapest University of Technology and Economics, Szt. Gellért tér 4, H-1111 Budapest, Hungary. E-mail: david.hunyadi89@gmail.com; Tel: +36-1-463-4141
bNMR Laboratory, Research Centre for Natural Sciences, Hungarian Academy of Sciences, Magyar tudósok körútja 2, H-1117 Budapest, Hungary
cInstitute of Materials and Environmental Chemistry, Research Centre for Natural Sciences, Hungarian Academy of Sciences, Magyar tudósok körútja 2, H-1117 Budapest, Hungary
dTechnical Analytical Chemistry Research Group of the Hungarian Academy of Sciences, Budapest University of Technology and Economics, Szt. Gellért tér 4, H-1111 Budapest, Hungary
First published on 3rd October 2017
Abstract
Previously the WO3–EDA hybrid material was obtained only from solvothermal reactions. In this study this hybrid was prepared by two novel methods: a solid–gas phase heterogeneous reaction, and a wet chemical process. In the case of the solid–gas phase reaction the effects of the composition, crystal structure, and the particle size of the WO3 powder and the presence of H2O vapor were studied, while in the case of the wet chemical process the effect of the solvent was investigated. The structure, composition, morphology and thermal decomposition of the as-prepared WO3–EDA hybrid were investigated by XRD, FTIR, solid-state NMR, elemental analysis, SEM, TEM and TG/DTA-MS measurements. In addition, its catalytic activity was tested in a Knoevenagel condensation model reaction. Based on the results the WO3–EDA empiric formula was proposed to replace the current WOx–EDA formula. From the solid–gas phase reaction WO3–EDA nanoplates were obtained for the first time. Furthermore, a new formation mechanism was proposed for the solid–gas phase formation of this hybrid material. The thermal decomposition of the hybrid resulted m-WO3 in air, and an amorphous tungsten oxide phase in nitrogen. During annealing, the evolved EDA transformed into a series of heterocyclic aromatic compounds in both atmospheres. The as-prepared hybrids had the same catalytic properties as the hybrids obtained previously from the solvothermal reactions.
1. Introduction
The tungsten oxides are widely used as catalysts,1–3 photocatalysts,4–8 gas sensors9–16 and as electrochromic coatings.17–20 The tungsten trioxide has several hydrated forms (WO3·xH2O), which have a layered structure.21–23 Their structure consists of sheets of corner-sharing WO6 octahedra, where the water molecules are intercalated between these sheets and the structure is maintained by hydrogen bonds.21–23 The layered structure gave the idea to several research groups to intercalate amines between these inorganic tungsten oxide sheets, forming organic–inorganic hybrid materials.23–29 They have great rigidity and thermal stability thanks to the inorganic frame, and have the flexibility and functionality of the organic part.30,31 These materials are used in water purification,26,32 catalysis,26 electronics27,33,34 and medicine.35Previously pyrazine,24 4,4-bipyridine,24,25 ethylenediamine (EDA) and other diamines23,26,27 were intercalated by hydrothermal and solvothermal reactions. Also WO3–pyridine was prepared by heating the reactants in a furnace.25 Several n-alkylamines (CxHx+1NH2, x = 4, 6, 8, 10, 12, 14) were intercalated by simply stirring the solutions at room temperature.28,29 For the hydrothermal and solvothermal reactions and for the other methods, generally, tungstic acids and Na2WO4 were used as W precursor.
The composition and structure of these materials were investigated in detail. The as-obtained hybrids have a layered structure, similar to WO3·xH2O. In the cases of some WO3–amine hybrids their composition and crystal structure were determined by single crystal XRD measurements.24,25
In the case of the WO3–EDA hybrid a single layer of EDA was intercalated between the tungsten oxide layers. The EDA was incorporated into the structure only in its neutral form (–NH2), as the amine functional groups formed hydrogen bonds with the apical O groups.23,26,36 However, the exact composition and the nature of the crystal structure of this hybrid were not fully determined before.
For the solvothermal reaction between W precursors and EDA two formation mechanisms were proposed previously. According to the solvent-coordination molecular template (SCMT) mechanism first the EDA molecules are adsorbed on the surface of the WO3 particles. Then the EDA molecules are incorporated into the neighboring WO6 octahedra layers through coordination, resulting in a layered structure.27 On the other hand, Li et al. proposed a phase transition–dissolution–nucleation–crystal growth mechanism. According to this proposal the m-WO3 transforms into tetragonal-WO3 (t-WO3), then this intermediate dissolves in the EDA and crystal nuclei of the WO3–EDA hybrids are formed. Continued dissolution induces further crystal growth.26
Previously our research group investigated the solid–gas phase reaction between WO3 powder, NH3 and H2O vapors, and prepared ammonium paratungstate, (NH4)10[H2W12O42]·4H2O (APT·4H2O) nanoparticles.37 These results gave the idea to use EDA vapor instead of NH3 to react with WO3 and prepare the WO3–EDA hybrid this way.
The main goal of this study was the investigation of the solid–gas phase heterogeneous reaction between tungsten oxide powder, EDA and H2O vapors with the goal of preparing the WO3–EDA hybrid. In addition, the same hybrid was prepared by a wet chemical process as-well to seek for alternative production methods. The effects of the reaction conditions on the products were examined: the composition, crystal structure and the particle size of the WO3 powder and the presence of H2O vapor in the case of the solid–gas phase reactions; while in the case of the wet chemical process different solvents were used.
The other goal was to determine the exact composition of the hybrid, and to gain more information on its crystal structure. For this, the as-prepared WO3–EDA hybrids were characterized by powder X-ray diffraction (XRD), Fourier transform infrared spectroscopy (FTIR), solid-state nuclear magnetic resonance (NMR), elemental analysis, transmission electron microscopy (TEM) and scanning electron microscopy (SEM) measurements.
Furthermore, the thermal decomposition was investigated both in air and nitrogen atmospheres by thermal analysis (TG/DTA-MS), since to the best to our knowledge the evolved gases and the decomposition intermediates of WO3–EDA were not studied in detail before. The evolved gases were further analyzed by gas chromatography-mass spectrometry (GC-MS) measurements due to their complexity.
In addition, the as-prepared WO3–EDA hybrids were used as solid catalysts in a Knoevenagel condensation model reaction. To see, whether the preparation method influenced its catalytic properties.
2. Experimental
2.1. Preparation of the WO3 starting materials
The WO3 powders were prepared from commercial ammonium paratungstate (APT·4H2O) and from hexagonal ammonium tungsten bronze (HATB, (NH4)0.33−xWO3−y). The APT was obtained from H. C. Starck GmbH, and the HATB was prepared by heating the APT at 400 °C in H2 for 6 h.38 The precursor tungsten oxides with different compositions (oxidized or partially reduced), structures (monoclinic, m-WO3 or hexagonal, h-WO3) and particle sizes (70–90 or 100–300 nm) were prepared by thermal decomposition from APT or HATB by controlling the annealing temperature and atmosphere.39 The details of preparations are summarized in Table 1.| Precursor Nr | Preparing WO3 precursor powders for the solid–gas phase reactions | Precursor powder structure | |||
|---|---|---|---|---|---|
| Starting material | Atmosphere | Temperature (°C) | Particle size (nm) | ||
| 1 | APT | Air | 600 | 100–300 | m-WO3 |
| 2 | APT | Nitrogen | 600 | 100–300 | m-WO3 partially reduced |
| 3 | HATB | Air | 600 | 70–90 | m-WO3 |
| 4 | HATB | Air | 470 | 70–90 | h-WO3 |
The tungsten oxides (1, 2) produced from APT consisted of ca. 100–300 nm particles (Fig. S1†), while the tungsten oxides (3, 4) obtained from HATB were built up by ca. 70–90 nm particles (Fig. S1c and d†).
2.2. Solid–gas phase reactions
The EDA was obtained from Sigma-Aldrich. The reactions between the tungsten oxide powder and the EDA and H2O vapors were carried out at room temperature in a sealed plastic box. WO3 powder and aqueous EDA solution were placed separately into the box, where the reagents were able to react only via the gas phase. For each reaction 500 mg WO3 powder and 20 ml saturated EDA solution were used, the total vapor pressure of the saturated solution was 1.72 kPa.40 For the investigation of the effect of the H2O vapor presence, a reaction was carried out between 20 mg WO3 powder (1) and anhydrous EDA. As a reference, a reaction was conducted between 20 mg WO3 powder (1) and 20 ml saturated EDA solution. The vapor pressure of the anhydrous EDA was 1.61 kPa.41 The yield of the reactions was calculated from the mass change of the powder during the reaction.2.3. Wet chemical process
400 mg m-WO3 (1), 10 ml EDA and 30 ml solvent were mixed in a glass flask, and then vigorously stirred. In the case of water, most of the solvent was evaporated by using a water bath (ca. 90–95 °C), and then the crystallized product which formed during the evaporation was filtered. In the case of using ethanol or acetone the WO3 powder did not dissolve. The suspension was stirred until a white precipitate formed. The precipitate was filtered directly after the reaction; thus, there was no need to evaporate the solvent. The filtered product was thoroughly washed with the used solvent to remove the excess EDA, and then dried in a lab stove for 24 h at 120 °C. The reaction without any solvent was carried out in a capped glass flask under argon atmosphere to protect the EDA from air and moisture. The temperature of the reactions, the reaction times and the used solvents are listed in Table 2. To calculate the yield of the reactions, the mass of the dried product was compared to the mass of the used WO3 powder.| Solvent | Temperature (°C) | Reaction time | Yield (%) |
|---|---|---|---|
| Water | 80 | 2 h | 94 |
| Ethanol | 80 | 1 day | 93 |
| Acetone | 60 | 3 days | 78 |
| — | 50 | 6 days | — |
2.4. Analytical methods used for the characterization
Powder XRD patterns were recorded on a PANalytical X'pert Pro MPD X-ray diffractometer using Cu Kα radiation.FTIR spectra were measured by an Excalibur Series FTS 3000 (Biorad) FTIR spectrophotometer in the range of 400–4000 cm−1 in KBr pellets.
The SEM images were recorded by a JEOL JSM-5500LV scanning electron microscope.
The TEM images were obtained by a FEI Morgagni 268D transmission electron microscope.
Solid-state magic angle spinning (MAS) spectra of samples were recorded on a Varian NMR system operating at a 1H frequency of 400 MHz with a Chemagnetics 4.0 mm narrow-bore double resonance probe. The spinning rate of the rotor was 10 kHz in all cases. For the one-dimensional 13C CP MAS (cross-polarization magic angle) 2000 transients were recorded with SPINAL-64 decoupling with strength of 83 kHz. The sample was measured with CP MAS (with 2 ms of contact time) with a recycle delay of 30 s was used, which is 5 times larger than T1H. For the combined rotation and multiple-pulse spectroscopy (CRAMPS) 1H spectra the DUMBO-1 sequence was used.42 13C and 1H spectra were deconvoluted with the DMFIT software.43 Two dimensional FSLG HETCOR 1H–13C spectra44 were recorded with contact time of 30 and 300 μs to detect only the directly bonded C–H pairs and the close connectives respectively. The FSLG scaling factor was 0.53. FSLG HETCOR spectra were recorded with 128 transients and 384 increments in the t1 dimension with 2 s of recycle delay. The temperature of all the measurements was 25 °C. Adamantane was used as external chemical shift reference (38.55 and 29.50 ppm on the 13C and 1.8 ppm on the 1H scale). The 90° pulse lengths were 3 μs for both the proton and the carbon channels for all the NMR experiments.
Elemental analysis was performed to determine the elemental composition of the as-prepared WO3–EDA hybrids with a Vario EL III elemental analyser.
TG/DTA measurements were performed on an STD 2960 Simultaneous DTA/TGA (TA Instruments Inc.) thermal analyzer using a heating rate of 10 °C min−1 and Pt crucibles. The furnace was purged either with air or nitrogen (130 ml min−1). Evolved gas analytical (EGA) curves were recorded by a Thermostar GSD 300 (Balzers Instruments) quadrupole mass spectrometer (MS). A mass range between m/z = 1–64 was monitored through 64 channels in Multiple Ion Detection Mode (MID) with a measuring time of 0.5 s per channel. Further details of the TG/DTA-MS setup are described in other studies.45,46
For further analysis and confirmation of the evolved gases the WO3–EDA samples were decomposed in a TGA 2050 (TA Instruments Inc.) Thermogravimetric Analyzer using a heating rate of 10 °C min−1 and Pt crucibles. The furnace was purged either with air or nitrogen atmospheres (80 ml min−1). The evolved gases were trapped in a mixture of n-hexane/methanol (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3), and then they were analyzed with a Shimadzu GC-MS-QP2010 gas chromatograph-mass spectrometer with a capillary column (Rxi-1MS, 30 m × 0.32 mm × 1.0 μm).
:3), and then they were analyzed with a Shimadzu GC-MS-QP2010 gas chromatograph-mass spectrometer with a capillary column (Rxi-1MS, 30 m × 0.32 mm × 1.0 μm).
The products of the Knoevenagel condensation model reaction were analyzed with the same gas chromatograph-mass spectrometer with a different capillary column (Rxi-5MS, 30 m × 0.32 mm × 1.0 μm).
2.5. Catalysis of the Knoevenagel condensation reaction
The catalytic effects of the as-prepared WO3–EDA hybrids produced from both the solid–gas phase reaction and the wet chemical process were tested in a Knoevenagel condensation model reaction. Benzaldehyde (0.5 mmol), ethyl α-cyanoacetate (0.5 mmol), toluene (4 ml) and WO3–EDA nanoplates/nanowires or m-WO3 (1) were added to a capped glass vessel. The mixture was vigorously stirred for 24 h at room temperature.26 The WO3–EDA catalyst was filtered, washed with ethanol, dried in a lab stove at 120 °C overnight and reused in the next reaction.3. Results and discussion
3.1. Synthesis of the WO3–EDA hybrid
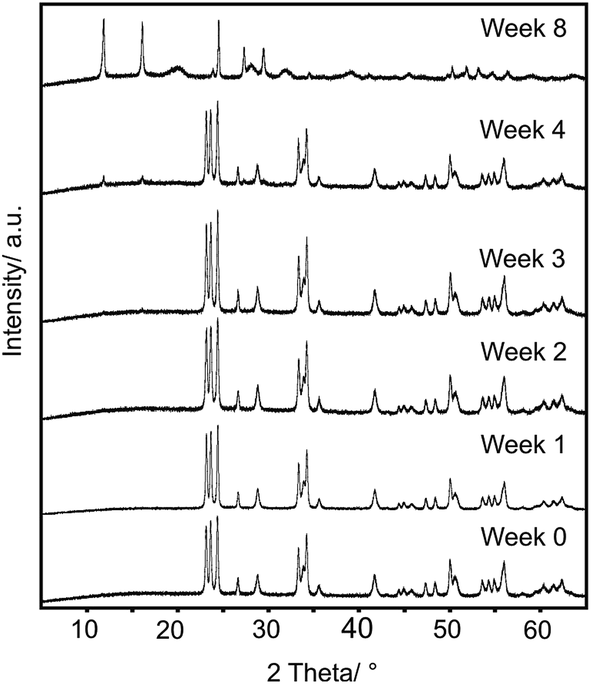 | ||
| Fig. 1 XRD patterns of intermediate samples obtained from precursor 1 at different reaction times. | ||
 | ||
| Fig. 2 FTIR spectra of intermediate samples obtained from precursor 1 at different reaction times. | ||
According to Li et al. the solvothermal reaction between m-WO3 and EDA takes place through a tetragonal-WO3 intermediate, which was confirmed by in situ synchrotron-radiation XRD (SR-XRD) measurements. The solvothermal reaction between APT·4H2O and EDA also have an unknown intermediate phase, which is probably a kind of tungstate-EDA complex.26 In our study, neither the XRD patterns, nor the FTIR spectra did not show any intermediates; the WO3–EDA hybrid formed directly from the starting materials.
The composition, crystal structure and the particle size of the WO3 powder had no effects on the product. According to the XRD patterns (Fig. 1 and S2†) there was no significant difference in the position of the peaks, only their relative intensities differed slightly. This indicated that the reaction did not depend on the properties of the WO3 powder.
The amount of the WO3 powder (1) was decreased from 500 mg to 20 mg for the reactions where the influence of H2O vapor was examined. In the presence of H2O vapor the most intense peak of the product (11.88°) appeared after 1 day. The WO3 powder (1) transformed completely into the hybrid after 2 weeks (Fig. S3a†). However, in the absence of H2O vapor the structure started to change after 4 weeks as the peak of the product appeared at 11.88° in the XRD patterns (Fig. S3b†). These results showed that the reaction time was decreased by decreasing the amount of the WO3 powder. Furthermore, the H2O vapor may have some catalytic role in the reaction.
The absorbed H2O was visible in the FTIR spectra even after one day (Fig. 2). Furthermore, the TEM measurements (see in Chapter 3.2.) revealed that on the surface of the particles a thin layer was present, presumably H2O and excess EDA. Thermal analytical measurements (see in Chapter 3.4.) showed that the mass of the WO3–EDA hybrid slowly decreased until ca. 300 °C. The XRD patterns of the decomposition intermediates proved that until ca. 300 °C only adsorbed H2O and excess EDA evolved, as the structure remained intact. Based on the mass loss values and the DTA peaks most of the film evaporated below 100 °C. However, some of the film was still present above 100 °C, indicating a stronger bond to the WO3–EDA particles, which would explain why the layer was visible during the TEM measurements.
The reaction did not depend on the properties of the WO3 powder, as there was no significant difference in the position position of the peaks, only their relative intensities differed slightly in the XRD patterns of the products (Fig. 1 and S2†). Furthermore, the morphology of the hybrid resembled the morphology of the precursor WO3 (Fig. S1†) indicating that the reaction may have occurred through the surface of the WO3 particles. This suggested that EDA had a crucial role in the reaction. All of these results supported our proposed formation mechanism.
 | ||
| Fig. 3 XRD patterns of the WO3–EDA hybrids obtained from different methods and solvents. | ||
Similar to the solvothermal reactions26,27 a reaction was carried out in EDA without any solvent. After 6 days the WO3 powder was still not dissolved. Based on the XRD pattern (Fig. 3) the structure of the m-WO3 (PDF 43-1035) was a little distorted but remained intact, the peaks of the product were not detected. The results indicated that for the reaction at atmospheric pressure and between 60 and 80 °C a solvent was necessary, in contrast to the solvothermal reactions. During the solvothermal reactions probably the higher pressure and temperature (140–180 °C) accelerate the intercalation of the EDA, while at milder conditions the solvent helps the intercalation.
3.2. Morphology of the as-prepared WO3–EDA hybrids
The WO3–EDA hybrid obtained from the solid–gas phase reactions (Fig. 4a and b) consisted of ca. 100–200 nm particles, which aggregated into 100–200 μm blocks. According to the TEM images, which gave more detail (Fig. 4c and d), the product consisted of 120–150 nm long and 50 nm wide plates. This result is remarkable, because previously only WO3–EDA hybrid nanowires and nanobelts were reported.23,26,27 The morphology of the as-obtained hybrid resembled the morphology of the precursor WO3 (Fig. S1†) indicating that the reaction may occurred through the surface of the WO3 particles. Furthermore, on the surface of the nanoplates a thin film could be seen (Fig. 4c), presumably a mixture of water and excess EDA. | ||
| Fig. 4 SEM (A, B) and TEM (C, D) images of the WO3–EDA hybrid obtained from the solid–gas phase reaction. | ||
The WO3–EDA hybrid crystallized from water (Fig. 5a and b) consisted of wires, which aggregated into 10–200 μm blocks. According to the TEM images (Fig. 5c and d) the nanowires were 20–40 nm wide and 500–1000 nm long. The morphology of the hybrid was the same, when ethanol or acetone was used as solvent (Fig. S4†).
 | ||
| Fig. 5 SEM (A, B) and TEM (C, D) images of the WO3–EDA hybrid crystallized from water. | ||
3.3. Composition and structure of the WO3–EDA hybrid
The as-prepared WO3–EDA hybrid was thoroughly investigated by FTIR, solid-state NMR and elemental analysis measurements to gain more information about its composition and structure. The solid-state NMR and elemental analysis measurements were carried out by using the WO3–EDA hybrid obtained from the wet chemical process using ethanol solvent, due to the large sample quantity requirements.In the FTIR spectrum (Fig. 2) all peaks could be assigned either to the EDA, or to the inorganic WO3 framework.23,27,36,47 The N–H deformation and stretching vibrations of the –NH2 group were visible at 1612 cm−1 and 3120–3310 cm−1 (3130, 3207 and 3238 cm−1). The peaks in the region of 2840–2970 cm−1 (2895, 2963 cm−1) were related to the C–H stretching vibrations. The –CH2– deformation vibrations appeared at 1280–1520 cm−1 and at 860 cm−1, while the bands at 1061 and 450 cm−1 were assigned to the C–N stretching and bending vibrations. The bending vibration of the N–C–C–N skeleton could be seen at 537 cm−1. The peaks at 885 and 950 cm−1 were assigned to the W![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O vibrations, while the bands at 580–900 cm−1 (585, 724, 817, 841 and 860 cm−1) were explained by the W–O vibrations. The absence of a broad peak at 2100 cm−1 (–NH3+ vibration) revealed that the EDA was incorporated into the structure only in its neutral form. The absence of the O–H vibrations showed that there was no water in the structure.
O vibrations, while the bands at 580–900 cm−1 (585, 724, 817, 841 and 860 cm−1) were explained by the W–O vibrations. The absence of a broad peak at 2100 cm−1 (–NH3+ vibration) revealed that the EDA was incorporated into the structure only in its neutral form. The absence of the O–H vibrations showed that there was no water in the structure.
One- and two-dimensional solid-state NMR measurements were carried out to investigate the structure of the WO3–EDA hybrid (Fig. 6). All the 1H and 13C signals could be assigned to the EDA. 13C CPMAS spectrum was composed of two signals with equal intensity, suggesting two –CH2– positions with the same population. These two signals could be originated either from two separate EDA molecules which have different positions in the crystal structure or from the same molecule in which the two –CH2– groups have different positions. Deconvolution of the 1H CRAMPS spectrum resulted three signals with chemical shifts of 4.9, 4.2 and 3.3 ppm, with integral values of 4:2:2 respectively. Two-dimensional 1H–13C HETCOR NMR spectra were recorded with short contact time (30 μs) to assign the directly bonded C–H pairs and with longer contact time (300 μs) to identify the longer C–H connectivity. Both the carbon signals show cross peaks with their two hydrogen atoms at shorter contact time suggesting that the hydrogen atoms are not equivalent in a –CH2– group. 1H signal with chemical shift of 4.9 ppm gave cross peak only with longer contact time, so this signal could be attributed to the NH2 groups. The CRAMPS spectra can be integrated only with restrictions, but in this case the presence of the water molecules in the crystal structure could be excluded.
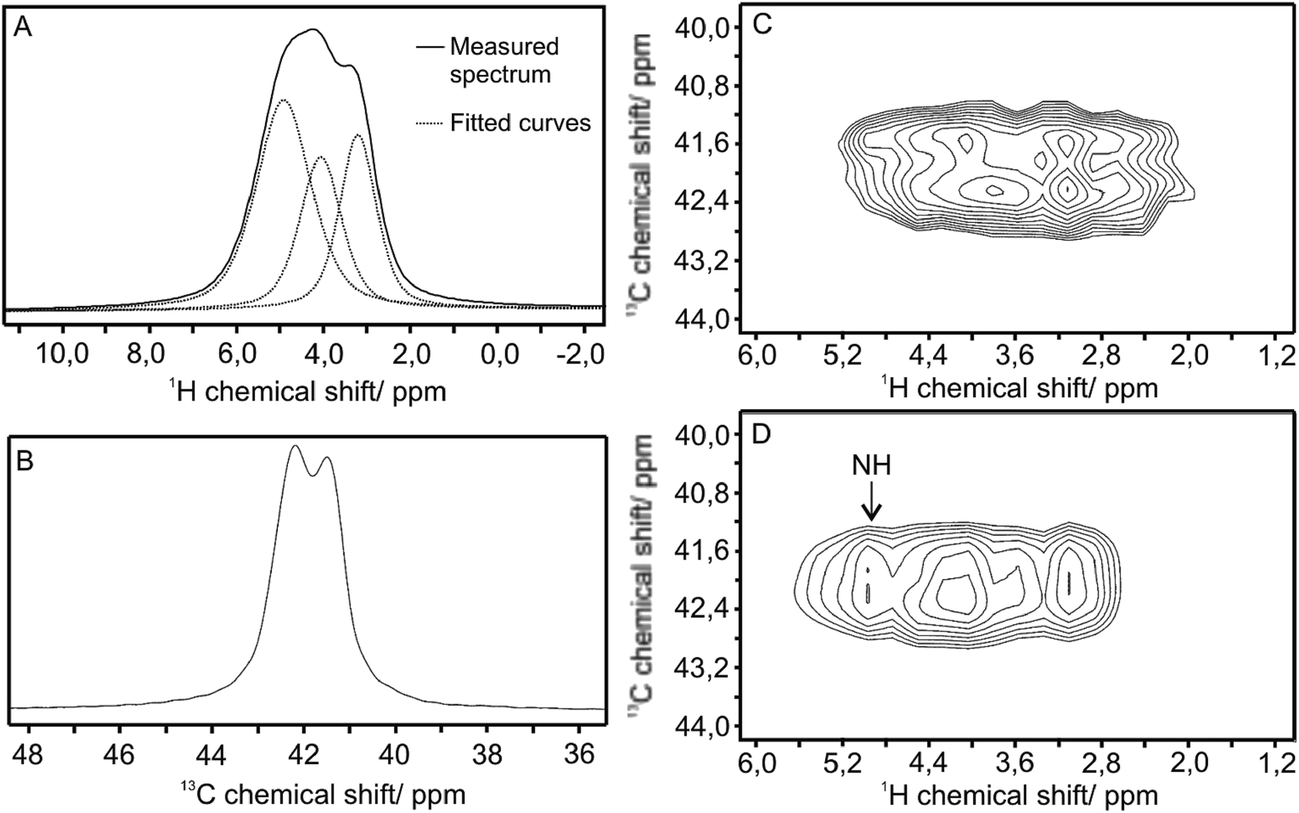 | ||
| Fig. 6 Solid-state NMR spectra of the WO3–EDA hybrid: 1H CRAMPS (A). 13C CP MAS (B). 1H–13C HETCOR with 30 μs (C) and 300 μs contact time (D). | ||
The results of the elemental analysis showed that the as-prepared WO3–EDA hybrid consisted of 7.39% C and 9.37% N. These results and the 62.64% W content, calculated from the TG results in air, indicated that the C:N:W ratio is 1.96:1.81:1, thus the EDA:W ratio is 1:1. According to previous X-ray photoelectron spectroscopy (XPS) results26,27 the W is mainly in the VI oxidation state (W6+), hence we propose the WO3–EDA empiric formula instead of the previously used WOx–EDA formula.
3.4. Thermal decomposition of the WO3–EDA hybrid
To the best to our knowledge, previous thermal analytical measurements were used only to determine the organic content of the WO3–EDA hybrids,23,26,27 while the evolved gases and the decomposition intermediates were not characterized in detail before. Due to the large sample quantity requirements of these measurements, the WO3–EDA hybrid obtained from the wet chemical process (using ethanol solvent) was used for the study of the thermal decomposition and for preparing the decomposition intermediates. In addition, the TG and DTA curves of the WO3–EDA hybrids obtained from the different methods was compared. The TG and DTA curves of the WO3–EDA hybrid obtained from the solid–gas phase reactions are presented in Fig. S5 and S6.† | ||
| Fig. 7 TG/DTA and evolved gas analytical MS ion current curves of the WO3–EDA hybrid in nitrogen. | ||
 | ||
| Fig. 8 XRD patterns of the thermal decomposition products of the WO3–EDA in nitrogen (A) and in air (B). | ||
 | ||
| Fig. 9 FTIR spectra of the thermal decomposition products of the WO3–EDA in nitrogen (A) and in air (B). | ||
The structure of the hybrid remained intact until 300 °C, then in the second decomposition step (300–360 °C) the structure collapsed, resulting in an amorphous phase (Fig. 8). This decomposition was accompanied by an endothermic peak at 333 °C. Based on the final mass loss (Fig. 7) and the FTIR spectrum (Fig. 9), this amorphous phase was tungsten oxide.
During the second decomposition step several evolved gases could be identified. Some of the organic fragments ignited with the traces of oxygen, hence a small amount of H2O and CO2 were detected. From the –NH2 groups a small amount of NH3 evolved. Instead of evaporation and decomposition the EDA transformed into a series of heterocyclic aromatic compounds. Due to the complexity of these compounds and the overlap between their masses, GC-MS measurements were carried out to identify these gases, and to determine their relative quantity (Fig. S7†). The results of these measurements and the assignation of the ion masses are in Table 3 for both nitrogen and air atmospheres. Based on the results, in nitrogen the EDA transformed mostly to 3-methylpyridine, 2-ethylpyrazine and 2-methylpyridine. 2,3-Diethylpyrazine, pyrazine, 2-methylpyrazine, 2,3-dimethylpyrazine, pyrrole and pyridine were detected as-well in smaller quantities.
| Name | Ion mass | Area% | |
|---|---|---|---|
| Nitrogen | Air | ||
| Pyrrole | 67+ | 3.7 | 3.2 |
| Pyridine | 79+ | 2.9 | 6.3 |
| Pyrazine | 80+ | 7.9 | 45.6 |
| 2-Methylpyridine | 93+ | 12.8 | 5.9 |
| 3-Methylpyridine | 93+ | 29.2 | 4.5 |
| 2-Methylpyrazine | 94+ | 5.6 | 15.2 |
| 2-Ethylpyrazine | 108+ | 23.1 | 15.3 |
| 2.3-Dimethylpyrazine | 108+ | 5.1 | 4.0 |
| 2.3-Diethylpyrazine | 136+ | 9.0 | — |
The thermal decomposition sequence of the WO3–EDA hybrid obtained from the solid–gas phase reaction was essentially the same in nitrogen (Fig. S5†). The mass losses were slightly greater (3.8 and 21.8% instead of 1.2 and 19.6%), as water and excess EDA were adsorbed on the surface of the particles (Fig. 4c).
 | ||
| Fig. 10 TG/DTA and evolved gas analytical MS ion current curves of the WO3–EDA hybrid in air. | ||
In the second decomposition step (280–330 °C) an amorphous phase was obtained (Fig. 8). According to the FTIR spectrum (Fig. 9) this intermediate still had some organic content (broad peaks at 1600 and 3100–3300 cm−1). The W–O bonds (below 1000 cm−1) changed significantly compared to the WO3–EDA hybrid. Some of the evolved gases ignited, resulting in H2O and CO2. This burning generated enough heat to overturn the endothermic DTA peak at 284 °C to an exothermic peak (312 °C). NH3 evolution was detected as-well. Although, while in the case of heating ammonium tungstates and ammonium thiotungstates in air at this temperature the evolved NH3 burnt into nitrous oxides;48–50 in this case no such products were detected.
Most of the EDA transformed into a series of heterocyclic aromatic compounds in air as-well (Fig. S8†). In contrast to the results in nitrogen, the main product was pyrazine in air. The quantities of 2-methylpyrazine and pyridine were larger than in nitrogen, while the amounts of 2-ethylpyrazine, 2-methylpyridine and 3-methylpyridine were smaller. The quantity of pyrrole and 2,3-dimethylpyrazine were ca. the same as in nitrogen, and 2,3-diethylpyrazine was not detected in air. Based on these results in nitrogen mainly pyridine derivates, and in air mostly pyrazine derivates were the main products, while 2-ethylpyrazine was present in great quantities in both atmospheres (Table 3).
In the third decomposition step (340–440 °C) from the amorphous phase m-WO3 started to crystallize (Fig. 8). The FTIR spectrum still showed some organic content (broad peak at 1600 cm−1), while the peaks of m-WO3 appeared in the region below 1000 cm−1. In this decomposition step only CO2 was detected, i.e. some of the remained organic content combusted. This combustion and the crystallization of the m-WO3 were both exothermic processes, which explained the exothermic peak at 398 °C in the DTA curve.
In the fourth decomposition step (470–520 °C) the m-WO3 crystallization continued (Fig. 8), and the final decomposition product was identified as m-WO3 (PDF 43-1035). Similar to the third decomposition step only the evolution of CO2 was detected; in this step all of the remaining organic content combusted. In the FTIR spectrum (Fig. 9) the broad peak at 1600 cm−1 disappeared completely, confirming that there was no organic content present after this decomposition step. The combustion and the crystallization of the m-WO3 were accompanied by an exothermic peak at 499 °C. The observed mass loss (20.0%) corresponded to the theoretical mass loss (20.6%) confirming the WO3–EDA formula.
In the case of the WO3–EDA hybrid obtained from the solid–gas phase reaction (Fig. S6†) the mass loss in the first decomposition step was slightly greater (2.8% instead of 1.0%), due to the absorbed water and excess EDA. The mass loss in the second decomposition step was the same (9.3% in both cases). The combustion of the remaining organic content took place at a higher temperature, resulting in an additional exothermic peak at 564 °C. Because of this in the third and fourth decomposition steps the mass losses were somewhat different (5.4 and 5.2% instead of 8.9 and 3.8%).
As reference, without the inorganic frame the EDA completely evaporated until 150 °C in both atmospheres (Fig. S9†). Based on the GC-MS results only EDA evolved, none of the heterocyclic aromatic compounds was detected. Previously in the case of several Co–EDA and Ni–EDA complexes the thermal decomposition took place between 200 and 300 °C, and in this temperature range EDA and its fragments were detected by MS measurements.51–53 It is known that at higher temperatures amines can recombine into heterocyclic compounds.54 In our case the cause of the recombination of the EDA was probably the higher stability of the WO3–EDA (the decomposition started above 300 °C), and not the presence of W atoms and WO3.
3.5. Catalytic effects of the WO3–EDA hybrid
It was previously described that the WO3–EDA hybrid could act as a solid base catalyst in a Knoevenagel condensation reaction, due to its amino groups.26 The conversion of the model reactions were ca. 90% both in the cases of the WO3–EDA hybrid26 and the SBA-15, and MCM-48.55–57 However, the WO3–EDA hybrid has a higher stability, than the amino-modified silica gel catalysts, making it a promising catalyst.In our study the same model reaction was carried out to see, how the preparation method influenced the hybrids catalytic properties. The as-prepared hybrids had the same efficiency as the hybrid obtained from the solvothermal reaction, and the modified silica gels. The conversion was 92.0% when WO3–EDA hybrid nanoplates obtained from the solid–gas phase reaction were used. In the case of WO3–EDA hybrid nanowires prepared by the wet chemical process the conversion was 98.7%. For reference m-WO3 (1) was used, in this case the conversion was only 1.0%. Furthermore, the recycling of the catalyst was studied (Table 4.) as-well. In each cycle the conversion slightly decreased; however, even in the third cycle the conversion was above 90%. This decrease was caused probably by to the losses during the filtration.
| Cycle Nr | Conversion (%) |
|---|---|
| 1 | 98.7 |
| 2 | 93.4 |
| 3 | 91.1 |
4. Conclusions
Previously the WO3–EDA hybrid material was obtained only from solvothermal reactions. In this study this hybrid was prepared by two novel methods at milder reaction conditions: a solid–gas phase heterogeneous reaction (room temperature, atmospheric pressure), and a wet chemical process (60–80 °C, atmospheric pressure). The effects of the reaction conditions on the products were examined in detail: the composition (oxidized or partially reduced), crystal structure (monoclinic or hexagonal), particle size (100–300 nm or 70–90 nm) of the WO3 powder during the solid–gas phase reactions; while the effect of the solvent was investigated in the case of the wet chemical process.The composition, crystal structure and the particle size of the tungsten oxide had no significant effects on the product, in each solid–gas phase reaction the same product was obtained. Unlike in the case of the solvothermal reactions, no intermediates were detected during the reactions. The WO3 precursors completely transformed to the hybrid after 8 weeks. Based on our results we proposed a novel forming mechanism for the solid–gas phase reactions.
In the case of the wet chemical process water, ethanol and acetone were used as solvent. Using ethanol solvent was the most beneficial, as the most ordered structure with a high yield was obtained by using this solvent.
The two methods yielded the same products, the only difference was in their morphology. From the solid–gas phase reaction WO3–EDA hybrid nanoplates were obtained for the first time, as previously only nanowires and nanobelts were reported. The wet chemical process yielded nanowires, similar to the previous solvothermal reactions.
The as-prepared WO3–EDA hybrid were thoroughly examined by solid-state NMR and elemental analysis measurements to determine its composition, and to gain more information on its crystal structure. Two different positions of –CH2– groups could be identified in the crystal structure by NMR measurements; also within the –CH2– group the two hydrogen atoms were not equivalent. Based on the elemental analysis and the TG results in air, the WO3:EDA ratio was 1:1, thus the WO3–EDA empiric formula was suggested.
The evolved gases and the decomposition intermediates of the WO3–EDA hybrid were characterized in detail for the first time, as previous thermal analytical measurements were used only to determine the organic content of the hybrid. In nitrogen atmosphere the decomposition product of WO3–EDA was an amorphous tungsten oxide phase, while in air m-WO3 was obtained. According to the GC-MS measurements the EDA transformed into a series of heterocyclic aromatic compounds in both atmospheres.
In addition, catalytic properties of the hybrid was tested in a Knoevenagel model reaction. The as-prepared hybrids had the same efficiency as the hybrids obtained from the solvothermal reaction, and the modified silica gels.
Conflicts of interest
There are no conflicts to declare.References
- W. Xie and T. Wang, Biodiesel production from soybean oil transesterification using tin oxide-supported WO3 catalysts, Fuel Process. Technol., 2013, 109, 150–155 CrossRef CAS.
- W. Xie and D. Yang, Transesterification of soybean oil over WO3 supported on AlPO4 as a solid acid catalyst, Bioresour. Technol., 2012, 119, 60–65 CrossRef CAS PubMed.
- W. Xie and P. Hu, Production of Structured Lipids Containing Medium-Chain Fatty Acids by Soybean Oil Acidolysis Using SBA-15-pr-NH2–HPW Catalyst in a Heterogeneous Manner, Org. Process Res. Dev., 2016, 20, 637–645 CrossRef CAS.
- I. M. Szilágyi, B. Fórizs, O. Rosseler, Á. Szegedi, P. Németh, P. Király, G. Tárkányi, B. Vajna, K. Varga-Josepovits, K. László, A. L. Tóth, P. Baranyai and M. Leskelä, WO3 photocatalysts: influence of structure and composition, J. Catal., 2012, 294, 119–127 CrossRef.
- C. T. Lin and T. H. Tsai, Solution volume effect of photodegradation by 1-D WO3 nanorods via microwave-assisted solvothermal heating under the UV irradiation, Asian J. Chem., 2013, 25, 7098–7102 CAS.
- A. B. D. Nandiyanto, O. Arutanti, T. Ogi, F. Iskandar, T. O. Kim and K. Okuyama, Synthesis of spherical macroporous WO3 particles and their high photocatalytic performance, Chem. Eng. Sci., 2013, 101, 523–532 CrossRef CAS.
- B. X. Liu, J. S. Wang, H. Y. Li, J. S. Wu, M. L. Zhou and T. Y. Zuo, Facile synthesis of hierarchical hollow mesoporous Ag/WO3 spheres with high photocatalytic performance, J. Nanosci. Nanotechnol., 2013, 13, 4117–4122 CrossRef CAS PubMed.
- E. Karacsonyi, L. Baia, A. Dombi, V. Danciu, K. Mogyorosi, L. C. Pop, G. Kovacs, V. Cosoveanu, A. Vulpoi, S. Simon and Z. Pap, The photocatalytic activity of TiO2/WO3/noble metal (Au or Pt) nanoarchitectures obtained by selective photodeposition, Catal. Today, 2013, 208, 19–27 CrossRef CAS.
- I. M. Szilágyi, S. Saukko, J. Mizsei, A. L. Tóth, J. Madarász and G. Pokol, Gas sensing selectivity of hexagonal and monoclinic WO3 to H2S, Solid State Sci., 2010, 12, 1857–1860 CrossRef.
- I. M. Szilágyi, L. Wang, P. I. Gouma, C. Balázsi, J. Madarász and G. Pokol, Preparation of hexagonal WO3 from hexagonal ammonium tungsten bronze for sensing NH3, Mater. Res. Bull., 2009, 44, 505–508 CrossRef.
- C. Balázsi, L. Wang, E. O. Zayim, I. M. Szilágyi, K. Sedlackova, J. Pfeifer, A. L. Tóth and P. I. Gouma, Nanosize hexagonal tungsten oxide for gas sensing applications, J. Eur. Ceram. Soc., 2008, 28, 913–917 CrossRef.
- I. M. Szilágyi, S. Saukko, J. Mizsei, P. Király, G. Tárkányi, A. L. Tóth, A. Szabó, K. Varga-Josepovits, J. Madarász and G. Pokol, Controlling the composition of nanosize hexagonal WO3 for gas sensing, Mater. Sci. Forum, 2008, 589, 161–165 CrossRef.
- L. Wang, J. Pfeifer, C. Balázsi, I. M. Szilágyi and P. I. Gouma, Nanostructured hexagonal tungsten oxides for ammonia sensing, Proceedings of SPIE - The International Society for Optical Engineering Nanosensing: Materials, Devices, and Systems III, 2007, vol. 6769, p. 67690E Search PubMed.
- C. Balázsi, K. Sedlackova, J. Pfeifer, A. L. Tóth, E. A. Zayim, I. M. Szilágyi, L. S. Wang, K. Kalyanasundaram and P. I. Gouma, Synthesis and examination of hexagonal Tungsten oxide nanocrystals for electrochromic and sensing applications, NATO Science for Peace and Security Series C: Environmental Security; Sensors for Environment, Health and Security, 2009, pp. 77–91 Search PubMed.
- J. Kukkola, M. Mohl, A. R. Leino, J. Maklin, N. Halonen, A. Shchukarev, Z. Konya, H. Jantunen and K. Kordas, Room temperature hydrogen sensors based on metal decorated WO3 nanowires, Sens. Actuators, B, 2013, 186, 90–95 CrossRef CAS.
- Y. D. Zhang, W. W. He, H. X. Zhao and P. J. Li, Template-free to fabricate highly sensitive and selective acetone gas sensor based on WO3 microspheres, Vacuum, 2013, 95, 30–34 CrossRef CAS.
- A. Subrahmanyam and A. Karuppasamy, Optical and electrochromic properties of oxygen sputtered tungsten oxide (WO3) thin films, Sol. Energy Mater. Sol. Cells, 2007, 91, 266–274 CrossRef CAS.
- S. M. Durrani, E. Khawaja, A. Al-Shukri and M. Al-Kuhaili, Dielectric/Ag/dielectric coated energy-efficient glass windows for warm climates, Energ. Build, 2004, 36, 891–898 CrossRef.
- G. Leftheriotis, E. Koubli and P. Yianoulis, Combined electrochromic-transparent conducting coatings consisting of noble metal, dielectric and WO3 multilayers, Sol. Energy Mater. Sol. Cells, 2013, 116, 110–119 CrossRef CAS.
- E. Koubli, S. Tsakanikas, G. Leftheriotis, G. Syrrokostas and P. Yianoulis, Optical properties and stability of near-optimum WO3/Ag/WO3 multilayers for electrochromic applications, Solid State Ionics, 2015, 272, 30–38 CrossRef CAS.
- J. T. Szymanski and A. C. Roberts, The crystal structure of tungstite, WO3·H2O, Can. Mineral., 1984, 22, 681–688 CAS.
- M. F. Daniel, B. Desbat, J. C. Lassegues, B. Gerand and M. Figlarz, Infrared and Raman study of WO3 tungsten trioxides and WO3, xH2O tungsten trioxide hydrates, J. Solid State Chem., 1987, 67, 235–247 CrossRef CAS.
- S. V. Chong, B. Ingham and J. L. Tallon, Novel materials based on organic-tungsten oxide hybrid systems I: synthesis and characterization, Curr. Appl. Phys., 2004, 4, 197–201 CrossRef.
- B. Yan, Y. Xu, N. K. Goh and L. S. Chia, Hydrothermal synthesis and crystal structures of two novel hybrid open-frameworks and a two-dimensional network based on tungsten(VI) oxides, Chem. Commun., 2000, 21, 2169–2170 RSC.
- J. W. Johnson, A. J. Jacobsen, S. M. Rich and J. F. Brody, New Layered Compounds with Transition-Metal Oxide Layers Separated by Covalently Bound Organic Ligands. Molybdenum and Tungsten Trioxide-Pyridine, J. Am. Chem. Soc., 1981, 103, 5246–5247 CrossRef CAS.
- W. Li, F. Xia, J. Qu, P. Li, D. Chen, Z. Chen, Y. Yu, Y. Lu and R. A. Caruso, Versatile inorganic-organic hybrid WOx-ethylenediamine nanowires: synthesis, mechanism and application in heavy metal ion adsorption and catalysis, Nano Res., 2014, 7, 903–916 CrossRef CAS.
- X. Hu, Q. Ji, J. P. Hill and K. Ariga, Large-scale synthesis of WOx-EDA nanobelts and their application as photoswitches, CrystEngComm, 2011, 13, 2237–2241 RSC.
- D. Chen and Y. Sugahara, Tungstate-Based Inorganic-Organic Hybrid Nanobelts/Nanotubes with Lamellar Mesostructures: Synthesis, Characterization, and Formation Mechanism, Chem. Mater., 2007, 19, 1808–1815 CrossRef CAS.
- D. Chen, T. Li, L. Yin, X. Hou, X. Yu, Y. Zhang, B. Fan, H. Wang, X. Li, R. Zhang, T. Hou, H. Lu, H. Xu, J. Sun and L. Gao, A comparative study on reactions of n-alkylamines with tungstic acids with various W-O octahedral layers: Novel evidence for the “dissolution-reorganization” mechanism, Mater. Chem. Phys., 2011, 125, 838–845 CrossRef CAS.
- M. R. Gao, W. T. Yao, H. B. Yao and S. H. Yu, Synthesis of unique ultrathin lamellar mesostructured CoSe2-amine (protonated) nanobelts in a binary solution, J. Am. Chem. Soc., 2009, 131, 7468–7487 Search PubMed.
- H. B. Yao, M. R. Gao and S. H. Yu, Small organic molecule templating synthesis of organic-inorganic hybrid materials: their nanostructures and properties, Nanoscale, 2010, 2, 322–334 Search PubMed.
- L. Yu, R. Z. Zou, Z. Y. Zhang, G. S. Song, Z. G. Chen, J. M. Yang and J. Q. Yu, A Zn2GeO4-ethylenediamine hybrid nanoribbon membrane as a recyclable adsorbent for the highly efficient removal of heavy metals from contaminated water, Chem. Commun., 2011, 47, 10719–10721 RSC.
- J. Lehmann, Spin qubits with electrically gated polyoxometalate molecules, Nat. Nanotechnol., 2007, 2, 312–317 CrossRef CAS PubMed.
- Y. Zhang, G. M. Dalpian, B. Fluegel, S. H. Wei, A. Mascarenhas, X. Y. Huang, J. Li and L. W. Wang, Novel approach to tuning the physical properties of organic-inorganic hybrid semiconductors, Phys. Rev. Lett., 2006, 96, 026405 CrossRef PubMed.
- J. T. Rhule, Polyoxometalates in Medicine, Chem. Rev., 1998, 98, 327–358 CrossRef CAS PubMed.
- B. Ingham, S. V. Chong and J. L. Tallon, Layered Tungsten Oxide-Based Organic-Inorganic Hybrid Materials: An Infrared and Raman Study, J. Phys. Chem. B, 2005, 109, 4936–4940 CrossRef CAS PubMed.
- D. Hunyadi, I. M. Szilágyi, A. L. Tóth, E. Drotár, T. Igricz and G. Pokol, Investigating the solid-gas phase reaction between WO3 powder, NH3 and H2O vapors to prepare ammonium paratungstate, Inorg. Chim. Acta, 2016, 444, 29–35 CrossRef CAS.
- I. M. Szilágyi, F. Hange, J. Madarász and G. Pokol, In situ HT-XRD Study on the Formation of Hexagonal Ammonium Tungsten Bronze by Partial Reduction of Ammonium Paratungstate Tetrahydrate, Eur. J. Inorg. Chem., 2006, 17, 3413–3418 CrossRef.
- I. M. Szilágyi, J. Madarász, G. Pokol, P. Király, G. Tárkányi, S. Saukko, J. Mizsei, A. L. Tóth, A. Szabó and K. Varga-Josepovits, Stability and Controlled Composition of Hexagonal WO3, Chem. Mater., 2008, 20, 4116–4125 CrossRef.
- N. Chiali-Baba Ahmed, L. Negadi, I. Mokbel and J. Jose, Phase equilibrium properties of binary aqueous solutions containing ethanediamine, 1,2-diaminopropane, 1,3-diaminopropane, or 1,4-diaminobutane at several temperatures, J. Chem. Thermodyn., 2011, 43, 719–724 CrossRef.
- T. Boublik, V. Fried and E. Hala, The Vapour Pressures of Pure Substances, Elsevier, 2nd edn, Amsterdam, 1984 Search PubMed.
- A. Lesage, D. Sakellariou, S. Hediger, B. Eléna, P. Charmont, S. Steuernagel and L. Emsley, Experimental aspects of proton NMR spectroscopy in solids using phase-modulated homonuclear dipolar decoupling, J. Magn. Reson., 2003, 163, 105–113 CrossRef CAS PubMed.
- D. Massiot, F. Fayon, M. Capron, I. King, S. Le Calve, B. Álonso, J. O. Durand, B. Bujoli, Z. H. Gan and G. Hoatson, Modelling one- and two-dimensional solid-state NMR spectra, Magn. Reson. Chem., 2002, 40, 70–76 CrossRef CAS.
- B. J. van Rossum, H. Förster and H. J. M. de Groot, High-Field and High-Speed CP-MAS13C NMR Heteronuclear Dipolar-Correlation Spectroscopy of Solids with Frequency-Switched Lee-Goldberg Homonuclear Decoupling, J. Magn. Reson., 1997, 124, 516–519 CrossRef CAS.
- R. L. Prasad, A. Kushwaha, I. M. Szilágyi and L. Kótai, Solid-state thermal degradation behaviour of 1-D coordination polymers of Ni(II) and Cu(II) bridged by conjugated ligand, J. Therm. Anal. Calorim., 2013, 114, 653–664 CrossRef CAS.
- I. M. Szilágyi and A. Deák, Structural and thermal study of asymmetric α-dioxime complexes of Co(III) with Cl and methyl-pyridines, Polyhedron, 2010, 10, 2185–2189 CrossRef.
- A. Sabatini and S. Califano, Infra-red spectra in polarized light of ethylenediamine and ethylenediamine-d4, Spectrochim. Acta, 1960, 16, 677–688 CrossRef CAS.
- D. Hunyadi, I. Sajó and I. M. Szilágyi, Structure and thermal decomposition of ammonium metatungstate, J. Therm. Anal. Calorim., 2014, 116, 329–337 CrossRef CAS.
- J. Madarász, I. M. Szilágyi, F. Hange and G. Pokol, Comparative evolved gas analyses (TG-FTIR, TG/DTA-MS) and solid state (FTIR, XRD) studies on thermal decomposition of ammonium paratungstate tetrahydrate (APT) in air, J. Anal. Appl. Pyrolysis, 2004, 72, 197–201 CrossRef.
- D. Hunyadi, A. L. Vieira Machado Ramos and I. M. Szilágyi, Thermal decomposition of ammonium tetrathiotungstate, J. Therm. Anal. Calorim., 2015, 120, 209–215 CrossRef CAS.
- S. Dash, P. K. Ajikumar, M. Kamruddin and A. K. Tyagi, Temperature programmed decomposition of cobalt ethylene diamine complexes, Thermochim. Acta, 1999, 334, 141–148 CrossRef CAS.
- K. S. Rejitha and S. Mathew, Thermoanalytical investigations of tris(ethylenediamine)nickel(II) oxalate and sulphate complexes TG-MS and TR-XRD studies, J. Therm. Anal. Calorim., 2010, 102, 931–939 CrossRef CAS.
- K. S. Rejitha, T. Ichikawa and S. Mathew, Investigations on the thermal behaviour of [Ni(NH3)6](NO3)2 and [Ni(en)3](NO3)2 using TG-MS and TR-XRD under inert condition, J. Therm. Anal. Calorim., 2012, 107, 887–892 CrossRef CAS.
- K. Pielichowski, Thermal Degradation of Polymeric Materials, Smithers Rapra Press, 2005 Search PubMed.
- E. Angeletti, C. Canepa, G. Martinetti and P. Venturello, Amino groups immobilized on silica gel: an efficient and reusable heterogeneous catalyst for the Knoevenagel condensation, J. Chem. Soc., Perkin Trans. 1, 1989, 1, 105–107 RSC.
- X. Wang, K. S. K. Lin, J. C. C. Chan and S. Cheng, Direct synthesis and catalytic applications of ordered large pore aminopropyl-functionalized SBA-15 mesoporous materials, J. Phys. Chem. B, 2005, 109, 1763–1769 CrossRef CAS PubMed.
- S. G. Wang, Amino groups immobilized on MCM-48: an efficient heterogeneous catalyst for the Knoevenagel reaction, Catal. Commun., 2003, 4, 469–470 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra10120a |
| This journal is © The Royal Society of Chemistry 2017 |