
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
How side-chain substituents and substrates influence mechanochromic luminescence: case study with pyrene†
Zongzheng Qian,
Weiting Deng,
Xuepeng Zhang,
Hui Miao* and
Guoqing Zhang *
*
Hefei National Laboratory for Physical Sciences at the Microscale, University of Science and Technology of China, 96 Jinzhai Road, Hefei, Anhui 230026, China. E-mail: gzhang@ustc.edu.cn
First published on 3rd October 2017
Abstract
Many organic molecules exhibit reversible, force-induced emission change known as mechanochromic luminescence (ML) and can potentially be used as mechanosensors. Here we provide an example on how ML behaviours can be modulated by side-chain substituents which serve as the directing group, as well as substrates which may disrupt the function of the directing group.
The photoluminescence (PL) properties of molecular solids, such as emission wavelength, lifetime, quantum yield and coherence, show critical reliance on intermolecular arrangements.1–4 For a crystalline solid with a given fashion of molecular packing, a corresponding, dominant solid-state PL arises owing to delocalized excitation unique to the specific arrangement.5–7 After mechanical stimuli, various excitonic splittings which result in new emission bands and excited-state energy transfers leading to amplification of low-energy emission can conspicuously alter the PL properties of the solids.8–10 The phenomenon is usually referred to as mechanochromic luminescence (ML).11–14 In the past decade, numerous examples of ML dyes have been reported and mechanisms carefully studied.15–22 Despite the success in identifying increasingly diverse molecules with such properties in the solid state, a pressing issue remains as how to put these ML dyes into practical application. The challenge facing ML application has two major aspects: (1) the ML behaviours of molecular solids, usually dictated by the substituent groups, are unpredictable, indicating that there is a lack of basis in design rationale; (2) the substrate effects, which can significantly alter the ML properties, are largely unknown while the real application requires that sensing be done on a specific substrate. As a result, ML is still in the theoretical phase and confined in the laboratory.
In this communication, we present the first case study in the ML field, employing one of the most studied dyes, pyrene (Fig. 1), to provide some preliminary clues for the design principles in terms of the effect of substituents and substrates. The rationale for the molecules used in the study is as follows: (1) the conditions for producing monomer and excimer emissions, respectively, are very well understood,23–27 which allows one to easily correlate fluorescence with intermolecular arrangements of the pyrene derivatives; (2) three types pyrene dyes (linear, 1P1F, wedge-shaped, 1P2F and tweezers-shaped, 2P1F) are synthesized and we predict that only the linear structure has a high probability of exhibiting ML because the tweezers are bound to form intramolecular excimers while the pyrenes are likely to be pushed together in the wedge-shaped molecules to form intermolecular excimers; (3) non-fluorinated and fluorinated pyrene derivatives were synthesized to interact with different substrates and we predict that the ML recovery is likely to be hindered on substrates with strong interactions with the dye molecules.
 | ||
| Fig. 1 Syntheses and chemical structures of alkyl- and fluoro-substituted pyrene derivatives. | ||
The syntheses for these pyrene derivatives are shown in Fig. 1 and the products are verified with 1H-NMR and mass spectroscopy, respectively (ESI†). The physical properties of the non-fluorinated and fluorinated pyrene dyes were compared: the most dramatic difference is that all three alkyl derivatives (1P1A, 2P1A and 1P2A) are transparent liquid at room temperature while the fluorinated counterparts (1P1F, 2P1F and 1P2F) are crystals with 2P1F exhibiting a crystal-liquid borderline situation at room temperature. This is not surprising since the interactions among alkyl chains and/or pyrenes are rather weak in the ground state. On the other hand, the attraction forces between fluorinated carbon chains are considerably stronger.
When dissolved in solutions, all six pyrene derivatives exhibit identical absorption spectra characteristic of pyrene (Fig. S1†). The steady-state emission spectra, however, are different depending on their molecular architectures (Fig. 2). The four linear and wedge-shaped molecules with a single pyrene show strong monomer fluorescence (λem = 376 nm) with slight variation in the vibrational progression known to be affected by polarity difference. The tweezers-like 2P1A and 2P1F show predominant excimer fluorescence (λem = 483 nm) due to the close proximity of the two pyrene moieties. In the solid state, the only dye that exhibits monomer-like fluorescence (λem = 404 nm, τFL = 0.14 ns and ΦFL = 0.039) is the linear, fluorinated 1P1F molecule, while the other five are dominated by excimer fluorescence (λem = 471–486 nm, τFL = 18.69–46.46 ns, Table 1). At this stage, it is rather safe to presume that 1P1F is the only molecule with ML properties. We have previously elucidated that the main mechanism for ML involves two steps, i.e., generation of low-energy emitting centres and exciton migration from higher-energy emitting centres.9 In the case of pyrene derivatives, the lowest emitting trap is the excimeric (λem ∼ 488 nm) configurations where the two pyrene molecules stack on top of each other.28 Consequently, 1P1F is the only molecule that does not adopt the excimer geometry in the solid state. For the alkyl-substituted pyrenes, their liquid nature at room temperature lends mobility and thus the key ingredient for excimer formation; while the two fluorinated counterparts (1P2F, λem = 474 nm, ΦFL = 0.071 and 2P1F, λem = 486 nm) form pre-arranged excimers.29,30
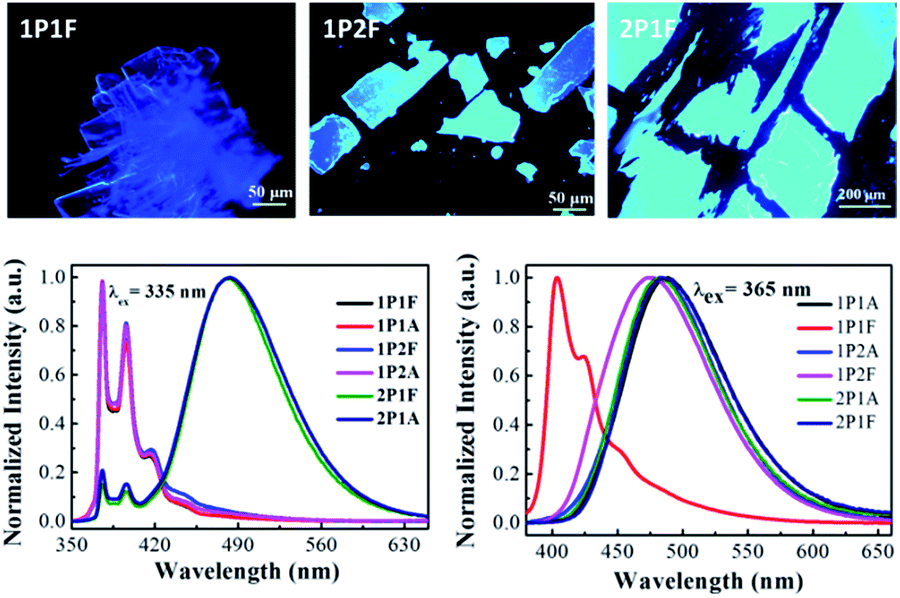 | ||
| Fig. 2 Top: fluorescence microscopy images showing solid-state morphologies of fluorinated pyrene derivatives 1P1F, 1P2F and 2P1F. Bottom: steady-state fluorescence emission spectra of all six pyrene dyes in dichloromethane solution (left) and in the solid-state (right) at room temperature. | ||
| λFLa (nm) | τFLb (ns)/% | |
|---|---|---|
| a Steady-state fluorescence emission maximum (λex = 370 nm).b Fluorescence emission lifetime components and weights. | ||
| 1P1A | 488 | 0.18/36.47 |
| 39.5/63.53 | ||
| 1P1F | 404 | 0.14/100 |
| 1P2A | 484 | 0.21/44.18 |
| 33.31/55.82 | ||
| 1P2F | 474 | 39.53/42.24 |
| 73.07/53.37 | ||
| 5.91/4.37 | ||
| 2P1A | 484 | 4.86/10.55 |
| 42.21/89.45 | ||
| 2P1F | 486 | 13.67/11.02 |
| 50.52/88.98 | ||
Fig. 3a shows the single-crystal structure and molecular packing for 1P1F. As expected, no excimeric configurations are found in the crystal; instead, the pyrene rings loosely stack and are offset from the ring centres by the equivalent of more than one benzene ring. Consequently, the overlapping area of adjacent rings is less than 50% the size of pyrene. Incidentally, from the space-fill model in Fig. 3a inset, it is clear that the main directing force for packing stems from the strong interactions among fluorinated alkyl chains,31,32 which are in short contacts, instead of pyrene stacking. Fig. 3b shows that the deep-blue fluorescence of 1P1F crystalline solids can be altered to emit in the green region typical of pyrene excimer when smeared by a cotton swab on a piece of weighing paper under UV light. The corresponding steady-state emission spectra are provided in Fig. 3c, where the emission maximum of the crystal is red-shifted from 404 to 484 nm after smearing. The measured fluorescence lifetime is also increased from 0.18 ns to 51 ns, indicating a typical excimer emission after the application of mechanical force. The phenomenon has been well-investigated in many other systems,1–22,33 in which force induces a crystalline-to-amorphous phase transition and the increased molecular rotational freedom allows for excimer/dimer formation. The observed ML is likely to be a competitive effect between pyrene packing and F–F interactions in fluorinated carbon chains, where a similar mechanism was previously reported from the Weder group.34,35
 | ||
| Fig. 3 (a) Structure and molecular packing of 1P1F determined by single-crystal X-ray diffraction, inset: space-fill model showing two adjacent 1P1F molecules with strong interactions between fluorinated alkyl chains. (b) Photo showing mechanochromic luminescence of 1P1F on weighing paper and (c) their corresponding fluorescence emission spectra. The reversible ML behaviour of 1P1F solid film is illustrated in (d–f). (g) X-ray diffraction patterns of 1P1F crystalline powders before (black line) and after (red line) pressing with a cotton swab. (h) Differential scanning calorimetry (DSC, 20 °C min−1) heating traces for 1P1F crystals before pressing (black line) and after pressing (the red line represents the 1st heating trace and the blue line represent the 2nd heating trace after cooling). | ||
Fig. 3d–f manifests the reversible ML properties of 1P1F solid on weighing paper under UV light (λex = 365 nm). After thoroughly distributing 1P1F on weighing paper (1.5 mg on 5 × 5 cm2) via smearing with a latex glove, the paper is briefly thermally annealed (∼10 s) in an oven pre-heated at 80 °C. A light touch of a cotton swab can induce the fluorescence switch from blue (λem = 462 nm) to green-blue (λem = 484 nm), visually. The green-blue emission slowly reverts (15–20 min) back to a more blue-shifted emission overtime at room temperature or immediately when heated at 80 °C. These observations are consistent with previously reported pyrene36–38 and BF2dbm39,40 systems, that smearing-caused amorphous phase generates low-energy emitting traps, through which high-energy excitons migrate and emit.9 Thermal annealing leads to molecular rearrangement partially to the original packing. However, since certain exciton traps cannot be entirely removed via simple thermal treatment, exciton migration is still prevalent and efficient, which explains the difference in fluorescence between the crystalline solids and recovered solid film. Examination of the smeared or recovered solid films at low temperature can supressed such migration and reveal the main emission species.9
To examine the proposed mechanism that has been seen in the studies from Nakano41 and Fraser.42 We performed X-ray diffraction (XRD) measurements for 1P1F crystalline solids before and after mechanical pressing. The results are presented in Fig. 3g and Table 2. Although the fluorescence emission has been largely altered from monomer-like to excimer-like, the peaks in the XRD pattern do not disappear for the pressed samples. Instead, we observed substantial intensity decrease for peaks at a smaller 2θ angle (11.2°) and broadening for all the peaks (Table 2). This observation suggests that while the majority of the solid remains in the crystalline phase, an observable portion of the ordered molecular packing have been destroyed to become amorphous, which is again consistent with our previous conclusion that energy migration from ordered crystalline region to amorphous, low-energy emitting traps can be responsible for the ML phenomenon at room temperature.9 The DSC traces from Fig. 3g also corroborate the assertion, where one additional exothermic peak at 69.4 °C could be recorded for the freshly pressed sample of 1P1F, a sign of existence of force-induced defects in substantial quantities, apart from the melting peak at 91 °C. Thus far, we have been able to demonstrate that linear substitution has a higher probability for the pyrene dye to adopt non-excimer configurations.
| 2θcalc (degree) | Pristine sample | Pressed sample |
|---|---|---|
| 11.26 | 0.24 | 0.29 |
| 12.46 | 0.23 | 0.30 |
| 18.84 | 0.28 | 0.39 |
| 20.99 | 0.25 | 0.38 |
| 21.92 | 0.24 | 0.30 |
| 24.90 | 0.38 | 0.41 |
| 25.56 | 0.32 | 0.37 |
| 38.44 | 0.30 | 0.34 |
| 41.34 | 0.35 | 0.40 |
Finally, we show that the choice of substrate can have a tremendous effect in the ML behaviours of the dye. Fig. 4 shows the ML properties of 1P1F on polytetrafluoroethene (PTFE) and polyethene (PE) substrates, respectively. Since 1P1F is heavily fluorinated, it is expected that the interaction between 1P1F and PTFE can disrupt the ordering or directing function of the fluorinated alkyl chains so that the randomly distributed pyrene moieties has a strong proclivity to the formation of excimer. As can be seen from the left panel of Fig. 4, indeed, no ML has been observed on PTFE. That cooling the solid film in liquid nitrogen does not alter the fluorescence behaviour indicates lack of ordered region, or intermolecular interactions, in the solid state. On the other hand, when a PE substrate was used, thermally reversible ML could once again be noted (Fig. 4, right panel). Similar to the observation on weighing paper, the fluorescence emission maxima of 1P1F switch in between 462 nm and 484 nm. For the smeared region, lowering temperature reveals that most excitons formed prior to migration are in fact in the blue region, which suggests that the fluorinated alkyl chains still function as the directing group for ML.
 | ||
| Fig. 4 Comparison of mechanochromic luminescence properties of 1P1F on polytetrafluoroethene (PTFE, left panel) and polyethene (PE, right panel). | ||
In conclusion, we designed and successfully synthesized three types of fluorinated and non-fluorinated carbon-chain derivatives of pyrene dyes and examined their mechanochromic luminescence properties in the solid state, with an emphasis on the role of substituents and substrates. Based on our design rationale, we predict that only one of the six molecules has a high probability to exhibit ML behaviour. Experimental results agree with the prediction and show that the fluorinated linear structure is more favourable for ML. The substrate effects were also explored and it can be concluded that strong interactions between the dye and the substrate can cost ML properties by randomizing dye distribution on a molecular level. Weak dye–substrate interactions and strong dye–dye interactions are perhaps the best scenario to design a functional ML system. These preliminary results can be employed as a general strategy to guide rational ML design in future works.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are very thankful to the Key Research Fund from University of Science and Technology of China (WK2340000068 to G. Z.), Anhui Provincial Natural Science Foundation (1708085MB38 to X. Z.) and General Financial Grant from the China Postdoctoral Science Foundation (2016M602029 to X. Z.).Notes and references
- A. Lavrenova, D. W. Balkenende, Y. Sagara, S. Schrettl, Y. C. Simon and C. Weder, J. Am. Chem. Soc., 2017, 139, 4302–4305 CrossRef CAS PubMed.
- X. Luo, J. Li, C. Li, L. Heng, Y. Q. Dong, Z. Liu, Z. Bo and B. Z. Tang, Adv. Mater., 2011, 23, 3261–3265 CrossRef CAS PubMed.
- L. Maggini and D. Bonifazi, Chem. Soc. Rev., 2012, 41, 211–241 RSC.
- Y. Ooyama and Y. Harima, J. Mater. Chem., 2011, 21, 8372 RSC.
- N. Harada, S. Karasawa, T. Matsumoto and N. Koga, Cryst. Growth Des., 2013, 13, 4705–4713 CAS.
- J. Mei, J. Wang, A. Qin, H. Zhao, W. Yuan, Z. Zhao, H. H. Y. Sung, C. Deng, S. Zhang, I. D. Williams, J. Z. Sun and B. Z. Tang, J. Mater. Chem., 2012, 22, 4290–4298 RSC.
- T. Seki, Y. Takamatsu and H. Ito, J. Am. Chem. Soc., 2016, 138, 6252–6260 CrossRef CAS PubMed.
- S. Karasawa, R. Hagihara, Y. Abe, N. Harada, J.-i. Todo and N. Koga, Cryst. Growth Des., 2014, 14, 2468–2478 CAS.
- X. Sun, X. Zhang, X. Li, S. Liu and G. Zhang, J. Mater. Chem., 2012, 22, 17332 RSC.
- S.-J. Yoon and S. Park, J. Mater. Chem., 2011, 21, 8338 RSC.
- H. Ito, T. Saito, N. Oshima, N. Kitamura, S. Ishizaka, Y. Hinatsu, M. Wakeshima, M. Kato, K. Tsuge and M. Sawamura, J. Am. Chem. Soc., 2008, 130, 10044–10045 CrossRef CAS PubMed.
- D. A. Davis, A. Hamilton, J. Yang, L. D. Cremar, D. Van Gough, S. L. Potisek, M. T. Ong, P. V. Braun, T. J. Martinez, S. R. White, J. S. Moore and N. R. Sottos, Nature, 2009, 459, 68–72 CrossRef CAS PubMed.
- Y. Sagara and T. Kato, Nat. Chem., 2009, 1, 605–610 CrossRef CAS PubMed.
- Y. Sagara, S. Yamane, M. Mitani, C. Weder and T. Kato, Adv. Mater., 2016, 28, 1073–1095 CrossRef CAS PubMed.
- S. C. B. Mannsfeld, B. C.-K. Tee, R. M. Stoltenberg, C. V. H.-H. Chen, S. Barman, B. V. O. Muir, A. N. Sokolov, C. Reese and Z. Bao, Nat. Mater., 2010, 9, 859–864 CrossRef CAS PubMed.
- Y. Lee and R. Eisenberg, J. Am. Chem. Soc., 2003, 125, 7778–7779 CrossRef CAS PubMed.
- H. Li, X. Zhang, Z. Chi, B. Xu, W. Zhou, S. Liu, Y. Zhang and J. Xu, Org. Lett., 2011, 13, 556–559 CrossRef CAS PubMed.
- A. L. Balch, Angew. Chem., Int. Ed., 2009, 48, 2641–2644 CrossRef CAS PubMed.
- T. Butler, A. S. Mathew, M. Sabat and C. L. Fraser, ACS Appl. Mater. Interfaces, 2017, 9, 17603–17612 CAS.
- T. Butler, W. A. Morris, J. Samonina-Kosicka and C. L. Fraser, ACS Appl. Mater. Interfaces, 2016, 8, 1242–1251 CAS.
- Z. Chi, X. Zhang, B. Xu, X. Zhou, C. Ma, Y. Zhang, S. Liu and J. Xu, Chem. Soc. Rev., 2012, 41, 3878–3896 RSC.
- Y. Sagara and T. Kato, Angew. Chem., Int. Ed., 2011, 50, 9128–9132 CrossRef CAS PubMed.
- G. Jones and V. I. Vullev, Org. Lett., 2001, 3, 2457–2460 CrossRef CAS PubMed.
- F. M. Winnik, Chem. Rev., 1993, 93, 587–614 CrossRef CAS.
- C. Lowe and C. Weder, Adv. Mater., 2002, 14, 1625–1629 CrossRef CAS.
- Y. Sagara, T. Mutai, I. Yoshikawa and K. Araki, J. Am. Chem. Soc., 2007, 129, 1520–1521 CrossRef CAS PubMed.
- Y. Sagara, T. Komatsu, T. Ueno, K. Hanaoka, T. Kato and T. Nagano, Adv. Funct. Mater., 2013, 23, 5277–5284 CrossRef CAS.
- K. M. Chan, D. K. Kolmel, S. Wang and E. T. Kool, Angew. Chem., Int. Ed., 2017, 56, 6497–6501 CrossRef CAS PubMed.
- S. Ito, K. Ikeda, S. Nakanishi, Y. Imai and M. Asami, Chem. Commun., 2017, 53, 6323–6326 RSC.
- F. Lu, T. Takaya, K. Iwata, I. Kawamura, A. Saeki, M. Ishii, K. Nagura and T. Nakanishi, Sci. Rep., 2017, 7, 3416 CrossRef PubMed.
- D. F. Eaton and B. E. Smart, J. Am. Chem. Soc., 1990, 112, 2821–2823 CrossRef CAS.
- V. Percec, G. Johansson, G. Ungar and J. Zhou, J. Am. Chem. Soc., 1996, 118, 9855–9866 CrossRef CAS.
- E. Nagata, T. Ara and H. Nakano, Dyes Pigm., 2017, 141, 48–52 CrossRef CAS.
- Y. Sagara, K. Kubo, T. Nakamura, N. Tamaoki and C. Weder, Chem. Mater., 2017, 29, 1273–1278 CrossRef CAS.
- Y. Sagara, A. Lavrenova, A. Crochet, Y. C. Simon, K. M. Fromm and C. Weder, Chem.–Eur. J., 2016, 22, 4374–4378 CrossRef CAS PubMed.
- X. Meng, G. Qi, X. Li, Z. Wang, K. Wang, B. Zou and Y. Ma, J. Mater. Chem. C, 2016, 4, 7584 RSC.
- Z. Ma, M. Teng, Z. Wang and X. Jia, Tetrahedron Lett., 2013, 54, 6504–6506 CrossRef CAS.
- M. J. Teng, X. R. Jia, S. Yang, X. F. Chen and Y. Wei, Adv. Mater., 2012, 24, 1255–1261 CrossRef CAS PubMed.
- G. Zhang, J. Lu, M. Sabat and C. L. Fraser, J. Am. Chem. Soc., 2010, 132, 2160–2162 CrossRef CAS PubMed.
- G. Zhang, J. P. Singer, S. E. Kooi, R. E. Evans, E. L. Thomas and C. L. Fraser, J. Mater. Chem., 2011, 21, 8295 RSC.
- E. Nagata, S. Takeuchi, T. Nakanishi, Y. Hasegawa, Y. Mawatari and H. Nakano, ChemPhysChem, 2015, 16, 3038–3043 CrossRef CAS PubMed.
- T. Butler, F. Wang, M. Sabat and C. L. Fraser, Mater. Chem. Front., 2017, 1, 1804–1817 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Details of synthesis and characterizations, HRMS, NMR and UV-vis spectra. CCDC 1569185. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7ra09453a |
| This journal is © The Royal Society of Chemistry 2017 |