
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Task-specific acidic ionic liquid-catalyzed efficient synthesis of β-enaminolactones from alkynoates and β-amino alcohols†
Lu Chen *,
Bin Chen,
Fuying Zhao,
Yibiao Li,
Bin Li* and
Min Zhang
*,
Bin Chen,
Fuying Zhao,
Yibiao Li,
Bin Li* and
Min Zhang
School of Chemical & Environmental Engineering, Wuyi University, Jiangmen, Guangdong Province 529090, China. E-mail: wyuchemcl@126.com; andonlee@163.com
First published on 12th June 2017
Abstract
By employing task-specific acidic ionic liquid as an efficient catalyst, a new method for the straightforward synthesis of β-enaminolactones has been demonstrated. A series of alkynoates in combination with various β-amino alcohols was efficiently converted into the desired products in good to excellent yields upon isolation. The skeleton of the seven-membered ring is generated via tandem intermolecular hydroamination and intramolecular transesterification processes. The developed synthetic protocol furnishes the desired products in a step- and atom-economic fashion with the advantages of high yields, broad substrate scope, good functional tolerance, and operational simplicity, offering an important basis for the construction of β-enaminolactones.
β-Amino acids constitute a highly significant branch in organic chemicals, which have been found to possess diverse bioactivities and are employed as useful building blockings for the preparation of β-lactam antibiotics and heterocycles. In addition, β-amino acid derivatives frequently occur in numerous biologically active natural products.1 Among various compounds investigated to date, β-enaminolactone derivatives, the precursors of enantiopure β-amino acids, are the main structural components of many bioactive natural products that have attracted significant attention.2
Owing to the abovementioned interesting functions of β-enaminolactone, several synthetic protocols have been nicely demonstrated during the past few decades to access these compounds.3–8 For instance, Abarbri and coworkers3 first reported the reaction of ethyl perfluorobut-2-ynoate with amino alcohols to generate 5-(perfluoroalkyl)-3,4-dihydro-2H-[1,4]oxazepin-7-ones via the intermolecular Michael addition and lactone formation. In 2005, Dechoux's group reported an efficient method for the synthesis of β-enaminolactones4 via the condensation of acetonedicarboxylate with β-amino alcohols followed by an intramolecular cyclization step. However, during this synthesis, the reaction intermediates needed to be isolated and it required excess NaH to achieve the cyclization; moreover, an additional neutralization manipulation by adding NH4Cl/H2O was essential to obtain the desired products after the completion of the reaction. From the viewpoint of green chemistry, the development of efficient shortcuts for accessing β-enaminolactones from easily available feedstocks would be of high significance.
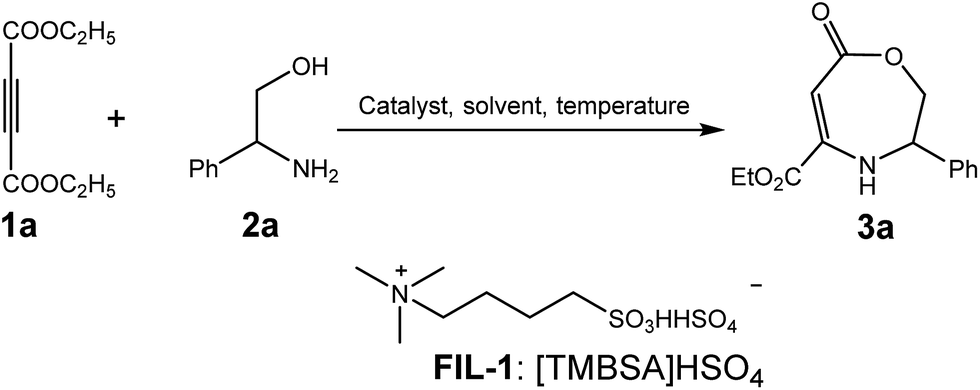
As a part of our program aimed at developing new synthetic methodologies for the construction of heterocycles,17 we initially had the idea to develop a ruthenium-catalyzed synthesis of ester-substituted pyrrole 3a′ from alkynoate 1a and β-amino alcohol 2a via dehydrogenative cyclization (Scheme 1, eqn (1)). However, we failed to obtain even traces of the anticipated product, and a small portion of β-enaminolactone 3a was obtained. Further investigations showed that the ruthenium catalyst was not essential for the product formation (3a), whereas the presence of 10 mol% of AlCl3 was able to improve the yield of 3a to 18% (eqn (2)). Upon a thorough investigation of this new observation, a straightforward method for the efficient synthesis of β-enaminolactones from alkynoates and β-amino alcohols using a task-specific sulfonic ionic liquid as the catalyst was realized and has been reported herein.
 | ||
| Scheme 1 The new observation leading to β-enaminolactone. | ||
Our initial investigation was to develop a more efficient reaction system by choosing the synthesis of β-enaminolactone 3a from diethyl but-2-ynedioate 1a and 2-amino-2-phenylethanol 2a as a model reaction. First, the reaction in t-amyl alcohol was performed at 120 °C for 12 h, and several conventional Lewis acid catalysts were tested (Table 1, entries 1–7); it was found that Cu(OTf)2 or Zn(OTf)2 shows good performance in affording the desired product 3a. However, in the absence of an acid catalyst, the desired product was not obtained (Table 1, entry 8), indicating that the acidic catalyst played a crucial role in the reaction. Moreover, attracted by the significant advantages, such as the designability, easy recovery, and reusability,9–15 of task-specific acidic ionic liquids, we evaluated the utility of a sulfonic-functionalized ionic liquid16 (TSFIL-1: [TMBSA]HSO4) as a catalyst. Gratifyingly, this catalyst exhibited an excellent activity in the production of product 3a (entries 9–11), and 10 mol% catalyst loading was essential to afford a satisfactory yield (entry 9). Further, changes in the reaction temperatures led to diminished product yields (Table 1, entries 12 and 13), implying that a lower temperature significantly decreased the reaction rate, whereas increasing this parameter would lower the product stability. However, use of toluene, DMF, DMAc, DMSO, and 1,4-dioxane as the solvents proved to be totally ineffective or less effective for the formation of products when compared with t-amyl alcohol (Table 1, entries 14–18). Finally, [TMBSA]HSO4 was used as both the solvent and the catalyst and it only gave a 77% yield, which could be rationalized since a viscous ionic liquid was not beneficial for the interaction of substrates. Based on these results, the optimal reaction conditions were obtained and are indicated in entry 9 of Table 1.
|
||||
|---|---|---|---|---|
| Entry | Catalyst (mol%) | Temp. (°C) | Solvent (2.0 mL) | Yieldb (%) |
| a Reaction conditions: unless otherwise stated, the reaction mixture of dimethyl acetylenedicarboxylate (1) (0.5 mmol), 2-amino-2-phenylethanol (2) (0.6 mmol), catalyst loaded in different solvents (2.0 mL) was stirred at 120 °C for 12 h under atmospheric condition.b GC yield using n-hexadecane as an internal standard.c Under a N2 atmosphere.d No product detectives. | ||||
| 1c | AlCl3 (10) | 120 | t-Amyl alcohol | 18 |
| 2 | ZnCl2 (10) | 120 | t-Amyl alcohol | 35 |
| 3 | FeCl3 (10) | 120 | t-Amyl alcohol | 40 |
| 4 | Yb(OTf)3 (10) | 120 | t-Amyl alcohol | 40 |
| 5 | Cu(OTf)2 (10) | 120 | t-Amyl alcohol | 78 |
| 6 | Al(OTf)3 (10) | 120 | t-Amyl alcohol | 80 |
| 7 | Zn(OTf)2 (10) | 120 | t-Amyl alcohol | 84 |
| 8 | None | 120 | t-Amyl alcohol | —d |
| 9 | TSILs (10) | 120 | t-Amyl alcohol | 94 |
| 10 | TSILs (5) | 120 | t-Amyl alcohol | 92 |
| 11 | TSILs (15) | 120 | t-Amyl alcohol | 90 |
| 12c | TSILs (10) | 100 | t-Amyl alcohol | 36 |
| 13 | TSILs (10) | 140 | t-Amyl alcohol | 87 |
| 14 | TSILs (10) | 120 | Toluene | —d |
| 15 | TSILs (10) | 120 | DMF | —d |
| 16 | TSILs (10) | 120 | DMAc | 22 |
| 17 | TSILs (10) | 120 | DMSO | 56 |
| 18 | TSILs (10) | 120 | 1,4-Dioxane | 14 |
| 19 | TSILs (1 mL) | 120 | None | 77 |
With the availability of the optimized reaction conditions, we subsequently examined the generality of the synthetic protocol. A variety of alkynoates (1) in combination with β-amino alcohols (2) were tested. As shown in Table 2, all the reactions smoothly proceeded and furnished the desired products in good to excellent isolated yields. Diethyl but-2-ynedioate 1a reacted with 2-amino-2-phenylethanol 2a and 2-amino-3-phenylpropan-1-ol 2c to provide the products 3a and 3c in 86% and 94% yields, respectively (Table 2, entries 1 and 3). Moreover, even a steric-hindered substrate, such as 2-amino-1,2-diphenylethanol 2b, also underwent smooth transformation with 2-amino-2-phenylethanol 2a, affording the desired product in 78% yield (Table 2, entry 2). Similarly, the reactions of 1a with amino alcohols 2d and 2e produced the corresponding products 3d and 3e in excellent yields (Table 2, entries 4 and 5), respectively. Interestingly, the less reactive ethyl 3-phenylpropiolate 1c could also generate the desired coupling products in moderate to good yields, demonstrating that the developed chemistry was applicable for a broad substrate scope (Table 2, entries 6–10). Note that amino alcohols (2a and 2c) with a phenyl group or a benzyl substituent could afford higher yields (Table 2, entries 6 and 9) than those with an alkyl group (Table 2, entries 1 and 3–5). Moreover, amino alcohols with an isopropyl group gave a relatively lower yield (Table 2, entry 8), presumably because of the influence of its strong electron-donating effect, thus deactivating the ester group. On the other hand, the secondary alcohols such as 1-aminopropan-2-ol 2f reacted with alkynoate 1c to give the corresponding product 3j in 74% yield (Table 2, entry 10). Note that various functional groups such as 4-Cl, 4-Br, 4-F, and 4-CH3CO on the phenyl ring of alkynoates (1d–1g) were well tolerated, affording the corresponding products in good to excellent yields (Table 2, entries 12–14, 19 and 20). Similarly, electron-donating groups on the aryl ring of the substrates 2 were also compatible with the transformation (Table 2, entries 15–17). The retention of these functional groups would offer the potential for further molecular complexity via chemical transformation.
|
|||
|---|---|---|---|
| Entry | 1 | 2 | 3, yield (%) |
| a Reaction conditions: unless otherwise stated, the reaction mixture of dimethyl acetylenedicarboxylate (1) (0.5 mmol), 2-amino-2-phenylethanol (2) (0.6 mmol), and [TMBSA]HSO4 (10 mol%) in t-amyl alcohol (2.0 mL) was stirred at 120 °C for 12 h under atmospheric condition. | |||
| 1 | 1a: R1 = Et, R2 = CO2Et | 2a: R3 = Ph, R4 = H | 3a, 86 |
| 2 | 1a: R1 = Et, R2 = CO2Et | 2b: R3 = Ph, R4 = Ph | 3b, 78 |
| 3 | 1a: R1 = Et, R2 = CO2Et | 2c: R3 = PhCH2, R4 = H | 3c, 94 |
| 4 | 1b: R1 = CH3, R2 = CO2CH3 | 2a: R3 = Ph, R4 = H | 3d, 80 |
| 5 | 1b: R1 = CH3, R2 = CO2CH3 | 2c: R3 = PhCH2, R4 = H | 3e, 90 |
| 6 | 1c: R1 = Et, R2 = Ph | 2a: R3 = Ph, R4 = H | 3f, 68 |
| 7 | 1c: R1 = Et, R2 = Ph | 2d: R3 = CH3, R4 = H | 3g, 78 |
| 8 | 1c: R1 = Et, R2 = Ph | 2e: R3 = (CH3)2CH, R4 = H | 3h, 58 |
| 9 | 1c: R1 = Et, R2 = Ph | 2c: R3 = PhCH2, R4 = H | 3i, 68 |
| 10 | 1c: R1 = Et, R2 = Ph | 2f: R3 = H, R4 = CH3 | 3j, 74 |
| 11 | 1d: R1 = Et, R2 = 4-ClPh | 2d: R3 = CH3, R4 = H | 3k, 84 |
| 12 | 1e: R1 = Et, R2 = 4-BrPh | 2d: R3 = CH3, R4 = H | 3l, 89 |
| 13 | 1d: R1 = Et, R2 = 4-ClPh | 2e: R3 = (CH3)2CH, R4 = H | 3m, 60 |
| 14 | 1e: R1 = Et, R2 = 4-BrPh | 2e: R3 = (CH3)2CH, R4 = H | 3n, 62 |
| 15 | 1h: R1 = Et, R2 = 4-CH3Ph | 2d: R3 = CH3, R4 = H | 3o, 72 |
| 16 | 1i: R1 = Et, R2 = 4-CH3OPh | 2d: R3 = CH3, R4 = H | 3p, 70 |
| 17 | 1h: R1 = Et, R2 = 4-CH3Ph | 2e: R3 = (CH3)2CH, R4 = H | 3q, 56 |
| 18 | 1i: R1 = Et, R2 = 4-CH3OPh | 2e: R3 = (CH3)2CH, R4 = H | 3r, 50 |
| 19 | 1f: R1 = Et, R2 = 4-FPh | 2d: R3 = CH3, R4 = H | 3s, 70 |
| 20 | 1g: R1 = Et, R2 = 4-CH3COPh | 2d: R3 = CH3, R4 = H | 3t, 75 |

To gain insight into the reaction information, we performed the control experiments. It was found that hydroamination between alkynoate 1a and amino alcohol 2a completed in 5 minutes without any catalyst or additive. Then, the resulting enamine intermediate (3a–l) under standard conditions furnished the cyclization product 3a in an almost quantitative yield (Scheme 2). This result, in combination with the fact that in the absence of a catalyst, product 3a cannot be formed (Table 1, entry 8), indicates that the acidic catalyst plays a crucial role in the activation of the ester group. These findings suggest that the product formation is initiated by fast intermolecular hydroamination followed by [TMBSA]HSO4-catalyzed intramolecular transesterification.
 | ||
| Scheme 2 Control experiments. | ||
Finally, we demonstrated the utility of the developed new method. The reaction of the diethyl but-2-ynedioate 1a with enantiopure amino alcohol (S)-(+)-2a′ produced the enantiopure enaminolactone (S)-(+)-3a′ in an excellent yield with retention of the chiral configuration (Scheme 3). This result shows that the task-specific acidic ionic liquid-catalyzed protocol is also applicable for the preparation of chiral β-enaminolactones from chiral amino alcohols.
 | ||
| Scheme 3 Synthesis of chiral β-enaminolactone. | ||
In summary, by employing a task-specific sulfonic ionic liquid as the catalyst, we demonstrated an environmentally friendly and straightforward approach for the versatile synthesis of β-enaminolactones from readily available amino alcohols and alkynoates for the first time. The synthetic protocol proceeds via tandem intermolecular hydroamination and intramolecular transesterification processes; moreover, it furnished the desired products in a step- and atom-economic fashion with the advantages of high isolated yields, broad substrate scope, good functional tolerance, and operational simplicity, which offers an important basis for the construction of β-enaminolactones.
Acknowledgements
The authors are grateful to the funds of the Science Foundation for Young Teachers of Wuyi University (No: 2016zk03, 2014zk04 and No: 2015td01), the Scientific Research Foundation for the Returned Oversea as Chinese Scholar, State Education Ministry ([2015]1098), the Natural Science Foundation of Guangdong Province (No: 2014A030310211), and the Foundation of the Department of Education of Guangdong Province (No: 2016KTSCX144).Notes and references
- (a) O. W. Griffith, Annu. Rev. Biochem., 1986, 55, 855–878 CrossRef CAS PubMed; (b) E. Juaristi, D. Quintania and J. Escalante, Aldrichimica Acta, 1994, 27, 3–11 CAS; (c) J. S. Carey, D. Laffan, C. Thomson and M. T. Williams, Org. Biomol. Chem., 2006, 4, 2337–2347 RSC.
- (a) D. Seebach and J. L. Matthews, Chem. Commun., 1997, 2015–2022 RSC; (b) R. P. Cheng, S. H. Gellman and W. F. DeGrado, Chem. Rev., 2001, 101, 3219–3232 CrossRef CAS PubMed.
- S. Richard, G. Prie, A. Guignard, J. Thibonnet, J. L. Parrain, A. Duchene and M. Abarbri, Helv. Chim. Acta, 2003, 86, 726–732 CrossRef CAS.
- J. Alladoum and L. Dechoux, Tetrahedron Lett., 2005, 46, 8203–8205 CrossRef CAS.
- J. Alladoum, V. Toum, S. Hebbe, C. Kadouri-Puchot and L. Dechoux, Tetrahedron Lett., 2009, 50, 617–619 CrossRef CAS.
- Y. Ishida, N. Iwahashi, N. Nishizono and K. Saigo, Tetrahedron Lett., 2009, 50, 1889–1892 CrossRef CAS.
- A. K. Singh, R. Chawla and L. D. S. Yadav, Synthesis, 2012, 44, 2353–2358 CrossRef CAS.
- Y. Tian, X. Wang, Q. Xiao, C. Sun and D. Yin, J. Org. Chem., 2014, 79, 9678–9685 CrossRef CAS PubMed.
- M. Petkovic, K. R. Seddon, L. P. N. Rebelo and C. S. Pereira, Chem. Soc. Rev., 2011, 40, 1383–1403 RSC.
- N. Isambert, M. M. S. Duque, J.-C. Plaquevent, Y. Génisson, J. Rodriguez and T. Constantieux, Chem. Soc. Rev., 2011, 40, 1347–1357 RSC.
- R. Sheldon, Chem. Commun., 2001, 23, 2399–2407 RSC.
- T. Welton, Chem. Rev., 1999, 99, 2071–2083 CrossRef CAS PubMed.
- J. P. Hallett and T. Welton, Chem. Rev., 2011, 111, 3508–3576 CrossRef CAS PubMed.
- T. Welton, Coord. Chem. Rev., 2004, 248, 2459–2477 CrossRef CAS.
- A. S. Amarasekara, Chem. Rev., 2016, 116, 6133–6183 CrossRef CAS PubMed.
- (a) S. G. Lee, Chem. Commun., 2006, 1049–1063 RSC; (b) G. Y. Zhao, T. Jiang, H. X. Gao, B. X. Han, J. Huang and D. H. Sun, Green Chem., 2004, 6, 75–77 RSC; (c) D. Q. Xu, J. Wu, S. P. Luo, J. Zhang, J. Y. Wu, X. H. Du and Z. Y. Xu, Green Chem., 2009, 11, 1246 Search PubMed.
- (a) C. B. Bheeter, L. Chen, J.-F. Soulé and H. Doucet, Catal. Sci. Technol., 2016, 6, 2005–2049 RSC; (b) L. Chen, X. Luo and Y. Li, Monatsh. Chem., 2017, 148, 957–961 CrossRef CAS; (c) J. Cai, S. Huang, R. He, L. Chen, D. Chen, S. Jiang, B. Li and Y. Li, Org. Biomol. Chem., 2017, 15, 333–337 RSC; (d) Y. Li, L. Cheng, B. Li, S. Jiang, L. Chen and Y. Shao, ChemistrySelect, 2016, 5, 1092–1095 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: General experimental information, experimental procedure for product synthesis, full characterization data, 1H and 13C NMR spectra of all products. See DOI: 10.1039/c7ra04028h |
| This journal is © The Royal Society of Chemistry 2017 |