
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Computational design to improve catalytic activity of cephalosporin C acylase from Pseudomonas strain N176†
Ye Tiana,
Zhaobin Xua,
Xiaoqiang Huanga and
Yushan Zhu *ab
*ab
aDepartment of Chemical Engineering, Tsinghua University, Beijing 100084, PR China. E-mail: yszhu@tsinghua.edu.cn; Tel: +86-10-62784572
bMOE Key Lab of Industrial Biocatalysis, Tsinghua University, Beijing 100084, PR China
First published on 12th June 2017
Abstract
Engineering enzymes with high catalytic activities using enzyme design in silico and a limited number of experimental evaluations is the new trend for the discovery of highly efficient biocatalysts. In this report, a double mutant (M31βF/H70βS) cephalosporin C acylase from the Pseudomonas strain N176 was used as the starting template, and a computational enzyme design strategy aided the identification of a quadruple mutant (M31βF/H70βS/F58βN/I176βT) that exhibited a 2.7-fold increase in catalytic efficiency (Vmax/Km) when compared with that of the template. The time-course results confirmed that the quadruple mutant was a promising enzyme to catalyze the hydrolysis of cephalosporin C to produce 7-amino cephalosporanic acid in one-step under industrial conditions.
Introduction
7-Amino cephalosporanic acid (7-ACA) is an important β-lactam nucleus for the preparation of many widely used semi-synthetic β-lactam antibiotics,1 and it is usually produced by hydrolyzing natural cephalosporin C (CPC), as shown in Fig. 1. Currently, no natural enzyme with the ability to convert CPC to 7-ACA that meets the requirement of industry-accepted catalytic efficiency has been found.2 As a compromise, a two-step enzymatic process is widely used in the industrial production of 7-ACA. Here, cephalosporin C is first oxidized to α-ketoadipyl-7-ACA by D-amino acid oxidase (DAAO, EC 1.4.3.3), with consumption of molecular oxygen and production of hydrogen peroxide as a by-product. Subsequently, α-ketoadipyl-7-ACA is converted to glutaryl-7-aminocephalosporanic acid (GL-7-ACA) spontaneously and GL-7-ACA is then hydrolyzed to 7-ACA by cephalosporin acylases (EC 3.5.1.93), which is also called GL-7-ACA acylase. The main drawback of the two-step enzymatic approach lies in the by-product hydrogen peroxide, which negatively affects the stability and decreases the half-life of the DAAO and GL-7-ACA acylase. The resulting increase in cost cannot entirely satisfy industrial production, and a one-step enzymatic process is actively pursued by cephalosporin antibiotics manufacturers.3 | ||
| Fig. 1 Reaction scheme of cephalosporin C hydrolysis catalyzed by cephalosporin C acylase. CPC: cephalosporin C; 7-ACA: 7-aminocephalosporanic acid. | ||
The primary substrate of cephalosporin acylases (EC 3.5.1.93) is GL-7-ACA and their activity towards the hydrolysis of CPC is very low, only 4% or less when compared with activity towards GL-7-ACA.4,5 As the structural differences between CPC and GL-7-ACA lie only on their side chains, various cephalosporin acylases have been engineered to promote their catalytic activities towards CPC.4–7 Aramori et al.8 found a cephalosporin acylase gene from the Pseudomonas strain N176, which catalyzes the conversion of CPC to 7-ACA with very weak activity. Ishii et al.9 reported a mutation M31βF of N176 that contributed to a 1.7-fold increase in catalytic activity. By virtue of combining site-saturation and site-directed mutagenesis technologies, Pollegioni et al.3 screened multiple mutants of N176, and two additional mutations H57βS and H70βS were identified to increase the activity by 4-fold based on the single mutant M31βF of N176. Recently, the crystal structure of cephalosporin acylase N176 was reported by Golden et al.10 and various mutants of N176 with promoted activities were identified by Conti et al.11 based on site-directed mutagenesis and molecular docking. However, the activities of the reported CPC acylase variants are still unsuitable for use at an industrial level.
Computational enzyme design methods have been developed to produce protein catalysts with novel catalytic machinery for non-biological reactions or improved specificity for unnatural substrates, which greatly extends the biotechnological applications of native enzymes.12–22 For instances, Gordon et al.16 computationally designed an α-gliadin peptidase which exhibited a kcat/Km of 568 M−1 s−1, representing a 116-fold greater proteolytic activity for a model gluten tetrapeptide than the native template enzyme. Siegel et al.20 designed a formolase using four iterations of computational design and experimental evaluations, led to a variant with 26-fold higher activity than the starting point with only 121 designs in total evaluated. Recently, Wijma et al.21 developed the catalytic selectivity by a computational design (CASCO) strategy based on RosettaDesign and high-throughput molecular dynamics simulations. Based the CASCO strategy, this study obtained highly stereoselective mutants of limonene epoxide hydrolase from a small set of only 37 variants that were tested experimentally. However, it is still a great challenge to obtain mutants with improved catalytic activities for enzyme design based only on computational predictions. Using the crystal structure of cephalosporin acylase N176 and our computational protein design program PRODA (PROtein Design Algorithmic package),23–27 we have identified several variants of N176 with improved stability.27 To obtain variants with higher catalytic efficiencies towards CPC, the computational enzyme design protocol was used in this study to predict effective mutations of N176.
Materials and methods
Structure data
The atomic coordinates for cephalosporin acylase from Pseudomonas strain N176 were taken from the 2.13 Å crystal structure (PDB ID: 4HSR).10 4HSR differs from N176 acylase by the presence of the M31βF substitution. Hydrogen atoms were added by PRODA using the topology parameters of the all-atom force field CHARMM 22 (ref. 28) and water molecules were stripped from the crystal structure.Computational design
The computational design method has been described in detail in our earlier work for stability redesign.27 For the active pocket redesign of N176, 19 active sites (M158α, L161α, M165α, F31β, Y32β, L47β, V49β, H57β, F58β, C67β, V68β, L154β, I176β, H178β, L470β, S471β, Y473β and F475β) were allowed to assume 18 natural amino acid types, with Pro and Lys excluded. Thirty-eight positions can change conformations only within their natural amino acid type, these positions interact with the active sites directly but contact with the substrate indirectly, as shown in ESI Table S1.† The side chain conformations for the designed positions were taken from the backbone-independent rotamer library that includes 11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 810 rotamers.29 The rotamers for serine, threonine and tyrosine are expanded, as described in our earlier work,25 and the side chain conformation of the design sites from the 4HSR structure is further added to the library for the calculation. All other side chains and the main chain were kept fixed. The atomic parameters and internal coordination parameters for amino acids are taken from the all-atom force-field CHARMM 22.28 The catalytic geometrical constraints between TS and active sites, as well as the placing rules, have been provided in detail in the ESI† of our former work,24,25 and the planar form of the central amide bond of cephalosporin C has been converted to the tetrahedral intermediate form of the transition state (TS). The small molecule library of the TS for CPC contains 6779 rotamers. The atomic parameters and partial atomic charges for cephalosporin C are constructed from the model compounds of the CHARMM 22 force field. Pairwise energy functions and global minimum energy sequences have also been described in our earlier work.24,25 The relative changes in binding energies (ΔΔGBind) for the top-ranked sequences are calculated as the differences between the binding energy (ΔGBind) of the complexed enzyme-TS system of each mutant and that of the starting point. The relative changes in folding energies (ΔΔGFold) were calculated by taking the difference between the complexed enzyme-TS system of each mutant and that of the starting point. All computations stated above were performed by PRODA.
810 rotamers.29 The rotamers for serine, threonine and tyrosine are expanded, as described in our earlier work,25 and the side chain conformation of the design sites from the 4HSR structure is further added to the library for the calculation. All other side chains and the main chain were kept fixed. The atomic parameters and internal coordination parameters for amino acids are taken from the all-atom force-field CHARMM 22.28 The catalytic geometrical constraints between TS and active sites, as well as the placing rules, have been provided in detail in the ESI† of our former work,24,25 and the planar form of the central amide bond of cephalosporin C has been converted to the tetrahedral intermediate form of the transition state (TS). The small molecule library of the TS for CPC contains 6779 rotamers. The atomic parameters and partial atomic charges for cephalosporin C are constructed from the model compounds of the CHARMM 22 force field. Pairwise energy functions and global minimum energy sequences have also been described in our earlier work.24,25 The relative changes in binding energies (ΔΔGBind) for the top-ranked sequences are calculated as the differences between the binding energy (ΔGBind) of the complexed enzyme-TS system of each mutant and that of the starting point. The relative changes in folding energies (ΔΔGFold) were calculated by taking the difference between the complexed enzyme-TS system of each mutant and that of the starting point. All computations stated above were performed by PRODA.
Gene cloning, plasmid construction and site-directed mutagenesis
The original gene was obtained from the nucleotide sequence for cephalosporin C acylase from Pseudomonas strain N176.8 The codon was optimized for expression in Escherichia coli (E. coli) and the gene was chemically synthesized by Takara Biotechnology Co. (Dalian, China). The synthesized gene was then subcloned into the pET28a(+) vector carrying resistance for kanamycin and the gene fragment coding for N176 cephalosporin C acylase (2336 bp) was ligated between the NcoI and XhoI sites of the pET28a(+) vector. Six codons for a hexa-histidine tag were introduced to the 3′-end of the N176 gene. Mutations were generated by site-directed mutagenesis using the polymerase chain reaction (PCR) and the mutations introduced were confirmed by automated DNA sequencing.Enzyme expression and purification
The recombinant pET28a(+)-N176 plasmids encoding various N176 mutants were first transferred to the host BL21(DE3) E. coli strain. Cells harboring the recombinant plasmids were grown overnight at 37 °C in LB medium containing 50 μg mL−1 kanamycin. The culture was inoculated into 500 mL × 2 LB medium with 50 μg mL−1 kanamycin and incubated at 37 °C until the OD600 nm of the cell suspension reached 0.6. Protein expression was then induced by adding isopropyl-β-D-thiogalactopyranoside (IPTG) to a final concentration of 0.5 mM and incubated at 28 °C for another 20 h. Cells were harvested by centrifugation at 10000g for 10 min at 4 °C.
Cells were resuspended in Tris–HCl buffer (100 mM Tris, pH 8.0) and lysed by sonication. The resulting insoluble fraction was removed by centrifugation. The supernatant was loaded onto a Ni-NTA column equilibrated with binding buffer A (100 mM Tris, pH 8.0, containing 500 mM NaCl and 20 mM imidazole) and the column was then washed with binding buffer B (100 mM Tris, pH 8.0, containing 500 mM NaCl and 50 mM imidazole) to remove cell proteins. The target enzyme was eluted by elution buffer (100 mM Tris, pH 8.0, containing 500 mM NaCl and 250 mM imidazole). Subsequently, fractions containing purified enzyme were concentrated with a 10k Amicon® Ultra-0.5 centrifugal filter (Millipore, Billerica, MA) and desalted twice to remove the imidazole and NaCl. The purity of the target enzyme was >90%, as indicated by SDS-PAGE analysis. The concentration of the purified enzymes was determined by the Bradford method30 using bovine serum albumin (BSA) as a standard reagent. The absorbance from each enzyme sample with the addition of Bradford reagent was measured at 595 nm.
Enzyme activity and kinetic assay
The catalytic activities and kinetic parameters were determined by HPLC analysis (LC-20AT, Shimadzu, Japan). 0.5 mL of enzyme (in 0.1 M Tris buffer, pH 8.0) was incubated with 0.5 mL CPC (pH 8.0, a linear gradient with the maximum 2% w/v) for 8 min at 37 °C. The reaction was terminated with 3 mL stop solution (15 mM NaOH, 15% acetate acid) and then 5 μL of this mixture was analyzed by HPLC at 280 nm with a flow rate of 0.8 mL min−1. The column temperature was maintained at 30 °C. Elution buffer contained 15% methanol, 7.5% acetonitrile and 1% acetate acid (v/v). The kinetic parameters (Vmax, Km and Vmax/Km) were obtained by fitting data to the Michaelis–Menten equation using the Lineweaver–Burk method. The time course of bioconversion was determined by HPLC. 0.5 mL of enzyme (in 0.1 M Tris buffer, pH 8.0) was incubated with 0.5 mL CPC, and the experimental conditions corresponded to 10 g L−1 (24 mM) CPC, 50 mg L−1 purified enzyme variants. The reaction temperature was set at 37 °C.Results and discussion
Identification of a starting point for computational design
The triple mutant M31βF/H57βS/H70βS reported by Pollegioni et al.3 is a very successful variant, which increased the catalytic activity by four-fold when compared with that of the single mutant M31βF.9 Therefore, this triple mutant was initially chosen as the starting template for computational design. The computed binding geometry between the transition state (TS) of CPC and the active site of the triple mutant (M31βF/H57βS/H70βS) is presented in Fig. S1.† We observed that the side chain of CPC adopts a high intrinsic energy conformation even though the mutation H57βS has made the active pocket of N176 larger to accommodate the bulky amino adipyl group of CPC. Single-point mutation prediction using PRODA based on the triple mutant (M31βF/H57βS/H70βS) indicated that the mutation F58βN could facilitate an extended conformation of the side chain of CPC because this mutation would further increase the size of the active pocket of N176. We added this mutation F58βN to obtain the quadruple mutant (M31βF/H57βS/F58βN/H70βS). The computed binding geometry of this quadruple mutant is shown in Fig. S2,† and its calculated free energies and kinetic parameters are provided in Table 1. Fig. S2† shows the formation of an excellent hydrogen-bonding network between the quadruple mutant and the side chain of CPC. However, the catalytic efficiency (Vmax/Km = 0.083 U mg−1 mM−1) of the quadruple mutant (M31βF/H57βS/F58βN/H70βS) decreased more than 60% when compared with that of the triple mutant (Vmax/Km = 0.227 U mg−1 mM−1). According to the crystal structure of N176 (PDB ID: 4HSR), residues H57β and F58β connect a long loop (V49β to H57β) with a β-strand (F58β to H60β) inside the active pocket. Thus, mutation of both H57β and F58β may destabilize the scaffold and even lead to partial collapse of the local backbone structure. Therefore, the mutation H57βS was removed from the triple mutant to yield the double mutant (M31βF/H70βS), and this mutant was taken as the final starting template for computational enzyme design. The catalytic efficiency (Vmax/Km = 0.151 U mg−1 mM−1) of this double mutant is given in Table 1, where this mutant is labeled as M1. The computed binding geometry between mutant M1 and CPC is shown in Fig. 2(a).| Variants | Mutations | PRODA ΔΔGBind (kcal mol−1) | PRODA ΔΔGFold (kcal mol−1) | Vmax (U mg−1) | Km (mM) | Vmax/Km (U mg−1 mM−1) |
|---|---|---|---|---|---|---|
| a The mutants were firstly reported by Pollegioni et al. (2005), but the corresponding kinetic parameters here were determined by this work.b The mutant was firstly reported by Conti et al. (2014), but the corresponding kinetic parameters here were determined by this work. | ||||||
| M1a | M31βF/H70βS | 0.00 | 0.00 | 1.84 | 12.25 | 0.151 |
| M2 | M31βF/H57βS/F58βN/H70βS | −7.77 | 21.36 | 0.82 | 9.90 | 0.083 |
| M3a | M31βF/H57βS/H70βS | −7.63 | 14.86 | 2.87 | 12.65 | 0.227 |
| M4 | M31βF/F58βN/H70βS | −1.83 | 12.90 | 2.26 | 9.88 | 0.228 |
| M5 | M31βF/F58βN/H70βS/I176βQ | −7.68 | 19.91 | 0.69 | 12.42 | 0.056 |
| M6 | M31βF/F58βN/H70βS/I176βT | −2.31 | 14.59 | 3.31 | 8.12 | 0.408 |
| M7b | M31βF/H57βS/H70βS/L154βY | −8.05 | 26.56 | 2.41 | 7.40 | 0.325 |
 | ||
| Fig. 2 Computed binding geometry between the TS of CPC and the active sites of the (a) triple mutant M3 (M31βF/H57βS/H70βS) and (b) double mutant M1 (M31βF/H70βS). The protein structure of N176 is shown as cartoon, where α- and β- chains are colored in wheat and green, respectively. Residues are labeled with black text, the O, N, C and S atoms are colored in red, slate, gray and orange, respectively. The hydrogen bonds are shown by dotted green lines, and the distances between donors and acceptors are shown in Å. | ||
Computational selection of variants with improved catalytic activities
The design scheme for sequence optimization is shown in Table S1.† A total of 312 sequences containing single or double mutations based on the starting template were predicted by PRODA, and the binding energy changes (ΔΔGBind) and folding energy changes (ΔΔGFold) of those mutants were calculated and shown in Tables S4–S6.† The single mutation F58βN showed a ΔΔGBind = −1.83 kcal mol−1 and the corresponding mutant was labeled as M4 (M31βF/F58βN/H70βS) in Table 1. The computed binding geometry between mutant M4 and CPC is shown in Fig. 2(b). Experimental results showed that the kinetic efficiency (Vmax/Km = 0.228 U mg−1 mM−1) of mutant M4 afforded a 1.5-fold increase in efficiency over the starting template. The kinetic efficiency increase of mutant M4 was attributed partly to the space enlargement of the introduced mutation F58βN, which allows the active pocket of N176 to accommodate the bulky side chain of CPC. Moreover, the mutation of a hydrophobic residue to a hydrophilic type has changed the electrostatic environment inside the active pocket of N176, and this mutation helps to stabilize the charged side chain of CPC during the binding process, as that shown in Fig. 2(b). To further increase the catalytic efficiency of N176 towards the hydrolysis of cephalosporin C, we examined the predicted mutants that included the mutation F58βN. Two double mutants F58βN/I176βQ and F58βN/I176βT, which are labeled as M5 and M6 in Table 1, were selected from the generated sequences because their calculated binding free energies decreased by −7.68 kcal mol−1 and −2.31 kcal mol−1, respectively. The computed binding geometry between the mutant M5 and CPC is presented in Fig. 3(a). A perfect hydrogen-bonding network formed between the amide group of the mutated residue Q176β and the amino adipyl group of CPC. However, I176βQ turned out to be a false positive, with only 25% of catalytic activity (Vmax/Km = 0.056 U mg−1 mM−1) retained. The overdesign of the hydrogen-bonding network in mutant M5 was attributed to the molecular mechanics models for protein design always softening the van der Waals repulsion to mitigate the adverse effect of conformation discretion.31 The experimental results of mutant M6 proved that I176βT was an ideal mutation, with a 2.7-fold increase in catalytic efficiency (Vmax/Km = 0.408 U mg−1 mM−1) when compared with that of the starting template. From the predicted binding geometry shown in Fig. 3(b), an additional hydrogen bond was observed to form between the carboxyl group of CPC and the hydroxyl group of the mutated residue Tβ176. Moreover, a possible water molecule can be accommodated perfectly between the amino group of CPC and the amide group of N58β, thereby constructing a water-mediated hydrogen bond. These additional interactions improve the local electrostatic environment to stabilize the TS of CPC. But, the water-mediated hydrogen bonding interaction still cannot be treated by the current energy function of PRODA. Therefore, the calculated binding energy change of M6 is higher than those of mutants M2, M3, M5 and M7 shown in Table 1 though its catalytic efficiency is the best.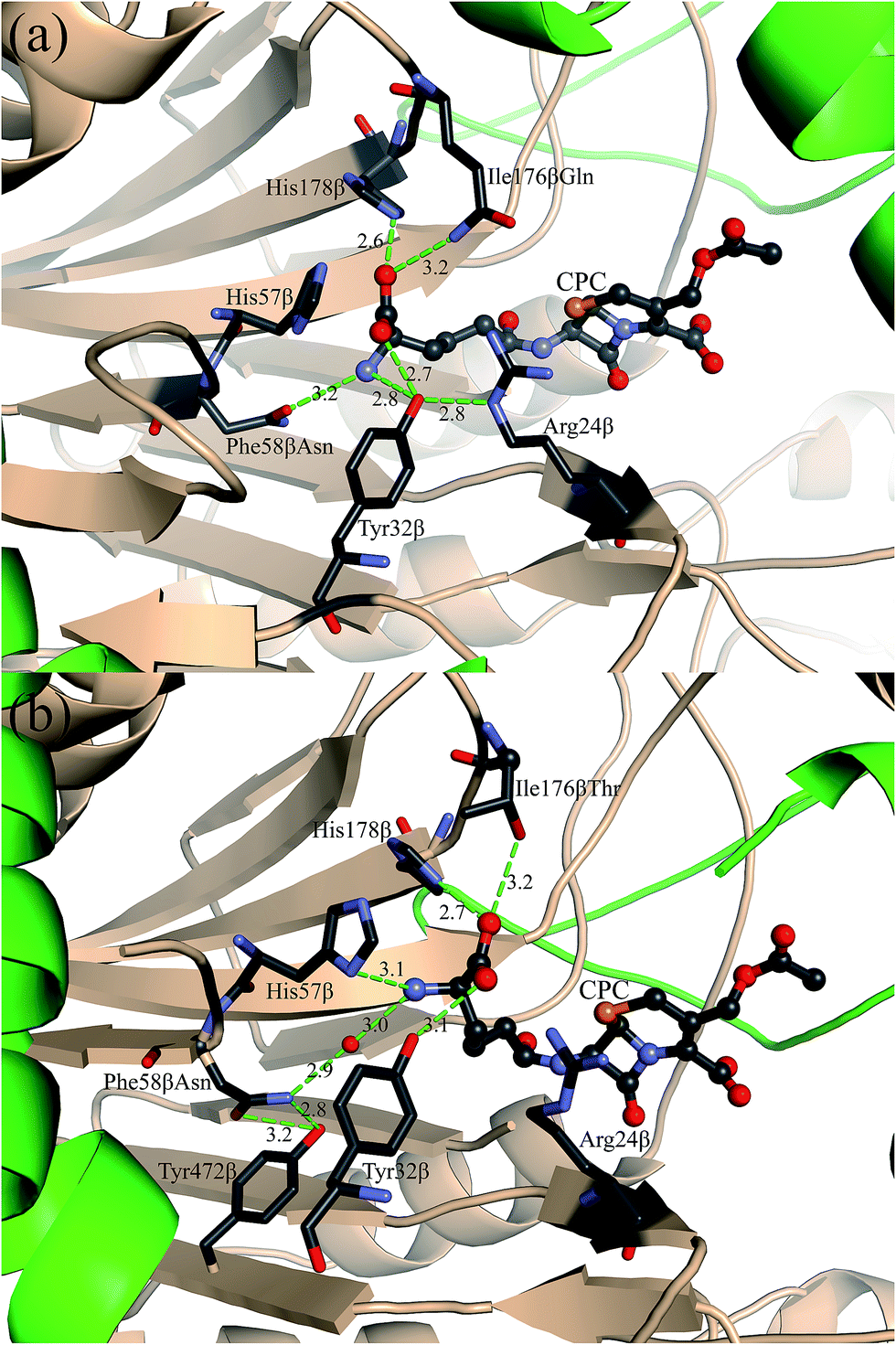 | ||
| Fig. 3 Computed binding geometry between the TS of CPC and the active site of the quadruple mutant (a) M5 (M31βF/F58βN/H70βS/I176βQ) and (b) M6 (M31βF/F58βN/H70βS/I176βT). The protein structure of N176 is shown as cartoon, where α- and β- chains are colored in wheat and green, respectively. The predicted geometries of CPC is shown as ball and stick models, while the residues are shown as stick models. CPC and all residues are labeled with black text, and the inserted water molecule is labeled with Wat. The O, N, C and S atoms are colored in red, slate, gray and orange, respectively. The hydrogen bonds are shown by dotted green lines, and the distances between donors and acceptors are shown in Å. | ||
Evaluation of mutants using cephalosporin C bioconversion
To evaluate the production abilities of the best CPC acylase variants obtained in this study and those reported in the literature, bioconversion time course experiments using the enzymes in the free form were performed under conditions resembling those used in industrial application. The quadruple mutant (M31βF/H57βS/H70βS/L154βY) reported by Conti et al.11 is considered to be the variant with highest conversion of CPC to 7-ACA under industrial conditions. This mutant is labeled M7 in Table 1 and its catalytic efficiency (Vmax/Km = 0.325 U mg−1 mM−1) was higher than that of M4, but lower than that of M6. The time course results for mutants M4 and M6 designed in this research, and mutants M3 and M7 reported in the literature are presented in Fig. 4, and the HPLC chromatographs of bioconversion reactions at 5 h catalyzed by these mutants are shown in Fig. S3.† The long conversion time of CPC catalyzed by mutant M4 is higher than that catalyzed by mutant M7; however, the kinetic efficiency of M7 is higher than that of M4. This may arise from substrate and product inhibition effects under industrial conditions for mutant M7. The results presented in Fig. 4 suggest that the highest production of 7-ACA was obtained using mutant M6, which is the quadruple mutant (M31βF/F58βN/H70βS/I176βT) designed in this research, and the time course experimental results are consistent with the kinetic parameters shown in Table 1, because the kinetic efficiency of mutant M6 was the highest among all mutants tested. | ||
| Fig. 4 Time course of 7-ACA production by variants of (■) M3 (M31βF/H57βS/H70βS), (●) M4 (M31βF/F58βN/H70βS), (▲) M6 (M31βF/F58βN/H70βS/I176βT) and (★) M7 (M31βF/H57βS/H70βS/L154βY). The experimental conditions are: 0.1 M Tris–HCl buffer (pH 8.0), 10 g L−1 (24 mM) CPC, 50 μg mL−1 purified enzyme variants. The reaction temperature is set at 37 °C. The error bars are smaller than the symbol used when not shown. | ||
The natural template of cephalosporin C acylase from N176 was extensively investigated using various directed evolution technologies before and after its crystal structure was measured, hundreds of mutants were screened experimentally to identify variants with increased catalytic activities.3,10,11 However, the best mutant M6 (M31βF/F58βN/H70βS/I176βT) which was computationally designed in this research was not found by directed evolution methods. The computational success of identifying mutant M6 was attributed mainly to the efficient search in the huge sequence and conformation space modeled by PRODA. The extensive conformation sampling for the computational design of cephalosporin C acylase described in the method section has arrived at a hyper-scale combinatorial optimization problem, whose computational complexity even reached 1.43 × 10170. The number of combinations of this virtual screening is far larger than that of the wet-lab based high-throughput screening. Finally, this combinatorial optimization problem was effectively addressed by the DEE/LP/MILP (Dead End Elimination/Linear Programming/Mixed-Integer Linear Programming) combined algorithm based on the accurate molecular modeling of the protein-ligand interactions in PRODA,23–27 which led to the discovery of the best mutant M6 (M31βF/F58βN/H70βS/I176βT).
Conclusion
In this research, we have shown that one cephalosporin C acylase mutants with improved catalytic activities was computationally predicted by PRODA, and its high catalytic efficiency was confirmed by measured kinetic parameters and time course experiments under industrial conditions. The results indicate that the identified quadruple mutant (M31βF/F58βN/H70βS/I176βT) is a promising cephalosporin C acylase that may aid the development of a one-step enzymatic process for CPC conversion to 7-ACA under industrial conditions.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (Grant Numbers: 21276136, 21476123), and the National High Technology Research and Development (863) Program of China (Grant Number: 2012AA021204). This work was also supported by a NSFC-DFG joint project (Grant number: 21311130420) between China and Germany for the international cooperation and exchange.References
- G. Volpato, R. C. Rodrigues and R. Fernandez-Lafuente, Curr. Med. Chem., 2010, 17, 3855–3873 CrossRef CAS PubMed.
- L. Pollegioni, E. Rosini and G. Molla, Appl. Microbiol. Biotechnol., 2013, 97, 2341–2355 CrossRef CAS PubMed.
- L. Pollegioni, S. Lorenzi, E. Rosini, G. L. Marcone, G. Molla, R. Verga, W. Cabri and M. S. Pilone, Protein Sci., 2005, 14, 3064–3076 CrossRef CAS PubMed.
- Y. Kim, K. Yoon, Y. Khang, S. Turley and W. G. J. Hol, Structure, 2000, 8, 1059–1068 CrossRef CAS PubMed.
- B. Oh, M. Kim, J. Yoon, K. Chung, Y. Shin, D. Lee and Y. Kim, Biochem. Biophys. Res. Commun., 2003, 310, 19–27 CrossRef CAS PubMed.
- Y. Kim and W. G. Hol, Chem. Biol., 2001, 8, 1253–1264 CrossRef CAS PubMed.
- H. C. Shin, J. Y. Jeon, K. H. Jung, M. R. Park and Y. Kim, WO 2005/014821, 2005.
- I. Aramori, M. Fukagawa, M. Tsumura, M. Iwami, T. Isogai, H. Ono, Y. Ishitani, H. Kojo, M. Kohsaka, Y. Ueda and H. Imanaka, J. Ferment. Bioeng., 1991, 72, 232–243 CrossRef CAS.
- Y. Ishii, Y. Saito, T. Fujimura, H. Sasaki, Y. Noguchi, H. Yamada, M. Niwa and K. Shimomura, Eur. J. Biochem., 1995, 230, 773–778 CrossRef CAS PubMed.
- E. Golden, R. Paterson, W. J. Tie, A. Anandan, G. Flematti, G. Molla, E. Rosini, L. Pollegioni and A. Vrielink, Biochem. J., 2013, 451, 217–226 CrossRef CAS PubMed.
- G. Conti, L. Pollegioni, G. Molla and E. Rosini, FEBS J., 2014, 281, 2443–2455 CrossRef CAS PubMed.
- D. N. Bolon and S. L. Mayo, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 14274–14279 CrossRef CAS PubMed.
- L. Jiang, E. A. Althoff, F. R. Clemente, L. Doyle, D. Röthlisberger, A. Zanghellini, J. L. Gallaher, J. L. Betker, F. Tanaka, C. F. Barbas, D. Hilvert, K. N. Houk, B. L. Stoddard and D. Baker, Science, 2008, 319, 1387–1391 CrossRef CAS PubMed.
- D. Röthlisberger, O. Khersonsky, A. M. Wollacott, L. Jiang, J. DeChancie, J. Betker, J. L. Gallaher, E. A. Althoff, A. Zanghellini, O. Dym, S. Albeck, K. N. Houk, D. S. Tawfik and D. Baker, Nature, 2008, 453, 190–195 CrossRef PubMed.
- J. B. Siegel, A. Zanghellini, H. M. Lovick, G. Kiss, A. R. Lambert, J. L. S. Clair, J. L. Gallaher, D. Hilvert, M. H. Gelb, B. L. Stoddard, K. N. Houk, F. E. Michael and D. Baker, Science, 2010, 329, 309–313 CrossRef CAS PubMed.
- S. R. Gordon, E. J. Stanley, S. Wolf, A. Toland, S. J. Wu, D. Hadidi, J. H. Mills, D. Baker, I. S. Pultz and J. B. Siegel, J. Am. Chem. Soc., 2012, 134, 20513–20520 CrossRef CAS PubMed.
- S. D. Khare, Y. Kipnis, P. Greisen Jr, R. Takeuchi, Y. Ashani, M. Goldsmith, Y. Song, J. L. Gallaher, I. Silman, H. Leader, J. L. Sussman, B. L. Stoddard, D. S. Tawfik and D. Baker, Nat. Chem. Biol., 2012, 8, 294–300 CrossRef CAS PubMed.
- F. Richter, R. Blomberg, S. D. Khare, G. Kiss, A. P. Kuzin, A. J. T. Smith, J. Gallaher, Z. Pianowski, R. C. Helgeson, A. Grjasnow, R. Xiao, J. Seetharaman, M. Su, S. Vorobiev, S. Lew, F. Forouhar, G. J. Kornhaber, J. F. Hunt, G. T. Montelione, L. Tong, K. N. Houk, D. Hilvert and D. Baker, J. Am. Chem. Soc., 2012, 134, 16197–16206 CrossRef CAS PubMed.
- S. Bjelic, L. G. Nivón, N. Çelebi-Ölçüm, G. Kiss, C. F. Rosewall, H. M. Lovick, E. L. Ingalls, J. L. Gallaher, J. Seetharaman, S. Lew, G. T. Montelione, J. F. Hunt, F. E. Michael, K. N. Houk and D. Baker, ACS Chem. Biol., 2013, 8, 749–757 CrossRef CAS PubMed.
- J. B. Siegel, A. L. Smith, S. Poust, A. J. Wargacki, A. Bar-Even, C. Louw, B. W. Shen, C. B. Eiben, H. M. Tran, E. Noor, J. L. Gallaher, J. Bale, Y. Yoshikuni, M. H. Gelb, J. D. Keasling, B. L. Stoddard, M. E. Lidstrom and D. Baker, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 3704–3709 CAS.
- H. J. Wijma, R. J. Floor, S. Bjelic, S. J. Marrink, D. Baker and D. B. Janssen, Angew. Chem., Int. Ed., 2015, 54, 3726–3730 CrossRef CAS PubMed.
- I. V. Pavlidis, M. S. Weiß, M. Genz, P. Spurr, S. P. Hanlon, B. Wirz, H. Iding and U. T. Bornscheuer, Nat. Chem., 2016, 8, 1076–1082 CrossRef CAS PubMed.
- Y. Lei, W. Luo and Y. Zhu, Protein Sci., 2011, 20, 1566–1575 CrossRef CAS PubMed.
- X. Huang, K. Han and Y. Zhu, Protein Sci., 2013, 22, 929–941 CrossRef CAS PubMed.
- Y. Tian, X. Huang and Y. Zhu, J. Mol. Model., 2015, 21, 191 CrossRef PubMed.
- X. Huang, J. Xue, M. Lin and Y. Zhu, PLoS One, 2016, 11, e0156559 Search PubMed.
- Y. Tian, X. Huang, Q. Li and Y. Zhu, Appl. Microbiol. Biotechnol., 2017, 101, 621–632 CrossRef CAS PubMed.
- A. D. MacKerell, D. Bashford, M. Bellott, R. L. Dunbrack, J. D. Evanseck, M. J. Field, S. Fischer, J. Gao, H. Guo, S. Ha, D. Joseph-McCarthy, L. Kuchnir, K. Kuczera, F. T. K. Lau, C. Mattos, S. Michnick, T. Ngo, D. T. Nguyen, B. Prodhom, W. E. Reiher, B. Roux, M. Schlenkrich, J. C. Smith, R. Stote, J. Straub, M. Watanabe, J. Wiorkiewicz-Kuczera, D. Yin and M. Karplus, J. Phys. Chem. B, 1998, 102, 3586–3616 CrossRef CAS PubMed.
- Z. Xiang and B. Honig, J. Mol. Biol., 2001, 311, 421–430 CrossRef CAS PubMed.
- M. M. Bradford, Anal. Biochem., 1976, 72, 248–254 CrossRef CAS PubMed.
- M. J. O'Meara, A. Leaver-Fay, M. D. Tyka, A. Stein, K. Houlihan, F. DiMaio, P. Bradley, T. Kortemme, D. Baker, J. Snoeyink and B. Kuhlman, J. Chem. Theory Comput., 2015, 11, 609–622 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra04597b |
| This journal is © The Royal Society of Chemistry 2017 |