
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Heteroaryl iminothioindoxyl (HA-ITI) photoswitches via regioselective aza-Wittig synthesis: unifying red-shifted absorption, large E/Z band separation, and tunable thermal recovery
Jialei
Chen-Wu
 a,
Carlos
Benitez-Martin
*bc,
José A.
González-Delgado
a,
Flip
de Jong
d,
Eduard
Fron
de,
Gert
Steurs
df,
Antonio J.
Martínez-Martínez
a,
Francisco
Nájera
gh,
Morten
Grøtli
*b,
Johan
Hofkens
*di,
Joakim
Andréasson
*c and
Uwe
Pischel
*a
a,
Carlos
Benitez-Martin
*bc,
José A.
González-Delgado
a,
Flip
de Jong
d,
Eduard
Fron
de,
Gert
Steurs
df,
Antonio J.
Martínez-Martínez
a,
Francisco
Nájera
gh,
Morten
Grøtli
*b,
Johan
Hofkens
*di,
Joakim
Andréasson
*c and
Uwe
Pischel
*a
aCIQSO – Center for Research in Sustainable Chemistry and Department of Chemistry, University of Huelva, Campus de El Carmen s/n, E-21071 Huelva, Spain. E-mail: uwe.pischel@diq.uhu.es
bDepartment of Chemistry and Molecular Biology, University of Gothenburg, 40530 Göteborg, Sweden. E-mail: carlos.benitez-martin@gu.se; grotli@chem.gu.se
cChemistry and Chemical Engineering, Chemistry and Biochemistry, Chalmers University of Technology, 41296 Göteborg, Sweden. E-mail: a-son@chalmers.se
dKU Leuven, Department of Chemistry, Celestijnenlaan 200F, B-3001 Leuven, Belgium. E-mail: johan.hofkens@kuleuven.be
eKU Leuven, Core Facility for Advanced Spectroscopy, Celestijnenlaan 200F, B-3001 Leuven, Belgium
fKU Leuven, Core Facility for Liquid-State NMR Spectroscopy, Celestijnenlaan 200F, B-3001 Leuven, Belgium
gDepartamento de Química Orgánica, Universidad de Málaga, Andalucía-Tech Campus Teatinos s/n, Málaga ES-29071, Spain
hInstituto de Investigación Biomédica de Málaga y Plataforma en Nanomedicina–IBIMA, Plataforma Bionand, Parque Tecnológico de Andalucía, Málaga ES-29590, Spain
iMax Planck Institute for Polymer Research, Ackermannweg 10, 55128 Mainz, Germany
First published on 24th December 2025
Abstract
Herein we introduce heteroaryl-substituted iminothioindoxyl (HA-ITI) photoswitches featuring N-heterocyclic moieties based on indole or benzimidazole. A regioselective aza-Wittig reaction of thioisatin, previously unexplored for the synthesis of imino-based photoswitches, enabled the obtention of HA-ITI derivatives and provides an alternative and versatile synthetic access to similar analogues. HA-ITIs undergo blue light-induced Z → E photoisomerization, followed by very fast thermal E → Z back isomerization at room temperature (µs to ms timescale; resolved by transient absorption spectroscopy). Irradiation at low temperature (<200 K) provided clear proof for the reversible T-type switching of HA-ITI. This behavior was corroborated by the changes in the UV/vis absorption spectra, with λabs values around 470 nm for the thermodynamically more stable Z form and ca. 525–550 nm for the metastable E isomer. Additional compelling evidence for the Z → E photoisomerization was obtained by NMR spectroscopy of samples irradiated in situ at low temperature. The combination of NMR data, single-crystal X-ray structures, and density functional theory calculations allowed the identification of both inter- and intramolecular interactions (chalcogen and hydrogen bonding), which are present in the Z and E isomers. Balancing these interactions dictates the differential performance of the switches, resulting in the significant kinetic stabilization, from µs to ms, of the E isomer in the benzimidazole-containing photoswitch. These findings establish HA-ITIs as a modular photoswitch platform with highly desirable and tunable photochemical features, thereby broadening the synthetic and conceptual landscape of heteroaryl photoactive systems.
Introduction
Light as an external stimulus of molecular systems gains its attractiveness from the possibility of spatiotemporal control, i.e., when and where light is applied. In this respect, the use of reversibly actuating photoswitches is regarded as a prime choice. Photoswitches are in many cases toggled between two states, which are commonly characterized by significant differences in their physicochemical properties, such as electronic conjugation, dipole moment, or acid/base behavior, among others. This is observed, for example, in diarylethenes,1,2 spiropyrans,3 fulgides4 or donor–acceptor Stenhouse adducts.5–7 These photoswitches operate based on ring opening/closing processes, often implying electrocyclic reactions. However, pronounced structural modification upon photoswitching is more inherent for E/Z isomerization, with azobenzenes being an archetypal example.8 Whether involving electronic variations, geometrical changes, or both simultaneously, the usefulness of photoswitches is evidenced by many applications including molecular systems for data storage and processing,9–12 sensing,13 imaging,14–18 catalysis,19 photopharmacology,20–22 or light-responsive release.23–29 In the particular context of E/Z photoswitches, the recent years have seen a growing interest in the diversification of this toolbox. This has led to the systematic investigation of hemi(thio)indigo-derived systems30–33 and their related Schiff-base variations,34–37 hydrazones,38–44 or imines.45–50 Furthermore, the integration of heterocyclic moieties in E/Z photoswitches, such as in azaheteroarenes,51–55 was shown to be beneficial toward the fine-tuning of optical properties and bistability. However, combining fast T-type thermal recovery with large E/Z band separation and red-shifted absorption features remains a largely unmet challenge in the design of imine-based photoswitches.Recent works by the groups of Newhouse56 and Dube31 are of particular importance for the present investigation (Fig. 1a). They have shown that the integration of N-heteroaryl moieties into hemithioindigos (HTI) leads to significant modulation of the reversible switching by photonic stimuli, that is, their P-type photoswitching behavior. The affected photochemical properties include the red-shift of the absorption spectra and the E/Z band separation (data for selected examples can be found in the SI; Table S1). These attributes enable the application of low-energy photons, which are potentially less harmful in biological environments, and improve the selective addressability of both isomeric forms. In parallel, Feringa, Buma, and Szymański have demonstrated that the aryl-substituted iminothioindoxyl (ITI) platform (Fig. 1b), which is chemically related to the HTI photoswitch family, behaves as a T-type photoswitch (i.e., the back isomerization is thermally activated) with large band separation and fast thermal E → Z conversion.34 Fast thermal recycling is particularly advantageous for applications where real-time responsiveness is targeted by the photoswitches.57 Inspired by these seminal findings, we envisaged that installing N-heteroaryl residues onto the ITI platform would yield a new class of robust and tunable T-type reversible photoswitches (heteroaryl iminothioindoxyls, HA-ITIs), joining fast thermal switching and substantial E/Z absorption band separation together with red-shifted spectral features (Fig. 1c). HA-ITIs are accessed by a so far unexplored regioselective aza-Wittig reaction, which circumvents the use of typically unstable nitroso reagents for the incorporation of N-heteroaryl units (indole and benzimidazole). This synthetic route additionally unlocks a broader chemical space for imino-based T-type photoswitches and related molecular architectures. Altogether, the herein obtained results establish the HA-ITI platform as a new design paradigm for photofunctional imino-based materials, pairing a synthetically versatile access route with fast, visible-light-responsive, and tunable switching.
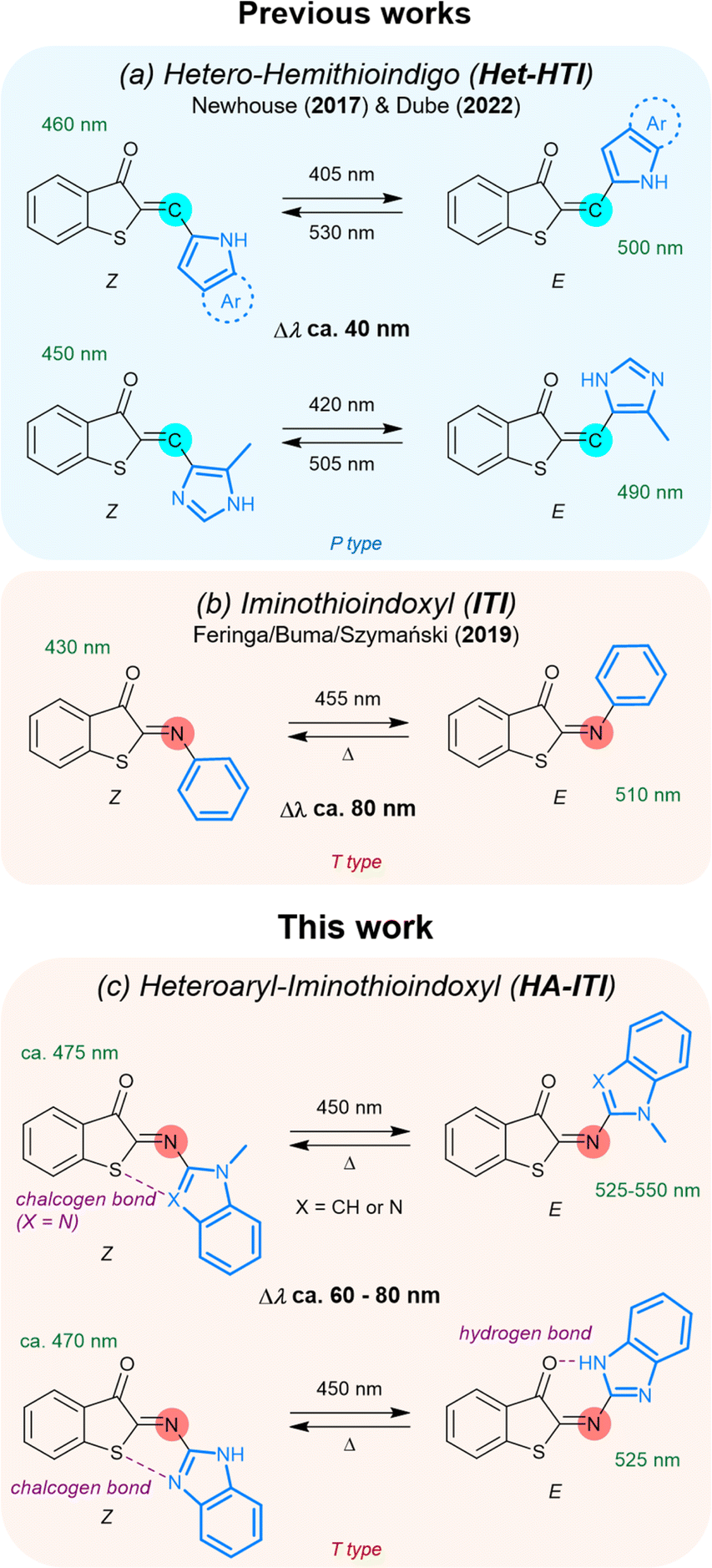 | ||
| Fig. 1 Comparison of heteroaryl-substituted iminothioindoxyl (HA-ITI) with related photoswitches from the previous literature. (a) Hetero-hemithioindigo (Het-HTI) systems reported by the Newhouse (2017) and Dube groups (2022), incorporating various nitrogen-containing heterocycles, behave as P-type photoswitches that can be addressed by blue/green light and the spectral shift between Z and E isomers (Δλ) is about 40 nm. (b) Iminothioindoxyl (ITI) systems reported by the Feringa/Buma/Szymański groups (2019) feature Z → E photoisomerization by irradiation at 455 nm and rapid thermal back-isomerization (T-type switching). The band separation between Z and E isomers (Δλ) is ca. 80–100 nm. (c) Light-induced Z → E photoisomerization of heteroaryl-substituted iminothioindoxyl (HA-ITI) reversible photoswitches. Representative examples show T-type switching behavior triggered by blue light and thermal back-isomerization. Substituents bearing nitrogen-containing heterocycles (e.g., indole, benzimidazole) facilitate supramolecular interactions in the Z or E isomer, including intramolecular chalcogen bonding (Z isomer) and hydrogen bonding (E isomer). The photoswitches display pronounced UV/vis absorption shifts upon isomerization (Δλ ≈ 60–80 nm). Wavelengths refer to absorption maxima of the corresponding isomers. | ||
Results and discussion
In the following, we will describe the synthesis and structural characterization of several HA-ITI photoswitches. Evidence of reversible Z ⇌ E T-type photoswitching is provided by irradiation at low temperatures, monitored by UV/vis absorption and 1H NMR spectroscopy. These findings are further supported by transient absorption spectroscopy at room temperature and theoretical calculations.Synthesis and characterization of HA-ITI photoswitches
It has been reported that ITI structures can be prepared by two strategies: (a) the base-catalyzed condensation of nitroso derivatives with thioindoxyl58 and (b) a [4 + 1] cycloaddition involving 2-mercaptobenzaldehydes and isocyanides.59 We focused initially on strategy (a), which had been employed successfully by the Feringa, Buma, and Szymański groups.34 However, the reaction with the relatively unstable 2-nitrosoindole was not successful in our case, and only decomposition products were obtained after workup.Hence, we changed our approach and decided to develop an alternative strategy for preparing the photoswitches 6–8 (Scheme 1). We found that an aza-Wittig reaction between benzo[b]thiophene-2,3-dione (thioisatin) and the aza-ylides 3–5 (ref. 60 and 61) (Scheme 1) showed a remarkable selectivity for the reaction at the carbonyl groups neighboring the sulfur atom, providing the desired photoswitches in moderate to good isolated yields (15–65%). The regioselectivity is analogous to the observations made by Jørgensen and Albrecht for conventional Wittig reactions of thioisatin.62 Importantly, this regioselective aza-Wittig transformation of thioisatin has not been described before for any photoactive imine system and therefore provides a synthetically distinct entry point compared to previously reported ITI derivatives obtained from nitroso intermediates. Because the final products were obtained in high purity and sufficient quantity, no further efforts were undertaken to optimize the reaction conditions. The final products were characterized by 1H and 13C NMR spectroscopy as well as by high-resolution mass spectrometry (HRMS). The results confirmed the chemical identity and purity of the compounds (see the SI; Fig. S2–S22).
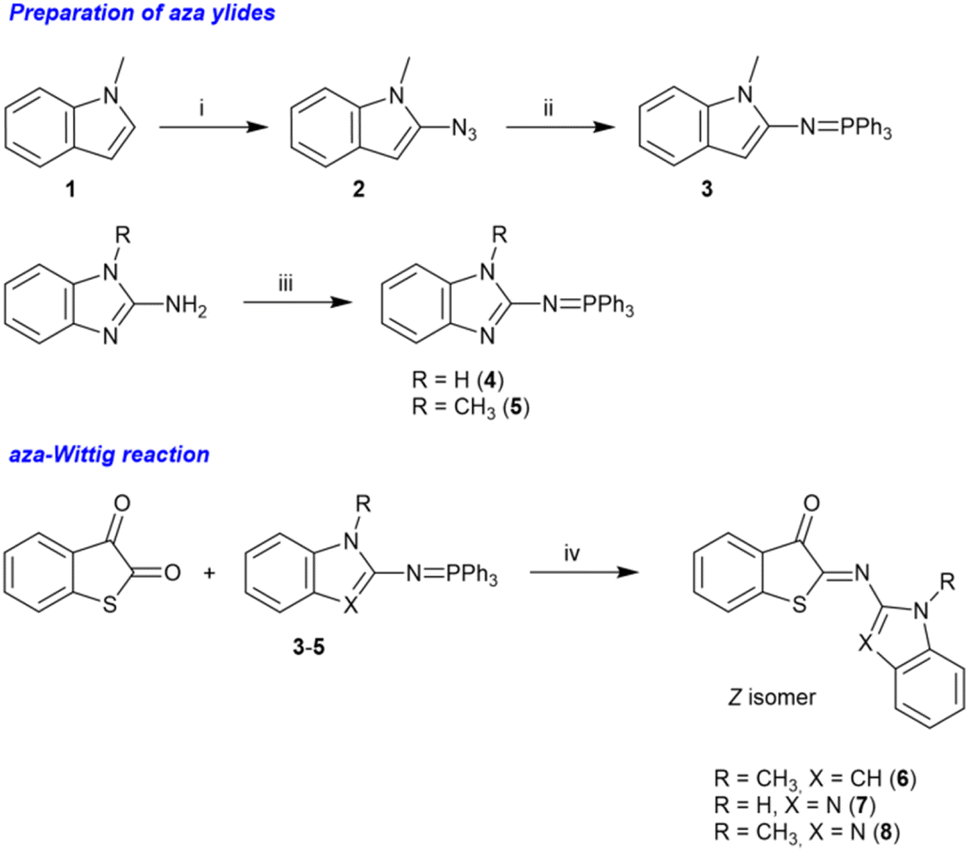 | ||
| Scheme 1 Synthesis of HA-ITI photoswitches 6–8. Reagents and reaction conditions: (i) TsN3, n-BuLi, Et2O, reflux, 70% yield; (ii) PPh3, Et2O, 0 °C, 30% yield; (iii) Ph3PBr2, toluene, Et3N, reflux, 72–80% yield; (iv) toluene, 90 °C, overnight, 15–65% yield. | ||
Furthermore, single crystals of X-ray diffraction quality were obtained by slow diffusion of pentane into a solution of the photoswitch in dichloromethane (6) or in tetrahydrofuran (7 or 8). From the resolved structures (Fig. 2 and S47–S49 in the SI), it can be safely concluded that the thermodynamically stable isomers are of Z configuration. This fits with general observations made previously for ITI and Het-HTI photoswitches,31,34,37,56 and also matches with the theoretical calculations (see below).
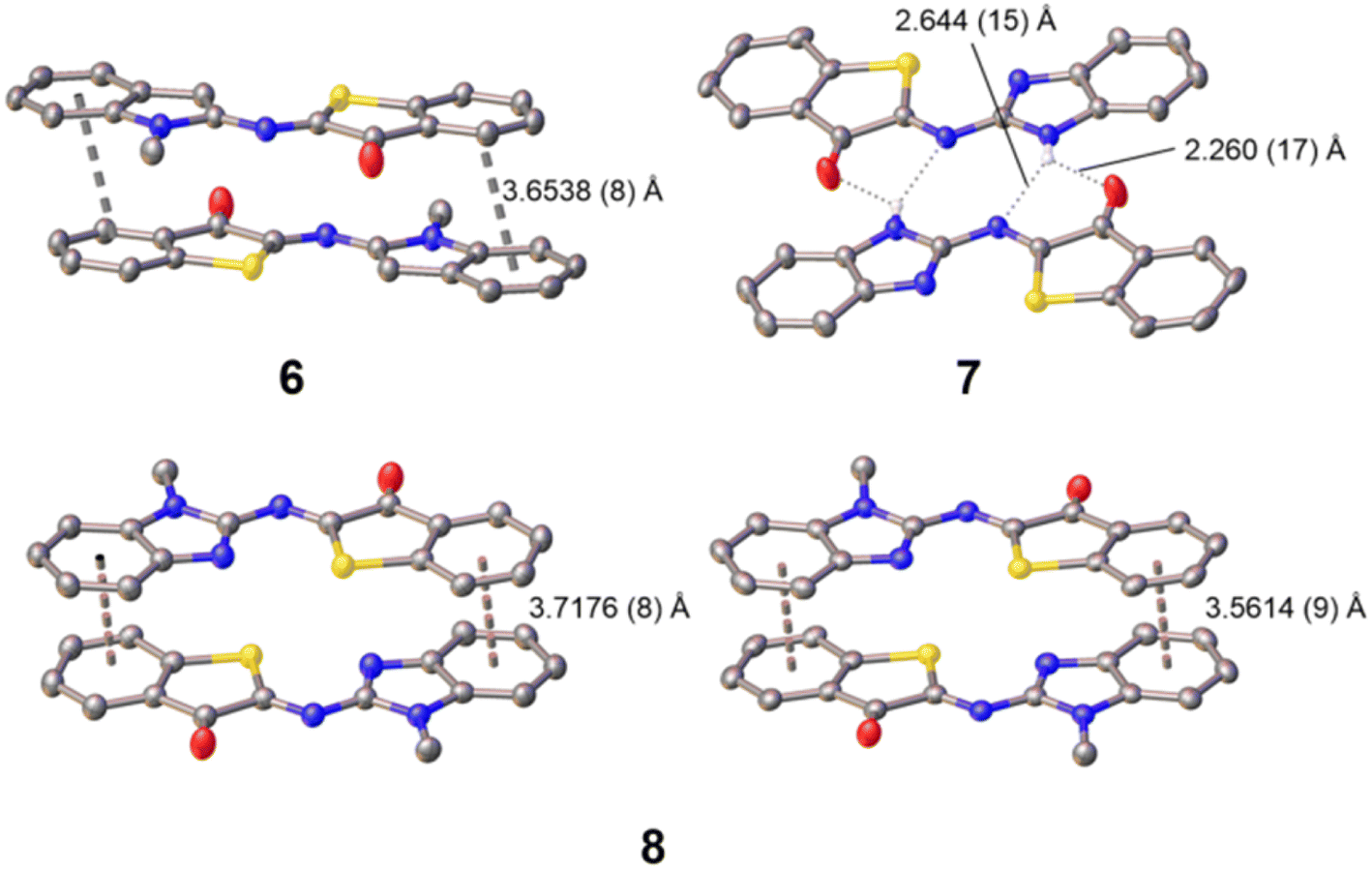 | ||
| Fig. 2 Single-crystal X-ray structures of 6–8 and their intermolecular supramolecular assembly through π-stacking (6 and 8) and H-bonding (7). More details can be found in the SI – Fig. S47–S49 and Tables S5–S7. CCDC: 6 – 2431430, 7 – 2431431; 8 – 2431432). | ||
Notably, among the several possible conformers, Z-6 crystallized with the methyl group pointing away from the sulfur atom, likely due to steric reasons and the occurrence of a close S⋯H–C (indole) interaction (S⋯H distance of 2.4736 (5) Å). For Z-7 and Z-8, it is worth noting that the rotamer with the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N (heterocycle) nitrogen orienting toward the sulfur atom is the one preferred out of all the possible rotamers of the benzimidazole moiety. Both S and N approach closely to a distance of 2.6735 (12) Å (Z-7) and 2.7652 (10) (Z-8; average for two independent molecules found in the asymmetric unit cell), pointing to the occurrence of S⋯N chalcogen bonding. Similar observations were recently made for an imidazole-derived Het-HTI.31 As for Z-6, this means that the N–CH3 group in Z-8 is pointing away from the sulfur, thereby avoiding a steric clash. All three photoswitches show intermolecular interactions. Z-7 forms pre-organized “head-to-tail” dimers, held together by complementary intermolecular hydrogen bonds of N–H with OC [O⋯H distance of 2.260 (17) Å] and NC [N⋯H distance of 2.644 (15) Å], but no further higher-order organization by π–π stacking is observed. Finally, the nearly planar Z-6 and Z-8 form π–π stacked “head-to-tail” dimers with an anti orientation of the carbonyl groups. In fact, for Z-8 two non-equivalent π–π dimers with slightly different packing are observed (Fig. 2). The inter-planar distance is ca. 3.6–3.7 Å. It should be noted that none of the dimers propagates further, and no π-chains are formed.
N (heterocycle) nitrogen orienting toward the sulfur atom is the one preferred out of all the possible rotamers of the benzimidazole moiety. Both S and N approach closely to a distance of 2.6735 (12) Å (Z-7) and 2.7652 (10) (Z-8; average for two independent molecules found in the asymmetric unit cell), pointing to the occurrence of S⋯N chalcogen bonding. Similar observations were recently made for an imidazole-derived Het-HTI.31 As for Z-6, this means that the N–CH3 group in Z-8 is pointing away from the sulfur, thereby avoiding a steric clash. All three photoswitches show intermolecular interactions. Z-7 forms pre-organized “head-to-tail” dimers, held together by complementary intermolecular hydrogen bonds of N–H with OC [O⋯H distance of 2.260 (17) Å] and NC [N⋯H distance of 2.644 (15) Å], but no further higher-order organization by π–π stacking is observed. Finally, the nearly planar Z-6 and Z-8 form π–π stacked “head-to-tail” dimers with an anti orientation of the carbonyl groups. In fact, for Z-8 two non-equivalent π–π dimers with slightly different packing are observed (Fig. 2). The inter-planar distance is ca. 3.6–3.7 Å. It should be noted that none of the dimers propagates further, and no π-chains are formed.
UV/vis absorption spectroscopy of the Z isomer
The HA-ITI photoswitches 6–8 in their Z form are characterized by a long-wavelength absorption band with a maximum at around 470 nm (Table 1 and Fig. 3). It is noteworthy that HA-ITI photoswitches show a considerable red-shift of about 40 nm compared to the ITI analogues.34 This hints at the involvement of the heterocyclic moiety in the molecular orbitals participating in the transition, which was verified in the theoretical calculations (see below). Based on the structural analogy with ITI photoswitches the long-wavelength absorption band is ascribed to a π, π* transition.34 As verified for Z-7, the spectral position of the absorption maximum is not significantly dependent on the solvent, featuring minor shifts no larger than 8 nm (λmax: 467 nm in acetonitrile and 475 nm in DMSO; the corresponding spectra can be found in Fig. S23 in the SI).| λ abs (nm) Za [ε (M−1cm−1)] | λ abs (nm) | τ (ms) E → Zd | E a (kJ mol−1) | |||
|---|---|---|---|---|---|---|
| E | E | E → Ze | E → Zf | |||
| a Absorption maximum of the Z isomer, determined by conventional UV/vis absorption spectroscopy in MTHF. b Absorption maximum of the E isomer, determined by low-temperature UV/vis absorption spectroscopy. c Absorption maximum of the E isomer, determined by transient absorption spectroscopy. d Time constant for the E → Z thermal back isomerization at 298 K, determined by transient absorption spectroscopy in MTHF. e Arrhenius activation energy for the E → Z thermal back isomerization in MTHF; extracted from low-temperature irradiation experiments with UV/vis absorption monitoring. f Arrhenius activation energy for the E → Z thermal back isomerization in tetrahydrofuran-d8 (THF-d8); extracted from low-temperature irradiation experiments with monitoring by 1H NMR spectroscopy. | ||||||
| 6 | 474 [20200] | 525 | ∼545 | 0.73 | 30.1 | 59.1 |
| 444 [14400] | ||||||
| 389 [7400] | ||||||
| 7 | 472 [16100] | 525 | 530 | 23.6 | 33.0 | 43.6 |
| 450 [14100] | ||||||
| 379 [10400] | ||||||
| 8 | 476 [16400] | 550 | ∼560 | 0.67 | 46.2 | 51.4 |
| 454 [14500] | ||||||
| 381 [10600] | ||||||
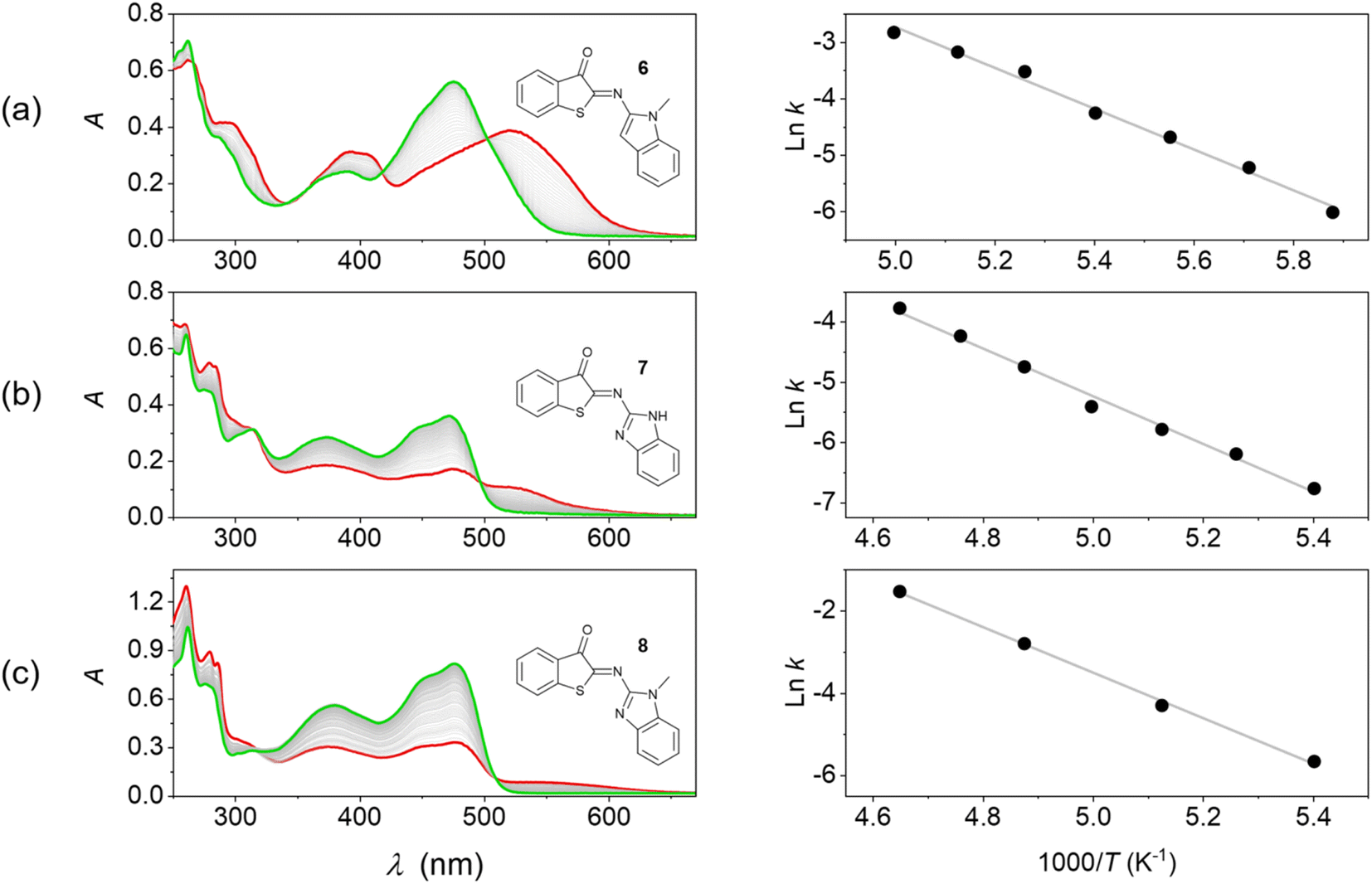 | ||
| Fig. 3 Thermal E → Z isomerization of HA-ITI photoswitches monitored by UV/vis absorption spectroscopy and Arrhenius analysis. (Left) Spectra of the thermal back isomerization of (a) 6 (at 170 K), (b) 7 (at 185 K), and (c) 8 (at 185 K) in MTHF. The green spectra correspond to the fully recovered signatures of the Z forms, while the red spectra correspond to the signature of the photothermal stationary state mixture (generated by irradiation at 375 nm). (Right) Corresponding Arrhenius plots. | ||
Photoswitching monitored by UV/vis absorption spectroscopy at low temperature
In a first attempt to test the photoswitching function of Z-6, Z-7, and Z-8, their 2-methyltetrahydrofuran (MTHF) solutions were subjected to irradiation with light at λ > 455 nm at room temperature. However, no changes were observed in the absorption spectra under such conditions. It is well known that imine-based photoswitches can undergo thermal back isomerization due to the possibility of inversion at the imine nitrogen atom, which was also observed for the ITI photoswitch platform.34 If the back isomerization is very fast at room temperature, this can explain the lack of observation of the E isomer under conventional experimental conditions. Therefore, we resorted to irradiation experiments (λexc = 375 nm) at low temperature (T < 220 K in MTHF). Indeed, under these conditions the thermally activated E → Z isomerization was slowed down sufficiently as to allow the observation of photoswitching (Fig. 3).In all cases the long-wavelength absorption of the Z form at ca. 470 nm diminished and new bathochromically displaced spectral features at wavelengths above 500 nm were detected, which are attributed to the formation of the corresponding E isomer. A substantial band separation of the Z and E isomers is a property that is often sought after, as it facilitates photoisomerization reactions in mainly one direction. The previously reported ITI photoswitches reach significant values of 80–100 nm,34 while our HA-ITI photoswitches feature a comparable band separation of 60–80 nm.
The spectral changes reverse in the dark and are characterized by several well-defined isosbestic points (Fig. 3), confirming the uniformity of the E → Z back isomerization process. Monitoring the kinetics and fitting them to monoexponential decays revealed lifetimes of several hundreds of seconds at low temperature (e.g., 414 s at 170 K for E-6, 885 s at 185 K for E-7, and 300 s at 185 K for E-8); Fig. S24–S38 in the SI.
The Arrhenius analysis of the temperature-dependent kinetic data revealed activation energies of 30.1, 33.0, and 46.2 kJ mol−1 for the E → Z isomerization of 6, 7, and 8, respectively. These numbers are in broad agreement with observations made for ITI photoswitches (Ea ∼55–60 kJ mol−1).34 Albeit associated with a large error, the extrapolation of these data provides a rough estimate of the expected timescale at room temperature on the order of tens of milliseconds. This is in accordance with the lack of observation of macroscopic conversion in conventional irradiation experiments at 298 K. More accurate kinetic data for the E → Z thermal back isomerization at room temperature were obtained from transient absorption studies and are shown in Table 1.
Study of the photoswitching by 1H NMR spectroscopy at low temperature
Having established the T-type photoswitching of HA-ITI by means of monitoring the changes in the UV/vis absorption spectra, we attempted to gain complementary insights by performing 1H NMR spectroscopic characterization of HA-ITI E- and Z-isomers at low temperature. For this purpose, samples of the HA-ITI photoswitches in THF-d8 were irradiated at 450 nm directly in the NMR tube at temperatures of 180 K, 190 K, and 200 K. It is worth emphasizing that the compounds feature only a few diagnostic protons that could help to unambiguously establish the occurrence of the Z → E photoisomerization. However, in the case of HA-ITI photoswitch 6, the proton at the 3-position of the N-methylindole moiety offers a unique opportunity for E/Z assignment.In Fig. 4a, the structures of the Z form and the metastable E form of 6 are shown. Each of the configurational isomers may exist as rotamer I or II, among many other possible conformations. A simple visual inspection of the structures indicates that steric clashes involving the methyl group will likely disfavor rotamer II of the Z form and rotamer I of the E form. Hence, on the one hand, Z-6 is expected to be present as rotamer I, as it is the case in the solid-state structure that was established by X-ray crystallography (Fig. 2). On the other hand, the photogenerated E-6 is more likely present as rotamer II, where the proton at the 3-position of the N-methylindole part is close to the carbonyl group, lying in the nodal plane of the CO. This should yield a pronounced deshielding of this proton.63
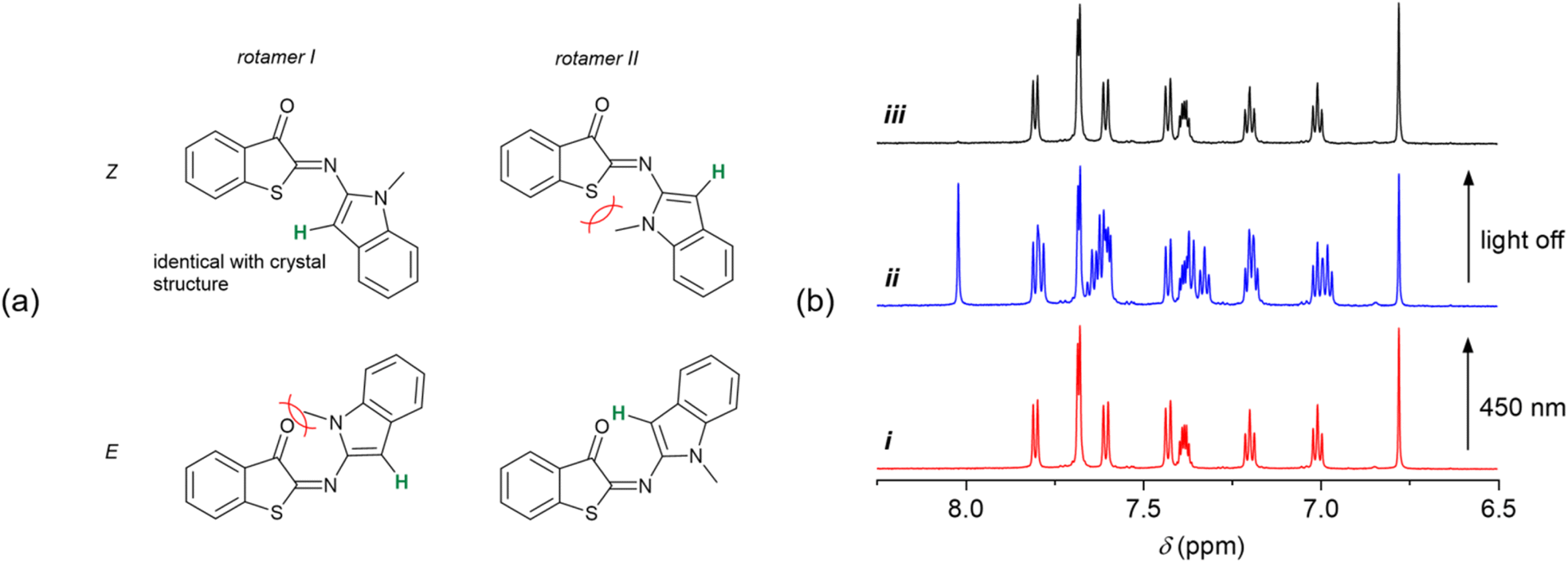 | ||
| Fig. 4 (a) Possible rotamers of the Z and E forms of HA-ITI photoswitch 6. The proton highlighted in green was used as diagnostic tool for rationalizing the formation of the E isomer upon irradiation. (b) Partial 1H NMR spectra of 6 in THF-d8 at 190 K, demonstrating the reversible T-type photoswitching. (i) Z-6 before irradiation, (ii) photothermal stationary state upon irradiation with 450-nm light (Z/E ca. 50/50), (iii) spectrum after thermal back isomerization in the dark, corresponding to Z-6. | ||
Importantly, these intuitively derived conformational preferences of photoswitch 6 are confirmed by the NMR experiment at 190 K. In Fig. 4b, the aromatic region of the 1H NMR spectra of HA-ITI 6 is shown (i) before, (ii) during continuous irradiation at 450 nm, and (iii) after switching off the light source. The signal at 6.78 ppm in the spectrum of Z-6 is ascribed to the proton at the 3-position of the N-methylindole moiety. Upon irradiation significant changes are observed, of which the most indicative is the emergence of a signal at 8.03 ppm. This signal is assigned to the aforementioned proton in the E form of the photoswitch. This pronounced deshielding by 1.25 ppm would only be expected in the E form of 6, thereby lending strong support for the Z → E photoisomerization. The integration of the signals yields ca. 50% Z → E conversion in the photothermal stationary state at 190 K. As expected, this value is even more favorable at lower temperature (180 K), i.e., ca. 78% Z → E conversion. For a higher temperature (200 K) the considerably faster thermal back isomerization yields a much lower Z → E conversion of ca. 10%; Fig. S40 in the SI. Upon irradiation of the other two HA-ITI photoswitches 7 and 8, pronounced changes were observed in their 1H NMR spectra; Fig. S41–S46 in the SI. However, the unambiguous assignment of these changes to the formation of the corresponding E isomers proved to be challenging, especially for HA-ITI 8. In the case of compound 7, the NH proton was considered a potential diagnostic marker. Interestingly, irradiation of Z-7 induced a notable upfield shift of this signal, from 12.79 to 11.67 ppm (Fig. S43 in the SI), which is contrary to expectations based on proximity to the nodal plane of the carbonyl π orbital or involvement in N–H⋯OC hydrogen bonding in the E isomer.
This unexpected behavior may be rationalized by steric constraints and less ideal alignment of the NH in E-7 and/or the initial presence of an intermolecular hydrogen-bonded dimer of the Z isomer, analogous to the arrangement observed in the solid-state structure of Z-7. In such dimer the NH proton signal may be more downfield shifted than in the case of intramolecular hydrogen bonding of NH in E-7. Despite the absence of definitive spectroscopic markers for E-7 and E-8, the robust evidence for Z → E photoisomerization in the structurally related HA-ITI 6 strongly suggests the occurrence of an analogous transformation in compounds 7 and 8. The photothermal stationary state distribution at 190 K points to significant Z → E photoisomerization for 7 (35%) and near-quantitative conversion for 8 (94%); data at other temperatures can be found in Tables S3 and S4 in the SI. It should be noted that a direct comparison of the conversion yields between the different photoswitches is complicated by the difficulty to guarantee exactly equal irradiation geometries and conditions in our setup (Fig. S1 in the SI). Importantly, all photoswitches demonstrated complete thermal reversibility upon cessation of irradiation (Fig. 4b).
Transient absorption spectroscopy (TAS)
TAS at the micro- to millisecond timescale enabled monitoring of the thermal E → Z back isomerization at room temperature (Fig. 5). These experiments were performed in MTHF for a better comparison with the low-temperature steady-state irradiations (Fig. 3).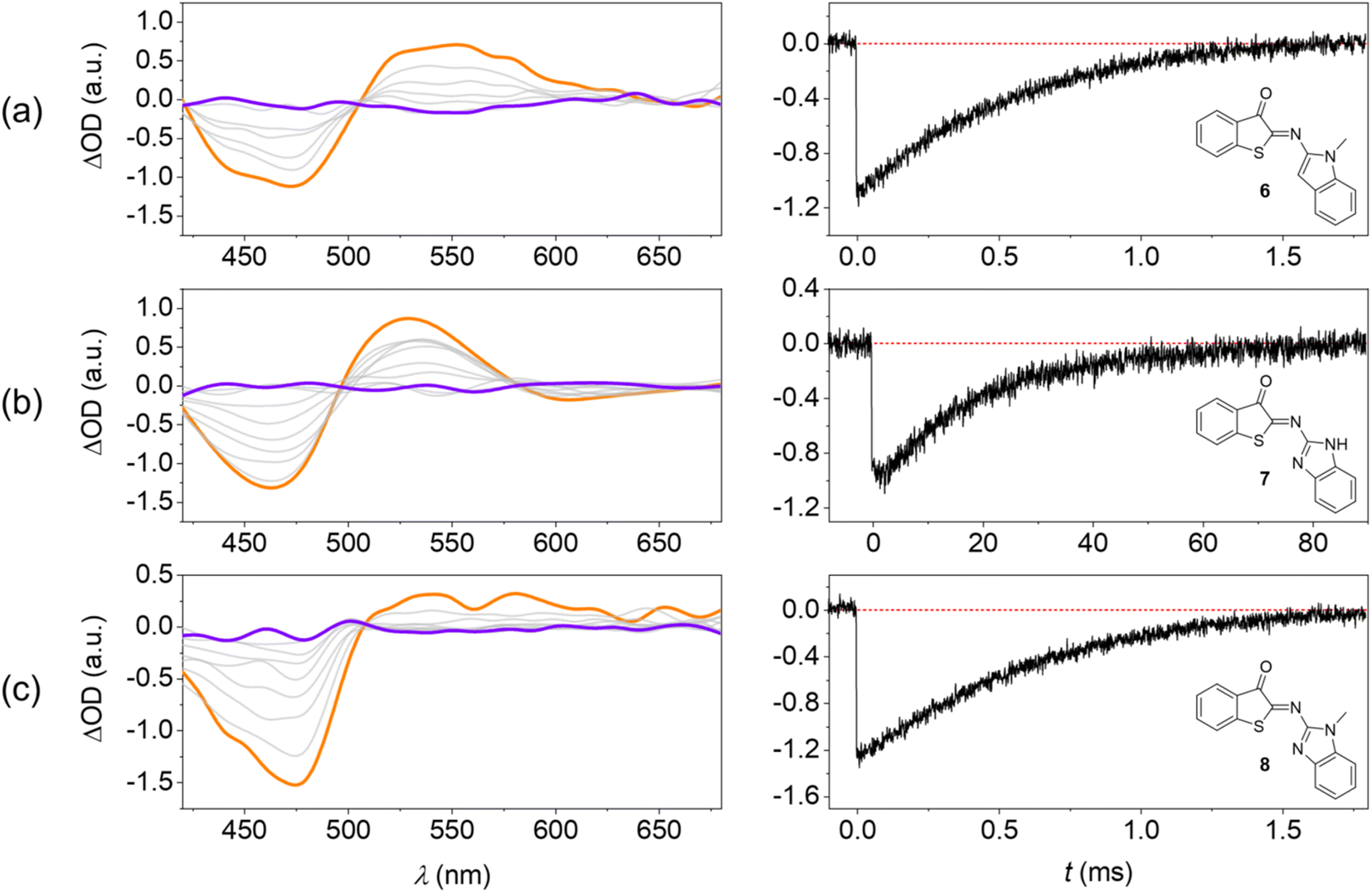 | ||
| Fig. 5 Transient absorption spectroscopy confirms fast thermal E → Z isomerization of HA-ITI photoswitches at room temperature. (Left) Transient absorption spectra of (a) 6, (b) 7, and (c) 8 in MTHF at room temperature; excitation at 355 nm, orange: spectrum after laser flash, purple: spectrum after complete ground-state recovery. (Right) Recovery kinetics of the Z form at 460 nm. | ||
The photoswitches show two transients: (i) a negative signal that coincides with the inverted long-wavelength absorption band in the 400–500 nm range and (ii) a positive signal in the 500–650 nm spectral window. The spectra cross at an isosbestic point at ca. 500 nm. On the one hand, signal (i) is unambiguously assigned to the ground-state bleaching (GSB) of the Z isomer. On the other hand, the spectral fingerprint of the transient (ii) coincides with the corresponding changes in the low-temperature irradiations and is therefore assigned to the E isomer.
Notably, the lifetime of the E isomer (τE→Z) in the thermal E → Z back isomerization at room temperature is readily determined from the recovery kinetics of the GSB (Fig. 5). For the photoswitches 6 and 8 this value was obtained as 730 µs and 670 µs, respectively. For HA-ITI 7, a decay two orders of magnitude slower is observed (τE→Z = 23.6 ms). The reason for this significant difference is assigned to the stabilization of E-7 by intramolecular N–H⋯OC hydrogen bonding. It is noteworthy that ITI photoswitches typically display lifetimes of tens of milliseconds for the E form. Recent attempts to control the kinetics of the thermal back-isomerization by means of electronic factors have shown that longer lifetimes (close to 100 ms) can be achieved.37 However, faster switching reaching the sub-millisecond regime was not attained by this strategy. Interestingly, the HA-ITI platform provides such characteristics, which may be particularly useful in applications where very fast turnover is an advantage. In addition, by drawing on tailored supramolecular interactions, such as in E-7, the lifetime can be tuned at will. Likely, the combination with electronic factors would allow for further extension of the lifetime range.
Unfortunately, the considerable spectral overlap of the E isomer with the Z isomer hampers the accurate determination of the quantum yield of the Z → E transformation of the HA-ITI photoswitches 6–8. However, it is certainly not unreasonable to assume similar efficiencies as observed for ITI34 or Het-HTI56 switches.
Theoretical calculations
To obtain additional insight into the relative stability of the Z and E isomers and their optical properties, density functional theory (DFT) calculations were performed. The ground-state structures were optimized at the M06-2X/6-311+G(d,p) level,64,65 including the solvation model based on density (SMD)66 for MTHF. This combination of exchange-correlation functional and solvation model was employed to describe ITI derivatives, based on its good accuracy for thermochemistry.34,37 For all investigated HA-ITI photoswitches 6–8, the Z isomer is more stable than the E isomer by approximately 21.3–25.9 kJ mol−1. This configurational preference is in full agreement with the observations made for the X-ray crystallographic structures (Fig. 2). The Z isomers of all the photoswitches are planar, as well as the E isomers of 6 and 7. For the E isomer of compound 8 a distorted structure with a dihedral angle of 110° between the ITI and benzimidazole ring systems was identified.The optical properties of the Z and E isomers in MTHF (Table 2) were calculated by means of time-dependent density-functional theory [SMD(MTHF)/mPW1PW91/6-311+G(d,p)].67 The S0 → S1 transition, corresponding to the lowest-energy absorption band, implies mainly the frontier molecular orbitals (FMOs) HOMO and LUMO. As a general observation, the HOMO is preferentially located on the N-heterocyclic moiety, while the LUMO can be found on the thioindoxyl residue (Fig. 6a, S50 and S52 in the SI). The calculated energies for the S0 → S1 transition are in very good agreement with the experimental values, deviating maximally by 0.14 eV. It is worth noting that, as observed for ITIs, the photophysical properties of our derivatives are not significantly dependent on the solvent (see above) despite featuring an intermediate, in some cases pronounced, charge-transfer excited state. This notion suggests that HA-ITIs are not subjected to the classical limitations of time-dependent density-functional theory, in analogy to ITIs.34,68 Thus, the use of the mPW1PW91 functional, having a certain exchange-correlation character and better reproducing the experimental results, was preferred.
| Oscillator strength f | FMO contribution | E calc (eV) (S0 → S1) | E exp (eV) (S0 → S1) | |
|---|---|---|---|---|
| Z-6 | 0.6682 | HOMO → LUMO (99%) | 2.48 | 2.62 |
| E-6 | 0.6097 | HOMO → LUMO (99%) | 2.27 | 2.27 |
| Z-7 | 0.5699 | HOMO → LUMO (92%) | 2.62 | 2.63 |
| E-7 | 0.5146 | HOMO → LUMO (95%) | 2.28 | 2.34 |
| Z-8 | 0.5932 | HOMO → LUMO (93%) | 2.60 | 2.60 |
| E-8 | 0.0924 | HOMO → LUMO (93%) | 2.10 | 2.21 |
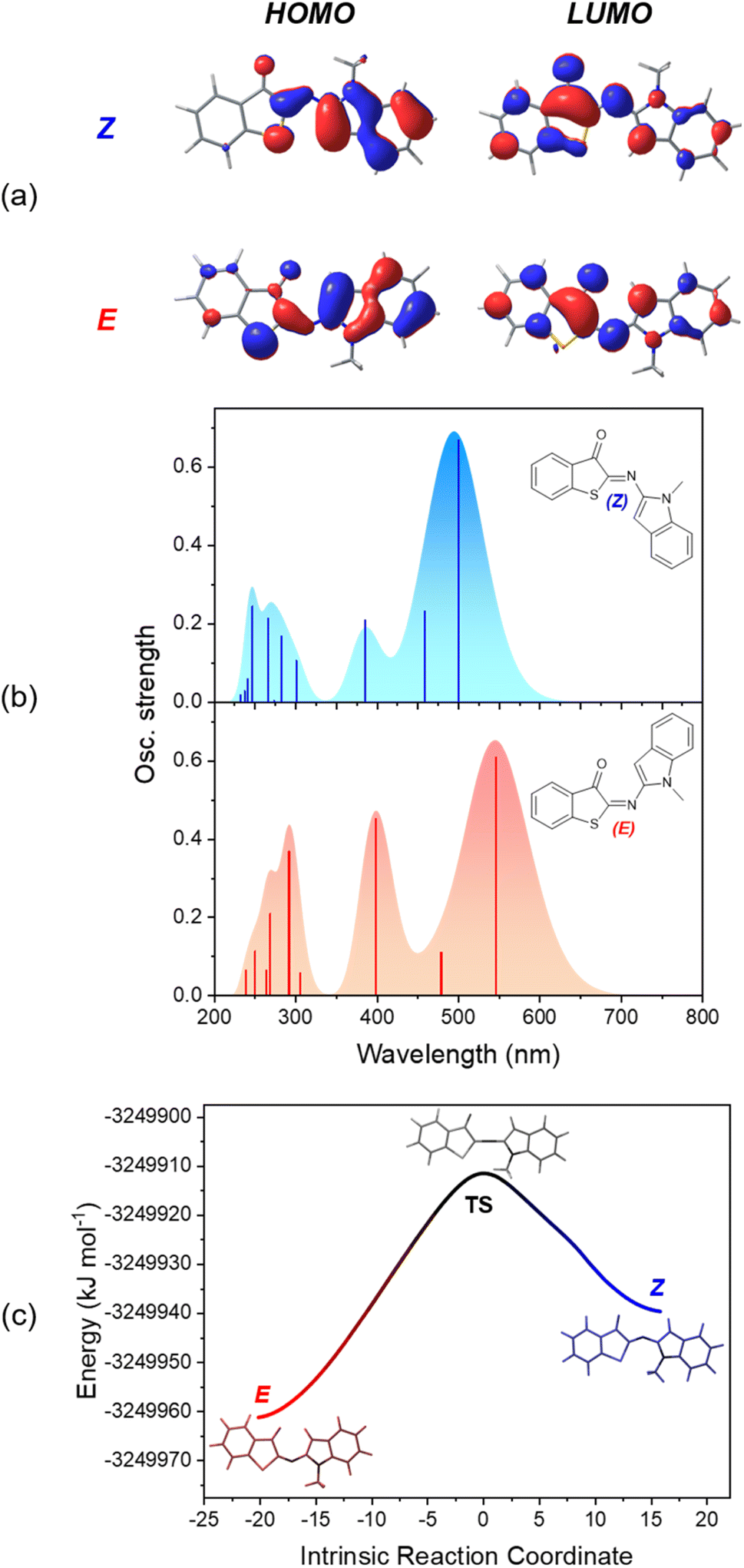 | ||
| Fig. 6 (a) HOMO and LUMO density plots for Z- and E-6. (b) Calculated absorption spectra of both isomers (Z-6: blue, E-6: red). (c) Calculated E → Z ground-state isomerization path for photoswitch 6. Note that the herein obtained rotamer of the Z isomer easily converts to the structure that was obtained for the crystal by rotation around the C–N σ bond (activation energy of ca. 3.8 kJ mol−1; Fig. S56 in the SI). | ||
The data confirm the experimental notion that the HA-ITI platform functions as a positive photoswitch, with the thermodynamically less stable E isomer absorbing at longer wavelengths than the Z isomer (Fig. 6b, S51 and S53 in the SI). Notably, the comparatively small calculated oscillator strength of the E isomer of compound 8 accounts for its relatively weak absorption signal observed in both TAS and low-temperature irradiation experiments (Fig. 3 and 5).
Finally, the reaction path for the thermal E → Z isomerization was mapped [SMD(MTHF)/M06-2X/6-311+G(d,p)]; Fig. 6c for photoswitch 6 and Fig. S54 and S55 in the SI for data for 7 and 8. The intrinsic reaction coordinate is correlated with the change of the CN–C inversion angle. In the transition state an angle of ca. 175–180° is observed for all three photoswitches. These calculations enabled us to extract activation energies for the E → Z isomerization, which were found to be in qualitative agreement with the experimentally determined values (Table 1): 49.8 kJ mol−1 for 6, 57.4 kJ mol−1 for 7, and 49.0 kJ mol−1 for 8. It is noteworthy that the calculated activation energies reflect also the enhanced lifetime of the E-7. This was not observed for the experimentally determined values, which are subjected to a higher uncertainty.
Conclusions
The integration of N-heterocyclic moieties, such as indoles or benzimidazoles, with iminothioindoxyl (ITI) photoswitches enables the pinpointed spectral tuning of the absorption properties of the Z and E forms, while maintaining the T-type character with fast thermal back isomerization. Specifically, the examples of heteroaryl iminothioindoxyl (HA-ITI), which were developed in this work and prepared by a so far unexplored aza-Wittig reaction strategy, show significantly red-shifted absorption spectra compared to ITI or hemithioindigo photoswitches. Importantly, a separation of the E/Z absorption bands of 60–80 nm was observed. Irradiation of the thermodynamically stable Z form with blue light generates the metastable E form, which reverts on a µs to ms timescale in a thermally activated process with relatively low activation energies (30–60 kJ mol−1). The occurrence of intraswitch supramolecular interactions such as N⋯S chalcogen bonding and CO⋯H–C as well as CO⋯H–N hydrogen bonding interactions dictates switch-specific variations of performance characteristics. Especially interesting is the case of photoswitch 7, in which CO⋯H–N interaction occurs in the metastable E form. This enabled us to design a switch that shows ms thermal recovery of the Z isomer as opposed to the µs time scale for 6 and 8. To our knowledge, HA-ITIs are the first class of imine-based photoswitches to exhibit both fast T-type switching and a red-shifted, strongly resolved E/Z absorption profile in the visible range. These results not only expand the design space of imine-based photoswitches, but also establish HA-ITIs as promising candidates for integration into light-controlled molecular devices, optical logic circuits, or responsive therapeutic systems, among others.
Author contributions
J. C.-W.: data curation, formal analysis, investigation, methodology, validation, visualization, writing – review & editing. C. B.-M.: conceptualization, data curation, formal analysis, investigation, methodology, validation, visualization, writing – review & editing. J. A. G.-D.: formal analysis, investigation, methodology, visualization, writing – review & editing. F. d. J.: formal analysis, investigation, methodology, visualization, writing – review & editing. E. F.: formal analysis, investigation, methodology, visualization. A. J. M.-M.: formal analysis, investigation, methodology. F. N.: formal analysis, investigation, methodology. M. G.: funding acquisition, project administration, resources, supervision, writing – review & editing. J. H.: funding acquisition, project administration, resources, supervision. J. A.: funding acquisition, project administration, resources, supervision, writing – review & editing. U. P.: funding acquisition, project administration, resources, supervision, writing – original draft.Conflicts of interest
There are no conflicts to declare.Data availability
The datasets generated during and/or analyzed during the current study are available in the KU Leuven RDR repository, https://doi.org/10.48804/LXAEID. All data is available from the corresponding author upon reasonable request.CCDC 2431430–2431432 contain the supplementary crystallographic data for this paper.69a–c
Supplementary information (SI) is available. See DOI: https://doi.org/10.1039/d5sc08074f.
Acknowledgements
The authors are grateful for financial support from the European Innovation Council and SMEs Executive Agency (EISMEA) for the EIC PATHFINDER project 101098934 - 4 for 2. J. A. thanks the Swedish Research Council VR (project 2021-05311). A. J. M.-M. is indebted to the Spanish Ministry of Science, Innovation, and Universities (MCIU/AEI/10.13039/501100011033) and the European Regional Development Fund ERDF (grants PID2022-142270OB-I00 and CNS2022-136087). A. J. M.-M. also received funding from the University of Huelva (EPIT1442023). The NMR facilities at KU Leuven were supported by the Research Foundation Flanders (FWO) through infrastructure grant I002720N. Furthermore, we acknowledge the computer resources provided by the SCBI (Supercomputing and Bioinformatics Centre) of the University of Málaga. E. F., F. d. J., and J. C.-W. are grateful to Prof. em. Mark Van der Auweraer for stimulating discussions on several aspects of the work. Open Access funding provided by the Max Planck Society.References
- M. Irie, T. Fukaminato, K. Matsuda and S. Kobatake, Photochromism of Diarylethene Molecules and Crystals: Memories, Switches, and Actuators, Chem. Rev., 2014, 114, 12174–12277 CrossRef CAS.
- J. Zhang and H. Tian, The Endeavor of Diarylethenes: New Structures, High Performance, and Bright Future, Adv. Opt. Mater., 2018, 6, 1701278 Search PubMed.
- G. Berkovic, V. Krongauz and V. Weiss, Spiropyrans and Spirooxazines for Memories and Switches, Chem. Rev., 2000, 100, 1741–1753 Search PubMed.
- Y. Yokoyama, Fulgides for memories and switches, Chem. Rev., 2000, 100, 1717–1739 CrossRef CAS PubMed.
- M. M. Lerch, W. Szymański and B. L. Feringa, The (photo)chemistry of Stenhouse photoswitches: guiding principles and system design, Chem. Soc. Rev., 2018, 47, 1910–1937 RSC.
- M. Clerc, S. Sandlass, O. Rifaie-Graham, J. A. Peterson, N. Bruns, J. Read de Alaniz and L. F. Boesel, Visible light-responsive materials: the (photo)chemistry and applications of donor-acceptor Stenhouse adducts in polymer science, Chem. Soc. Rev., 2023, 52, 8245–8294 RSC.
- C. A. Reyes, A. Karr, C. A. Ramsperger, A. Talim, G. K, H. J. Lee and E. Picazo, Compartmentalizing Donor−Acceptor Stenhouse Adducts for Structure−Property Relationship Analysis, J. Am. Chem. Soc., 2025, 147, 10–26 Search PubMed.
- H. M. Dhammika Bandara and S. C. Burdette, Photoisomerization in different classes of azobenzene, Chem. Soc. Rev., 2012, 41, 1809–1825 RSC.
- J. Andréasson, U. Pischel, S. D. Straight, T. A. Moore, A. L. Moore and D. Gust, All-photonic multifunctional molecular logic device, J. Am. Chem. Soc., 2011, 133, 11641–11648 CrossRef.
- P. Remón, M. Bälter, S. M. Li, J. Andréasson and U. Pischel, An all-photonic molecule-based D flip-flop, J. Am. Chem. Soc., 2011, 133, 20742–20745 CrossRef.
- D. Gust, J. Andréasson, U. Pischel, T. A. Moore and A. L. Moore, Data and signal processing using photochromic molecules, Chem. Commun., 2012, 48, 1947–1957 RSC.
- M. Bälter, S. M. Li, J. R. Nilsson, J. Andréasson and U. Pischel, An All-Photonic Molecule-Based Parity Generator/Checker for Error Detection in Data Transmission, J. Am. Chem. Soc., 2013, 135, 10230–10233 CrossRef.
- M. Natali and S. Giordani, Molecular switches as photocontrollable ‘‘smart’’ receptors, Chem. Soc. Rev., 2012, 41, 4010–4029 RSC.
- C. Benitez-Martin, S. Li, A. Dominguez-Alfaro, F. Najera, E. Pérez-Inestrosa, U. Pischel and J. Andréasson, Toward Two-Photon Absorbing Dyes with Unusually Potentiated Nonlinear Fluorescence Response, J. Am. Chem. Soc., 2020, 142, 14854–14858 CrossRef CAS.
- G. Naren, W. Larsson, C. Benitez-Martin, S. Li, E. Perez-Inestrosa, B. Albinsson and J. Andréasson, Rapid amplitude-modulation of a diarylethene photoswitch: en route to contrast-enhanced fluorescence imaging, Chem. Sci., 2021, 12, 7073–7078 Search PubMed.
- D. Kim, A. Aktalay, N. Jensen, K. Uno, M. L. Bossi, V. N. Belov and S. W. Hell, Supramolecular Complex of Photochromic Diarylethene and Cucurbit[7]uril: Fluorescent Photoswitching System for Biolabeling and Imaging, J. Am. Chem. Soc., 2022, 144, 14235–14247 Search PubMed.
- M. Olesińska-Mönch and C. Deo, Small-molecule photoswitches for fluorescence bioimaging: engineering and applications, Chem. Commun., 2023, 59, 660–669 RSC.
- K.-X. Qin, Y.-S. Su, M.-Q. Zhu and C. Li, Recent Progress of Photoswitchable Fluorescent Diarylethenes for Bioimaging, ChemBioChem, 2024, 25, e202400326 CrossRef CAS PubMed.
- R. S. Stoll and S. Hecht, Artificial Light-Gated Catalyst Systems, Angew. Chem., Int. Ed., 2010, 49, 5054–5075 CrossRef CAS.
- W. A. Velema, W. Szymański and B. L. Feringa, Photopharmacology: beyond proof of principle, J. Am. Chem. Soc., 2014, 136, 2178–2191 Search PubMed.
- K. Hüll, J. Morstein and D. Trauner, In Vivo Photopharmacology, Chem. Rev., 2018, 118, 10710–10747 Search PubMed.
- P. Kobauri, F. J. Dekker, W. Szymański and B. L. Feringa, Rational Design in Photopharmacology with Molecular Photoswitches, Angew. Chem., Int. Ed., 2023, 62, e202300681 CrossRef CAS.
- M. Han, R. Michel, B. He, Y.-S. Chen, D. Stalke, M. John and G. H. Clever, Light-Triggered Guest Uptake and Release by a Photochromic Coordination Cage, Angew. Chem., Int. Ed., 2013, 52, 1319–1323 Search PubMed.
- J. del Barrio, S. T. J. Ryan, P. G. Jambrina, E. Rosta and O. A. Scherman, Light-Regulated Molecular Trafficking in a Synthetic Water-Soluble Host, J. Am. Chem. Soc., 2016, 138, 5745–5748 Search PubMed.
- S. T. J. Ryan, J. del Barrio, R. Suardíaz, D. F. Ryan, E. Rosta and O. A. Scherman, A Dynamic and Responsive Host in Action: Light-Controlled Molecular Encapsulation, Angew. Chem., Int. Ed., 2016, 55, 16096–16100 CrossRef CAS.
- J. Chen-Wu, P. Máximo, P. Remón, A. J. Parola, N. Basílio and U. Pischel, Phototransduction in a supramolecular cascade: a mimic for essential features of the vision process, Chem. Commun., 2023, 59, 3431–3434 Search PubMed.
- A. Ghosh, L. Slappendel, B.-N. T. Nguyen, L. K. S. von Krbek, T. K. Ronson, A. M. Castilla and J. R. Nitschke, Light-Powered Reversible Guest Release and Uptake from Zn4L4 Capsules, J. Am. Chem. Soc., 2023, 145, 3828–3832 Search PubMed.
- E. Benchimol, J. Tessarolo and G. H. Clever, Photoswitchable coordination cages, Nat. Chem., 2024, 16, 13–21 CrossRef CAS PubMed.
- M. Colaço, J. Ewert, J.-S. von Glasenapp, U. Pischel, R. Herges and N. Basílio, Diazocines as Guests of Cucurbituril Macrocycles: Light-Responsive Binding and Supramolecular Catalysis of Thermal Isomerization, J. Am. Chem. Soc., 2025, 147, 734–745 CrossRef.
- C. Petermayer and H. Dube, Indigoid photoswitches: visible light responsive molecular tools, Acc. Chem. Res., 2018, 51, 1153–1163 CrossRef CAS.
- V. Josef, F. Hampel and H. Dube, Heterocyclic Hemithioindigos: Highly Advantageous Properties as Molecular Photoswitches, Angew. Chem., Int. Ed., 2022, 61, e202210855 CrossRef CAS.
- A. Gernet, A. El Rhaz and L. Jean, Easily Accessible Substituted Heterocyclic Hemithioindigos as Bistable Molecular Photoswitches, Chem. – Eur. J., 2023, 29, e202301160 CrossRef CAS.
- M. Sacherer, F. Hampel and H. Dube, Diaryl-hemiindigos as visible light, pH, and heat responsive four-state switches and application in photochromic transparent polymers, Nat. Commun., 2023, 14, 4382 CrossRef CAS PubMed.
- M. W. H. Hoorens, M. Medved', A. D. Laurent, M. Di Donato, S. Fanetti, L. Slappendel, M. Hilbers, B. L. Feringa, W. J. Buma and W. Szymański, Iminothioindoxyl as a molecular photoswitch with 100 nm band separation in the visible range, Nat. Commun., 2019, 10, 2390 Search PubMed.
- S. Crespi, N. A. Simeth, M. Di Donato, S. Doria, C. N. Stindt, M. F. Hilbers, F. L. Kiss, R. Toyoda, S. Wesseling, W. J. Buma, B. L. Feringa and W. Szymański, Phenylimino Indolinone: A Green-Light-Responsive T-Type Photoswitch Exhibiting Negative Photochromism, Angew. Chem., Int. Ed., 2021, 60, 25290–25295 Search PubMed.
- M. Medved', M. W. H. Hoorens, M. D. Donato, A. D. Laurent, J. Fan, M. Taddei, M. Hilbers, B. L. Feringa, W. J. Buma and W. Szymański, Tailoring the optical and dynamic properties of iminothioindoxyl photoswitches through acidochromism, Chem. Sci., 2021, 12, 4588–4598 Search PubMed.
- M. E. Boëtius, M. W. H. Hoorens, M. Ošťadnický, A. D. Laurent, M. Di Donato, A. C. A. van Wingaarden, M. F. Hilbers, B. L. Feringa, W. J. Buma, M. Medved and W. Szymański, Getting a molecular grip on the half-lives of iminothioindoxyl photoswitches, Chem. Sci., 2024, 15, 14379–14389 RSC.
- M. N. Chaur, D. Collado and J.-M. Lehn, Configurational and Constitutional Information Storage: Multiple Dynamics in Systems Based on Pyridyl and Acyl Hydrazones, Chem. – Eur. J., 2011, 17, 248–258 CrossRef CAS.
- G. Vantomme, S. Jiang and J.-M. Lehn, Adaptation in Constitutional Dynamic Libraries and Networks, Switching between Orthogonal Metalloselection and Photoselection Processes, J. Am. Chem. Soc., 2014, 136, 9509–9518 CrossRef CAS PubMed.
- D. J. van Dijken, P. Kovaříček, S. P. Ihrig and S. Hecht, Acylhydrazones as Widely Tunable Photoswitches, J. Am. Chem. Soc., 2015, 137, 14982–14991 Search PubMed.
- H. Qian, S. Pramanik and I. Aprahamian, Photochromic Hydrazone Switches with Extremely Long Thermal Half-Lives, J. Am. Chem. Soc., 2017, 139, 9140–9143 Search PubMed.
- B. Shao, M. Baroncini, H. Qian, L. Bussotti, M. Di Donato, A. Credi and I. Aprahamian, Solution and Solid-State Emission Toggling of a Photochromic Hydrazone, J. Am. Chem. Soc., 2018, 140, 12323–12327 Search PubMed.
- B. Shao, H. Qian, Q. Li and I. Aprahamian, Structure Property Analysis of the Solution and Solid-State Properties of Bistable Photochromic Hydrazones, J. Am. Chem. Soc., 2019, 141, 8364–8371 CrossRef CAS PubMed.
- Q. Qi, Y. Liu, V. Puranik, S. Patra, Z. Svindrych, X. Gong, Z. She, Y. Zhang and I. Aprahamian, Photoswitchable Fluorescent Hydrazone for Super-Resolution Cell Membrane Imaging, J. Am. Chem. Soc., 2025, 147, 16404–16411 Search PubMed.
- L. Greb and J. M. Lehn, Light-driven molecular motors: Imines as four-step or two-step unidirectional rotors, J. Am. Chem. Soc., 2014, 136, 13114–13117 Search PubMed.
- J. Wu and J. L. Greenfeld, Photoswitchable Imines Drive Dynamic Covalent Systems to Nonequilibrium Steady States, J. Am. Chem. Soc., 2024, 146, 20720–20727 Search PubMed.
- J. Wu, L. Kreimendahl, S. Tao, O. Anhalt and J. L. Greenfield, Photoswitchable imines: aryliminopyrazoles quantitatively convert to long-lived Z-isomers with visible light, Chem. Sci., 2024, 15, 3872–3878 RSC.
- J. Wu and J. L. Greenfield, Harnessing light to drive a non-photoresponsive reaction out of equilibrium, Chem, 2025, 11, 102579 CAS.
- J. Wu, L. Kreimendahl and J. L. Greenfeld, Enhancing the Photoswitching Properties of N-Alkyl Imines, J. Am. Chem. Soc., 2025, 147, 17549–17554 Search PubMed.
- J. Wu, L. Kreimendahl and J. L. Greenfield, Switching Sides: Regiochemistry and Functionalization Dictate the Photoswitching Properties of Imines, Angew. Chem., Int. Ed., 2025, 64, e202415464 CrossRef CAS.
- C. E. Weston, R. D. Richardson, P. R. Haycock, A. J. P. White and M. J. Fuchter, Arylazopyrazoles: Azoheteroarene Photoswitches Offering Quantitative Isomerization and Long Thermal Half-Lives, J. Am. Chem. Soc., 2014, 136, 11878–11881 Search PubMed.
- L. Stricker, E.-C. Fritz, M. Peterlechner, N. L. Doltsinis and B. J. Ravoo, Arylazopyrazoles as Light-Responsive Molecular Switches in Cyclodextrin-Based Supramolecular Systems, J. Am. Chem. Soc., 2016, 138, 4547–4554 CrossRef CAS.
- J. Calbo, C. E. Weston, A. J. P. White, H. S. Rzepa, J. Contreras-García and M. J. Fuchter, Tuning Azoheteroarene Photoswitch Performance through Heteroaryl Design, J. Am. Chem. Soc., 2017, 139, 1261–1274 CrossRef CAS PubMed.
- Y. Xu, C. Gao, J. Andréasson and M. Grøtli, Synthesis and Photophysical Characterization of Azoheteroarenes, Org. Lett., 2018, 20, 4875–4879 CrossRef CAS PubMed.
- S. Crespi, N. A. Simeth and B. König, Heteroaryl azo dyes as molecular photoswitches, Nat. Rev., 2019, 3, 133–146 CAS.
- J. E. Zweig and T. R. Newhouse, Isomer-Specific Hydrogen Bonding as a Design Principle for Bidirectionally Quantitative and Redshifted Hemithioindigo Photoswitches, J. Am. Chem. Soc., 2017, 139, 10956–10959 Search PubMed.
- J. Garcia-Amorós, M. Díaz-Lobo, S. Nonell and D. Velasco, Fastest Thermal Isomerization of an Azobenzene for Nanosecond Photoswitching Applications under Physiological Conditions, Angew. Chem., Int. Ed., 2012, 51, 12820–12823 Search PubMed.
- R. Pummerer, Ü. Isatin-anile and D. des Thionaphtenchinons, Chem. Ber., 1910, 43, 1370–1376 Search PubMed.
- T. Soeta, S. Shitaya, T. Okuno, S. Fujinami and Y. Ukaji, Efficient synthesis of benzothiophenes by [4+1] cycloaddition of 2-mercaptobenzaldehyde derivatives with isocyanides, Tetrahedron, 2016, 72, 7901–7905 CrossRef CAS.
- P. Molina, A. Lorenzo and E. Aller, Iminophosphorane-Mediated Annulation of 1,3,5-Triazine to Benzimidazole: Synthesis of 1,3,5-Triazino[1,2-a]benzimidazoles, Synthesis, 1992, 297–302 Search PubMed.
- C. Bonini, M. Funicello and P. Spagnolo, Novel α-Carboline Synthesis Using Tandem aza-Wittig-Electrocyclization Reaction of Indol-2-yl Phosphorane with Enone, Synlett, 2006, 1574–1576 CAS.
- J. Stiller, D. Kowalczyk, H. Jiang, K. A. Jørgensen and Ł. Albrecht, Novel Organocatalytic Activation of Unmodified Morita-Baylis-Hillman Alcohols for the Synthesis of Bicyclic α-AlkylideneKetones, Chem. – Eur. J., 2014, 20, 13108–13112 CrossRef CAS PubMed.
- H. Günther, NMR Spectroscopy - Basic Principles, Concepts, and Applications in Chemistry, Wiley-VCH, Weinheim, 2013 Search PubMed.
- N. Mardirossian and M. Head-Gordon, Thirty years of density functional theory in computational chemistry: an overview and extensive assessment of 200 density functionals, Mol. Phys., 2017, 115, 2315–2372 CrossRef CAS.
- Y. Zhao and D. G. Truhlar, The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other functionals, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS.
- C. Adamo and V. Barone, Exchange functionals with improved long-range behavior and adiabatic connection methods without adjustable parameters: The mPW and mPW1PW models, J. Chem. Phys., 1998, 108, 664–675 CrossRef CAS.
- A. D. Laurent and D. Jacquemin, TD-DFT benchmarks: A review, Int. J. Quantum Chem., 2013, 113, 2019–2039 CrossRef CAS.
- (a) CCDC 2431430: Experimental Crystal Structure Determination, 2025, DOI:10.5517/ccdc.csd.cc2mm37m; (b) CCDC 2431431: Experimental Crystal Structure Determination, 2025, DOI:10.5517/ccdc.csd.cc2mm38n; (c) CCDC 2431432: Experimental Crystal Structure Determination, 2025, DOI:10.5517/ccdc.csd.cc2mm39p.
| This journal is © The Royal Society of Chemistry 2026 |