
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Mass transport-dependent in situ Raman detection in CO/CO2 electrolysis
Wen
Yan
a,
Hangyu
Bu
a,
Xinjuan
Du
a,
Beining
Xu
a,
Jia
Liu
*b and
Ming
Ma
 *a
*a
aSchool of Chemical Engineering and Technology, Xi'an Jiaotong University, Xi'an 710049, People's Republic of China. E-mail: mingma@xjtu.edu.cn
bInstrument Analysis Center, Xi'an Jiaotong University, Xi'an 710049, People's Republic of China. E-mail: liujia6j@xjtu.edu.cn
First published on 16th December 2025
Abstract
In situ Raman spectroscopy has been widely employed in the CO2/CO electroreduction field to extract mechanistic insights into reaction pathways toward product formation. However, most of the previous in situ Raman studies are based on H-type spectroelectrochemical cells, which constrain the mass transport to the catalyst surface, potentially affecting the coverage of key intermediates relevant to the Raman signals and even distorting the mechanistic understanding. Here, we present a systematic comparison for the in situ Raman detection of intermediates during CO2/CO reduction between an H-type spectroelectrochemical cell and a GDE-type spectroelectrochemical flow cell. We found that cell configurations exert a minimal influence on the in situ Raman detection and analysis of surface-adsorbed intermediates during CO2 reduction, which is likely linked to the high solubility of CO2 in aqueous media. In contrast, during CO reduction, the severe CO mass transport limitations in H-type spectroelectrochemical cells significantly lower the formation and coverage of key intermediates, resulting in in situ Raman detection and analysis results that are distinct from those obtained using GDE-type spectroelectrochemical flow cells. Thereby, circumventing mass transport limitation is crucial for in situ Raman tests to unveil the underlying mechanism of electrolysis.
Introduction
Electrocatalytic CO2/CO reduction to high-value fuels and chemicals is widely recognized as a promising strategy for achieving a sustainable carbon cycle.1–4 To date, Cu remains the only monometallic catalyst that is capable of directly converting CO2/CO into valuable oxygenates and hydrocarbons.5,6 However, its limited selectivity toward specific multi-carbon (C2+) product at commercially-relevant current densities poses a significant challenge for practical applications.7,8 Previous studies have shown that the dynamic evolution of *CO intermediates on the Cu catalyst surface, including adsorption configurations,9–11 surface coverage,12,13 and C–C coupling pathways,14,15 is crucial in governing the selectivity for C2+ products. To better understand the reaction mechanism of C2+ product formation and achieve controllable C2+ selectivity, many attempts have focused on the detection of the transient evolution of reaction key intermediates and the dynamic restructuring of active sites during electrocatalysis.16 In this context, in situ spectroscopic techniques are urgently needed to monitor the microscopic interfacial processes at the catalyst surface in real-time.9,17–22In situ infrared (IR) and Raman spectroscopy, which are two widely employed techniques for probing molecular vibrational characteristics, have been extensively used to investigate the dynamic behavior of surface species and reaction mechanisms in electrocatalysis.18,20,23–30 Among the two techniques, in situ IR spectroscopy with its high signal-to-noise ratio and excellent temporal resolution can precisely track the dynamic evolution of reaction intermediates,18,25,27,28,30 but its low sensitivity in the low-wavenumber region significantly restricts the detection of characteristic vibrational signals associated with key intermediates, such as adsorbed CO and metal–carbon bonds.10,31,32 In contrast, in situ Raman spectroscopy is capable of overcoming this limitation owing to its high sensitivity to low-wavenumber vibrations, and it has been broadly applied to probe the key intermediates and reaction paths associated with the formation of final products and to gain a better understanding of the interfacial reaction processes during CO2/CO electrolysis.23,24,29,33
In recent years, the CO2/CO electrolysis field has progressed from H-cells to flow electrolyzers with gas diffusion electrodes (GDEs).14,34–36 However, to the best of our knowledge, the majority of in situ Raman studies have still been performed in H-type spectroelectrochemical cells.23,24,32,33,37–39 The thick mass transfer boundary layer in H-type spectroelectrochemical cells significantly restricts reactant (i.e. CO2/CO) transport to the catalyst surface, which may influence the intermediates coverage that is linked to the signal for in situ Raman spectroscopy experiments,40 particularly in the case of CO reduction due to the extremely low CO solubility in electrolytes.36 Thereby, the poor mass transport of reactants in traditional H-type spectroelectrochemical cells may inadvertently influence the detection of key intermediates and the related mechanistic analysis. To circumvent mass transport limitations and get better mechanistic insights into the formation and coverage of key intermediates, GDE-type spectroelectrochemical flow cells should be employed for the measurement of in situ Raman spectroscopy. However, the specific discrepancy in the in situ Raman detection of CO2/CO reduction intermediates between H-type spectroelectrochemical cells and GDE-type spectroelectrochemical flow cells remains unclear.
Herein, we demonstrate a systematic comparison for the in situ Raman detection of intermediates in CO2/CO electrolysis with and without mass transport limitations. We found that although cell configurations do not play an important role in the in situ Raman detection and analysis of surface-adsorbed intermediates in CO2 reduction, CO electrolysis in commonly used H-type spectroelectrochemical cells provided in situ Raman detection and analysis results that are significantly distinct from those acquired in GDE-type spectroelectrochemical flow cells, which include intermediates related to C2+ products. Further analysis reveals that the CO mass transport constraints during CO reduction in H-type spectroelectrochemical cells significantly reduce the formation and coverage of key intermediates, inadvertently influencing in situ Raman detection results.
Results and discussion
Catalyst characterization and in situ Raman tests
The Cu catalysts were deposited on the microporous layers of GDEs (Fig. 1a) using direct current magnetron sputtering at an argon pressure of 0.5 Pa (Fig. S1). The vacuum deposition technique enables the fabrication of catalyst layers with high purity and excellent reproducibility. Fig. 1b shows that the deposited Cu electrocatalyst had a rough surface, consisting of densely packed Cu nanoparticles with an average diameter of ∼100 nm. The rough surface morphology can significantly enhance the local electromagnetic field, thereby inducing a surface-enhanced effect that substantially amplifies the intensity of the in situ Raman signal when using the incident light with a wavelength of 785 nm.33,37 Additionally, all catalysts used in this study were Cu films with a uniform thickness of approximately 200 nm (Fig. S2). | ||
| Fig. 1 Morphology characterization and cell design. SEM images of (a) microporous layers of a GDE (Sigracet 39 BB) and (b) Cu catalyst layers coated on a GDE. (c) Schematic illustrations of an H-type spectroelectrochemical cell (left) and a GDE-type spectroelectrochemical flow cell (right) for in situ Raman spectroscopy. (d) Comparison of mass transport in two representative spectroelectrochemical configurations: an H-type cell (left) and a GDE-based flow cell (right). The thickness of the mass-transport boundary layer in the two cells was obtained from ref. 34 and 42. | ||
To explore the discrepancy in the in situ Raman detection of CO2/CO reduction intermediates with and without mass transport limitations, in situ Raman spectroscopy was performed using two spectroelectrochemical cell configurations (Fig. 1c, S4 and S5) with distinct mass transport characteristics: an H-type spectroelectrochemical cell and a GDE-based spectroelectrochemical flow cell. Specifically, the conventional H-type spectroelectrochemical cell consists of two compartments (the left scheme of Fig. 1c), namely the catholyte and anolyte compartments, which are separated by an anion exchange membrane (AEM). During in situ Raman experiments, CO/CO2 saturated electrolytes were continuously circulated through both the catholyte and anolyte compartments using two peristaltic pumps. The extremely thick diffusion layer for gas reactants in H-type spectroelectrochemical cells leads to significantly sluggish mass transport in the electrolyte (the left scheme of Fig. 1d).34,41 In contrast, a custom-designed spectroelectrochemical flow cell with a GDE, where the mass-transport of gas reactant can be accelerated significantly, was employed for in situ Raman experiments. As shown in the right scheme of Fig. 1c, the GDE-based spectroelectrochemical flow cell is composed of a catholyte and an anolyte chamber, separated by an AEM, and a gas chamber that allows gas flow into and out of the flow cell during electrolysis. The gas diffusion layer of the GDE, positioned between catholyte and gas chambers, allows gas reactants to access the catalyst surface via an extremely short diffusion layer (the right scheme of Fig. 1d).34,41
Additionally, all the CO2/CO reduction tests in both cells were based on a three-electrode configuration, in which a GDE loaded with Cu catalysts served as the working electrode, while an Ag/AgCl or Hg/HgO electrode and a graphite rod were used as the reference electrode and the counter electrode, respectively (Fig. 1c). To ensure reliable comparison of peak areas in the in situ Raman detection, all of the Raman spectra were acquired on the identical cathodic GDE under a constant electrolyte flow rate (14 mL min−1).
In situ Raman spectroscopy measurements during electrochemical CO2 reduction
To explore the impact of mass transport on the analysis of species adsorbed on Cu surfaces during electrocatalytic CO2 reduction, we performed in situ Raman spectroscopy in an H-type spectroelectrochemical cell and a GDE-type spectroelectrochemical flow cell, respectively. Additionally, it should be noted that when conducting CO2 electrolysis in GDE-type flow electrolyzers, carbonate formation near the cathodic GDE/catholyte interface could lead to a rapid variation in the composition, concentration and pH of the electrolyte for most commonly used electrolytes.35,42,43 The electrolyte variation over electrolysis may influence the formation and coverage of intermediates related to CO2 reduction. Herein, in order to circumvent the electrolyte variation over electrolysis and to readily compare the difference in situ Raman detection caused by the two distinct cell configurations, all the CO2 tests were carried out in 0.5 M CO2-saturated KHCO3 electrolyte (the ionic species and pH of the electrolyte can be maintained using a CO2-saturated bicarbonate electrolyte36,43).Fig. 2 presents a comparison of the in situ Raman spectra of surface species on Cu catalysts obtained from two spectroelectrochemical configurations with distinct mass transport conditions. In the low-frequency region, two characteristic peaks at 525 and 621 cm−1 attributed to surface Cu2O were observed in both cell configurations at the open-circuit potential (OCP),23,24 indicating the presence of a native oxide layer on Cu surfaces upon exposure to ambient air (Fig. 2a, d and S7). Once applying negative potentials (even as low as −0.3 V), the Raman peaks associated with the Cu2O layer disappeared, which is due to the electroreduction of Cu oxides to metallic Cu.44 This result suggests that all the in situ Raman spectra in this work were acquired on the metallic Cu surface at negative potentials.
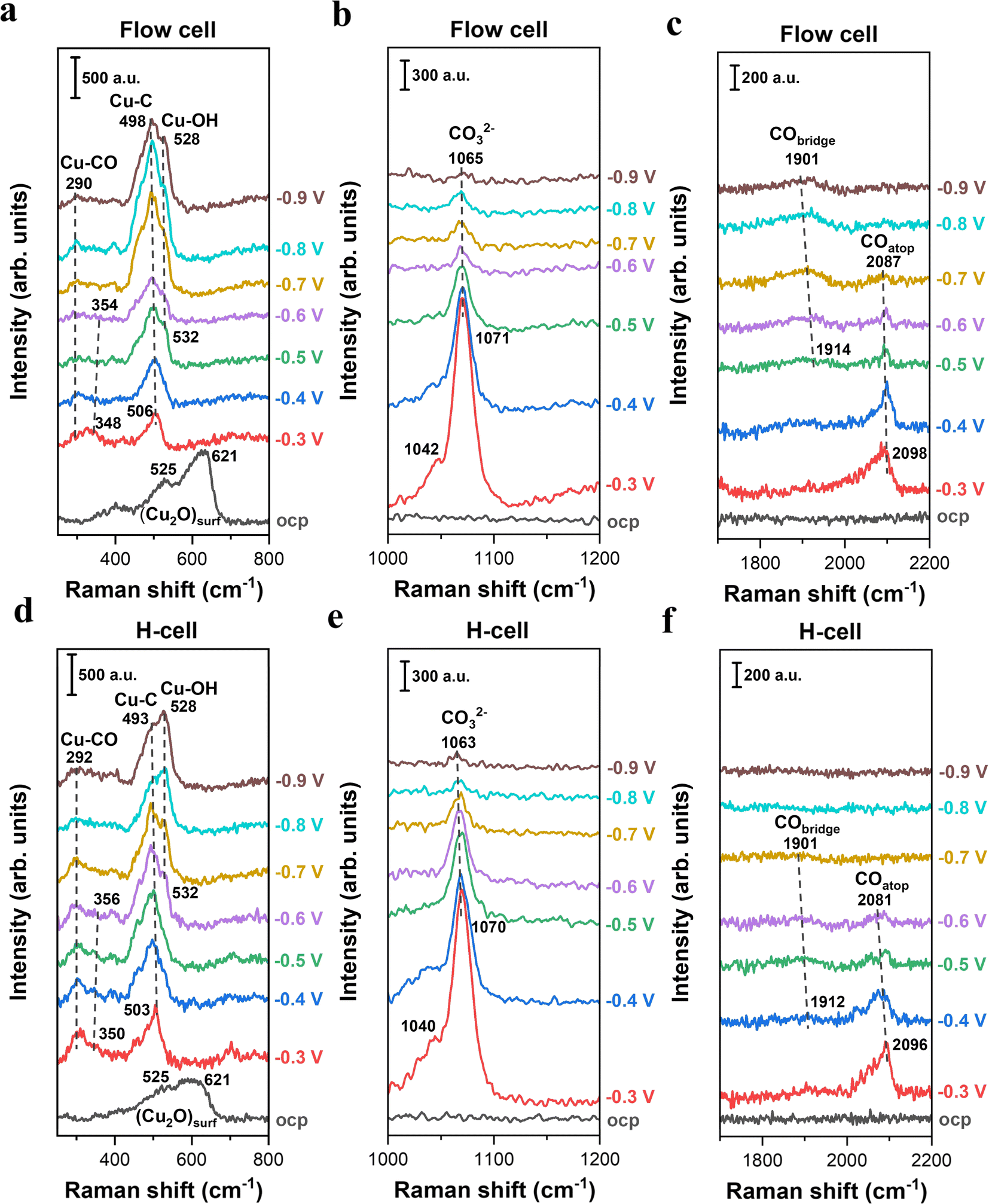 | ||
| Fig. 2 In situ Raman spectroscopy of electrochemical CO2 reduction on Cu catalysts in 0.5 M KHCO3 using two representative in situ Raman configurations. (a–c) Raman spectra on Cu catalysts as a function of applied potentials (vs. RHE, no iR correction) collected in the GDE-type spectroelectrochemical flow cell. (d–f) Raman spectra on Cu catalysts as a function of applied potentials (vs. RHE, no iR correction) collected in the H-type spectroelectrochemical cell. | ||
When using the GDE-type spectroelectrochemical flow cell, the broad band centered at ∼506 cm−1 (Fig. 2a) was observed in the low-frequency region under an applied potential of −0.3 V, which is attributed to Cu–C related intermediates.45–47 Regarding the Cu–C peak, there are two different theories: (i) the Cu–C peak reflects the complex C-related intermediates during CO2 conversion on the Cu surface;45 (ii) the Cu–C peak is likely correlated with the surface coverage of bridge-bonded CO (CObridge) at ∼1914 cm−1 (Fig. 2c).46,47 In this study, we found that the integrated area of the Cu–C peak initially increased when lowering the potentials from −0.3 V to −0.8 V, and subsequently diminished at more negative potentials than −0.8 V (Fig. S8). This trend directly follows the variation in the CObridge peak intensity as a function of potential (Fig. S9), which may suggest that the Cu–C peak is likely linked to the coverage of CObridge. As a comparison, we also found an obvious Cu–C peak under the use of an H-type spectroelectrochemical cell (Fig. 2d). Additionally, the variation trend of the Cu–C peak area as a function of potential also follows CObridge peak area in the H-cell (Fig. S9). These findings reveal that while there is a debate regarding the Cu–C peak in the field, both cells can provide the similar detection results of in situ Raman for the Cu–C peak and CObridge when performing CO2 electroreduction.
It is well-known that CO is the key intermediate in the formation of hydrocarbons and oxygenates on the Cu surface.14,36,48 As expected, we found that the C–O stretching peak from 2098 cm−1 to ∼2096 cm−1 in the flow-cell (Fig. 2c) and the H-cell (Fig. 2f) at −0.3 V corresponds to adsorbed CO in the atop configuration (COatop).10,36 Meanwhile, the Cu–CO rotation (∼290 cm−1) and the Cu–C stretching (∼350 cm−1) features were also detected in both cell configurations (Fig. 2a and d). Importantly, it has been demonstrated that the COatop (Fig. 3a) contributes to the further conversion of CO intermediates into hydrocarbons.10,31 Thus, to uncover the mass transport effect, it is essential to compare the coverage of COatop during CO2 electroreduction between the flow-cell and the H-cell. Previous work has indicated that the integrated band area is directly proportional to the CO coverage on the Cu surface.9,49 Thus, to better illustrate the COatop coverage in the two cases, the peak area of the COatop at various potentials was quantified by integrating the Gaussian-fitted Raman spectra (Fig. 3b). As shown in Fig. 3b, we found the roughly equal integral area of the COatop peak between the H-type spectroelectrochemical cell and the GDE-type spectroelectrochemical flow cell when applying a fixed potential. This observation implies that the cell configuration has a minimal effect on COatop coverage during CO2 reduction.
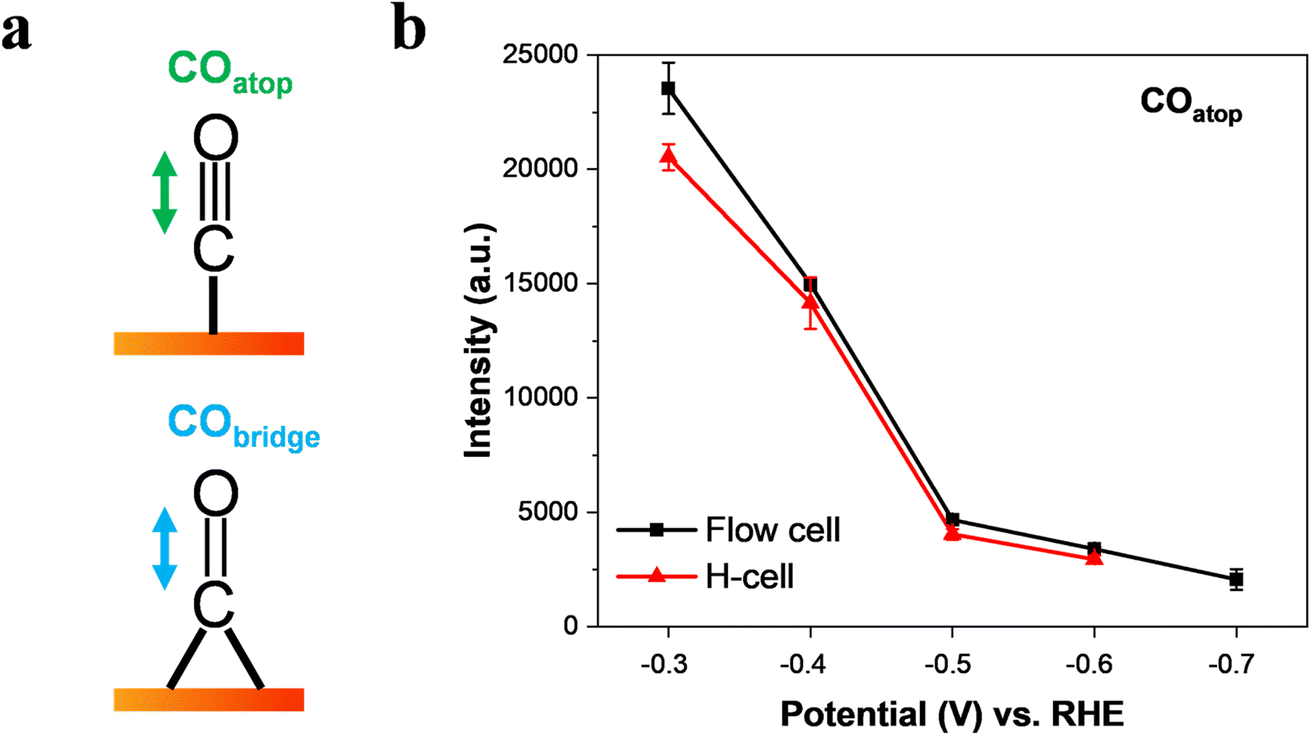 | ||
Fig. 3 Intensity of the C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) O stretch band of COatop as a function of applied potential in CO2-saturated 0.5 M KHCO3. (a) Schematic view of the CO stretching mode of COatop and CObridge on a Cu surface. (b) Effect of potential on the intensity of the COatop band on Cu catalysts collected in a GDE-type spectroelectrochemical flow cell and an H-type spectroelectrochemical cell. The data were obtained by Gaussian peak fitting of the Raman spectra from Fig. 2c and f. The error bars represent the standard deviation from at least three independent measurements. O stretch band of COatop as a function of applied potential in CO2-saturated 0.5 M KHCO3. (a) Schematic view of the CO stretching mode of COatop and CObridge on a Cu surface. (b) Effect of potential on the intensity of the COatop band on Cu catalysts collected in a GDE-type spectroelectrochemical flow cell and an H-type spectroelectrochemical cell. The data were obtained by Gaussian peak fitting of the Raman spectra from Fig. 2c and f. The error bars represent the standard deviation from at least three independent measurements. | ||
In addition to the intermediates related to CO2 reduction products, the symmetric stretching mode of the C–O bond in carbonate was observed as a strong band of ∼1071 cm−1 in both cell configurations (Fig. 2b and e) under relatively less negative potentials. The carbonate band redshifted from 1071 to 1065 cm−1 as the applied potential decreased from −0.3 to −0.9 V. This redshift corresponds to a Stark tuning rate of 10 cm−1 V−1 (Fig. S10), which is in reasonable agreement with the previous work (12 cm−1 V−1).33 Additionally, the Cu–OH stretching mode at ∼532 cm−1 (Fig. 2a and d) suggests the presence of surface-adsorbed OH species.47,50
Based on the above results in this section, we therefore conclude that cell configurations could not significantly affect the in situ Raman detection and analysis of surface-adsorbed intermediates during CO2 electroreduction, and even an H-type spectroelectrochemical cell should be able to provide a reliable detection of in situ Raman for CO2 reduction, particularly for the relatively low reaction rates. This conclusion is likely linked to the relatively high solubility of CO2 in aqueous media and relatively low reaction rates.51
In situ Raman spectroscopy measurements during electrochemical CO reduction
CO2 generates carbonate or bicarbonate through a buffering reaction in alkaline electrolytes, resulting in substantial alterations in ionic composition and pH of the electrolyte.35,42,43 In contrast, CO does not participate in buffering reactions with OH− and thus does not affect the ionic composition or pH of the electrolyte. Additionally, CO serves as a key intermediate for C–C coupling toward C2+ products. Thereby, CO reduction has been widely utilized as a significant method for gaining mechanistic insights into the formation of C2+ products.14,36,48 In recent years, to get better understanding of the reaction mechanisms of C2+ product formation, adsorbed surface species on Cu surfaces from CO reduction have been intensively explored via in situ Raman spectroscopy.24,31,36–39,52 However, to the best of our knowledge, all of the previous in situ Raman studies based on CO reduction have been operated in H-type spectroelectrochemical cells (with the one exception of CO reduction36).24,31,37–39,52 The extremely low solubility of CO in electrolytes further exacerbates the CO mass transport limitations in H-type spectroelectrochemical cells,36 which severely constrains CO supply to the catalyst surface, significantly influencing the formation and coverage of intermediates that lead to reaction pathways toward final products. The variation in the coverage of intermediates is closely correlated to the detection and analysis of Raman signals during CO reduction, which may inevitably affect the understanding of the underlying reaction mechanisms.To uncover the influence of mass transport on the spectral signals of surface species during electrocatalytic CO reduction, we conducted in situ Raman spectroscopy on Cu catalysts in an H-type spectroelectrochemical cell and a GDE-type spectroelectrochemical cell, respectively. Here, in order to circumvent the effect of electrolyte variation and to maintain the ionic species and pH of electrolyte over electrolysis,43 0.5 M CO-saturated KOH was employed in both cell configurations. From the in situ Raman spectra, the presence of a surface Cu2O peak was observed at OCP in both cell configurations; however, the Cu2O peak disappeared upon applying the negative potentials (Fig. 4a, c and S13). These observations are consistent with the discoveries during CO2 reduction, indicating that all the in situ Raman spectra in CO reduction were also obtained on the metallic Cu surface at negative potentials.
 | ||
| Fig. 4 In situ Raman spectroscopy of electrochemical CO reduction on Cu catalysts in 0.5 M KOH using two representative in situ Raman configurations. (a and b) Raman spectra on Cu catalysts as a function of applied potentials (vs. RHE, no iR correction) collected in the GDE-type spectroelectrochemical flow cell. (c and d) Raman spectra on Cu catalysts as a function of applied potentials (vs. RHE, no iR correction) collected in the H-type spectroelectrochemical cell. | ||
In light of the critical role of *CO in the C–C coupling process, further investigation into the Raman band of *CO was conducted in both cell configurations. When employing the GDE-type spectroelectrochemical flow cell for in situ Raman measurements, a weak C–O stretching band centered at ∼2099 cm−1 was observed in the high-wavenumber region under an applied potential of −0.2 V (Fig. 4b). As the potential decreased from −0.2 to −0.7 V, we found that the C–O stretching peak in the high-wavenumber region intensified, indicative of an increased CO coverage on the Cu surface. When a more negative potential (<−0.7 V) was applied, the intensity of the C–O stretching peak weakened, which is likely linked to significant consumption of adsorbed CO on the Cu surface caused by the accelerated CO reduction rate. As a comparison, the C–O stretching band was only observed at relatively less negative potentials in the H-type spectroelectrochemical cell (Fig. 4d), but disappeared at more negative potentials, which reflects a low CO coverage on the Cu surface. To obtain a better analysis of the discrepancy in CO coverage between the two cell configurations, the area of the C–O stretching peak at various potentials was quantified by integrating the Gaussian-fitted Raman spectra (Fig. S15). Notably, we found that a substantially higher CO coverage on the Cu surface when using the GDE-type spectroelectrochemical flow cell compared to the H-cell (Fig. S15). These findings show that the difference in mass transport between the GDE-type spectroelectrochemical flow cell and the H-type spectroelectrochemical cell (Fig. 1d) has a significant effect on CO coverage on the catalyst surface when performing CO electrolysis. Additionally, the discrepancy in the line shape of the C–O stretching peak was observed between the GDE-type spectroelectrochemical flow cell (Fig. 4b) and the H-type spectroelectrochemical cell (Fig. 4d), which may be due to distinct CO mass transport that leads to different distributions of CO adsorption sites on the catalyst surface (Fig. S16).
Previous studies suggest that dynamic dipole coupling of CO can introduce a nonlinear relationship between integrated intensity and adsorbate coverage.36,53 Thus, to more accurately track changes in CO coverage associated with C2+ products, the intensity ratio between the Cu–C stretching and Cu–CO rotation peaks was monitored in prior work.12,36 Herein, we also further analyzed the intensity ratio between the Cu–C stretching and Cu–CO rotation peaks (Fig. S17). When employing a GDE-type spectroelectrochemical flow cell (i.e. without mass transport limitation) for in situ Raman measurements, the application of −0.1 V resulted in the emergence of weak bands at ∼286 and 348 cm−1 (Fig. 4a), attributable to the restricted rotation of adsorbed CO and Cu–C stretching of adsorbed CO,12,45,54 respectively. As the applied potential decreased from −0.1 to −0.7 V, the integrated area of the Cu–C stretching of adsorbed CO increased progressively, reaching a peak at −0.7 V (Fig. 4a and S18). Notably, we found that the potential-dependent evolution of the intensity ratio between the Cu–C stretching of adsorbed CO and Cu–CO rotation (Fig. S18) closely parallels the variation in the C–O stretching peak (Fig. S15), suggesting that the intensity of the C–O stretching peak may serve as a reliable indicator of CO coverage associated with C2+ products on the Cu surface. In contrast, when conducting CO electrolysis in the H-type spectroelectrochemical cell, we did not observe any signals for Cu–C stretching of adsorbed CO and Cu–CO rotation bands across all tested potential ranges (Fig. 4c). This result indicates that CO mass transport limitation in the commonly used H-type spectroelectrochemical cell could significantly influence the detection and analysis of Raman signals related to C–C bond formation. Specifically, CO mass transport limitation results in extremely low CO coverage, which significantly lowers the formation of intermediates that leads to C–C bond formation. Notably, the distinct Raman signals observed in the GDE-type spectroelectrochemical flow cell and the H-type spectroelectrochemical cell are fully consistent with the corresponding differences in electrocatalytic CO reduction performance on Cu surfaces operated in the two different cell configurations (Fig. S19).
Additionally, in the H-type spectroelectrochemical cell, a more pronounced Cu–OH stretching peak was observed compared to that in the GDE-type spectroelectrochemical flow cell (Fig. 4a, c and S20). This may be linked to the fact that only a small amount of CO is adsorbed on the Cu catalyst surface under CO mass transport limitation (H-cell), which results in most of the active sites being occupied by OH species (i.e. high OH coverage). This corresponds to a much stronger Cu–OH stretching peak in the H-cell than that in the flow cell.
All the above results in this section manifest that cell configurations could significantly influence the in situ Raman detection and analysis of surface-adsorbed intermediates when performing CO reduction. In particular, severe CO mass transport limitation in the commonly used H-type spectroelectrochemical cells substantially reduces the formation and coverage of intermediates related to C2+ products, which may significantly affect the detection and analysis of Raman signals during CO reduction and the mechanistic insights for C2+ product formation.
Conclusions
In summary, to uncover the impact of mass transport on the spectral signals and analysis of surface species during electrocatalytic CO2/CO reduction, in situ Raman spectroscopy was performed using the commonly used H-type spectroelectrochemical cells and GDE-type spectroelectrochemical flow cells, respectively. Our results show that cell configurations have a negligible impact on the in situ Raman detection and analysis of surface-adsorbed intermediates during CO2 reduction, which indicates that even the commonly used H-type spectroelectrochemical cells can provide reliable in situ Raman results for CO2 reduction. However, when employing CO reduction, we found that in situ Raman signals of surface-adsorbed intermediates in H-cells are significantly distinct from those acquired in GDE-type flow cells (e.g., intermediates related to C2+ products). Specifically, the pronounced CO mass transport limitations in commonly used H-type spectroelectrochemical cells markedly suppress the formation and coverage of intermediates associated with C2+ products, thereby affecting in situ Raman detection results during CO reduction. This work highlights the critical role of mass transport in the in situ Raman detection and analysis of surface-adsorbed intermediates, particularly for CO reduction, where mass transport limitations may significantly affect the mechanistic understanding.Author contributions
M. M. conceived the original idea of this work. M. M. and J. L. supervised the project. W. Y. synthesized the Cu catalysts, performed SEM and in situ Raman experiments. H. B. performed the electrocatalytic tests. M. M., W. Y. and J. L. analyzed the data and wrote the original manuscript. All authors contributed to discussion of the results and manuscript preparation.Conflicts of interest
The authors declare no competing financial interests.Data availability
All data needed to evaluate the conclusions in the paper are present in the paper and/or the supplementary information (SI). Additional data related to this paper may be requested from the authors. Supplementary information is available. See DOI: https://doi.org/10.1039/d5sc07260c.Acknowledgements
This work was supported by the National Natural Science Foundation of China (22179105 and 22403074) and “Young Talent Support Plan” of Xi’an Jiaotong University (awarded to M. M.). W. Y. was supported by the China Scholarship Council (CSC) scholarship. The authors would like to thank Zhe Zheng for assistance in the in situ Raman experiments.References
- Z. W. Seh, J. Kibsgaard, C. F. Dickens, I. B. Chorkendorff, J. K. Norskov and T. F. Jaramillo, Combining theory and experiment in electrocatalysis: Insights into materials design, Science, 2017, 355(6321), eaad4998 CrossRef.
- T. X. Yan, X. Y. Chen, L. Kumari, J. L. Lin, M. L. Li, Q. Fan, H. Y. Chi, T. J. Meyer, S. Zhang and X. B. Ma, Multiscale CO2 Electrocatalysis to C2+ Products: Reaction Mechanisms, Catalyst Design, and Device Fabrication, Chem. Rev., 2023, 123(17), 10530–10583 Search PubMed.
- M. Ma and B. Seger, Rational Design of Local Reaction Environment for Electrocatalytic Conversion of CO2 into Multicarbon Products, Angew. Chem., Int. Ed., 2024, 63(23), e202401185 CrossRef CAS.
- Y. Y. Birdja, E. Pérez-Gallent, M. C. Figueiredo, A. J. Göttle, F. Calle-Vallejo and M. T. M. Koper, Advances and challenges in understanding the electrocatalytic conversion of carbon dioxide to fuels, Nat. Energy, 2019, 4(9), 732–745 CrossRef CAS.
- S. Nitopi, E. Bertheussen, S. B. Scott, X. Liu, A. K. Engstfeld, S. Horch, B. Seger, I. E. L. Stephens, K. Chan, C. Hahn, J. K. Norskov, T. F. Jaramillo and I. Chorkendorff, Progress and Perspectives of Electrochemical CO2 Reduction on Copper in Aqueous Electrolyte, Chem. Rev., 2019, 119(12), 7610–7672 CrossRef CAS PubMed.
- K. P. Kuhl, E. R. Cave, D. N. Abram and T. F. Jaramillo, New insights into the electrochemical reduction of carbon dioxide on metallic copper surfaces, Energy Environ. Sci., 2012, 5(5), 7050–7059 RSC.
- M. Jouny, W. Luc and F. Jiao, General Techno-Economic Analysis of CO2 Electrolysis Systems, Ind. Eng. Chem. Res., 2018, 57(6), 2165–2177 CrossRef CAS.
- H. R. Jhong, S. C. Ma and P. J. A. Kenis, Electrochemical conversion of CO2 to useful chemicals: current status, remaining challenges, and future opportunities, Curr. Opin. Chem. Eng., 2013, 2(2), 191–199 Search PubMed.
- C. M. Gunathunge, V. J. Ovalle, Y. W. Li, M. J. Janik and M. M. Waegele, Existence of an Electrochemically Inert CO Population on Cu Electrodes in Alkaline pH, ACS Catal., 2018, 8(8), 7507–7516 CrossRef CAS.
- H. Y. An, L. F. Wu, L. D. B. Mandemaker, S. Yang, J. de Ruiter, J. H. J. Wijten, J. C. L. Janssens, T. Hartman, W. van der Stam and B. M. Weckhuysen, Sub-Second Time-Resolved Surface-Enhanced Raman Spectroscopy Reveals Dynamic CO Intermediates during Electrochemical CO2 Reduction on Copper, Angew. Chem., Int. Ed., 2021, 60(30), 16576–16584 Search PubMed.
- Z.-H. Zhao and D. Ren, Unravelling the Effect of Crystal Facet of Derived-Copper Catalysts on the Electroreduction of Carbon Dioxide under Unified Mass Transport Condition, Angew. Chem., Int. Ed., 2024, 64, e202415590 CrossRef PubMed.
- C. Zhan, F. Dattila, C. Rettenmaier, A. Bergmann, S. Kühl, R. García-Muelas, N. López and B. R. Cuenya, Revealing the CO Coverage-Driven C-C Coupling Mechanism for Electrochemical CO2 Reduction on Cu2O Nanocubes via Operando Raman Spectroscopy, ACS Catal., 2021, 11(13), 7694–7701 Search PubMed.
- J. Gao, H. Zhang, X. Y. Guo, J. S. Luo, S. M. Zakeeruddin, D. Ren and M. Gratzel, Selective C-C Coupling in Carbon Dioxide Electroreduction via Efficient Spillover of Intermediates As Supported by Operando Raman Spectroscopy, J. Am. Chem. Soc., 2019, 141(47), 18704–18714 CrossRef CAS PubMed.
- M. Ma, W. Deng, A. Xu, D. Hochfilzer, Y. Qiao, K. Chan, I. Chorkendorff and B. Seger, Local reaction environment for selective electroreduction of carbon monoxide, Energy Environ. Sci., 2022, 15(6), 2470–2478 RSC.
- W. Luc, X. B. Fu, J. J. Shi, J. J. Lv, M. Jouny, B. H. Ko, Y. B. Xu, Q. Tu, X. B. Hu, J. S. Wu, Q. Yue, Y. Y. Liu, F. Jiao and Y. J. Kang, Two-dimensional copper nanosheets for electrochemical reduction of carbon monoxide to acetate, Nat. Catal., 2019, 2(5), 423–430 CrossRef CAS.
- Y. P. Zhu, J. L. Wang, H. Chu, Y. C. Chu and H. M. Chen, In Situ Operando Studies for Designing Next-Generation Electrocatalysts, ACS Energy Lett., 2020, 5(4), 1281–1291 CrossRef CAS.
- G. Iijima, T. Inomata, H. Yamaguchi, M. Ito and H. Masuda, Role of a Hydroxide Layer on Cu Electrodes in Electrochemical CO2 Reduction, ACS Catal., 2019, 9(7), 6305–6319 Search PubMed.
- S. Zhu, B. Jiang, W. B. Cai and M. Shao, Direct Observation on Reaction Intermediates and the Role of Bicarbonate Anions in CO2 Electrochemical Reduction Reaction on Cu Surfaces, J. Am. Chem. Soc., 2017, 139(44), 15664–15667 CrossRef CAS PubMed.
- H. Sheng, M. H. Oh, W. T. Osowiecki, W. Kim, A. P. Alivisatos and H. Frei, Carbon Dioxide Dimer Radical Anion as Surface Intermediate of Photoinduced CO2 Reduction at Aqueous Cu and CdSe Nanoparticle Catalysts by Rapid-Scan FT-IR Spectroscopy, J. Am. Chem. Soc., 2018, 140(12), 4363–4371 CrossRef CAS PubMed.
- C. M. Gunathunge, X. Li, J. Y. Li, R. P. Hicks, V. J. Ovalle and M. M. Waegele, Spectroscopic Observation of Reversible Surface Reconstruction of Copper Electrodes under CO2 Reduction, J. Phys. Chem. C, 2017, 121(22), 12337–12344 CrossRef CAS.
- N. Heidary, K. H. Ly and N. Kornienko, Probing CO2 Conversion Chemistry on Nanostructured Surfaces with Operando Vibrational Spectroscopy, Nano Lett., 2019, 19(8), 4817–4826 CrossRef CAS PubMed.
- R. Kas, O. Ayemoba, N. J. Firet, J. Middelkoop, W. A. Smith and A. Cuesta, In-Situ Infrared Spectroscopy Applied to the Study of the Electrocatalytic Reduction of CO2: Theory, Practice and Challenges, ChemPhysChem, 2019, 20(22), 2904–2925 CrossRef CAS PubMed.
- N. Bodappa, M. Su, Y. Zhao, J. B. Le, W. M. Yang, P. Radjenovic, J. C. Dong, J. Cheng, Z. Q. Tian and J. F. Li, Early Stages of Electrochemical Oxidation of Cu(111) and Polycrystalline Cu Surfaces Revealed by in Situ Raman Spectroscopy, J. Am. Chem. Soc., 2019, 141(31), 12192–12196 CrossRef CAS PubMed.
- Y. Zhao, X. Chang, A. S. Malkani, X. Yang, L. Thompson, F. Jiao and B. Xu, Speciation of Cu Surfaces During the Electrochemical CO Reduction Reaction, J. Am. Chem. Soc., 2020, 142(21), 9735–9743 CAS.
- M. Dunwell, Q. Lu, J. M. Heyes, J. Rosen, J. G. Chen, Y. Yan, F. Jiao and B. Xu, The Central Role of Bicarbonate in the Electrochemical Reduction of Carbon Dioxide on Gold, J. Am. Chem. Soc., 2017, 139(10), 3774–3783 CrossRef CAS PubMed.
- J. C. Dong, X. G. Zhang, V. Briega-Martos, X. Jin, J. Yang, S. Chen, Z. L. Yang, D. Y. Wu, J. M. Feliu, C. T. Williams, Z. Q. Tian and J. F. Li, In situ Raman spectroscopic evidence for oxygen reduction reaction intermediates at platinum single-crystal surfaces, Nat. Energy, 2019, 4(1), 60–67 CrossRef CAS.
- X. Chang, A. Malkani, X. Yang and B. Xu, Mechanistic Insights into Electroreductive C-C Coupling between CO and Acetaldehyde into Multicarbon Products, J. Am. Chem. Soc., 2020, 142(6), 2975–2983 CrossRef CAS PubMed.
- N. J. Firet and W. A. Smith, Probing the Reaction Mechanism of CO2 Electroreduction over Ag Films via Operando Infrared Spectroscopy, ACS Catal., 2017, 7(1), 606–612 CrossRef CAS.
- D. A. Zhang, X. Liu, Y. Zhao, H. Zhang, A. V. Rudnev and J. F. Li, In situ Raman spectroscopic studies of CO2 reduction reactions: from catalyst surface structures to reaction mechanisms, Chem. Sci., 2025, 16(12), 4916–4936 RSC.
- E. Pérez-Gallent, M. C. Figueiredo, F. Calle-Vallejo and M. T. M. Koper, Spectroscopic Observation of a Hydrogenated CODimer Intermediate During CO Reduction on Cu(100) Electrodes, Angew. Chem., Int. Ed., 2017, 56(13), 3621–3624 Search PubMed.
- X. X. Chang, S. Vijay, Y. R. Zhao, N. J. Oliveira, K. R. Chan and B. J. Xu, Understanding the complementarities of surface-enhanced infrared and Raman spectroscopies in CO adsorption and electrochemical reduction, Nat. Commun., 2022, 13(1), 2656 Search PubMed.
- Y. Zhao, X. G. Zhang, N. Bodappa, W. M. Yang, Q. Liang, P. M. Radjenovica, Y. H. Wang, Y. J. Zhang, J. C. Dong, Z. Q. Tian and J. F. Li, Elucidating electrochemical CO2 reduction reaction processes on Cu(hkl) single-crystal surfaces by in situ Raman spectroscopy, Energy Environ. Sci., 2022, 15(9), 3968–3977 RSC.
- I. V. Chernyshova, P. Somasundaran and S. Ponnurangam, On the origin of the elusive first intermediate of CO2 electroreduction, Proc. Natl. Acad. Sci. U. S. A., 2018, 115(40), E9261–E9270 CrossRef CAS.
- T. Burdyny and W. A. Smith, CO2 reduction on gas-diffusion electrodes and why catalytic performance must be assessed at commercially-relevant conditions, Energy Environ. Sci., 2019, 12(5), 1442–1453 RSC.
- M. Ma, E. L. Clark, K. T. Therkildsen, S. Dalsgaard, I. Chorkendorff and B. Seger, Insights into the carbon balance for CO2 electroreduction on Cu using gas diffusion electrode reactor designs, Energy Environ. Sci., 2020, 13(3), 977–985 RSC.
- W. Yan, T. T. Wu, J. Liu, Z. Zheng and M. Ma, Mass Transport-Dependent C-C Bond Formation for CO Electroreduction with Alkali Cations, J. Am. Chem. Soc., 2025, 147(11), 9990–10001 CrossRef CAS PubMed.
- X. X. Chang, Y. R. Zhao and B. J. Xu, pH Dependence of Cu Surface Speciation in the Electrochemical CO Reduction Reaction, ACS Catal., 2020, 10(23), 13737–13747 CrossRef CAS.
- J. Y. Zang, W. T. Ye, Q. L. Liu, J. H. Meng and W. X. Yang, Plasmonic-Promoted Formation of Surface Adsorbed Stochastic CO during Electrochemical CO2 and CO Reduction on Cu at Extreme Low Overpotentials, J. Am. Chem. Soc., 2025, 147(12), 10260–10267 CrossRef CAS PubMed.
- J. Li, X. X. Chang, H. C. Zhang, A. S. Malkani, M. J. Cheng, B. J. Xu and Q. Lu, Electrokinetic and in situ spectroscopic investigations of CO electrochemical reduction on copper, Nat. Commun., 2021, 12(1), 3264 Search PubMed.
- E. L. Clark, J. Resasco, A. Landers, J. Lin, L. T. Chung, A. Walton, C. Hahn, T. F. Jaramillo and A. T. Bell, Standards and Protocols for Data Acquisition and Reporting for Studies of the Electrochemical Reduction of Carbon Dioxide, ACS Catal., 2018, 8(7), 6560–6570 CrossRef CAS.
- L. C. Weng, A. T. Bell and A. Z. Weber, Modeling gas-diffusion electrodes for CO2 reduction, Phys. Chem. Chem. Phys., 2018, 20(25), 16973–16984 RSC.
- M. Ma, S. Kim, I. Chorkendorff and B. Seger, Role of ion-selective membranes in the carbon balance for CO2 electroreduction via gas diffusion electrode reactor designs, Chem. Sci., 2020, 11(33), 8854–8861 RSC.
- Z. Zheng, Y. F. Yao, W. Yan, H. Y. Bu, J. F. Huang and M. Ma, Mechanistic Insights into the Abrupt Change of Electrolyte in CO2 Electroreduction, ACS Catal., 2024, 14(8), 6328–6338 CrossRef CAS.
- S. B. Scott, T. V. Hogg, A. T. Landers, T. Maagaard, E. Bertheussen, J. C. Lin, R. C. Davis, J. W. Beeman, D. Higgins, W. S. Drisdell, C. Hahn, A. Mehta, B. Seger, T. F. Jaramillo and I. Chorkendorff, Absence of Oxidized Phases in Cu under CO Reduction Conditions, ACS Energy Lett., 2019, 4(3), 803–804 CrossRef CAS.
- H. Y. An, J. de Ruiter, L. F. Wu, S. Yang, F. Meirer, W. van der Stam and B. M. Weckhuysen, Spatiotemporal Mapping of Local Heterogeneities during Electrochemical Carbon Dioxide Reduction, JACS Au, 2023, 3(7), 1890–1901 Search PubMed.
- J. de Ruiter, V. R. M. Benning, S. Yang, B. J. den Hartigh, H. Wang, P. T. Prins, J. M. Dorresteijn, J. C. L. Janssens, G. Manna, A. V. Petukhov, B. M. Weckhuysen, F. T. Rabouw and W. van der Stam, Multiscale X-ray scattering elucidates activation and deactivation of oxide-derived copper electrocatalysts for CO2 reduction, Nat. Commun., 2025, 16(1), 373 CrossRef CAS PubMed.
- M. Moradzaman and G. Mul, In Situ Raman Study of Potential-Dependent Surface Adsorbed Carbonate, CO, OH, and C Species on Cu Electrodes During Electrochemical Reduction of CO2, ChemElectroChem, 2021, 8(8), 1478–1485 CrossRef CAS.
- M. Jouny, W. Luc and F. Jiao, High-rate electroreduction of carbon monoxide to multi-carbon products, Nat. Catal., 2018, 1(10), 748–755 CrossRef CAS.
- C. M. Gunathunge, V. J. Ovalle and M. M. Waegele, Probing promoting effects of alkali cations on the reduction of CO at the aqueous electrolyte/copper interface, Phys. Chem. Chem. Phys., 2017, 19(44), 30166–30172 Search PubMed.
- Y. F. Cao, Z. Chen, P. H. Li, A. Ozden, P. F. Ou, W. Y. Ni, J. Abed, E. Shirzadi, J. Q. Zhang, D. Sinton, J. Ge and E. H. Sargent, Surface hydroxide promotes CO2 electrolysis to ethylene in acidic conditions, Nat. Commun., 2023, 14(1), 2387 CrossRef CAS PubMed.
- R. Wiebe and V. L. Gaddy, The solubility of carbon dioxide in water at various temperatures from 12 to 40° and at pressures to 500 atmospheres: Critical phenomena, J. Am. Chem. Soc., 1940, 62(4), 815–817 CrossRef CAS.
- F. Shao, J. K. Wong, Q. H. Low, M. Iannuzzi, J. G. Li and J. G. Lan, In situ spectroelectrochemical probing of CO redox landscape on copper single-crystal surfaces, Proc. Natl. Acad. Sci. U. S. A., 2022, 119(29), e2118166119 CrossRef CAS.
- E. Borguet and H. L. Dai, Site-specific properties and dynamical dipole coupling of CO molecules adsorbed on a vicinal Cu(100) surface, J. Chem. Phys., 1994, 101(10), 9080–9095 CrossRef CAS.
- D. Ren, J. Gao, S. M. Zakeeruddin and M. Grätzel, New Insights into the Interface of Electrochemical Flow Cells for Carbon Dioxide Reduction to Ethylene, J. Phys. Chem. Lett., 2021, 12(31), 7583–7589 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2026 |