
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSymmetry-guided monomer design enables the combinatorial synthesis and targeted screening of polyesters
Xiaojie
Feng†
a,
Xiaoying
He†
b,
Jiayi
Zhu
b,
Li-Hong
Lin
c,
Qiaoyan
Shang
b,
Zheng-Hong
Luo
 c,
Yin-Ning
Zhou
*c and
Fangyou
Yan
*a
c,
Yin-Ning
Zhou
*c and
Fangyou
Yan
*a
aSchool of Chemical Engineering and Materials Science, Tianjin University of Science and Technology, Tianjin 300457, P.R. China. E-mail: yanfangyou@tust.edu.cn
bSchool of Marine and Environmental Science, Tianjin University of Science and Technology, Tianjin 300457, P.R. China
cState Key Laboratory of Synergistic Chem-Bio Synthesis, School of Chemistry and Chemical Engineering, Shanghai Jiao Tong University, Shanghai 200240, People's Republic of China. E-mail: zhouyn@sjtu.edu.cn
First published on 15th December 2025
Abstract
The rational design of polyester materials plays a crucial role in the development of functional polymers with tailored properties. In this work, we introduce a novel symmetry-guided molecular design strategy, which is a symmetry-aware, parameter-controlled design paradigm that both broadens and rationalizes the accessible chemical space of functional molecules. By introducing the concept of a pairwise atomic symmetry index (PASI) metric and applying targeted modifications to small molecules, a library of 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 614 diacids and 9983 diols is constructed, enabling a systematic and unexplored expansion of the chemical space of polyesters. The combinatorial pairing of these diacids and diols leads to the generation of over 100 million polyester structures. High-throughput prediction of the glass transition temperature (Tg) by the Tg-QSPR model aligns well with the typical thermal behavior in polyester materials. To validate the design methodology, a two-level verification process is performed. The predicted Tg values are first examined using molecular dynamics (MD) simulations and subsequently confirmed by differential scanning calorimetry experiments. The calculated Tg values show good agreement with both MD simulations (average absolute error (AAE) of 17.54 °C) and experimental measurements (AAE of 16.45 °C). These results further confirm the reliability and robustness of the proposed approach. This study not only provides an effective strategy for the large-scale generation of a polyester library and screening of property targeted polyesters, but also carries broader chemical implications beyond polyester design, offering potential insights for the development of functional molecules.
614 diacids and 9983 diols is constructed, enabling a systematic and unexplored expansion of the chemical space of polyesters. The combinatorial pairing of these diacids and diols leads to the generation of over 100 million polyester structures. High-throughput prediction of the glass transition temperature (Tg) by the Tg-QSPR model aligns well with the typical thermal behavior in polyester materials. To validate the design methodology, a two-level verification process is performed. The predicted Tg values are first examined using molecular dynamics (MD) simulations and subsequently confirmed by differential scanning calorimetry experiments. The calculated Tg values show good agreement with both MD simulations (average absolute error (AAE) of 17.54 °C) and experimental measurements (AAE of 16.45 °C). These results further confirm the reliability and robustness of the proposed approach. This study not only provides an effective strategy for the large-scale generation of a polyester library and screening of property targeted polyesters, but also carries broader chemical implications beyond polyester design, offering potential insights for the development of functional molecules.
Introduction
Materials science is the cornerstone of human civilization, playing a fundamental and pioneering role in the development of science and technology. Polyester materials are a significant branch of polymer engineering and are widely used in various fields of life and production due to their excellent thermal stability, mechanical properties, and biodegradability. Examples include polymer electrolytes,1–4 polymer membranes,5–7 polymer dielectrics for flexible electronics,8,9 self-healing polymers,10–12 and high-temperature resistant materials.13–16 However, the growing demand for high-performance and sustainable polyesters poses increasing challenges for traditional trial-and-error methods.The emergence of polymer informatics has opened new opportunities for the expansion of polymers.17–21 The rapid development of polymer informatics has led to research into the computer-aided design of high-performance polymers. Researchers can explore the relationship between the structure and properties (e.g., thermal performances,22–27 transfer performances,28–33 electrical performances,34–36 mechanical performances,37–40 and optical performances41–43) of novel polymers to meet the needs of different fields.
Several frameworks have been developed to generate and evaluate polymers. Existing frameworks, such as the Open Macromolecular Genome (OMG)44 and Small Molecules into Polymers (SMiPoly),45 provide collections of commercially available or literature-derived monomers and define canonical polymerization pathways, enabling the construction of virtual polymer libraries. Generative models based on the Variational Autoencoder (VAE) framework46 are accessible to the inverse design of polymers with targeted topologies and properties. High-throughput and data-driven strategies are also applied to accelerate the discovery of functional polymers. For example, Yu et al.47 constructed a virtual space of over 100000 polyimides and identified nine promising candidates for high-temperature energy storage through computational screening and molecular dynamics (MD) simulations. Similarly, He et al.48 generated over 95000 polyester candidates by combining diacids and diols and experimentally validated a quantitative structure–property relationship (QSPR) model for glass transition temperature (Tg).
Despite these advances, most current strategies rely mainly on retrosynthetic or combinatorial approaches, and systematic monomer design remains underexplored. To address this gap, we introduce a symmetry-guided monomer design strategy leveraging the pairwise atomic symmetry index (PASI) to guide the generation of novel monomers. By explicitly incorporating atomic-level symmetry constraints, this approach enables systematic exploration of polyester chemical space, providing a conceptual framework for rational polyester design that complements existing generative polymer methodologies.
Building on this conceptual framework, we apply the PASI-guided strategy to develop a practical monomer design workflow. In this study, we focus on small-molecule modification and use the Tg of polyesters as a case study to broaden the chemical space and enable targeted screening of polyesters (Fig. 1). First, small molecules for designing diacids and diols are obtained by systematically modifying the collected organic molecules. Second, the concept of PASI is introduced for the first time to address the issue of symmetry in atomic pairs within molecules. Guided by the PASI theory and incorporating the modified fragments, diacids and diols are designed systematically. Subsequently, a library of hypothetical polyesters is generated through the enumeration of all possible diacid–diol combinations. To validate the design methodology, the Tg-QSPR model48 is used to conduct a high-throughput screening of the virtual polyester library. Also, mechanistic or chemical insights are also provided according to the distribution of polyester Tg values along with their chemical structures. MD simulations and experimental validation are then performed. This design strategy not only enhances the efficiency of polyester design but also provides innovative ideas and methods for discovering polymer materials.
 | ||
| Fig. 1 Schematic illustration of this work. (a) Modification and screening of the initial molecular structures. (b) Calculation of PASI, generation of a virtual polyester library. (c) High-throughput screening of virtual polyesters, molecular dynamics, experimental synthesis, and characterization. | ||
Results and discussion
Modification and screening of the initial molecular structures
First, the structures of more than 20000 organic molecules are collected from the National Institute of Standards and Technology (NIST) database.49 Owing to the complex structural features of some organic molecules, the organic structures are systematically modified according to the following rules (Fig. 1a): (1) exclusion of the charged species and cis–trans isomers; (2) removal of halogen atoms from organic molecules; (3) removal of metal atoms from organic molecules; (4) removal of intrinsic functional groups, including carboxyl, hydroxyl, ester, and amino groups; and (5) removal of duplicate small molecules. Furthermore, based on an analysis of the polyester database derived from PoLyInfo50 (see the SI for details; Fig. S1), the modified molecules (H-suppressed structures) are further screened according to the following principles (Fig. 1a): (1) the total number of heavy atoms, defined as non-hydrogen atoms including carbon (C), nitrogen (N), oxygen (O), phosphorus (P), and sulfur (S), in small molecules <80; (2) the molecular weight of small molecules <1000; (3) the maximum step (i.e., the longest topological distance) of small molecules <50; (4) the number of C atoms in small molecules <50; (5) the number of O atoms in small molecules <10; and (6) the number of N atoms in small molecules <10.
The synthetic accessibility score (SAscore) metric is used to assess the synthetic difficulty of a compound during the chemical synthesis process by analyzing its structural features.51 Synthetic accessibility analysis enables researchers to screen and design substances more effectively, thereby enhancing the success rate and efficiency of novel material development. Generally, compounds with a lower SAscore are more readily synthesized, requiring relatively simpler reaction conditions and fewer synthetic steps. To reduce the synthetic complexity, 4116 small molecules with SAscores of less than 4.0 are selected for the subsequent design of polyester monomers. Detailed distribution information is provided in Fig. S2.
Pairwise atomic symmetry index (PASI)
Analysis of existing polyester monomers reveals that in most diacids and diols, the hydroxyl and carboxyl groups are located at symmetric positions. Based on this observation, the concept of the PASI is introduced to quantify the degree of symmetry between two atoms in a molecule (see the SI for details; Algorithm S1). This facilitates the design of monomers with symmetry. The specific steps are as follows:(1) Calculate the following for atom i: the topological distance (D)52 between atom i and all atoms; the branched degree (bra); the sum of bond orders (∑bds), and the product of bond orders (∏bds). Additionally, record the atomic number (Z) and the number of bonded hydrogens (#H).
(2) The attribute tuples (D, Z, bra, ∑bds, ∏bds, #H) are sorted in ascending order following a lexicographic comparison scheme. Specifically, D is compared first; if entries have the same D value, Z is compared next, and the comparison proceeds sequentially through the remaining attributes. It should be noted that these attributes are treated as a set of parallel equivalence conditions rather than a weighted linear combination.
(3) Calculate the PASI between atoms i and j, as described in eqn (1).
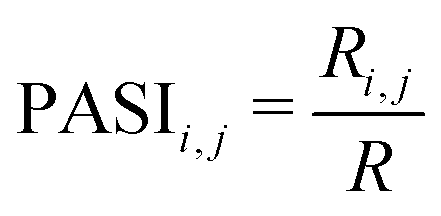 | (1) |
A representative example is provided to illustrate the PASI (Fig. 2). First, the atomic information (Z, bra, ∑bd, ∏bds, and #H) for all atoms is obtained to construct the atomic information matrix. The D values between the atom and all other atoms are then computed, forming the initial matrix. Each matrix is sorted in ascending lexicographic order according to the sequence (Z, bra, ∑bd, ∏bds, and #H). Finally, the sorted matrices of two atoms are compared, and the ratio of identical rows to the total number of rows is defined as the PASI between the two atoms. In this example, atoms a and b have identical matrices, giving a PASI of 1.0, whereas atoms a and c share no identical rows, resulting in a PASI of 0. This example demonstrates how PASI quantitatively captures topological equivalence based on parallel atomic attributes.
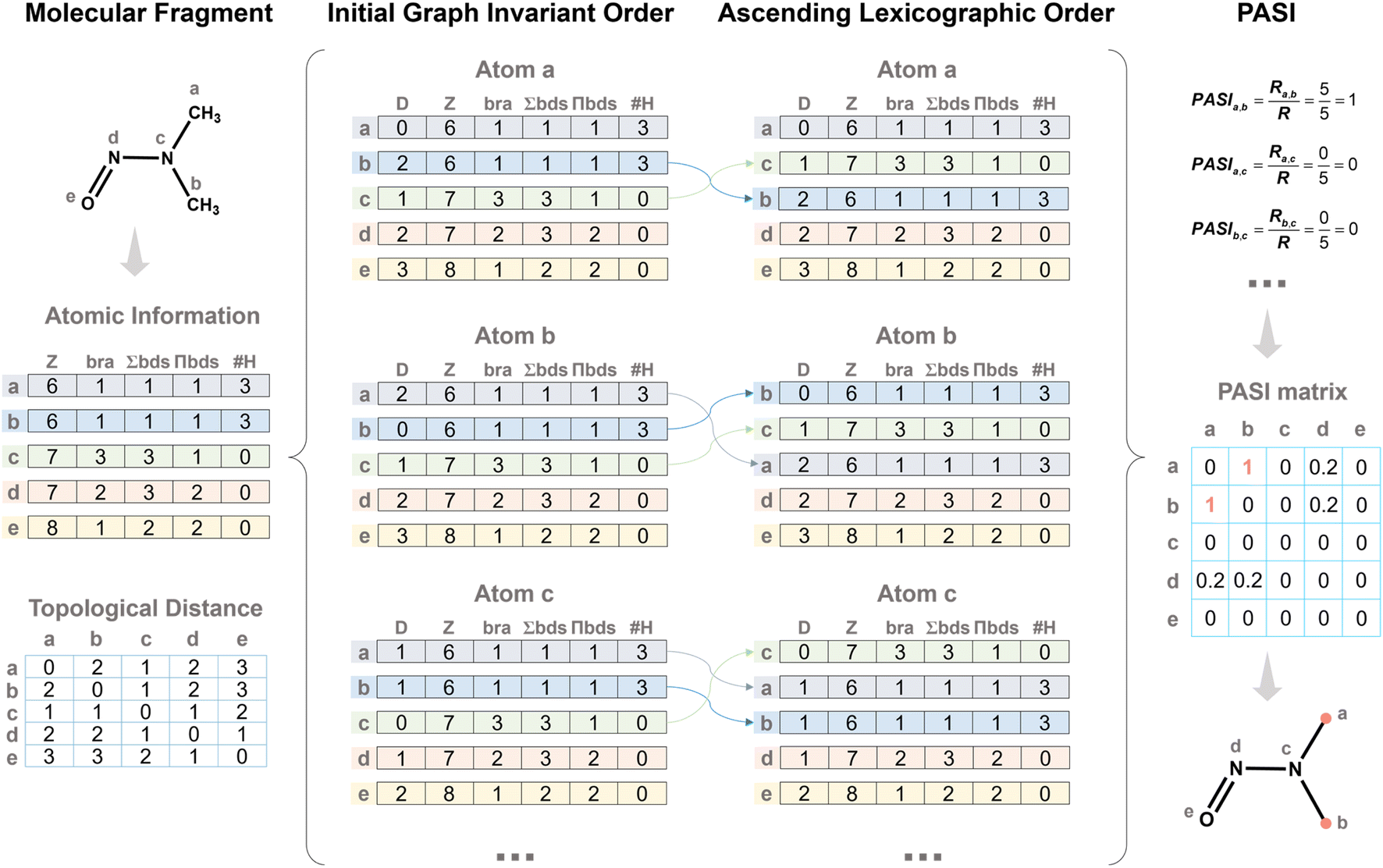 | ||
| Fig. 2 Representative example illustrating the PASI. | ||
Chemical space analysis
The virtual polyesters are produced by the condensation of diacids and diols. Analysis of the monomers (reported by He et al.48) derived from decomposing polyesters in the PolyInfo database shows that the majority of reactive functional groups are located at positions with PASI = 1.0 (Fig. 3). Specifically, 155 out of 267 diacids (58.05%) and 256 out of 358 diols (71.51%) have their –COOH and –OH groups located at PASI = 1.0, respectively. To prevent combinatorial explosion and ensure the simplicity and synthetic feasibility of the designed molecules, only atom pairs with a PASI of 1.0 (perfect symmetry) are used to introduce carboxyl and hydroxyl groups to generate diacid and diol molecules. In addition, it is stipulated that the maximum step between symmetric points with a PASI of 1.0 should be greater than 0.3 times the maximum step of the small molecule to prevent the monomer molecules from having excessively long side chains. Finally, a total of 10614 diacids and 9983 diols are successfully designed by introducing carboxyl and hydroxyl groups at the symmetric positions. Comprehensive details are provided in the SI (Data.xlsx). The diacids are labeled as A1-A10614, while the diols are marked as B1-B9983. Although constraining the design to PASI = 1.0 reduces the design space, this is an intentional and adjustable choice. The PASI enables quantitative control of atomic-level topological symmetry, allowing the design space to be flexibly expanded or contracted according to the application.
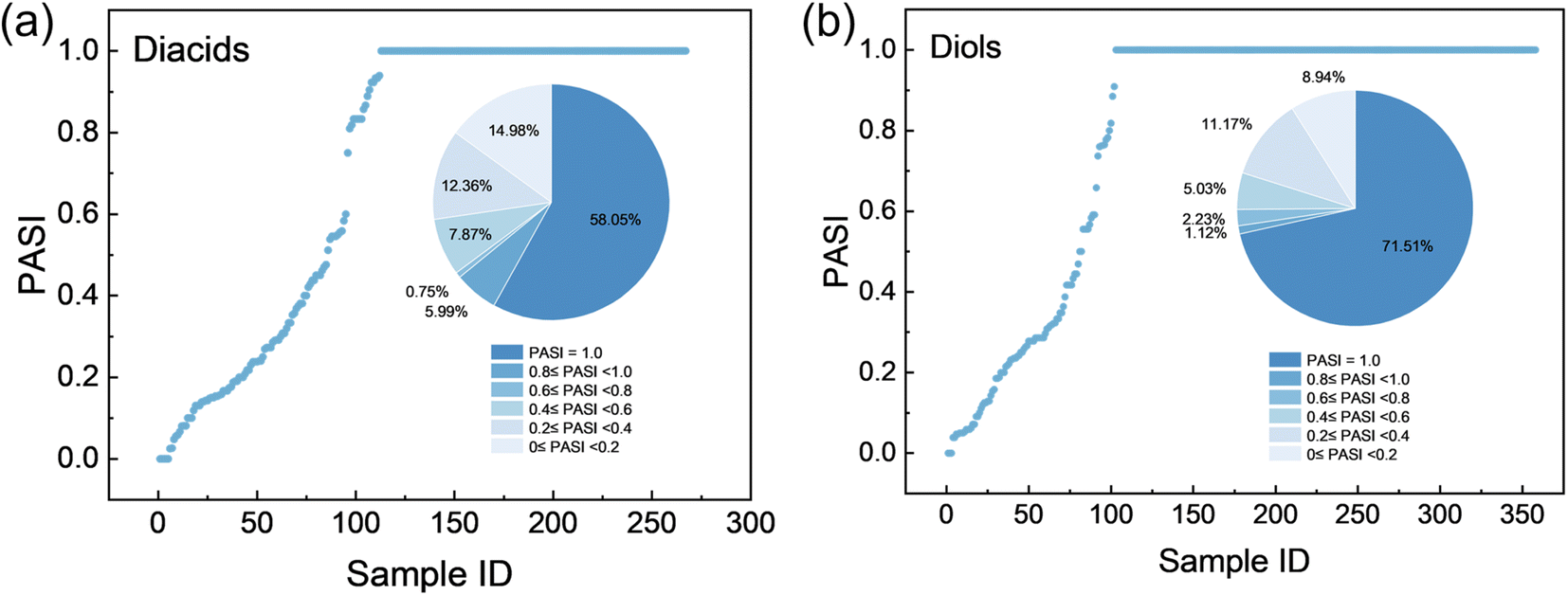 | ||
| Fig. 3 Distributions of PASI values for (a) diacid and (b) diol sites in the monomer dataset (sourced from He et al.48). | ||
In terms of data scale and chemical diversity, the diacid and diol monomers included in several representative frameworks are compared, as summarized in Table 1. SMiPoly45 collected 1083 small molecules extracted from the literature, including 81 diacids and 63 diols, whereas OMG44 screened 3.1 million molecules from the eMolecules database and identified 1911 diacids and 6581 diols. In this work, the PASI-guided design strategy generates 10614 diacids and 9983 diols. Fig. 4a illustrates the visualization of the Morgan fingerprint feature (radius = 2, fpSize = 2048) for diacids and diols, respectively, obtained using the t-distributed stochastic neighbor embedding (t-SNE) algorithm.53–55 Compared to the existing methods, this work spans a broader chemical space. It highlights that our method introduces a symmetry-aware, parameter-controlled design paradigm that both broadens and rationalizes the accessible chemical space.
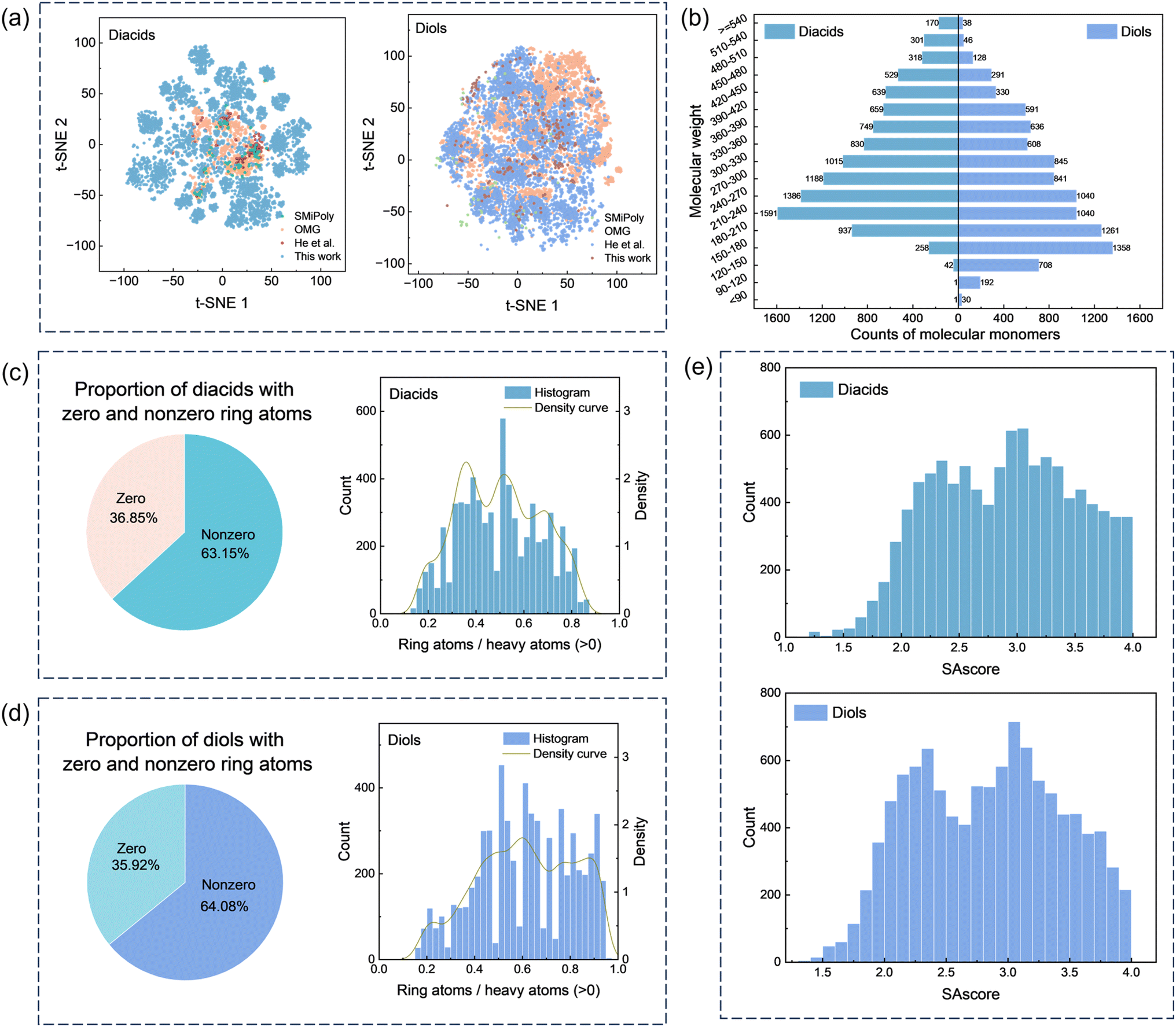 | ||
| Fig. 4 Information on the designed diacids and diols. (a) Chemical space visualization of the designed diacids and diols in datasets OMG,44 SMiPoly,45 He et al.,48 and this work. (b) Counts of the designed diacids and diols across different molecular weight ranges. (c) Distribution of ring atom ratios in the designed diacid molecules. (d) Distribution of ring atom ratios in the designed diol molecules. (e) Distribution histogram of the SAscore of the designed diacids and diols. | ||
Additionally, the PASI-guided monomers were evaluated by searching the designed diacids and diols in the PubChem database (https://pubchem.ncbi.nlm.nih.gov/), which contains 122 million compounds. The results show that 77.9% of the designed diacids and 67.8% of the designed diols are not present in PubChem, indicating high novelty. These findings confirm that PASI-guided selection effectively explores previously unreported chemical space.
Furthermore, Fig. 4b shows the molecular weight distribution of the diacids and diols. The molecular weight of the diacids is primarily concentrated in the range of 150 to 540 g mol−1, while the molecular weight of the diols is mainly distributed between 120 and 480 g mol−1. Ring atom distributions (Fig. 4c and d) reveal that over 60% of monomers contain cyclic substructures, with the ratio of ring atoms to heavy atoms (RA/HA) values spanning a wide range. This variability allows systematic tuning of polyester properties. For example, lower RA/HA values enhance flexibility and processability, while higher RA/HA values improve rigidity and thermal stability. Fig. 4e shows that the SAscores of both the diacids and diols are concentrated between 1.7 and 4.0, indicating that the synthesis of the designed diacids and diols is acceptable and feasible under certain conditions and thus accelerate synthesis.
Finally, over 100 million virtual polyester molecules are successfully generated utilizing computational methods to identify characteristic functional groups, such as carboxyl (–COOH) and hydroxyl (–OH) groups, within the simplified molecular input line entry system (SMILES) of the monomer. In the future, one can also adopt symmetry constraints with extra expert knowledges as new design principles to control physicochemical properties of polymers (e.g., controlling chain rigidity or crystallinity).
High-throughput screening of polyesters
Employing the validated Tg-QSPR model48 (eqn (2)) to calculate the Tg of over 100 million virtual polyester molecules represented by Ai_Bj provides an initial assessment of their thermal stability.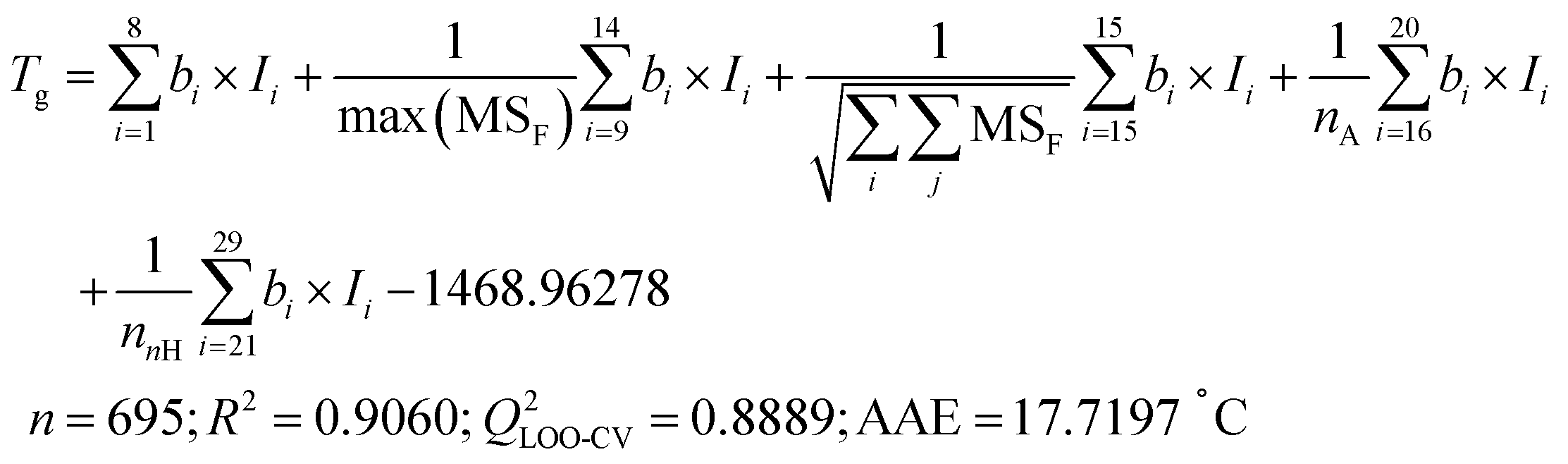 | (2) |
Fig. 5a presents the distribution histogram of the polyester Tg values. This distribution trend aligns with the typical thermal stability characteristics of polyester materials (He et al.48), suggesting that this design strategy is feasible and effective. It is worth noting that there are also Tg values beyond the plotted range (−200 °C to 400 °C): 0.0189% polyesters have Tg values below −200 °C, and 0.005% have values above 400 °C. Such extreme values are likely due to model-induced deviations when operating outside its applicable domain. Fig. 5b–d show representative polyester samples selected from different Tg ranges. Analysis of these structures reveals a clear trend that polyesters with higher Tg values typically contain a higher fraction of cyclic units (e.g., aromatic or alicyclic rings), whereas those with lower Tg values generally contain fewer ring units and often feature longer aliphatic chains. This behavior arises from the intrinsic rigidity of cyclic groups, which restricts local segmental mobility and consequently increases the Tg. In contrast, longer aliphatic chains increase conformational flexibility and enhance segmental mobility, ultimately leading to lower Tg values. In addition, several representative Tg values of commonly used commercial polyesters from open reports and model predictions are listed in Table S1 to provide a reference for the Tg range of the designed polyesters.
 | ||
| Fig. 5 Distribution of polyester Tg values and representative samples across different Tg ranges. (a) Distribution histogram of the polyester Tg values. (b) Several polyester samples with low Tg values (Tg < −20 °C). (c) Several polyester samples with medium Tg values (−20 ≤ Tg < 100 °C). (d) Several polyester samples with high Tg values (Tg > 100 °C). | ||
Molecular dynamics simulations
MD simulations are conducted to preliminarily assess the reliability of the screening results derived from the Tg-QSPR model. All molecular simulations were performed using the polymer consistent force field (PCFF)56,57 within the LAMMPS program.58,59 Details of the dynamics simulation procedure are provided in the SI.A total of 19 polyesters (Fig. 6a and b) were selected based on their predicted Tg values, which are randomly distributed within the range of −80 °C to 180 °C. This ensures that the MD validation covers a broad chemical space. The detailed MD simulation results are provided in Fig. S3 in the SI. Fig. 6c shows the correlation between the Tg values obtained from MD simulations and those predicted by the Tg-QSPR model. The shaded region represents the convex hull of the Tg-QSPR model. All MD data points fall within this convex hull, indicating that the MD predictions are consistent with the reasonable distribution domain of the Tg-QSPR model and further confirming the rationality of the design strategy. The maximum absolute error (AEmax, SI eqn (S1)) is 38.94 °C, and the average absolute error (AAE, SI eqn (S2)) is 17.54 °C, closely matching the model's AAE (17.72 °C). These findings suggest that the selected polyesters preliminarily exhibit the targeted thermal properties. It should be noted that, as not all PCFF parameters are directly available in LAMMPS, missing terms are generated using the automated conversion script insight2lammps.pl (https://www.MatSci.org). This process may result in minor deviations in bond-angle or torsional parameters, which can have a slight impact on the MD-predicted Tg values.
 | ||
| Fig. 6 Summary of MD simulations and experimental validation. (a) Polyester structures with only MD simulations. (b) Polyester structures with both MD simulations and experimental validation. (c) A comparison of the calculated Tg, MD-predicted Tg, and experimental Tg, with Dataset B sourced from He et al.48 | ||
Experimental synthesis and characterization of polyesters
To experimentally validate the Tg values predicted by the Tg-QSPR model and corroborated by MD simulations, a subset of the selected polyesters was synthesized and characterized by differential scanning calorimetry (DSC) measurements. For the experimentally relevant polyesters, the diacid or diol components were confirmed to be absent from the dataset reported by He et al.,48 ensuring that the resulting polyesters represent new structures. Details of the experimental procedures and the corresponding DSC curves are available in the SI (Fig. S4).Fig. 6c illustrates a comparison of the calculated Tg values (Calc.), MD-predicted Tg values, and experimental Tg values (Exp.). Similarly, all experimental data points fall within the convex hull. The AEmax between the Calc. and Exp. values is 36.02 °C, with an AAE of 16.45 °C. A similar consistency is observed between the MD and Exp. values (AEmax of 42.25 °C and AAE of 19.55 °C). These results demonstrate that the Tg-QSPR model produces consistent results with both experimental measurements and MD simulations. They further confirm the effectiveness of the proposed polyester design strategy, providing a reliable approach to the high-throughput screening and rational design of polyesters with the desired thermal properties.
Limitations and future directions
It should be noted that the PASI method primarily addresses molecular symmetry at the level of topological structures. Three-dimensional geometric symmetry and electronic symmetry are not considered in the current implementation. Additionally, this study focuses on the synthetic feasibility of the monomers, whereas a systematic evaluation of the polyesters' synthetic accessibility is not conducted. The synthetic accessibility of actual polyesters may still be influenced by factors such as melting point, boiling point, and monomer compatibility. These limitations point to important directions for future work, including the integration of three-dimensional and electronic symmetry information to further enhance the accuracy and reliability of polyester design, as well as the systematic assessment of polyester synthetic feasibility to accelerate their production.Conclusion
This work proposes a symmetry-aware, parameter-controlled design paradigm that both broadens and rationalizes the accessible chemical space of functional molecules. The PASI metric enables quantitative control over atomic-level topological symmetry, allowing the design space to be flexibly expanded or contracted according to the specific application. As a demonstration purpose, a rational framework for constructing symmetric diacid and diol monomers with PASI = 1.0 was illustrated through the modification of small molecules with SAscores <4.0, resulting in 10614 diacids and 9983 diols with SAscores ranging from 1.7 to 4.0.
Combinatorial enumeration of these designed diacids and diols generated over 100 million polyester structures, greatly enriching the diversity of candidate materials. A high-throughput evaluation of the Tg across the designed polymer library reveals a consistent trend with the typical thermal behavior observed in polyester materials. This statistical trend supports the effectiveness of the proposed monomer-design based methodology. Furthermore, the strategy was validated through a two-level verification process, in which the Tg values predicted by the Tg-QSPR model were first examined by MD simulations and subsequently confirmed by DSC experiments. The calculated Tg values show good agreement with both MD simulations (AEmax of 38.94 °C and AAE of 17.54 °C) and experimental measurements (AEmax of 36.02 °C and AAE of 16.45 °C). This consistency further confirms the reliability and robustness of the design approach that significantly expands the chemical space of polyesters. The expanded polyester library is expected to accelerate real-world polymer discovery and enable the development of high-performance materials for packaging, biomedical devices, and sustainable plastics.
It is worth emphasizing that diacids and diols, as highly reactive key intermediates, play an important role in the construction of complex organic molecules such as drug compounds and fine chemicals. Therefore, this strategy also carries broader chemical implications beyond polyester design, offering potential insights for the development of functional molecules.
Author contributions
F. Y. Y and Y.-N. Z. conceived the problem. X. J. F. and X. Y. H. carried out detailed studies. X. J. F., X. Y. H., J. Y. Z., F. Y. Y and Y.-N. Z. analyzed the problem and designed the method. X. J. F. L. H. L and Q. Y. S co-analyzed the results. X. J. F. wrote the manuscript and F. Y. Y and Y.-N. Z. made modifications. Z. H. L. provided strategic guidance. All authors contributed to useful discussions.Conflicts of interest
There are no conflicts to declare.Data availability
The Python codes supporting the findings of this study are publicly available from GitHub (https://github.com/FangyouYan/PairwiseAtomicSymmetryIndex). The repository includes the LAMMPS input files, force–field parameters, and Python scripts used for PASI calculation. In addition, the predicted Tg of the virtual polyesters are available on Zenodo (https://zenodo.org/records/17627401).Supplementary information (SI): additional results. See DOI: https://doi.org/10.1039/d5sc07720f.
Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (22578332, 22222807 and 22278319), Advanced Materials-National Science and Technology Major Project (2025ZD0619604), and Autonomous Project of State Key Laboratory of Synergistic Chem-Bio Synthesis (sklscbs202577).References
- X.-Y. Huang, C.-Z. Zhao, W.-J. Kong, N. Yao, Z.-Y. Shuang, P. Xu, S. Sun, Y. Lu, W.-Z. Huang, J.-L. Li, L. Shen, X. Chen, J.-Q. Huang, L. A. Archer and Q. Zhang, Nature, 2025, 646, 343–350 CrossRef CAS PubMed.
- R. K. Gautam, X. Wang and J. J. Jiang, Nat. Commun., 2025, 16, 8830 CrossRef CAS PubMed.
- S. Liu, W. Liu, D. Ba, Y. Zhao, Y. Ye, Y. Li and J. Liu, Adv. Mater., 2023, 35, 2110423 CrossRef CAS.
- J. Chen, C. He, X. Peng, J. Li, X. Xu, Y. Zhou, J. Shen, J. Sun, Y. Li and T. Zhao, Nat. Commun., 2025, 16, 8494 CrossRef CAS PubMed.
- M. Sandru, E. M. Sandru, W. F. Ingram, J. Deng, P. M. Stenstad, L. Deng and R. J. Spontak, Science, 2022, 376, 90–94 CrossRef CAS.
- C. Fan, H. Wu, J. Guan, X. You, C. Yang, X. Wang, L. Cao, B. Shi, Q. Peng, Y. Kong, Y. Wu, N. A. Khan and Z. Jiang, Angew. Chem., Int. Ed., 2021, 60, 18051–18058 CrossRef CAS.
- M. J. Baran, M. E. Carrington, S. Sahu, A. Baskin, J. Song, M. A. Baird, K. S. Han, K. T. Mueller, S. J. Teat, S. M. Meckler, C. Fu, D. Prendergast and B. A. Helms, Nature, 2021, 592, 225–231 CrossRef CAS.
- G. Lee, S. C. Jang, J. H. Lee, J.-M. Park, B. Noh, H. Choi, H. Kweon, D. H. Kim, H. Y. Kim, H.-S. Kim and K. J. Lee, Adv. Funct. Mater., 2024, 34, 2405530 CrossRef CAS.
- J.-H. Lee, K. Cho and J.-K. Kim, Adv. Mater., 2024, 36, 2310505 CrossRef CAS.
- J. Jing, B. Yao, W. Sun, J. Chen, J. Xu and J. Fu, Angew. Chem., Int. Ed., 2024, 63, e202410693 CAS.
- M. Chi, L. Sun, M. Nishiura, L. Huang, H. Zhang, Y. Higaki, S. Lee, K. Fukuda, Y. Zhao, T. Someya and Z. Hou, J. Am. Chem. Soc., 2025, 147, 23128–23135 CrossRef CAS.
- C. C. M. Sproncken, P. Liu, J. Monney, W. S. Fall, C. Pierucci, P. B. V. Scholten, B. Van Bueren, M. Penedo, G. E. Fantner, H. H. Wensink, U. Steiner, C. Weder, N. Bruns, M. Mayer and A. Ianiro, Nature, 2024, 630, 866–871 CrossRef CAS PubMed.
- T. Luo, C. Lu, J. Qi, C. Wang, F. Chu and J. Wang, Chem. Eng. J., 2024, 479, 147729 CrossRef CAS.
- W. Xu, C. Zhou, W. Ji, Y. Zhang, Z. Jiang, F. Bertram, Y. Shang, H. Zhang and C. Shen, Angew. Chem., Int. Ed., 2024, 63, e202319766 CrossRef CAS.
- Z. Xu, M. Zhao, Z. Yang, P. Wang, J. Liu, Y. Xie, Y. Wu, M. Gao, L. Li, X. Song and C. Dai, Adv. Funct. Mater., 2024, 34, 2405111 CrossRef CAS.
- R. Wang, Y. Zhu, S. Huang, J. Fu, Y. Zhou, M. Li, L. Meng, X. Zhang, J. Liang, Z. Ran, M. Yang, J. Li, X. Dong, J. Hu, J. He and Q. Li, Nat. Mater., 2025, 24, 1074–1081 CrossRef CAS PubMed.
- A. Jayaraman and B. Olsen, Macromolecules, 2024, 57, 7685–7688 CAS.
- L. Chen, G. Pilania, R. Batra, T. D. Huan, C. Kim, C. Kuenneth and R. Ramprasad, Mater. Sci. Eng.: R: Rep., 2021, 144, 100595 CrossRef.
- L. Gao, J. Lin, L. Wang and L. Du, Acc. Mater. Res., 2024, 5, 571–584 CrossRef CAS.
- W. Ge, R. De Silva, Y. Fan, S. A. Sisson and M. H. Stenzel, Adv. Mater., 2025, 37, 2413695 Search PubMed.
- P. L. Jacob, M. I. Parker, D. J. Keddie, V. Taresco, S. M. Howdle and J. Hirst, Chem. Sci., 2025 10.1039/D5SC05380C.
- S. Wu, Y. Kondo, M.-a. Kakimoto, B. Yang, H. Yamada, I. Kuwajima, G. Lambard, K. Hongo, Y. Xu, J. Shiomi, C. Schick, J. Morikawa and R. Yoshida, npj Comput. Mater., 2019, 5, 66 CrossRef.
- L. Tao, J. He, N. E. Munyaneza, V. Varshney, W. Chen, G. Liu and Y. Li, Chem. Eng. J., 2023, 465, 142949 CrossRef CAS.
- L. Tao, G. Chen and Y. Li, Patterns, 2021, 2, 100225 CrossRef CAS PubMed.
- S. Zhang, S. Du, L. Wang, J. Lin, L. Du, X. Xu and L. Gao, Chem. Eng. J., 2022, 448, 137643 CrossRef CAS.
- H. Qiu, J. Wang, X. Qiu, X. Dai and Z.-Y. Sun, Macromolecules, 2024, 57, 3515–3528 CrossRef CAS.
- J. Xu and T. Luo, npj Comput. Mater., 2024, 10, 74 CrossRef CAS.
- J. W. Barnett, C. R. Bilchak, Y. Wang, B. C. Benicewicz, L. A. Murdock, T. Bereau and S. K. Kumar, Sci. Adv., 2020, 6, eaaz4301 Search PubMed.
- M. Wang, Q. Xu, H. Tang and J. Jiang, ACS Appl. Mater. Interfaces, 2022, 14, 8427–8436 CrossRef CAS PubMed.
- J. Yang, L. Tao, J. He, J. R. McCutcheon and Y. Li, Sci. Adv., 2022, 8, eabn9545 CrossRef CAS PubMed.
- M. Yang, J.-J. Zhu, A. L. McGaughey, R. D. Priestley, E. M. V. Hoek, D. Jassby and Z. J. Ren, Environ. Sci. Technol., 2024, 58, 10128–10139 Search PubMed.
- B. K. Phan, K.-H. Shen, R. Gurnani, H. Tran, R. Lively and R. Ramprasad, npj Comput. Mater., 2024, 10, 186 CrossRef CAS.
- J. Xu, A. Suleiman, G. Liu, M. Perez, R. Zhang, M. Jiang, R. Guo and T. Luo, Cell Rep. Phys. Sci., 2024, 5, 102067 Search PubMed.
- L. Chen, C. Kim, R. Batra, J. P. Lightstone, C. Wu, Z. Li, A. A. Deshmukh, Y. Wang, H. D. Tran, P. Vashishta, G. A. Sotzing, Y. Cao and R. Ramprasad, npj Comput. Mater., 2020, 6, 61 Search PubMed.
- R. Wang, Y. Zhu, J. Fu, M. Yang, Z. Ran, J. Li, M. Li, J. Hu, J. He and Q. Li, Nat. Commun., 2023, 14, 2406 Search PubMed.
- P. Xu, T. Lu, L. Ju, L. Tian, M. Li and W. Lu, J. Phys. Chem. B, 2021, 125, 601–611 CrossRef CAS PubMed.
- X. Liang, X. Zhang, L. Zhang, L. Liu, J. Du, X. Zhu and K. M. Ng, Ind. Eng. Chem. Res., 2019, 58, 15542–15552 Search PubMed.
- Y. Hu, W. Zhao, L. Wang, J. Lin and L. Du, ACS Appl. Mater. Interfaces, 2022, 14, 55004–55016 CrossRef CAS.
- T. Yue, J. He, L. Tao and Y. Li, J. Chem. Theory Comput., 2023, 19, 4641–4653 CrossRef CAS.
- W. Guo, S. Chai, L. Zhang and J. Du, Chem. Ing. Tech., 2023, 95, 447–457 CrossRef CAS.
- S. Zhang, X. He, P. Xiao, X. Xia, F. Zheng, S. Xiang and Q. Lu, Adv. Funct. Mater., 2024, 34, 2409143 CrossRef CAS.
- A. Mishra, P. Rajak, A. Irie, S. Fukushima, R. K. Kalia, A. Nakano, K.-i. Nomura, F. Shimojo and P. Vashishta, Appl. Phys. Lett., 2023, 123, 121901 Search PubMed.
- J. Najeeb, S. S. A. Shah, M. H. Tahir, A. I. Hanafy, S. M. El-Bahy and Z. M. El-Bahy, Mater. Chem. Phys., 2024, 324, 129685 Search PubMed.
- S. Kim, C. M. Schroeder and N. E. Jackson, ACS Polym. Au, 2023, 3, 318–330 Search PubMed.
- M. Ohno, Y. Hayashi, Q. Zhang, Y. Kaneko and R. Yoshida, J. Chem. Inf. Model., 2023, 63, 5539–5548 CrossRef CAS.
- S. Jiang, A. B. Dieng and M. A. Webb, npj Comput. Mater., 2024, 10, 139 CrossRef CAS.
- M. Yu, Q. Jia, Q. Wang, Z.-H. Luo, F. Yan and Y.-N. Zhou, Chem. Sci., 2024, 15, 18099–18110 RSC.
- X. He, M. Yu, J.-P. Han, J. Jiang, Q. Jia, Q. Wang, Z.-H. Luo, F. Yan and Y.-N. Zhou, AIChE J., 2024, 70, e18409 CrossRef CAS.
- NIST Chemistry WebBook, NIST Standard Reference Database Number 69, https://webbook.nist.gov/chemistry/.
- M. Ishii, T. Ito, H. Sado and I. Kuwajima, Sci. Technol. Adv. Mater.: Methods, 2024, 4, 2354649 Search PubMed.
- P. Ertl and A. Schuffenhauer, J. Cheminform., 2009, 1, 8 CrossRef PubMed.
- J. Xiong, X. Feng, J. Xue, Y. Wang, H. Niu, Y. Gu, Q. Jia, Q. Wang and F. Yan, Digital Discovery, 2024, 3, 1842–1851 RSC.
- L. Van der Maaten and G. Hinton, J. Mach. Learn. Res., 2008, 9, 2579–2605 Search PubMed.
- L. Van Der Maaten, J. Mach. Learn. Res., 2014, 15, 3221–3245 Search PubMed.
- A. C. Belkina, C. O. Ciccolella, R. Anno, R. Halpert, J. Spidlen and J. E. Snyder-Cappione, Nat. Commun., 2019, 10, 5415 CrossRef.
- H. Sun, S. J. Mumby, J. R. Maple and A. T. Hagler, J. Am. Chem. Soc., 1994, 116, 2978–2987 Search PubMed.
- H. Sun, Macromolecules, 1995, 28, 701–712 CrossRef CAS.
- A. P. Thompson, H. M. Aktulga, R. Berger, D. S. Bolintineanu, W. M. Brown, P. S. Crozier, P. J. in 't Veld, A. Kohlmeyer, S. G. Moore, T. D. Nguyen, R. Shan, M. J. Stevens, J. Tranchida, C. Trott and S. J. Plimpton, Comput. Phys. Commun., 2022, 271, 108171 CrossRef CAS.
- L.-H. Lin, J.-J. Li, Y.-X. Pan, F. Yan, Z.-H. Luo and Y.-N. Zhou, ACS Appl. Mater. Interfaces, 2025, 17, 55347–55359 Search PubMed.
Footnote |
| † Both authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2026 |