
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCharacterization of the ketoreductase domain of pikromycin module 2
Eiji
Okamura
 a,
Kosuke
Ohsawa
bc,
Hidetoshi
Ban
b,
Yoshiyuki
Sugiyama
b,
Junko
Hashimoto
d,
Kei
Kudo
ef,
Masahito
Yoshida
g,
Kazuo
Shin-ya
ef,
Haruo
Ikeda†
h,
Shunji
Takahashi
*a and
Takayuki
Doi
*b
a,
Kosuke
Ohsawa
bc,
Hidetoshi
Ban
b,
Yoshiyuki
Sugiyama
b,
Junko
Hashimoto
d,
Kei
Kudo
ef,
Masahito
Yoshida
g,
Kazuo
Shin-ya
ef,
Haruo
Ikeda†
h,
Shunji
Takahashi
*a and
Takayuki
Doi
*b
aNatural Product Biosynthesis Research Unit, RIKEN Center for Sustainable Resource Science, Saitama 351-0198, Japan. E-mail: shunjitaka@riken.jp
bGraduate School of Pharmaceutical Sciences, Tohoku University, 6-3 Aza-aoba, Aramaki, Aoba-ku, Sendai 980-8578, Japan. E-mail: doi_taka@mail.pharm.tohoku.ac.jp
cFaculty of Pharmacy, Juntendo University, 6-8-1 Hinode, Urayasu 279-0013, Japan
dJapan Biological Informatics Consortium, Koto-ku, Tokyo, Japan
eNational Institute of Advanced Industrial Science and Technology (AIST), 2-4-7 Aomi, Koto-ku, Tokyo 135-0064, Japan
fBiotechnology Research Center, The University of Tokyo, 1-1-1 Yayoi, Bunkyo-ku, Tokyo 113-8657, Japan
gDepartment of Chemistry, Institute of Pure and Applied Sciences, University of Tsukuba, 1-1-1 Tennodai, Tsukuba, Ibaraki 305-8571, Japan
hKitasato Institute for Life Sciences, Kitasato University, 1-15-1 Kitasato, Minami-ku, Sagamihara, Kanagawa 252-0373, Japan
First published on 17th December 2025
Abstract
Polyketides represent a diverse class of natural products that serve as major sources of medicinal compounds. Their biosynthesis is generally catalyzed by multimodular polyketide synthase comprising functional domains, such as a β-ketosynthase, an acyltransferase, and an acyl carrier protein. These domains mediate the elongation of polyketide chains via decarboxylative Claisen-like condensation. A reductive loop comprising β-ketoreductase (KR), dehydratase (DH), and enoyl reductase domains converts the β-keto group into a hydroxy group, alkene, and alkane, respectively. Particularly, the KR domains are pivotal in determining the stereochemical configurations of the hydroxy and methyl groups on the macrolide backbone and are classified into A1, A2, B1, B2, and C types. In this study, we performed a reductive loop exchange using pikromycin PKS, PikAIII module 5 (PikAIII-M5) as a template. The PikAIII-M5 was derived from the pikromycin biosynthetic gene cluster of the pikromycin-producing Streptomyces sp. AM4900. Next, we constructed a chimeric enzyme by replacing the KR domain of PikAIII-M5 with a DH–KR di-domain derived from PikAI module 2 (PikAI-M2), followed by the artificial addition of a thioesterase domain derived from PikAIV module 6. Thereafter, we evaluated the enzymatic activity of the construct using various chemically synthesized N-acetylcysteamine substrate analogs. This demonstrated that the chimeric module enzyme catalyzed the formation of (2R,3R,4S)-3-hydroxy-2,4-dimethylheptanoic acid, indicating that the KR domain of PikAI-M2 is a B1-type. These findings offer insights into the unresolved classification of KR domains that do not strictly conform to the Caffrey motif.
Introduction
Macrolide antibiotics, which contain a macrolactone aglycone, as well as sugar moieties, are a class of biologically active polyketide products.1 Contemporary organic synthesis faces significant challenges in modifying functional groups and controlling the stereochemistry of macrolactone rings. To address these limitations, the biosynthetic engineering of natural products represents a valuable approach for accessing otherwise inaccessible chemical spaces within polyketide core scaffolds.2–5 Notably, the first macrolide antibiotic, pikromycin, was isolated in 1950.6,7 Pikromycin is biosynthesized by a modular type-I polyketide synthase (PKS), as illustrated in Fig. 1.8–10 Each module comprises β-keto synthase (KS), acyltransferase (AT), and acyl carrier protein (ACP) domains, which facilitate the elongation of the polyketide chain via decarboxylative Claisen-like condensation. Additional domains, such as β-ketoacyl-ACP reductase (KR), dehydratase (DH), and enoyl reductase (ER) domains, sequentially convert the β-keto group into hydroxy, alkene, and alkane functionalities, respectively. Further, a thioesterase (TE) in module 6 releases narbonolide, a 14-membered-ring biosynthetic precursor of pikromycin. Interestingly, the pikromycin PKS can also generate 10-deoxymethynolide, a 12-membered-ring biosynthetic precursor of methymycin (Fig. 1). These structural variations are valuable for assembling the macrolactone cores of polyketides through PKS engineering. The PikAIII module of pikromycin PKS, comprising KS–AT–KR–ACP domains, has been modified to produce the hybrid modular enzyme, PikAIII-M5–TE, a biocatalyst for homologation and lactonization reactions.11,12 The structural variations of macrolides depend on the stereochemistry of the functional groups on their macrolactone ring. In the PKS assembly line, the reductive loop, defined as the KR, KR–DH, or KR–DH–ER domain, plays a crucial role in determining the reduction degree of the β-carbonyl group formed by the decarboxylative Claisen-like condensation of the KS domain.13–15 Furthermore, the β-position hydroxylation of a carboxyl moiety by A- and B-type KRs generates L- and D-configured alcohols, respectively. The functions of KR domains are further categorized into two groups based on the final orientation of their α-substituents: A1/B1- and A2/B2-type KRs that generate D- and L-configured α-substituents, respectively.13,16 Dissimilar to these KR domains, C-type KRs lack reductase activity, although they can still function as epimerases.13 The functions of KRs can be distinguished based on their characteristic amino acid–sequence motifs.15,17 Although the LDD motif is conserved in B-type KR domains, it is absent in A-type KRs, which contain a W motif instead. The functions of KRs are further classified based on the presence or absence of the H (defining A1 or A2 types) and P (defining B1 or B2 types) motifs. Therefore, exchanging the KR-domain type within module enzymes represents a central strategy for controlling the stereochemistry of polyketide chains. This strategy generally relies on well-established sequence motifs, such as the Caffrey motif, to predict stereochemical outcomes.17 KR domains containing an XXD sequence within the LDD motif are summarized in Fig. S1. Based on structural analyses of various KR domains, only the third Asp residue of the LDD motif is required for classification as a B-type KR.18,19 This finding also suggests that XXD motifs such as SKD20 and VAD21 can be defined as a B-type KR domain and may function to orient the pantetheine arm of the substrate through hydrogen bonding. Indeed, CmiP2 KR has been experimentally demonstrated to be B-type. Additionally, the presence of a nearby proline residue classifies a KR as B2-type, whereas its absence classifies it as B1-type. The KR domain of pikromycin PKS module 2 (PikAI-M2) is an example of the XXD-type KR domain lacking the P motif, suggesting that it belongs to the B1-type. However, its classification should be validated by experimental evidence. Historically, the in vitro enzymatic activity of PikAI-M2 was indirectly demonstrated in a study exploring the substrate specificity of KS domains, where the PikAI-M2 functioned as the enzyme responsible for supplying the substrate for target modules enzymes.22 The use of malonyl-CoA as the extender substrate for PikAI-M2 in this experiment resulted in a polyketomethylene chain with no methyl group at the α-position. Consequently, it was not possible to determine the epimerase activity of the PikAI-M2 KR domain towards the α-methyl group. In addition, the stereochemical configuration at the β-carbon was elucidated through detailed enzymatic analyses based on the investigation of the enzymatic activity of the DH- or KR-domain-inactivated mutant, PikAI-M2,23 and by studying KR activity in PKSs using a novel set of chemical probes.24 The results revealed that the KR domain of PikAI-M2 was a B-type. Despite these extensive analyses, the stereochemical outcome at the α-position remains unresolved. As the two experiments were conducted using only malonyl–CoA as an extender substrate, it is still unclear whether this KR domain yields a B1- or B2-type product. In this study, to obtain experimental evidence of the stereochemistry of the α-methyl group introduced by the KR domain of PikAI-M2, we performed an in vitro enzymatic reaction using synthetic substrates.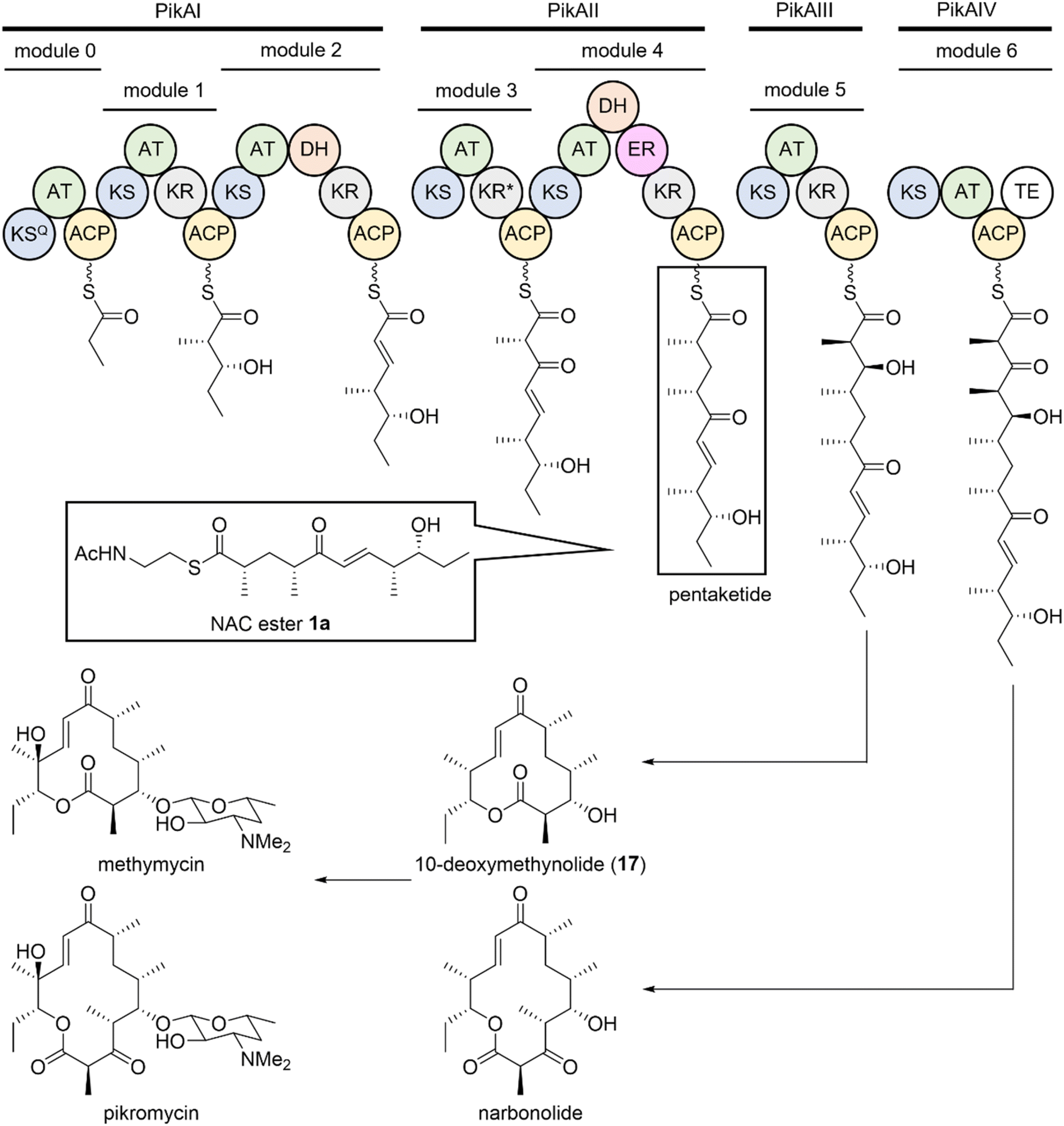 | ||
| Fig. 1 Pathway for pikromycin biosynthesis. ACP: acyl carrier protein, AT: acyltransferase, DH: dehydratase, ER: enoyl reductase, KR: β-ketoacyl-ACP reductase, KS: β-ketoacyl-ACP synthase, KSQ: KS-like domain (active motif, CSSSL, is converted into QSSSL), KR*: dysfunctional KR, and TE: thioesterase. | ||
As an initial step, we evaluated the enzymatic activity and substrate specificity of an E. coli–expressed recombinant enzyme, PikAIII-M5–TE, from Streptomyces sp. AM4900 (ref. 25 and 26) using various chemically synthesized N-acetylcysteamine (NAC) substrate analogs. To achieve the stereochemical inversion of the products generated by PikAIII-M5–TE, we constructed a chimeric module enzyme, PikAIII-M5ΔKR5::DH2-KR2-TE, by replacing the KR domain of PikAIII-M5–TE with the reductive loop (the DH–KR di-domain) from PikAI-M2. Analysis of the product revealed that the chimeric enzyme, when supplied with the NAC analog, S-(2-acetamidoethyl) (S)-2-methylpentanethioate, catalyzed the formation of (2R,3R,4S)-3-hydroxy-2,4-dimethylheptanoic acid. These results enabled us to elucidate stereochemistry of the α-position methyl group, classifying the KR domain of PikAI-M2 as a B1-type.
Results
Synthesis of N-acetylcysteamine substrates for enzymatic reactions
Our retrosynthetic analysis of the NAC ester 1a is illustrated in Scheme 1. Although the thioesterification of enone-bearing carboxylic acids has been reported without side reactions,27,28 accomplishing this with more electrophilic enones might be challenging owing to the 1,4-addition of thiols.29–33 Thus, we first condensed 2 with NAC before oxidizing the allylic alcohol in 2 into the corresponding enone. The allylic alcohol 2 was obtained via the Nozaki–Hiyama–Kishi (NHK) coupling of vinyl iodide 3 with aldehyde 4. | ||
| Scheme 1 Retrosynthetic analysis of 1a. | ||
The vinyl iodide 3 was prepared, as shown in Scheme 2. The Evans aldol reaction34 of the boron enolate (produced by the known N-acylated chiral oxazolidinone 5 (ref. 35)) with propanal yielded syn-enriched 6, with good yield (86%) and diastereoselectivity (dr > 95![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5). The resulting hydroxy group in 6 was protected by a tert-butyldimethylsilyl (TBS) group to yield the silyl ether 7 in a 92% yield. Further, the reductive cleavage of the chiral oxazolidinone by LiBH4 yielded the alcohol 8 in a 69% yield. Furthermore, the oxidation of the primary alcohol into an aldehyde with Dess–Martin periodinane,36,37 followed by Takai olefination,38,39 yielded 3 in a 47% yield over two steps. The trans geometry of the alkene in 3 was determined by 1H NMR spectroscopy through a large coupling constant (3JH,H = 14.5 Hz).
:5). The resulting hydroxy group in 6 was protected by a tert-butyldimethylsilyl (TBS) group to yield the silyl ether 7 in a 92% yield. Further, the reductive cleavage of the chiral oxazolidinone by LiBH4 yielded the alcohol 8 in a 69% yield. Furthermore, the oxidation of the primary alcohol into an aldehyde with Dess–Martin periodinane,36,37 followed by Takai olefination,38,39 yielded 3 in a 47% yield over two steps. The trans geometry of the alkene in 3 was determined by 1H NMR spectroscopy through a large coupling constant (3JH,H = 14.5 Hz).
 | ||
| Scheme 2 Synthesis of 3. | ||
After synthesizing 3, we performed the convergent synthesis of the NAC thioester 1avia NHK coupling, as depicted in Scheme 3. The optically active alcohol 9 (93% ee), which was obtained via the enzymatic desymmetrization of the corresponding meso-diol,40 was oxidized using Dess–Martin periodinane to yield 4 in a 53% yield. The NHK coupling of 4 with 2.0 equivalents of 3 in the presence of a catalytic amount of nickel(II) chloride and an excess amount of chromium(II) chloride yielded the allylic alcohol 10 in 77% yield as a 1:1 diastereomeric mixture. As the allylic alcohol moiety in 10 can be subsequently converted into an enone, these diastereomers were utilized without separation. The protection of the hydroxy group in 10 by a TBS group furnished the silyl ester 11a in a 94% yield. Following the deacetylation of 11avia basic methanolysis, the two-step oxidation of the resulting primary alcohol 12a yielded the carboxylic acid 2a in a 72% yield. Thioester 13a was obtained by coupling 2a with NAC using 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide (EDCI)·HCl/4-(dimethylamino)pyridine (DMAP) without epimerization at the α-position. The two TBS groups in 13a were removed by HF·pyridine/pyridine before the formation of the enone 1a. However, the resulting allylic alcohols (5R)-14a and (5S)-14b were unstable. The (5R)-14a isomer was readily cyclized to yield the six-membered-ring lactone (5R)-15a.41–43 The large coupling constant between the allylic and adjacent protons (3JH,H = 10.3 Hz) indicated the equatorial orientation of all substituents in (5R)-15a. Surprisingly, the six-membered lactone derived from (5S)-15b was not isolated, as it decomposed during the reaction or in vacuo solvent removal. Other acidic conditions for removing the TBS groups (aqueous HF/CH3CN and AcOH/THF/H2O) were also ineffective.
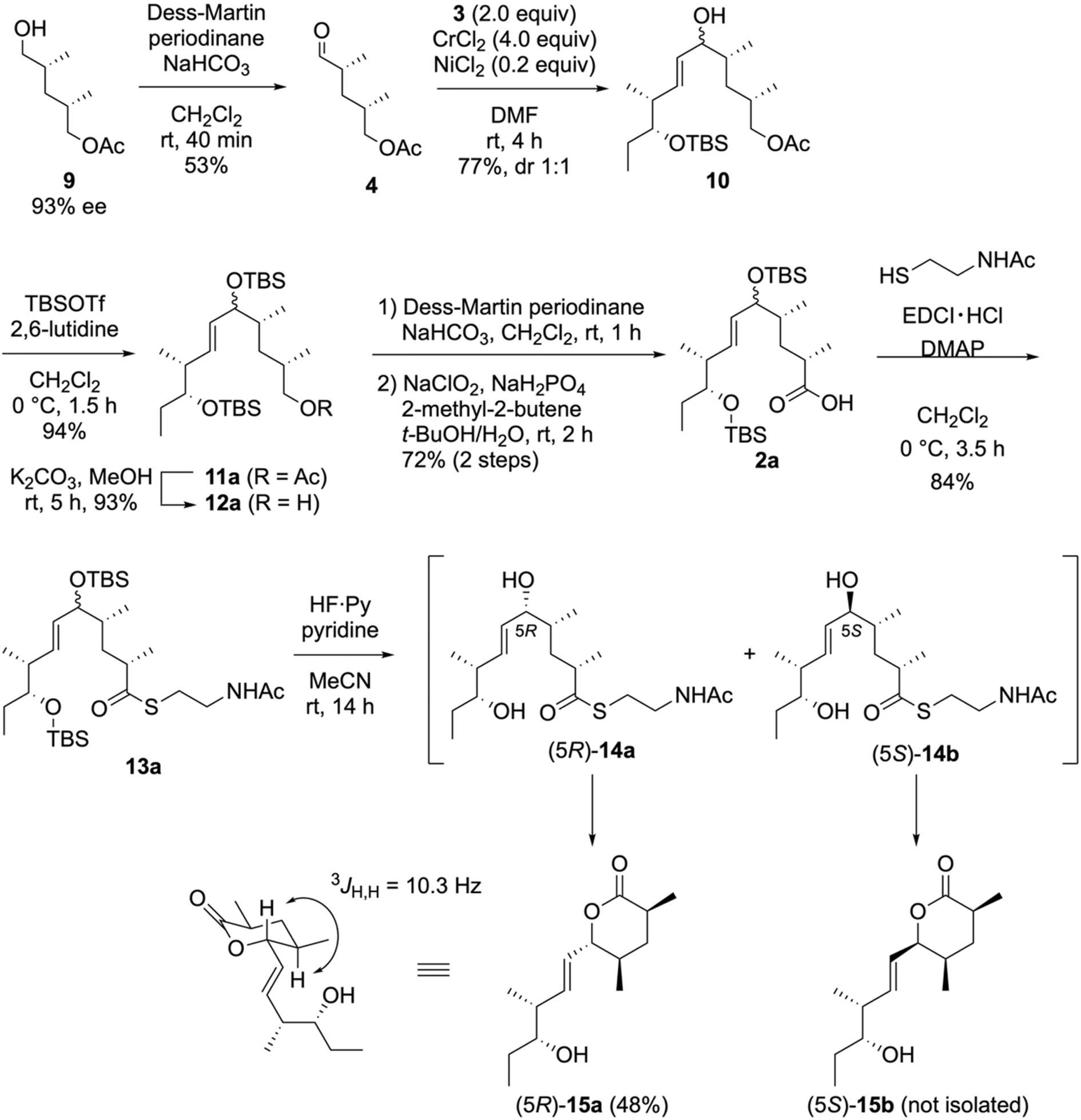 | ||
| Scheme 3 Synthesis of the NAC ester, 13a, and the accompanying undesired lactonization. | ||
To resolve the undesirable lactonization of 14a, we performed the direct conversion of the silyl ether into an enone (Scheme 4). Considering the chemoselective removal of the allylic silyl ether, we selected a triethylsilyl (TES) group as the allylic-alcohol-protecting group. Thus, the TES-protected 11b was synthesized by treating 10 with triethylsilyl trifluoromethanesulfonate (TESOTf)/2,6-lutidine. Further conversions, including the oxidation of a primary alcohol into a carboxylic acid and thioesterification with NAC, were performed in the same manner as for 13a, yielding the NAC ester 13b. Next, we removed the TES group in 13b and oxidized the resulting alcohols via a one-pot reaction with 2-iodoxybenzoic acid (IBX) in DMSO44,45 to yield the enone without forming a lactone. Finally, we synthesized 1a in an 82% yield by removing the TBS group using aqueous HF over two steps.
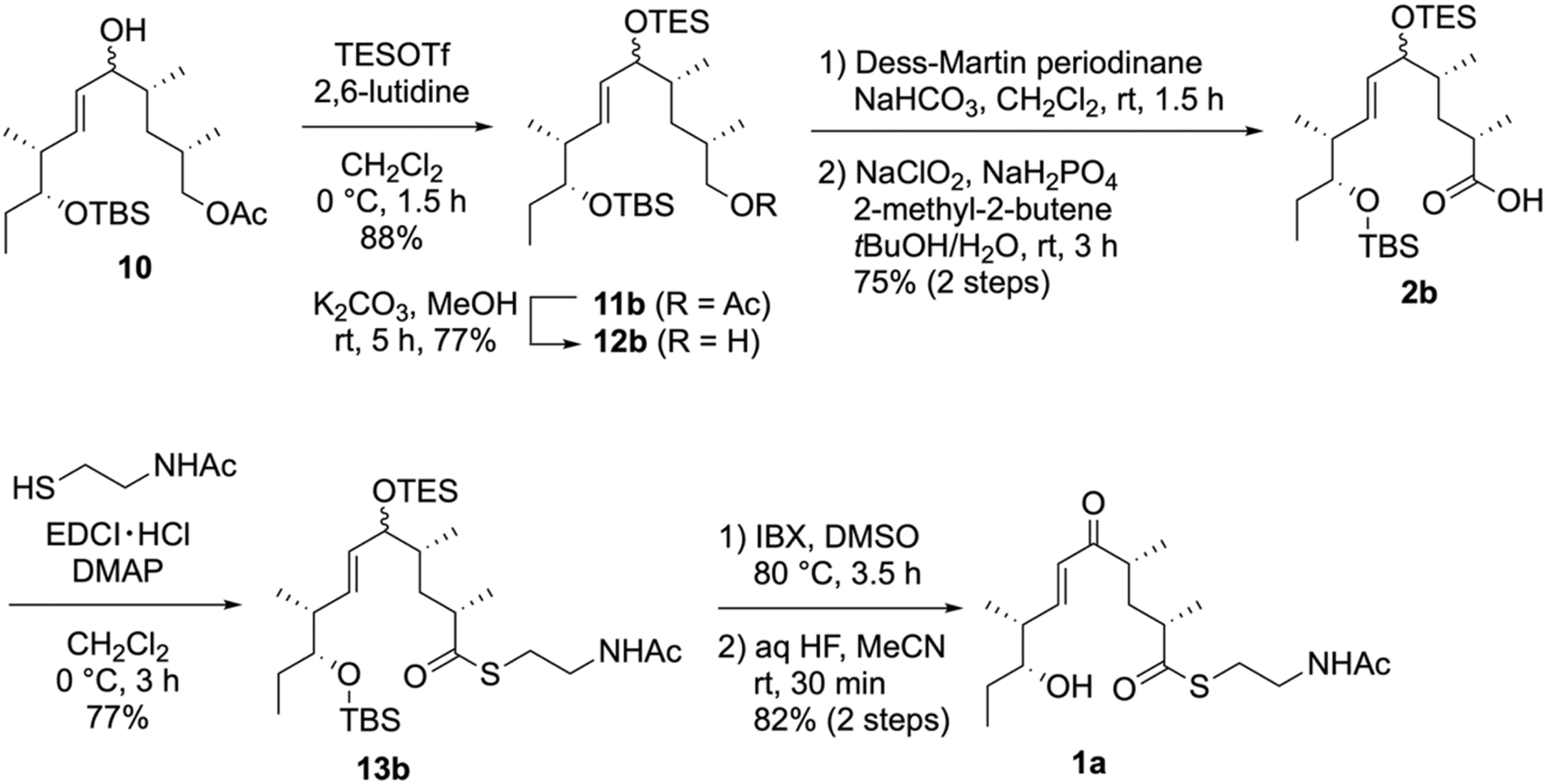 | ||
| Scheme 4 Synthesis of the NAC ester, 1a. | ||
Enzymatic conversion of the N-acetylcysteamine thioester into 10-deoxymethynolide
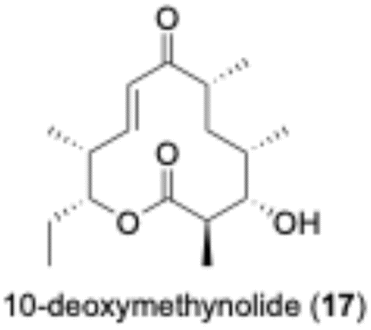
To confirm the function of the PikAIII-M5–TE chimera enzyme obtained from Streptomyces sp. AM4900 (Fig. S2–S4), we performed an enzymatic reaction using synthetic 1a and methylmalonyl–NAC (16), as shown in Fig. 2. We confirmed the conversion of substrate 1a ([M + H]+ = 358.2) into product 17 ([M + H]+ = 297.2) by ultra-performance liquid chromatography–mass spectrometry (UPLC–MS) analysis (Fig. 2). Further, the scale-up enzymatic reaction, isolation, and NMR analysis of 17 indicated that its structure corresponded to 10-deoxymethynolide.46 The absence of methylmalonyl–NAC (16) in the reaction mixture completely terminated the enzymatic reaction. | ||
| Fig. 2 Enzymatic reaction of the NAC ester, 1a, with recombinant pikromycin module 5–TE (PikAIII-M5–TE). (a) Enzymatic synthesis of 10-deoxymethynolide (17) using recombinant PikAIII-M5–TE. (b) UPLC–MS analysis of the PDA spectra (226 nm) of PikAIII-M5–TE. The red and black lines represent enzyme reactions in the presence and absence (negative control) of methylmalonyl–NAC (16), respectively. (c) Selected ion monitoring (SIM) spectra (left panel) and mass spectra (right panel) of 17 ([M + H]+ = 297.2) after the reaction in the presence of 16. (d) SIM spectra (left panel) and mass spectra (right panel) of 1a ([M + H]+ = 358.2) after the reaction in the absence of 16. | ||
Evaluation of substrate specificity of PikAIII-M5–TE
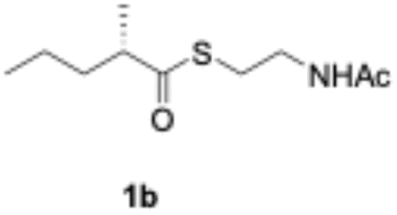
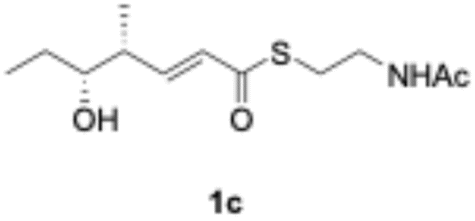
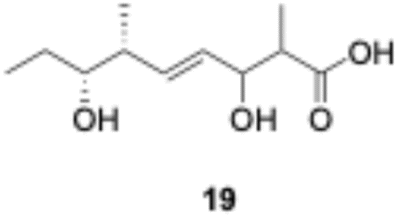
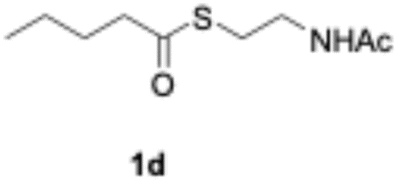
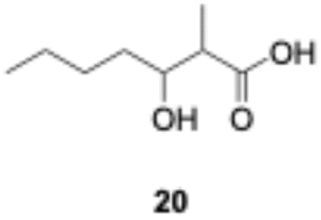
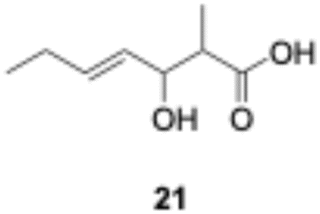
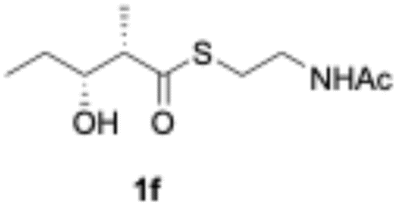
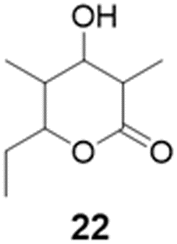
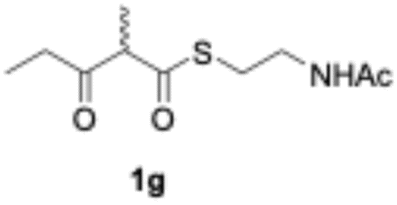
We explored PikAIII-M5–TE chimera enzyme as a template module enzyme for reductive loop exchange by first evaluating its primer substrate specificity using a set of NAC primer substrates, including physiological substrates (1a, 1c, and 1f) and analogous substrates (1b, 1d, 1e, and 1g), for various PKS modules, such as PikAI-M2, PikAII-M3, PikAII-M4, and PikAIII-M5 (Fig. 3A). As mentioned above, we obtained a reaction product from 1a ([M + H]+ = 297), and its structure was resolved to be 10-deoxymethynolide (17).47 Reaction products 18a, 19, 20, and 21, with m/z 173 [M − H]−, m/z 215 [M − H]−, m/z 159 [M − H]−, and m/z 157 [M − H]−, were detected from 1b, 1c, 1d, and 1e, respectively. In addition, triketide lactone product 22, with m/z 171.1 [M − H]−, were detected from 1f (Fig. S7 and Table 1). Conversely, no reaction product was detected from 1g (Fig. 3A and Table 1).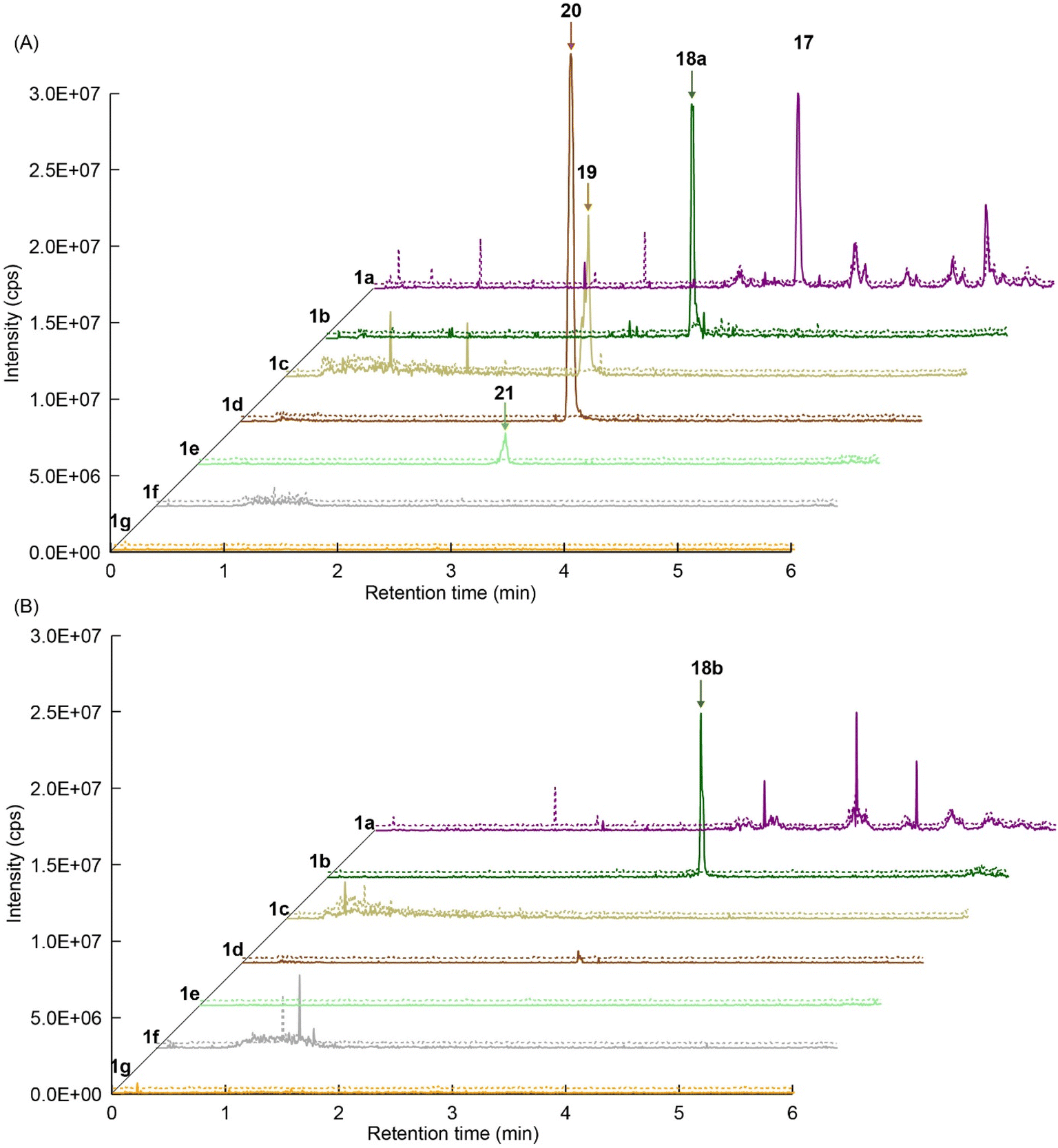 | ||
| Fig. 3 UPLC–MS analyses of reactions catalyzed by PikAIII-M5–TE (a) and PikAIII-M5ΔKR5::DH2-KR2 (b) using synthetic NAC primer substrates. The extracted-ion chromatograms were generated by filtering the data for masses within a tolerance of m/z ± 0.5 of predicted reaction products. The line numbers (Table 1) in each panel indicates reaction mixtures containing primer substrates as follows: 1a (purple); M5 substrate NAC, 1b (dark green); M5 analog substrate NAC 1, 1c (dark khaki); M3 substrate NAC, 1d (brown); M5 analog substrate NAC 2, 1e (light green); M3 analog substrate NAC, 1f (dark gray); M2 substrate NAC, 1g (orange); M2 analog substrate NAC. The retention times for the products are as follows: 17, 3.7 min; 18a, 3.2 min; 19, 2.6 min; 20, 2.9 min; 21, 2.7 min; 18b, 3.2 min. Dashed lines indicate reaction mixture without methylmalonyl-CoA. The total mass spectra in each retention time are summarized in Fig. S6. All analyses were performed in negative ion mode except for 1a, which was detected in the positive ion mode. | ||
| Substrate | Product | |
|---|---|---|
| Method A | Method B | |

|
|
ND |
|
|
|
|
|
ND |
|
|
ND |

|
|
ND |
|
|
ND |
|
ND | ND |
Evaluation of substrate specificity of PikAIII-M5-ΔKR5::DH2-KR2-TE
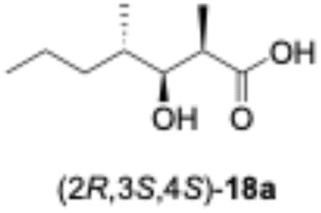
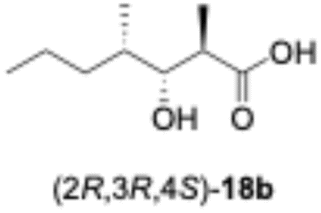
We predicted that exchanging the A1-type KR domain for a B-type KR domain would result in a stereochemical conversion at the β-position of the hydroxy group. Thus, we replaced the A1-type KR domain of PikAIII-M5–TE with the reductive loop of the DH2–KR2 di-domain from PikAI-M2 to generate the chimera module enzyme, PikAIII-M5ΔKR5::DH2-KR2-TE. To assess the activity of this module enzyme, it was heterologously expressed in E. coli and purified via Ni-affinity chromatography (Fig. S5). Thereafter, the enzymatic reaction product was analyzed using the primer substrate (Table 1, 1a–1g) and extender substrate (methylmalonyl–CoA). The result revealed that only the peak corresponding to the hydroxylated product, 18b, was detected in the reaction of 1b with PikAIII-M5-ΔKR5::DH2-KR2-TE (Fig. 3B).To determine the chemical structure of 18b, we performed a large-scale enzymatic reaction to isolate 18b, after which NMR revealed that the product 18b was (2R,3R,4S)-3-hydroxy-2,4-dimethylheptanoic acid (Fig. S8 and S9). The result indicated that the KR domain of PikAI-M2 was a B1-type.
Discussion
In the enzymatic reactions of PikAIII-M5–TE, we hypothesized that all substrates would be converted to reaction products, as PikAIII-M5–TE naturally accepts the polyketomethylene chain of 1a. We also hypothesized that other synthetic substrates could function as substrates since they exhibit smaller molecular sizes than 1a. However, contrary to our prediction, our MS analysis did not detect any reaction product from substrate 1g. Yin et al. reported that a chimeric module enzyme, which is a homolog of PikAIII-M5–TE, produced a triketide lactone.12 Among the four primer substrates exhibiting different configurations at the α- and β-positions: (2S,3S), (2S,3R), (2R,3S), and (2R,3R), the triketide lactone was produced from (2S,3R). The (2S,3R) stereoisomer was identical to 1f.12 Therefore, our results correlated with those of the literature. Although the stereochemistry of the β-functional group on the primer substrate was initially assumed to account for the reactivity of PikAIII-M5–TE reaction systems, our experimental results attributed the reactivity of PikAIII-M5–TE to the presence or absence of the β-keto group.The KR domain of PikAI-M2 was initially presumed to be type B based on sequence information,20,21 and the structure of its product also supported this classification.22 However, because the natural substrate is malonyl–CoA, it was not possible to obtain experimental evidence to distinguish between B1 and B2 types. In this study, experimental designs using methylmalonyl–CoA and synthetic substrates demonstrated that KR domain of PikAI-M2 is a B1 type. This finding represents the most significant result of our work, as KR-domain types have previously been determined only by detecting D-hydroxylated products. Notably, epimerization of the methyl group in a product synthesized by Claisen-like condensation of the KS domain on a module enzyme had not been reported experimentally. Our analysis of the chimeric module enzyme, PikAIII-M5ΔKR5::DH2-KR2-TE, therefore provides conclusive evidence that the KR domain of PikAI-M2 is a B1 type. Intriguingly, a database search revealed seven other KR domains that, like the PikAI-M2 KR domain, share the XXD motif and lack a P motif (Fig. S1).
The chimeric module enzyme produced the hydroxylated product (18b) from 1b but not the dehydrated product, consistent with prior reductive-loop swapping studies. Previous reductive-loop swap studies (DH–ER–KR) have shown that DH catalyzed dehydration is the rate-limiting step, and that this bottleneck can be rescued by selecting a compatible DH. These studies also suggested that DH domains generally exhibit high substrate specificity.48 In addition, the Pik-DH2 domain has been reported to display high substrate specificity.49 In our chimeric module enzyme, the DH2 domain must dehydrate the β-hydroxy intermediate generated after methylmalonyl-CoA incorporation. Therefore, our results likely reflect insufficient compatibility of DH2 with this noncognate intermediate. Consequently, dehydration would not proceed efficiently, leading to accumulation of the hydroxylated intermediate. Notably, substrate-reductive-loop incompatibility can manifest at different steps depending on the substrate presented to the reductive loop. In the case of the smaller substrate (1b), dehydration appears to be the primary bottleneck; however, when a bulkier substrate is supplied, failure may occur earlier in the reductive sequence. Consistent with this substrate-dependent shift, no product was detected in the reaction of the chimeric module enzyme with 1a, which we interpret this as follows. As shown in Fig. 3B, no reaction intermediates were detected when 1a was used, indicating that the KR2 reaction (and thus subsequent DH2 dehydration) did not proceed. Because the cognate PikAI-M2 substrate is relatively small, the active-site architecture of the KR2–DH2 chimera likely cannot accommodate the bulkier 1a substrate. Given that KR domains act as gatekeepers in reductive-loop processing,19 we infer that poor substrate compatibility at the KR2 step is the main reason for the lack of detectable product.
Although including chimeras bearing canonical B1- or B2-type KRs (or KR–DH pairs) as controls would further strengthen our comparisons, prior studies show that single-KR swaps often result in poor functional recovery, whereas larger domain replacements are more successful.20,48,50 Structurally, this trend is consistent with evidence that KR domains within DH/ER-containing reductive loops are stabilized through mutual interactions.51,52 A recent comprehensive study did report successful single exchanges of various KR domains, including B1-type KRs.53 However, all B1-type donor KRs were originally isolated within their native modules (i.e., not paired with an active DH domain), except for tylactone module 1, where the DH is inactive. Thus, excising a KR domain from an intact, active DH-KR reductive loop, such as the PikAI-M2 loop, remains untested,53 and is expected to disrupt stabilizing interactions, likely leading to inactivation. We therefore prioritized the KR2–DH2 replacement to evaluate KR2-dependent outcomes. Notably, because DH2 was not functionally expressed in this chimera, the observed product profile primarily reflected KR2 activity, allowing us to evaluate B1/B2-type behavior as intended. For more detailed future analyses, it will be important to assess the effects of single-domain swaps as controls. We plan to address this in future work.
Materials and methods
Chemical synthesis
:1) to afford the allylic alcohol 10 (180 mg, 449 µmol, 77%) as a colorless oil. The spectral data of the synthetic 10 were in good agreement with those of reported.551H NMR (400 MHz, CDCl3, 1:1 diastereomer mixture) δ 5.59–5.70 (m, 1H), 5.41–5.48 (m, 1H), 3.80–4.00 (m, 3H), 3.42–3.47 (m, 1H), 2.29–2.36 (m, 1H), 2.06 (s, 1.5H), 2.05 (s, 1.5H), 1.86–1.96 (m, 1H), 1.64–1.75 (m, 1H), 1.33–1.54 (m, 4H), 0.84–1.05 (m, 22H), 0.038–0.043 (m, 6H); 13C{1H} NMR (100 MHz, CDCl3, 1:1 diastereomer mixture) δ 171.20, 171.17, 135.8, 135.5, 130.7, 129.8, 77.1, 76.9, 76.5, 69.0, 41.2, 36.8, 36.7, 36.20, 36.19, 30.02, 30.0, 26.5, 26.4, 25.9, 20.9, 18.2, 18.1, 15.8, 15.5, 15.0, 9.4, 9.2, −4.3, −4.36, −4.45, −4.48; IR (neat) 3463, 2959, 2931, 2857, 1741, 1251, 1017, 836, 774 cm−1; HRMS[ESI] m/z calcd for C22H44O4SiNa [M + Na]+ 423.2901, found 423.2889.
:1) to afford the TBS ether 11a (233 mg, 450 µmol, 94%) as a colorless oil. 1H NMR (400 MHz, CDCl3, 1:1 diastereomer mixture) δ 5.56 (dd, 0.5H, J = 15.5, 7.7 Hz), 5.32–5.48 (m, 1.5H), 3.94–3.99 (m, 1H), 3.84–3.88 (m, 1H), 3.76–3.81 (m, 1H), 3.39–3.45 (m, 1H), 2.26–2.33 (m, 1H), 2.04 (s, 3H), 1.83–1.92 (m, 1H), 1.37–1.65 (m, 4H), 0.83–0.97 (m, 31H), −0.01–0.04 (m, 12H); 13C{1H} NMR (100 MHz, CDCl3, 1:1 diastereomer mixture) δ 171.3, 134.0, 131.4, 130.3, 77.5, 77.14, 77.09, 69.2, 69.1, 41.5, 41.3, 37.32, 37.29, 36.64, 36.58, 30.0, 26.51, 26.48, 25.94, 25.90, 21.0, 20.9, 18.19, 18.17, 18.11, 16.6, 16.5, 15.6, 15.5, 9.1, 9.0, −3.9, −4.1, −4.3, −4.4, −4.5, −4.8, −4.9; IR (neat) 2958, 2930, 2886, 2857, 1744, 1251, 1064, 1018, 836, 774 cm−1; HRMS[ESI] m/z calcd for C28H58O4Si2Na [M + Na]+ 537.3766, found 537.3762.
:1). 1H NMR (400 MHz, CDCl3, 1:1 diastereomer mixture) δ 5.56 (dd, 0.5H, J = 15.9, 7.3 Hz), 5.33–5.48 (m, 1.5H), 3.94–3.99 (m, 1H), 3.76–3.88 (m, 2H), 3.39–3.45 (m, 1H), 2.26–2.33 (m, 1H), 2.05 (s, 1.5H), 2.04 (s, 1.5H), 1.83–1.93 (m, 1H), 1.58–1.66 (m, 1H), 1.35–1.54 (m, 3H), 0.83–0.97 (m, 31H), 0.56 (q, 6H, J = 7.9 Hz), 0.05 (s, 3H), 0.04 (s, 3H); 13C{1H} NMR (100 MHz, CDCl3, 1:1 diastereomer mixture) δ 171.1, 134.1, 131.2, 130.2, 77.8, 77.2, 77.1, 69.14, 69.11, 41.5, 41.2, 37.4, 37.3, 36.5, 30.1, 30.0, 26.50, 26.48, 25.9, 20.9, 18.2, 18.1, 16.3, 15.7, 15.5, 9.2, 9.0, 6.9, 5.1, 5.0, −4.3, −4.49, −4.53; IR (neat) 2957, 2876, 1743, 1061, 1015, 835, 742 cm−1; HRMS[ESI] m/z calcd for C28H58O4Si2Na [M + Na]+ 537.3766, found 537.3755.
:1) to afford the alcohol 12a (237 mg, 620 µmol, 93%) as a colorless oil. 1H NMR (400 MHz, CDCl3, 1:1 diastereomer mixture) δ 5.54 (dd, 0.5H, J = 15.8, 7.6 Hz), 5.31–5.47 (m, 1.5H), 3.84–3.88 (m, 1H), 3.49–3.53 (m, 1H), 3.33–3.43 (m, 2H), 2.26–2.33 (m, 1H), 1.36–1.73 (m, 5H), 0.83–0.97 (m, 31H), 0.00–0.04 (m, 12H) (one proton (OH) was not observed); 13C{1H} NMR (100 MHz, CDCl3, 1:1 diastereomer mixture) δ 134.1, 133.9, 131.2, 130.3, 77.9, 77.3, 77.2, 77.1, 68.0, 67.8, 41.6, 41.3, 37.44, 37.43, 36.5, 36.0, 33.23, 33.20, 26.4, 26.3, 25.93, 25.91, 18.20, 18.18, 18.0, 17.9, 16.63, 16.58, 16.1, 16.0, 9.1, −3.9, −4.1, −4.31, −4.33, −4.4, −4.5, −4.79, −4.82; IR (neat) 3343, 2957, 2929, 2857, 1463, 1254, 1064, 1018, 835, 773 cm−1; HRMS[ESI] m/z calcd for C26H56O3Si2Na [M + Na]+ 495.3660, found 495.3648.
:1). 1H NMR (400 MHz, CDCl3, 1:1 diastereomer mixture) δ 5.57 (dd, 0.5H, J = 15.8, 7.4 Hz), 5.33–5.49 (m, 1.5H), 3.83–3.87 (m, 1H), 3.38–3.53 (m, 3H), 2.27–2.34 (m, 1H), 1.34–1.75 (m, 5H), 0.83–0.98 (m, 31H), 0.56 (q, 6H, J = 7.8 Hz), 0.05 (s, 3H), 0.04 (s, 3H) (one proton (OH) was not observed); 13C{1H} NMR (100 MHz, CDCl3, 1:1 diastereomer mixture) δ 134.3, 134.2, 130.9, 130.3, 78.2, 77.6, 77.1, 67.9, 67.6, 41.7, 41.2, 37.54, 37.47, 36.9, 35.8, 33.4, 33.3, 26.4, 26.3, 25.92, 25.91, 18.22, 18.15, 18.0, 16.53, 16.48, 16.3, 16.1, 9.3, 9.1, 6.9, 5.02, 4.99, −4.3, −4.46, −4.52; IR (neat) 3351, 2957, 2931, 1462, 1254, 1064, 1016, 836, 773 cm−1; HRMS[ESI] m/z calcd for C26H56O3Si2Na [M + Na]+ 495.3660, found 495.3647.
To a solution of the crude aldehyde in tert-BuOH (4.0 mL) and water (4.0 mL) were added 2-methyl-2-butene (1.2 mL, 11.3 mmol, 30 equiv), NaClO2 (102 mg, 1.13 mmol, 3.0 equiv) and NaH2PO4 (136 mg, 1.13 mmol, 3.0 equiv) at 0 °C under an argon atmosphere. After being stirred at room temperature for 2 h, the reaction mixture was diluted with EtOAc. The organic layer was separated, and the aqueous layer was extracted twice with EtOAc. The combined organic layers were washed with brine, dried over MgSO4, and filtered. The filtrate was concentrated in vacuo, and the resulting residue was purified by column chromatography on silica gel (eluted with n-hexane/EtOAc = 6:1) to afford the carboxylic acid 2a (165 mg, 340 µmol, 72% in 2 steps) as a colorless oil. 1H NMR (400 MHz, CDCl3, 1:1 diastereomer mixture) δ 5.55 (dd, 0.5H, J = 15.6, 7.6 Hz), 5.48 (dd, 0.5H, J = 15.6, 7.8 Hz), 5.33–5.40 (m, 1H), 3.84–3.90 (m, 1H), 3.42 (quin, 1H, J = 5.4 Hz), 2.51–2.63 (m, 1H), 2.25–2.34 (m, 1H), 1.82–1.92 (m, 1H), 1.55–1.64 (m, 1H), 1.36–1.49 (m, 2H), 1.19 (d, 1.5H, J = 6.8 Hz), 1.18 (d, 1.5H, J = 7.1 Hz), 1.06–1.14 (m, 1H), 0.83–0.97 (m, 27H), −0.01–0.04 (m, 12H) (one proton (COOH) was not observed); 13C{1H} NMR (100 MHz, CDCl3, 1:1 diastereomer mixture) δ 183.0, 182.9, 134.3, 134.1, 130.9, 130.2, 77.9, 77.4, 77.1, 41.5, 41.3, 37.9, 37.7, 37.4, 37.2, 36.9, 36.7, 26.53, 26.49, 25.91, 25.88, 18.20, 18.18, 18.14, 18.0, 16.54, 16.47, 15.2, 15.1, 9.1, 9.0, −4.0, −4.2, −4.3, −4.4, −4.5, −4.88, −4.90; IR (neat) 2958, 2929, 2857, 1708, 1462, 1254, 1062, 835, 773 cm−1; HRMS[ESI] m/z calcd for C26H54O4Si2Na [M + Na]+ 509.3453, found 509.3448.
:1). 1H NMR (400 MHz, CDCl3, 1:1 diastereomer mixture) δ 5.57 (dd, 0.5H, J = 15.6, 7.6 Hz), 5.33–5.50 (m, 1.5H), 3.83–3.89 (m, 1H), 3.40–3.45 (m, 1H), 2.53–2.63 (m, 1H), 2.27–2.33 (m, 1H), 1.82–1.92 (m, 1H), 1.55–1.63 (m, 1H), 1.34–1.48 (m, 2H), 1.19 (d, 1.5H, J = 6.8 Hz), 1.18 (d, 1.5H, J = 7.1 Hz), 1.06–1.13 (m, 1H), 0.83–0.97 (m, 27H), 0.56 (q, 6H, J = 8.2 Hz), 0.05 (s, 3H), 0.04 (s, 3H) (one proton (COOH) was not observed); 13C{1H} NMR (100 MHz, CDCl3, 1:1 diastereomer mixture) δ 183.0, 182.9, 134.4, 134.2, 130.8, 130.4, 78.0, 77.5, 77.1, 41.5, 41.2, 37.9, 37.8, 37.4, 37.3, 36.9, 36.7, 26.5, 25.9, 18.2, 18.12, 18.05, 16.4, 16.3, 15.3, 15.0, 9.2, 9.0, 6.9, 5.01, 5.00, −4.3, −4.45, −4.50; IR (neat) 2958, 2934, 2877, 1709, 1463, 1254, 1064, 1015, 835 cm−1; HRMS[ESI] m/z calcd for C26H54O4Si2Na [M + Na]+ 509.3453, found 509.3450.
:1) to afford the thioester 13a (150 mg, 256 µmol, 84%) as a colorless oil. 1H NMR (400 MHz, CDCl3, 1:1 diastereomer mixture) δ 5.78 (brs, 1H), 5.55 (dd, 0.5H, J = 15.5, 7.7 Hz), 5.47 (dd, 0.5H, J = 15.4, 7.6 Hz), 5.30–5.38 (m, 1H), 3.83–3.86 (m, 1H), 3.40–3.45 (m, 3H), 2.98–3.05 (m, 2H), 2.76–2.86 (m, 1H), 2.26–2.34 (m, 1H), 1.84–1.96 (m, 4H), 1.38–1.54 (m, 3H), 1.18 (d, 1.5H, J = 6.8 Hz), 1.17 (d, 1.5H, J = 7.1 Hz), 1.05–1.14 (m, 1H), 0.83–0.97 (m, 27H), −0.01–0.05 (m, 12H); 13C{1H} NMR (100 MHz, CDCl3, 1:1 diastereomer mixture) δ 204.4, 170.1, 134.34, 134.27, 130.8, 130.1, 77.7, 77.4, 77.1, 46.5, 46.4, 41.5, 41.3, 39.89, 39.87, 37.7, 37.5, 37.3, 37.2, 28.1, 26.5, 26.4, 25.92, 25.89, 23.2, 19.0, 18.8, 18.2, 18.1, 16.5, 16.4, 15.3, 15.2, 9.14, 9.09, −3.9, −4.1, −4.31, −4.33, −4.4, −4.5, −4.8, −4.9; IR (neat) 3285, 2957, 2930, 2857, 1691, 1657, 1462, 1254, 1062, 836, 774 cm−1; HRMS[ESI] m/z calcd for C30H61NO4SSi2Na [M + Na]+ 610.3752, found 610.3741.
:1). 1H NMR (400 MHz, CDCl3, 1:1 diastereomer mixture) δ 5.79 (brs, 1H), 5.56 (dd, 0.5H, J = 15.6, 7.6 Hz), 5.47 (dd, 0.5H, J = 15.7, 7.7 Hz), 5.30–5.39 (m, 1H), 3.81–3.85 (m, 1H), 3.40–3.45 (m, 3H), 2.96–3.05 (m, 2H), 2.77–2.87 (m, 1H), 2.27–2.33 (m, 1H), 1.88–1.96 (m, 4H), 1.34–1.56 (m, 3H), 1.18 (d, 1.5H, J = 6.6 Hz), 1.17 (d, 1.5H, J = 6.8 Hz), 1.05–1.12 (m, 1H), 0.83–0.97 (m, 27H), 0.56 (q, 6H, J = 8.1 Hz), 0.05 (s, 3H), 0.04 (s, 3H); 13C{1H} NMR (100 MHz, CDCl3, 1:1 diastereomer mixture) δ 204.34, 204.32, 170.2, 134.4, 134.3, 130.6, 129.9, 77.8, 77.4, 46.42, 46.36, 41.4, 41.1, 39.79, 39.77, 37.6, 37.5, 37.4, 37.1, 28.03, 28.02, 26.4, 25.9, 23.1, 18.9, 18.7, 18.1, 16.3, 16.2, 15.3, 15.1, 9.2, 9.0, 6.8, 5.0, 4.9, −4.4, −4.5, −4.6; IR (neat) 3290, 2957, 2876, 2857, 1691, 1656, 1461, 1254, 1102, 1061, 1015, 970, 836, 742 cm−1; HRMS[ESI] m/z calcd for C30H61NO4SSi2Na [M + Na]+ 610.3752, found 610.3735.
:1) to afford the lactone 15a (7.9 mg, 33.0 µmol, 48%) as a colorless oil. [α]22D −14 (c 0.68, CHCl3); 1H NMR (600 MHz, CDCl3) δ 5.74 (dd, 1H, J = 15.5, 7.7 Hz), 5.47 (dd, 1H, J = 15.5, 8.0 Hz), 4.28 (dd, 1H, J = 10.3, 8.0 Hz), 3.40–3.43 (m, 1H), 2.50–2.57 (m, 1H), 2.29–2.35 (m, 1H), 1.97 (ddd, 1H, J = 13.4, 6.2, 3.3 Hz), 1.76–1.83 (m, 1H), 1.52–1.59 (m, 1H), 1.34–1.41 (m, 2H), 1.29 (d, 3H, J = 7.1 Hz), 1.04 (d, 3H, J = 6.7 Hz), 0.96 (t, 3H, J = 7.4 Hz), 0.94 (d, 3H, J = 6.7 Hz) (one proton (OH) was not observed); 13C{1H} NMR (150 MHz, CDCl3) δ 174.1, 138.6, 127.9, 88.3, 76.3, 42.0, 37.3, 36.3, 34.2, 27.1, 17.2, 14.7, 10.3; IR (neat) 3447, 2963, 2933, 2876, 1729, 1458, 1207, 1167, 1089, 1004, 974 cm−1; HRMS[ESI] m/z calcd for C14H24O3Na [M + Na]+ 263.1618, found 263.1615.
To a solution of the crude enone in dry CH3CN (1.6 mL) was added 55% aqueous HF (0.8 mL) at 0 °C under an argon atmosphere. After being stirred at room temperature for 30 min, the reaction mixture was diluted with CH2Cl2, and quenched with saturated aqueous NaHCO3. The organic layer was separated, and the aqueous layer was extracted twice with CH2Cl2. The combined organic layers were washed with saturated aqueous brine, dried over Na2SO4, and filtered. The filtrate was concentrated in vacuo, and the resulting residue was purified by chromatography on silica gel (eluted with n-hexane/EtOAc = 1:7) to afford the alcohol 1a (54.0 mg, 151 µmol, 82% in 2 steps) as a colorless oil. The spectral data of the synthetic 1a were in good agreement with those of reported.27 [α]20D +31 (c 1.0, CH3OH) [lit. [α]D +36.5 (c 0.203, CH3OH)]; 1H NMR (600 MHz, CDCl3) δ 6.90 (dd, 1H, J = 16.0, 7.5 Hz), 6.16 (dd, 1H, J = 16.0, 0.9 Hz), 5.96 (brs, 1H), 3.53–3.56 (m, 1H), 3.45–3.51 (m, 1H), 3.38–3.43 (m, 1H), 2.98–3.06 (m, 2H), 2.80–2.84 (m, 1H), 2.71–2.76 (m, 1H), 2.45–2.50 (m, 1H), 2.13–2.18 (m, 1H), 1.98 (s, 3H), 1.92 (brd, 1H, J = 4.4 Hz), 1.50–1.56 (m, 1H), 1.38–1.47 (m, 2H), 1.18 (d, 3H, J = 6.9 Hz), 1.12 (d, 3H, J = 6.9 Hz), 1.10 (d, 3H, J = 6.9 Hz), 0.99 (t, 3H, J = 7.3 Hz); 13C{1H} NMR (150 MHz, CDCl3) δ 204.0, 202.8, 170.3, 150.1, 128.3, 75.8, 46.7, 42.2, 41.5, 39.4, 37.2, 28.5, 27.3, 23.2, 18.6, 16.9, 13.5, 10.4; IR (neat) 3309, 2969, 2933, 1686, 1660, 970 cm−1; HRMS[ESI] m/z calcd for C18H31NO4SNa [M + Na]+ 380.1866 found 380.1859.
Vector construction, protein expression, and purification
A recombinant plasmid pKU518pkmyP1_G14 containing entire gene cluster for pikromycin biosynthesis was selected from our BAC library of pikromycin-producing Streptomyces sp. AM4900 which was prepared by the same methods as described previously.56 Domain annotation was performed based on amino acid alignment by referred by previously works.10,57,58 Then, an expression vector of pHis10C-PikAIII-M5-TE (Fig. S2) containing PikAIII module 5 and thioesterase (TE) domain from module 6 were constructed as follows. Both coding regions were amplified by PCR using pKU518pkmyP1_G14 and primer sets (Table S1). Assembled gene encoding pikAIII (4.7 kb) and TE domain (1 kb) from pikAIV were cloned into the pET28b(+)vector, in which the C-terminal hexa-histidine tag was modified to deca-histidine tag (pHis10C vector). The expression vectors were introduced into NEB 10β Competent E. coli. The resulting transformants were selected on LB-Miller agar plate containing 50 µg mL−1 kanamycin at 37 °C for 12 h. The transformants were further cultured on LB-Miller medium containing 50 µg mL−1 kanamycin at 37 °C for 12 h. After plasmid isolation, the sequences of pHis10C-PikAIII-M5-TE were confirmed by sequencing.For in vivo phosphopantetheinylation of acyl carrier protein (ACP) domains on recombinant modular enzymes, pHis10C-PikAIII-M5-TE was co-expressed in E. coli with phosphopantetheinyl transferase pptA2 (sav1748) gene from Streptomyces avermitilis.59 Expression vector of the pptA2 gene was constructed as follows. The codon optimized pptA2 gene was prepared for efficient expression in E. coli (Integrated DNA Technologies Inc., Coralville, IA). Then, the pptA2 gene was cloned into NcoI and HindIII sites of pACYCDuet-1 vector.
Both pACYC-pptA2 and pHis10C-PikAIII-M5-TE vectors were used to transform Invitrogen™ One Shot™ BL21 Star™ (DE3) Chemically Competent E. coli. The resulting transformants were cultured on LB-Miller agar plate containing 50 µg mL−1 kanamycin and 34 µg mL−1 chloramphenicol at 37 °C for 16 h. The transformant was inoculated into 30 mL (15 mL × 6) of LB-Miller medium containing 50 µg mL−1 kanamycin and 34 µg mL−1 chloramphenicol and cultured at 28 °C overnight (final OD600 = 3.0). Then, overnight seed culture was inoculated into 900 mL (150 mL × 6) LB-Miller medium containing 25 µg mL−1 kanamycin and 34 µg mL−1 chloramphenicol (initial OD600 = 0.04), and cultured at 28 °C for 5 h until OD600 = 1.0 was achieved. Cooling of cell culture flasks in an ice bath (1 h) was followed by addition of 0.1 mM IPTG and cultured at 18 °C for 16 h. Cells were harvested by centrifugation at 4000×g at 4 °C for 20 min using an Allegra X-15R Benchtop Centrifuge (Beckman Coulter), and were suspended in 90 mL lysis buffer (50 mM HEPES-NaOH (pH 7.5) containing 10% [v/v] glycerol and 300 mM NaCl). Then, cell suspension containing 10 mM MgCl2 and 20 U mL−1 Cryonase Cold-active Nuclease (Takara Bio Inc.) was sonicated using BIORUPTOR 2 (Sonicbio Co., Ltd, Kanagawa, Japan) for 1 h (30 s on/30 s off, 60 cycles) on ice chilled water, and centrifuged at 20000×g at 4 °C for 30 min using an Sorvall Legend XFR (Thermo Fisher). The cleared lysate was applied to a chromatography system, BioLogic LP System + Model 2110 fraction collector (Bio-RAD) equipped with HisTrap HP (5 mL, Cytiva) for affinity purification under the following system at 4 °C. A, 200 mM potassium phosphate buffer (pH 7.5) containing 10% [v/v] glycerol; B, 200 mM potassium phosphate buffer (pH7.5) containing 10% [v/v] glycerol and 500 mM imidazole; C, lysis buffer. Column equilibrating, sample loading, and elution procedure were as follows. 100% C for 25 min (10 CV), loading of cleared lysate for 45 min (20 CV), 100% C for 25 min (10 CV), 5% B (95% A) for 25 min, 5% to 100% B (95% to 0% A) over 50 min, 100% B for 25 min; flow rate, 2 mL min−1 (Fig. S4).
Expression vector of reductive loop exchanged module enzymes, PikAIII-M5ΔKR5::DH2-KR2-TE was constructed as follows. Full length DNA of PikAIII-M5ΔKR5::DH2-KR2 gene sequence (Fig. S2) was obtained by artificial gene synthesis (Integrated DNA Technologies). Then, the PikAIII-M5ΔKR5::DH2-KR2 fragment was amplified by PCR using primer set (Table S1). TE domain from PikAIV module 6 was also amplified by PCR using primer set (Table S1) and template (pKU518pkmyP1_G14). These amplicons were assembled into NdeI and XhoI sites of the pET28b(+) modified vector (The C-terminal hexa-histidine tag coding region was modified to deca-histidine tag) by seamless cloning method using Gibson assemble master mix. Then, the assembled DNA was introduced into NEB 10β Competent E. coli. The transformants were grown on LB-Miller agar plate supplemented with 50 µg mL−1 kanamycin for 12 h at 37 °C and colonies were selected by colony directed PCR. The positive transformants were further grown in LB-Miller liquid medium supplemented with 50 µg mL−1 kanamycin for 12 h at 37 °C. The plasmids were recovered from the transformants by using QIAprep Spin Miniprep Kit and the inserted DNA sequences were verified by sequencing, obtaining pHis10C-PikAIII-M5ΔKR5::DH2-KR2-TE. For in vivo phosphopantetheinylation of ACP domains on recombinant module enzymes, the expression vectors for the module enzymes were co-expressed with phosphopantetheinyl transferase by the same expression system of PikAIII-M5-TE.
For coupling reaction in preparation of NMR samples, E. coli recombinant protein of malonyl-CoA ligases (MatB)60 were prepared template DNA of matB gene sequence was obtained by artificial gene synthesis with codon optimization for E. coli expression (GENEWIZ). The artificially synthesized DNA of matB gene were amplified by PCR using specific primer set as shown in Table S1. The amplified DNA fragment assembled into NdeI and HindIII sites of the pET28b(+) modified vector pHis8 (ref. 61) (the N-terminal hexa-histidine tag coding region was modified to octa-histidine tag) by seamless cloning method using In-Fusion method. After that, expression plasmid was constructed by same method to pACYC-pptA2 vector, excepting 50 µg mL−1 kanamycin was used as a selection marker in all cultivation steps, to obtain pHis8-matB. Excepting no co-expression with pptA2 gene, MatB recombinant protein were prepared by the same method for the module enzymes (Fig. S5).
Enzymatic reaction and UPLC-MS analysis of reaction product
A 25 µL reaction mixture (50 mM sodium phosphate buffer (pH 7.2) containing 400 mM citric acid, 10 mM NADPH, 7.5 mM methylmalonyl-NAC (16), 5 mM the NAC thioester 1a, and 42.7 nM PikAIII-M5-TE) were incubated at 25 °C for 72 h. Then, the reaction was quenched by addition of CH3CN (50% [v/v], final) and the mixture was centrifuged at 4 °C for 30 min. The samples (1 µL) was analyzed by UPLC (ACQUITY UPLC H-Class PLUS, Waters)-MS (QDa, Waters) system under the following conditions: mobile phase (A, water containing 0.1% formic acid; B, acetonitrile containing 0.1% formic acid), 5% B for 1 min, 5% to 100% B over 5 min, 100% B for 1 min, 100% to 5% B over 1 min; flow rate, 0.5 mL min−1, column; ACQUITY UPLC BEC C18 1.7 µm (Waters), column temperature; 40 °C.Substrate septicity analysis of primer substrate for PikAIII-M5-TE and PikAIII-M5-ΔKR5::DH2-KR2-TE was performed as follows: 12.5 µL reaction mixture (50 mM HEPES-NaOH (pH 7.5) containing 10% [v/v] glycerol, 300 mM NaCl, 1 mM NADPH, 1 mM methylmalonyl-CoA, and 1 mM primer substrates) was incubated for 1 h (2 nM PikAIII-M5-TE) or 12 h (28 nM PikAIII-M5ΔKR5::DH2-KR2-TE) at 25 °C. Then the mixture was acidified by an addition of hydrochloric acid (2 M, final) followed by extraction with equal volumes of ethyl acetate (3 times). Ethyl acetate phases were evaporated and dissolved in 100 µL methanol. After filtration, 4 µL of the sample was analyzed by UPLC (ACQUITY UPLC H-Class PLUS, Waters) -MS (API3200, AB Sciex) system under the following conditions: mobile phase (A, water containing 0.05% formic acid, B, acetonitrile), 5% B for 1 min, 5% to 100% B over 5 min, 100% B for 1 min, 100% to 5% B over 1 min; flow rate, 0.5 mL/min.
Enzyme reaction and isolation of reaction products for structure analysis
To determine the chemical structure of PikAIII-M5-TE reaction product, a total of 2.5 mL of the reaction (50 mM sodium phosphate buffer (pH 7.2) containing 400 mM citric acid, 10 mM NADPH, 7.5 mM methylmalonyl-NAC (16), 5 mM the NAC thioester 1a, and 42.7 nM PikAIII-M5-TE) was performed. Then, the 2.5 mL aqueous layer was extracted three times with 7.5 mL ethyl acetate. The organic layer was separated and dried under nitrogen flow. Then, 10 mL CH3OH were added to the residues, and the solution was analyzed by HPLC (2535 Quaternary Gradient Module, Waters) equipped with photodiode array detector (2998 Modular LC photodiode array detector, Waters) under the following conditions: elution rate, acetonitrile/water = 7:3 (isocratic); flow rate, 4 mL min; column, Pegasil ODS SP100 10Φ × 250 mm (Senshu Scientific Co. Ltd, Tokyo, Japan); column temperature, 20 °C (room temperature). A peak of 6.2 min was collected and the solvent was evaporated by a rotary evaporator to afford 10-deoxymethynolide (17). The spectral data of the synthetic compound 17 were in good agreement with those of reported.461H NMR (600 MHz, CDCl3) δ 6.74 (dd, 1H, J = 15.9, 5.5 Hz), 6.74 (dd, 1H, J = 15.9, 1.9 Hz), 5.01 (dd, 1H, J = 9.2, 6.1, 2.9 Hz), 3.56 (d, 1H, J = 10.5 Hz), 2.56–2.66 (m, 2H), 2.50–2.55 (m, 1H), 1.62–1.73 (m, 4H), 1.25–1.35 (m, 5H), 1.22 (d, 3H, J = 7.0 Hz), 1.12 (d, 3H, J = 7.0 Hz), 1.00 (d, 3H, J = 6.5 Hz), 0.91 (t, 3H, J = 7.6 Hz); 13C{1H} NMR (150 MHz, CDCl3) δ 204.9, 174.7, 147.1, 125.7, 78.3, 73.7, 45.2, 43.4, 38.0, 33.24, 33.21, 25.2, 17.7, 17.4, 16.4, 10.3, 9.6.
Enzymatic reaction products 18b were prepared as follows. 15 mL enzyme reaction mixture (10% [v/v] glycerol, 50 mM potassium phosphate buffer (pH 7.2), 250 mM citrate buffer (pH 7.2), 3 mM NADPH, 4 mM methylmalonate, 4 mM ATP, 0.5 mM coenzyme A, 10 mM MgCl2, 2.5 mM TCEP, 3.08 µM MatB, 2 mM 1b, and 2.18 µM PikAIII-M5ΔKR5::DH2-KR2-TE were incubated for 18 h at 25 °C. Each reaction mixture was directly applied to an ODS column and washed thoroughly with ultrapure water. Then, the bound products were eluted with 100% acetonitrile. The eluents were evaporated in vacuo and dissolved in methanol.
Author contributions
E. Okamura, K. Ohsawa. H. Ban, Y. Sugiyama, and J. Hashimoto carried out the experiments, analyzed the data. E. Okamura and K. Ohsawa wrote the original manuscript. K. Kudo, M. Yoshida, K. Shin-ya, and H. Ikeda analyzed the data and participated in the discussion. K. Shin-ya and H. Ikeda guided the project and edited the manuscript. S. Takahashi and T. Doi supervised the whole project & guided writing, reviewed, and edited the manuscript.Conflicts of interest
The authors declare no conflict of interest.Data availability
Reasonable requests for additional information can be made to the corresponding authors.Supplementary information (SI): 1H and 13C NMR spectra and additional experimental data. See DOI: https://doi.org/10.1039/d5sc07470c.
Acknowledgements
This study was supported by the Japan Agency for Medical Research and Development (AMED), Grant Number JP20ae0101045, MEXT Grant-in-Aid for Transformative Research Areas (A) Digitalization-driven Transformative Organic Synthesis (Digi-TOS), Grant Number JP24H01055, JSPS KAKENHI Grant-in-Aid for Scientific Research(C), Grant Number 25K08901, and MEXT Grant-in-Aid for Transformative Research Areas (A), Grant Number, 25H01597.References
- S. E. Rossiter, M. H. Fletcher and W. M. Wuest, Chem. Rev., 2017, 117, 12415–12474 Search PubMed.
- K. J. Weissman and P. F. Leadlay, Nat. Rev. Microbiol., 2005, 3, 925–936 CrossRef CAS PubMed.
- A. A. Koch, J. J. Schmidt, A. N. Lowell, D. A. Hansen, K. M. Coburn, J. A. Chemler and D. H. Sherman, Angew Chem. Int. Ed. Engl., 2020, 59, 13575–13580 CrossRef CAS PubMed.
- A. R. Narayan, G. Jimenez-Oses, P. Liu, S. Negretti, W. Zhao, M. M. Gilbert, R. O. Ramabhadran, Y. F. Yang, L. R. Furan, Z. Li, L. M. Podust, J. Montgomery, K. N. Houk and D. H. Sherman, Nat. Chem., 2015, 7, 653–660 CrossRef CAS PubMed.
- M. Klaus and M. Grininger, Nat. Prod. Rep., 2018, 35, 1070–1081 RSC.
- H. Brockmann and W. Henkel, Naturwissenschaften, 1950, 37, 138–139 CrossRef CAS.
- H. Brockmann and W. Henkel, Chem. Ber., 1951, 84, 284–288 CrossRef CAS.
- Y. Xue, L. Zhao, H. W. Liu and D. H. Sherman, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 12111–12116 CrossRef CAS.
- J. D. Kittendorf and D. H. Sherman, Bioorg. Med. Chem., 2009, 17, 2137–2146 CrossRef CAS PubMed.
- S. Dutta, J. R. Whicher, D. A. Hansen, W. A. Hale, J. A. Chemler, G. R. Congdon, A. R. Narayan, K. Hakansson, D. H. Sherman, J. L. Smith and G. Skiniotis, Nature, 2014, 510, 512–517 CrossRef CAS.
- D. A. Hansen, A. A. Koch and D. H. Sherman, J. Am. Chem. Soc., 2017, 139, 13450–13455 CrossRef CAS.
- Y. Yin, H. Lu, C. Khosla and D. E. Cane, J. Am. Chem. Soc., 2003, 125, 5671–5676 CrossRef CAS.
- J. F. Barajas, J. M. Blake-Hedges, C. B. Bailey, S. Curran and J. D. Keasling, Synth. Syst. Biotechnol., 2017, 2, 147–166 CrossRef.
- K. J. Weissman, Beilstein J. Org. Chem., 2017, 13, 348–371 CrossRef CAS PubMed.
- A. T. Keatinge-Clay, Nat. Prod. Rep., 2016, 33, 141–149 RSC.
- A. T. Keatinge-Clay, Nat. Prod. Rep., 2012, 29, 1050–1073 RSC.
- P. Caffrey, ChemBioChem, 2003, 4, 654–657 CrossRef CAS PubMed.
- C. Liu, M. Yuan, X. Xu, L. Wang, E. A. T. Keatinge-Clay, Z. Deng, S. Lin and J. Zheng, J. Struct. Biol., 2018, 203, 135–141 CrossRef CAS PubMed.
- E. E. Drufva, N. R. Spengler, E. G. Hix and C. B. Bailey, ChemBioChem, 2021, 22, 1122–1150 CrossRef CAS.
- S. K. Piasecki, J. Zheng, A. J. Axelrod, M. E. Detelich and A. T. Keatinge-Clay, Proteins, 2014, 82, 2067–2077 Search PubMed.
- D. Kawasaki, A. Miyanaga, T. Chisuga, F. Kudo and T. Eguchi, Biochemistry, 2019, 58, 4799–4803 CrossRef CAS.
- K. Watanabe, C. C. Wang, C. N. Boddy, D. E. Cane and C. Khosla, J. Biol. Chem., 2003, 278, 42020–42026 CrossRef CAS PubMed.
- J. Wu, T. J. Zaleski, C. Valenzano, C. Khosla and D. E. Cane, J. Am. Chem. Soc., 2005, 127, 17393–17404 CrossRef CAS PubMed.
- Y. Li, W. D. Fiers, S. M. Bernard, J. L. Smith, C. C. Aldrich and R. A. Fecik, ACS Chem. Biol., 2014, 9, 2914–2922 CrossRef CAS.
- R. Ueoka, J. Hashimoto, I. Kozone, T. Hashimoto, K. Kudo, N. Kagaya, H. Suenaga, H. Ikeda and K. Shin-ya, Biosci. Biotechnol. Biochem., 2021, 85, 890–894 CrossRef.
- S. Omura, H. Ikeda, H. Matsubara and N. Sakakane, J. Antibiot., 1980, 33, 1570–1572 CrossRef CAS PubMed.
- C. C. Aldrich, B. J. Beck, R. A. Fecik and D. H. Sherman, J. Am. Chem. Soc., 2005, 127, 8441–8452 CrossRef CAS PubMed.
- D. A. Hansen, C. M. Rath, E. B. Eisman, A. R. Narayan, J. D. Kittendorf, J. D. Mortison, Y. J. Yoon and D. H. Sherman, J. Am. Chem. Soc., 2013, 135, 11232–11238 CrossRef CAS PubMed.
- I. H. Gilbert, M. Ginty, J. A. Oneill, T. J. Simpson, J. Staunton and C. L. Willis, Bioorg. Med. Chem. Lett., 1995, 5, 1587–1590 CrossRef CAS.
- W. He, J. Wu, C. Khosla and D. E. Cane, Bioorg. Med. Chem. Lett., 2006, 16, 391–394 Search PubMed.
- M. C. Tang, C. R. Fischer, J. V. Chari, D. Tan, S. Suresh, A. Chu, M. Miranda, J. Smith, Z. Zhang, N. K. Garg, R. P. St Onge and Y. Tang, J. Am. Chem. Soc., 2019, 141, 8198–8206 CrossRef CAS.
- G. J. Wormer, N. L. Villadsen, P. Norby and T. B. Poulsen, Angew Chem. Int. Ed. Engl., 2021, 60, 10521–10525 Search PubMed.
- In the initial attempts for the synthesis of 1a, we performed the thioesterification of enone-bearing carboxylic acid. However, the reaction was complicated, producing a trace amount of the desired 1a with inseparable impurities..
- D. A. Evans, J. V. Nelson, E. Vogel and T. R. Taber, J. Am. Chem. Soc., 1981, 103, 3099–3111 CrossRef CAS.
- M. Dow, F. Marchetti, K. A. Abrahams, L. Vaz, G. S. Besra, S. Warriner and A. Nelson, Chem.–Eur. J., 2017, 23, 7207–7211 CrossRef CAS.
- D. B. Dess and J. C. Martin, J. Org. Chem., 1983, 48, 4155–4156 CrossRef CAS.
- D. B. Dess and J. C. Martin, J. Am. Chem. Soc., 1991, 113, 7277–7287 CrossRef CAS.
- K. Takai, K. Nitta and K. Utimoto, J. Am. Chem. Soc., 1986, 108, 7408–7410 CrossRef CAS.
- T. Okazoe, K. Takai and K. Utimoto, J. Am. Chem. Soc., 1987, 109, 951–953 CrossRef CAS.
- G. Q. Lin and W. C. Xu, Tetrahedron, 1996, 52, 5907–5912 CrossRef CAS.
- The formation of the 6-membered lactones by the 1,2-addition to the enone moiety has been reported in the macrolide synthesis, as mentioned in ref. 42 and 43..
- V. Velvadapu, T. Paul, B. Wagh, I. Glassford, C. DeBrosse and R. B. Andrade, J. Org. Chem., 2011, 76, 7516–7527 CrossRef CAS.
- H. S. Oh and H. Y. Kang, J. Org. Chem., 2012, 77, 1125–1130 CrossRef CAS.
- Y. K. Wu, J. H. Huang, X. Shen, Q. Hu, C. J. Tang and L. Li, Org. Lett., 2002, 4, 2141–2144 CrossRef CAS.
- N. C. Wilde, M. Isomura, A. Mendoza and P. S. Baran, J. Am. Chem. Soc., 2014, 136, 4909–4912 CrossRef CAS.
- R. Xuan, H. S. Oh, Y. Lee and H. Y. Kang, J. Org. Chem., 2008, 73, 1456–1461 CrossRef CAS PubMed.
- R. H. Lambalot and D. E. Cane, J. Antibiot., 1992, 45, 1981–1982 CrossRef CAS PubMed.
- A. Hagen, S. Poust, T. Rond, J. L. Fortman, L. Katz, C. J. Petzold and J. D. Keasling, ACS Synth. Biol., 2016, 5, 21–27 CrossRef CAS PubMed.
- Y. Li, G. J. Dodge, W. D. Fiers, R. A. Fecik, J. L. Smith and C. C. Aldrich, J. Am. Chem. Soc., 2015, 137, 7003–7006 CrossRef CAS.
- A. Zargar, R. Lal, L. Valencia, J. Wang, T. W. H. Backman, P. Cruz-Morales, A. Kothari, M. Werts, A. R. Wong, C. B. Bailey, A. Loubat, Y. Liu, Y. Chen, S. Chang, V. T. Benites, A. C. Hernandez, J. F. Barajas, M. G. Thompson, C. Barcelos, R. Anayah, H. G. Martin, A. Mukhopadhyay, C. J. Petzold, E. E. K. Baidoo, L. Katz and J. D. Keasling, J. Am. Chem. Soc., 2020, 142, 9896–9901 Search PubMed.
- A. T. Keatinge-Clay, Nat. Prod. Rep., 2012, 29, 1050–1073 Search PubMed.
- T. M. McCullough, A. Dhar, D. L. Akey, J. R. Konwerski, D. H. Sherman and J. L. Smith, Structure, 2023, 31, 1109–1120 CrossRef CAS PubMed.
- L. S. Keiser, P. P. Gatenil, Y. Zhu, K. Deng, L. Waldburger, J. W. Gin, Y. Chen, E. E. K. Baidoo, C. J. Petzold, N. Lanclos, T. R. Northen, E. Englund and J. D. Keasling, J. Am. Chem. Soc., 2025, 147, 42237–42252 CAS.
- S. V. Ley, M. N. Tackett, M. L. Maddess, J. C. Anderson, P. E. Brennan, M. W. Cappi, J. P. Heer, C. Helgen, M. Kori, C. Kouklovsky, S. P. Marsden, J. Norman, D. P. Osborn, M. Á. Palomero, J. B. J. Pavey, C. Pinel, L. A. Robinson, J. Schnaubelt, J. S. Scott, C. D. Spilling, H. Watanabe, K. E. Wesson and M. C. Willis, Chem.–Eur. J., 2009, 15, 2874–2914 CrossRef CAS PubMed.
- L. Venkatraman, C. C. Aldrich, D. H. Sherman and R. A. Fecik, J. Org. Chem., 2005, 70, 7267–7272 CrossRef CAS PubMed.
- T. Hashimoto, J. Hashimoto, I. Kozone, K. Amagai, T. Kawahara, S. Takahashi, H. Ikeda and K. Shin-ya, Org. Lett., 2018, 20, 7996–7999 CrossRef CAS.
- S. C. Tsai, H. Lu, D. E. Cane, C. Khosla and R. M. Stroud, Biochemistry, 2002, 41, 12598–12606 Search PubMed.
- J. R. Whicher, S. Dutta, D. A. Hansen, W. A. Hale, J. A. Chemler, A. M. Dosey, A. R. Narayan, K. Hakansson, D. H. Sherman, J. L. Smith and G. Skiniotis, Nature, 2014, 510, 560–564 CrossRef CAS.
- J. H. Kim, M. Komatsu, K. Shin-ya, S. Omura and H. Ikeda, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 6828–6833 CrossRef.
- A. J. Hughes and A. Keatinge-Clay, Chem. Biol., 2011, 18, 165–176 CrossRef CAS.
- J. M. Jez, J. L. Ferrer, M. E. Bowman, R. A. Dixon and J. P. Noel, Biochemistry, 2000, 39, 890–902 CrossRef CAS PubMed.
Footnote |
| † Present address: Technology Research Association for Next generation natural products chemistry, 2-4-7 Aomi, Koto-ku, Tokyo 135-0064, Japan. |
| This journal is © The Royal Society of Chemistry 2026 |