Preparation, characterization and properties of PPC-based fully biodegradable ternary blends for sustainable packaging application
Received
31st August 2025
, Accepted 18th December 2025
First published on 9th January 2026
Abstract
This study aimed to develop fully biodegradable ternary composite films based on poly(vinyl alcohol) (PVA), poly(propylene carbonate)-based polyurethane (PPC-PU), and poly(propylene carbonate) (PPC-X) for sustainable packaging applications. The films were prepared by melt blending pre-plasticized PVA1788 with PPC-PU and PPC-X at varying mass ratios, followed by hot-pressing. A comprehensive set of analyses-including mechanical tests, FTIR, SEM, DSC, TGA, gas permeability, water contact angle, and water absorption measurements-was conducted to evaluate the structure-property relationships. The results demonstrate that PPC-PU serves as both a compatibilizer and a toughener via hydrogen bonding and polar interactions with PPC-X and PVA. The optimal formulation, PVA/PPC-PU/PPC-X (30/30/40 wt%), exhibited a balanced performance: a tensile strength of 14.32 MPa, an elongation at break of 30.29%, a glass transition temperature of 44.6 °C, and a thermal degradation onset at 191 °C. The films showed moderate hydrophilicity, suitable for humidity regulation in fresh produce packaging. Notably, the composites displayed superior CO2 and oxygen barrier properties, with the 30/40/30 blend achieving a CO2 permeability of 5360 cm3 µm (m2 day)−1 and an O2 permeability of 1002 cm3 µm (m2 day)−1. However, water vapor permeability increased due to PVA's hydrophilicity. Overall, these ternary composites exhibit promising mechanical, thermal, and barrier properties, positioning them as viable and sustainable alternatives to conventional non-biodegradable plastics such as LDPE, PET, and PP, with significant potential to reduce petroleum dependence and promote carbon dioxide valorization.
1. Introduction
Plastic packaging industry accounts for 36% of global plastic production,1 with food packaging representing 40% of this market.2 Petroleum-based polymers such as polyethylene (PE), polypropylene (PP), polyamide (PA), poly(vinylidene chloride) (PVDC), polyethylene terephthalate (PET), and ethylene vinyl alcohol copolymer (EVOH) remain predominant in food packaging applications due to their robust mechanical properties and wide availability. However, their reliance on non-renewable fossil fuels and lack of biodegradability pose significant environmental challenges, as these materials persist in landfills for centuries. Gro-wing ecological concerns and societal demand for sustainability are driving the transition towards greener alternatives.3
Biodegradable polymers including cellulose, thermoplastic starch, polylactic acid (PLA), polyhydroxyalkanoates (PHAs), polycaprolactone (PCL), poly(1,4-butanediol Succinate) (PBS) and poly(butylene adipate-co-terephthalate) (PBAT) are recognized for their favorable environmental profile. A primary limitation for their use in high-barrier packaging, however, is their generally insufficient resistance to oxygen and water vapor transmission. This has spurred significant research into modification and surface coating technologies as a promising avenue for enhancing the performance of biodegradable films.3,4 Blending modification techniques involving the incorporation of metal oxides (e.g., zinc oxide, mesoporous silica, calcium carbonate, hydrotalcite, copper oxide),5–9 metal–organic frameworks (MOFs),10 natural minerals (e.g., graphene oxide, montmorillonite),11,12 or renewable natural resources (e.g., cellulose and its derivatives, lignin derivatives, bamboo flour, nanocellulose, zein, polydopamine, polycitric acid, polyaspartamide, rice husk)13–17 into polymer matrices such as PLA, PBAT, or PLA/PBAT blends have been shown to enhance the mechanical and barrier properties (against oxygen and water vapor) of the resulting composites. For instance, Mahsa Ansari et al. prepared a novel biodegradable nanocomposite film using a poly(lactic acid) (PLA)/poly(butylene adipate-co-terephthalate) (PBAT) matrix incorporated with zinc–aluminum layered double hydroxide (Zn–Al LDH) nanoparticles via melt compounding. They found that the nanocomposite containing 1 phr of nanoparticles exhibited the lowest water vapor transmission rate (WVTR) of 0.00244 g h−1 m−2, representing a 70% reduction compared to the unfilled material.5 Jissy Jacob et al. produced PLA composites with functionalized mesoporous silica (PLA/0.6SPA-MPS-ACN) using a solution casting method. Their results indicated that incorporating 5 wt% of the filler reduced the oxygen transmission rate (OTR) by 52%.8 Similarly, Zheng Cheng et al. developed a bamboo-plastic composite by mixing bamboo powder lignocellulose with PBAT resin granules (10 wt% bamboo content), followed by hot-pressing to form films. The resulting bamboo/PBAT bioplastic composite demonstrated a superior oxygen permeability of 1.48 × 10−2 cm3 m (m2 24 h 0.1 MPa)−1, while its water vapor permeability remained close to that of neat PBAT.13 Furthermore, Li Wu et al. fabricated PLA/poly(propylene carbonate) (PPC)/acetylated lignin (ACEL) films via melt blending and biaxial drawing, using PPC and ACEL as functional toughening components. The biaxially oriented films exhibited an exceptional oxygen permeability of 0.13 × 10−13 cm3 cm (cm2 s Pa)−1, surpassing the barrier performance of neat PLA.17 However, these improvements still remain at the experimental stage and fall significantly short of the performance requirements for high-barrier packaging materials. Moreover, advanced coating strategies deployed via wet deposition are pivotal for improving the barrier properties of polymers in sustainable packaging.18 Key approaches include organic/inorganic hybrid coatings, which provide tunable oxygen barriers but are often limited by mechanical fragility, and nanocomposite coatings, which offer excellent gas barrier performance yet raise questions regarding food-contact safety. While multilayer thin-film structures represent a viable approach, their economic feasibility depends on successful optimization. Critically, these innovations must align with circular economy principles, comply with evolving regulations such as the EU Packaging and Waste Regulation, and incorporate bio-based or biodegradable matrices to develop next-generation packaging that balances functionality with end-of-life sustainability.19,20 In summary, although these commercially applied polymer materials have achieved certain research progress, they still face several limiting factors for development. For instance, PBAT production relies entirely on non-renewable petroleum resources, and the material typically exhibits poor mechanical strength. Conversely, PLA is produced via microbial fermentation from agricultural feedstocks, which may compete with food resources, and it often suffers from high mechanical brittleness and high production costs.21
Consequently, ongoing research is dedicated to overcoming these drawbacks through the development of novel materials and modification strategies. An environmentally promising alternative, poly(propylene carbonate) (PPC), produced through the copolymerization of carbon dioxide and propylene oxide, was first commercialized by Empower Materials in 2010.22 However, unmodified PPC suffers from: low glass transition temperature (∼35 °C), limiting thermal stability, inadequate mechanical strength (tensile strength <10 MPa).23,24 Recent breakthroughs in PPC-P terpolymer synthesis (phthalate-modified) and PPC-X quadripolymers synthesis (cyclohexene carbonate phthalate-modified) by Sun Yat-sen University's research group, demonstrated enhanced thermal stability (Tg up to 54 °C for PPC-P/116 °C for PPC-X) and high mechanical strength (43 MPa for PPC-P/53.9 MPa for PPC-X), but retained suboptimal toughness (elongation at break below 5% for PPC-P/10% for PPC-X),25–27 which restricts its widespread use. Consequently, developing effective toughening strategies for PPC-P/PPC-X represents a critical research challenge. To date, only a limited number of studies have explored this area, primarily employing two approaches: (1) physical blending modification and (2) chemical chain extension or cross-linking. For instance, Zhang T. W. et al. from Meng's group prepared PPC-P/PBAT blends with 30–50 wt% PBAT and found that PBAT incorporation improved thermal stability and elongation at break but compromised barrier properties.28 Similarly, Deng J. T. et al. achieved an optimal property balance using reactive compatibilization with a dual-component system of methylene diphenyl diisocyanate (MDI) and glycerol (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio) at 2 wt% content in a PPC-P/PBAT (75 wt%:25 wt%) matrix. The chain extender significantly enhanced thermal stability and tensile elongation, though barrier performance deteriorated with increasing PBAT content. Notably, samples containing the chain extender exhibited superior barrier properties compared to unmodified blends at the same composition.29 Although both above-mentioned modification approaches improved the overall mechanical properties of PPC-P, the high PBAT incorporation ratio significantly compromised its gas barrier performance and negative carbon characteristics. Concurrently, no studies have yet reported the modification of PPC-X (tetrapolymer with analogous backbone structure to PPC-P, Scheme 1) in prior literature. Therefore, the goal was to identify a modifier capable of substantially improving the mechanical properties of PPC-X while maintaining its acceptable gas barrier characteristics, thereby obtaining a composite composed of biodegradable constituents with confirmed degradation potential.30
:1 molar ratio) at 2 wt% content in a PPC-P/PBAT (75 wt%:25 wt%) matrix. The chain extender significantly enhanced thermal stability and tensile elongation, though barrier performance deteriorated with increasing PBAT content. Notably, samples containing the chain extender exhibited superior barrier properties compared to unmodified blends at the same composition.29 Although both above-mentioned modification approaches improved the overall mechanical properties of PPC-P, the high PBAT incorporation ratio significantly compromised its gas barrier performance and negative carbon characteristics. Concurrently, no studies have yet reported the modification of PPC-X (tetrapolymer with analogous backbone structure to PPC-P, Scheme 1) in prior literature. Therefore, the goal was to identify a modifier capable of substantially improving the mechanical properties of PPC-X while maintaining its acceptable gas barrier characteristics, thereby obtaining a composite composed of biodegradable constituents with confirmed degradation potential.30
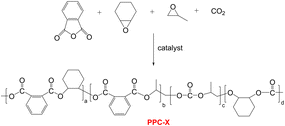 |
| | Scheme 1 Mechanistic scheme for PPC-X synthesis. | |
Poly(vinyl alcohol) (PVA), a semicrystalline polymer, is widely recognized for its superior oxygen barrier properties (OP < 0.1 cm3 mm [m2 day atm]−1) and complete biodegradability in food packaging applications. It is polymerized through vinyl acetate followed by hydrolysis with controlled saponification degrees ranging from 70% to 99%. The molecular structure of PVA features densely packed hydroxyl groups that establish robust hydrogen bonding networks, resulting in exceptional gas barrier performance.31 For instance, recent studies on EVOH/PVA blend systems (LDPE/EVOH-PVA/LDPE multilayers) have demonstrated that incorporating PVA (15vol%) via melt-blending significantly enhances oxygen barrier properties while preserving thermal-mechanical performance, attributed to improved morphological compatibility from structural similarity between PVA segments and EVOH matrices.32 Wu et al. studied bio-based PA610/PVA blends and found that the oxygen barrier properties of the composite blown films depended on composition. The optimal performance was achieved at critical PVA contents inversely related to the degree of polymerization (10 wt% for DP1400, 15 wt% for DP800, 20 wt% for DP500), where DSC/DMA/WAXD analyses confirmed molecular-level miscibility below these thresholds due to enhanced intermolecular interactions between PA610 and PVA. The results showed performance comparable to EVOH copolymer while maintaining single-phase thermal characteristics.33 Zeng et al. investigated the impact of film-forming methods including solution casting, blown film extrusion and extrusion casting on the structural and physical properties of PVA films. The results showed that both blown film and solution-cast films exhibited high tensile strength, excellent puncture resistance, and superior oxygen barrier properties.34 It is evident that PVA exhibits excellent oxygen barrier properties, and its incorporation into composite films significantly enhances oxygen barrier performance while preserving biodegradability. Thus, introducing PVA into PPC-X matrices is expected to further improve oxygen barrier characteristics. However, the poor miscibility between highly hydrogen-bonded PVA and weakly polar PPC-X necessitates a compatibilizer to enhance interfacial adhesion. Polycarbonate-based polyurethane (PPC-PU), synthesized from biodegradable low-molecular-weight polycarbonate polyols (PPCDL) via epoxide/CO2 copolymerization, serves as a promising candidate.35–37 This FDA-approved biodegradable elastomer for food-contact applications contains carbamate groups capable of strong hydrogen-bonding interactions, exhibiting superior mechanical elasticity, hydrolysis resistance, abrasion resistance, chemical stability, and biodegradability compared to conventional polyurethanes.30
In this study, interfacial engineering plays a critical role in balancing barrier properties and processability, aligning with polymer composite design principles. Polyvinyl alcohol (PVA) is incorporated to enhance oxygen barrier performance, while polycarbonate-based polyurethane (PPC-PU) serves as an interfacial compatibilizer and toughener, enabling controlled dispersion of PVA while preserving CO2 and O2 barrier properties. Further investigation is needed to determine the optimal component concentrations required to achieve these synergistic effects. In contrast to conventional packaging material preparation technologies, the proposed design strategy offers several key advantages: it achieves high CO2 utilization efficiency, thereby reducing dependence on petroleum-based resources; the resulting materials are free of metallic compounds, mitigating safety concerns associated with metal ion migration in food contact applications; and the approach demonstrates strong potential to significantly enhance gas barrier performance, contributing to the development of sustainable packaging solutions.
2. Experimental
2.1 Materials
Poly(vinyl alcohol) (PVA1788) was supplied by Chuangchun Chemical Co. (Jiangsu, China). Glycerol (99.9%) and poly(ethyleneg-lycol) (99%, Mw = 4000 g mol−1, denoted as PEG-4K) were obtained from Shanghai Aladdin Bio-Chem Technology Co., Ltd and used without further purification. Poly(propylene carbonate phthalate-ran-cyclohexene carbonate phthalate) (PPC-X) (Mw = 7.35 × 104 g mol−1, PDI = 1.73) was synthesized according to Scheme 1.27 and provided by Beijing Risun Science & Technologies Ltd (China). Poly(propylene carbonate)-based polyurethane (PPC-PU) (Mw = 5.28 × 104 g mol−1, PDI = 2.12) was synthesized according to Scheme 2.35 and supplied by Zhuhai Zhongguan Petrochemical Co., Ltd (China). Both polymers underwent standardized pretreatment involving vacuum drying at 50 °C for 8 hours to eliminate residual moisture prior to subsequent processing and characterization.
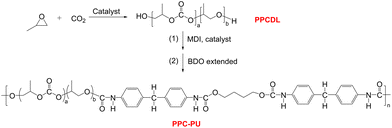 |
| | Scheme 2 Reaction mechanism and stepwise synthetic route of PPC-PU. | |
2.2 Preparation of PPC-PU/PVA/PPC-X composite films
2.2.1 Preplasticization of PVA.
The preplasticization of PVA was conducted as follows: PVA1788 powders were first dried at 60 °C for 8 h to eliminate residual moisture. Subsequently, PVA1788 (1000 g), glycerol (150 g), and PEG-4K (50 g) (1:0.15:0.05 wt%) were precisely weighed and fed into a premixing apparatus. The mixture was premixed at room temperature (25 ± 2 °C) for 5 min at 2000 rpm. The premixed blend was then transferred to a ceramic tray and placed in a constant-temperature oven at 80 °C for 10 h to enable thorough diffusion of glycerol and PEG-4K into the PVA1788 matrix, achieving effective preplasticization. This procedure ensures uniform plasticizer distribution within the polymer structure prior to further processing.
2.2.2 Preparation of ternary PVA/PPC-PU/PPC-X blends.
The PPC-PU/PVA/PPC-X blends were prepared by first drying all polymers at 70 °C for 8 h. Melt compounding was then carried out in a torque rheometer at 180 °C and a screw speed of 50 rpm for 7 minutes, using a range of mass ratios as specified in Table 1. Neat PPC-X, PPC-PU and preplasticized PVA1788 were also processed under the same conditions as control samples.
Table 1 Formulations of PPC-PU/PVA/PPC-X blends
| Entry |
PVAa (wt%) |
PPC-PU (wt%) |
PPC-X (wt%) |
(xPVA/yPPC-PU/zPPC-X, wt%) |
|
Pre-plasticized according to the procedure described in Section 2.2.1.
|
| 1 |
20 |
10 |
70 |
20/10/70 |
| 2 |
20 |
20 |
60 |
20/20/60 |
| 3 |
20 |
30 |
50 |
20/30/50 |
| 4 |
20 |
40 |
40 |
20/40/40 |
| 5 |
30 |
10 |
60 |
30/10/60 |
| 6 |
30 |
20 |
50 |
30/20/50 |
| 7 |
30 |
30 |
40 |
30/30/40 |
| 8 |
30 |
40 |
30 |
30/40/30 |
| 9 |
40 |
10 |
50 |
40/10/50 |
| 10 |
40 |
20 |
40 |
40/20/40 |
| 11 |
40 |
30 |
30 |
40/30/30 |
| 12 |
40 |
40 |
20 |
40/40/20 |
| 13 |
100 |
0 |
0 |
100/0/0 |
| 14 |
0 |
100 |
0 |
0/100/0 |
| 15 |
0 |
0 |
100 |
0/0/100 |
| 16 |
0 |
30 |
70 |
0/30/70 |
2.2.3 Preparation of PVA/PPC-PU/PPC-X composite films.
The prepared ternary blends were compression-molded into composite films using a flat-plate vulcanizing press at 165 °C under a pressure of 10 MPa for 5 minutes. From the resulting films, dumbbell-shaped specimens were cut for mechanical testing, and film samples were prepared for barrier property evaluation. All samples were fabricated using an identical protocol. The compositions are designated as xPVA/yPPC-PU/zPPC-X, where x, y, and z represent the weight percentages of PVA, PPC-PU, and PPC-X, respectively, per 100 parts of the blend.
2.3 Analysis method
Mechanical properties were assessed according to ISO 527-3:2018 using standard dumbbell specimens with nominal dimensions of 25 mm × 4 mm × 1 mm, which were die-cut from compression-molded sheets via a servo-hydraulic cutting press. Uniaxial tensile testing was conducted on an electromechanical universal testing frame (AMETEK LLOYD Instruments, China) at a crosshead displacement rate of 50 mm min−1 under ambient laboratory conditions of 23 ± 2 °C and 50 ± 5% RH. All specimens were conditioned for 24 hours in a climate-controlled chamber set to the same thermo-hygrometric parameters prior to testing to ensure reproducibility. Stress-strain data were recorded for no fewer than five replicates per material composition, and the average values of the measured parameters were reported as the tensile performance indices.
Thermogravimetric analysis (TGA) was performed on a NETZSCH TG 209F3 instrument (NETZSCH-Gerätebau GmbH, Germany) in a nitrogen atmosphere with a flow rate of 50 mL min−1. Samples were heated dynamically from 35 °C to 600 °C at a heating rate of 10 °C min−1. The isothermal thermogravimetric analysis (ITG) was performed under a nitrogen flow rate of 20 mL min−1, with heating from 50 °C to 200 °C at a rate of 10 °C min−1, followed by an isothermal hold at 200 °C for 35 minutes.
Thermal transitions of the samples were evaluated using differential scanning calorimetry (DSC Q2000, TA Instruments, New Castle, DE) via the following procedure: an initial heating from 25 °C to 160 °C at 10 °C min−1 under N2 at 50 mL min−1, followed by a 2-minute isothermal hold; rapid cooling to −70 °C at 10 °C min−1 using liquid nitrogen cooling; and a second heating to 160 °C at the same rate to remove thermal history. Glass transition temperatures (Tg) were determined from the second heating scan.
Fourier transform infrared (FTIR) spectroscopy was conducted on a Bruker Invenio S FTIR spectrometer (Bruker Optics China, Beijing) equipped with a diamond ATR accessory. The samples were prepared by compression molding at 150 °C in a hydraulic hot press to form uniform thin films with thicknesses between 5 and 8 μm. Spectra were acquired by averaging 32–64 scans at a resolution of 4 cm−1 across the mid-infrared region from 400 to 4000 cm−1. Background spectra were collected under ambient conditions and automatically subtracted from all sample measurements to minimize atmospheric contributions.
Morphological analysis of fracture surfaces was performed using cryogenic preparation methods. Specimens with a thickness of 1 mm were submerged in liquid nitrogen for 120 seconds to induce brittle fracture and subsequently dried at ambient conditions. A 30 nm gold layer was deposited using a high-performance ion sputtering system (108, Cressington Scientific Instruments, Watford, UK) with a deposition current of 25 mA for 30 seconds. High-resolution images were acquired using a Phenom Pro X desktop SEM (FUNA Scientific Instruments, Shanghai, China) operated at 15 kV under high vacuum conditions of 5 × 10−5 Torr and a working distance of 10 mm.
Gas barrier performance was quantitatively evaluated through standardized permeability measurements. Prior to testing, all samples underwent 48-hour conditioning at 23 ± 2 °C and 50 ± 5% RH. The thickness of all samples was measured in accordance with the standard procedure by a digital thickness gauge, which involved taking five measurements at the four corners and the geometric center, with the average value being used for the gas permeability test. The oxygen permeability coefficient (OP) were determined with an OX-TRAN 2/12R system (AMETEK MOCON, USA) following ISO 15105-1:2007 at 23 ± 0.5 °C. Similarly, the carbon dioxide permeability coefficient (CO2P) was measured using a PERMATRAN-C 4/30 analyzer (same manufacturer) under identical thermodynamic conditions. Water vapor permeability coefficient (WVP) was characterized with a MOCON 3/61 instrument (AMETEK MOCON) in accordance with ASTM F-1249 at 23 °C and 50% RH.
Water absorption was characterized in accordance with ASTM D570-22. Square specimens measuring 60 × 60 × 1 mm3 were prepared by compression molding with a flat vulcanizing press and then dried in a vacuum oven at 50 °C for 24 h to remove residual moisture. The initial dry mass (m1) of each sample was measured using a precision analytical balance with 0.1 mg resolution. All samples were fully immersed in deionized water maintained at 25.0 ± 0.5 °C for 24 h. After immersion, surface moisture was promptly blotted with lint-free filter paper and the saturated mass (m2) was determined within 30 seconds to minimize evaporation loss. The water absorption percentage was calculated as:
| Water absorption (wt%) = [(m2 − m1)/m1] × 100% |
Surface wettability was evaluated by static contact angle measurements following ASTM D7334-08. Square specimens (20 × 20 × 1 mm3) were ultrasonically cleaned at 40 kHz in analytical-grade isopropanol for 10 minutes and dried using a high-purity nitrogen jet (N2 ≥ 99.999%). Measurements were carried out on a commercial contact angle goniometer featuring a high-resolution CCD camera (5 μm per pixel) and a temperature-controlled stage set to 25.0 ± 0.5 °C. Ultrapure water droplets (5.0 μL, 18.2 MΩ cm) were dispensed using a precision syringe with a 0.5 mm flat-tipped needle under static conditions. Images were captured at 5 seconds after deposition to minimize evaporation effects. Contact angles were determined via Young–Laplace fitting using Attension Theta software with the Drop Analysis module, with five replicates performed at randomized locations (≥5 mm apart) to account for surface heterogeneity. All procedures were conducted under controlled ambient conditions (23 ± 2 °C, 50 ± 5% RH) to ensure reproducibility.
3. Results and discussion
3.1 Mechanical properties
The tensile properties of xPVA/yPPC-PU/zPPC-X blends with varying mass ratios were investigated. Neat PPC-X, PPC-PU, and plasticized PVA were used as reference materials and tested under identical conditions. As summarized in Fig. 1 and Table 2, neat PPC-X exhibited high tensile strength (37.52 MPa) but low ductility (3.76% elongation at break), characteristic of brittle fracture behavior. In contrast, PPC-PU demonstrated elastomeric properties with high fracture elongation (931.37%) but relatively low tensile strength (13.77 MPa). Plasticized PVA showed both high tensile strength (42.37 MPa) and high ductility (585.00% elongation at break).
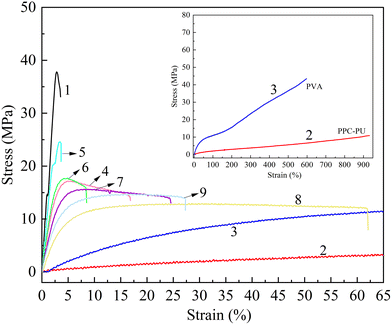 |
| | Fig. 1 Tensile stress-strain curves of typical PVA/PPC-PU/PPC-P composites (1: PPC-X; 2: PPC-PU; 3: preplasticized PVA. 4–9: xPVA/yPPC-PU/zPPC-X = 20/30/50, 30/10/60, 30/20/50, 30/30/40, 30/40/30, 40/30/30). | |
Table 2 Tensile properties of PVA/PPC-PU/PPC-X composites (xPVA/yPPC-PU/zPPC-X, wt%)
| Sample |
Tensile strength (mean ± SD) (MPa) |
Elongation at break (mean ± SD) (%) |
Yong's modulus (mean ± SD) (MPa) |
| PVA |
42.37 ± 0.50 |
585.00 ± 2.83 |
332.22 ± 14.14 |
| PPC-PU |
10.88 ± 0.22 |
931.89 ± 7.28 |
50.07 ± 2.82 |
| PPC-X |
37.52 ± 0.28 |
3.76 ± 0.28 |
1798.17 ± 28.28 |
| 20/10/70 |
25.56 ± 0.22 |
3.04 ± 0.06 |
1287.35 ± 15.21 |
| 20/20/60 |
19.74 ± 0.23 |
7.51 ± 0.28 |
1051.57 ± 16.32 |
| 20/30/50 |
16.08 ± 0.25 |
20.13 ± 0.71 |
737.10 ± 21.21 |
| 20/40/40 |
12.56 ± 0.24 |
51.42 ± 1.41 |
475.13 ± 14.14 |
| 30/10/60 |
25.04 ± 0.26 |
3.53 ± 0.28 |
1381.73 ± 22.57 |
| 30/20/50 |
18.39 ± 0.23 |
11.30 ± 0.71 |
856.26 ± 20.25 |
| 30/30/40 |
14.32 ± 0.22 |
30.29 ± 1.47 |
584.59 ± 19.80 |
| 30/40/30 |
11.20 ± 0.21 |
78.86 ± 2.83 |
283.63 ± 13.57 |
| 40/10/50 |
14.45 ± 0.25 |
4.93 ± 0.29 |
787.80 ± 22.26 |
| 40/20/40 |
13.61 ± 0.22 |
12.32 ± 0.57 |
659.86 ± 14.21 |
| 40/30/30 |
12.09 ± 0.20 |
24.03 ± 1.06 |
574.77 ± 21.23 |
| 40/40/20 |
8.56 ± 0.21 |
94.95 ± 1.44 |
79.77 ± 2.83 |
The incorporation of PVA and PPC-PU significantly improved the toughness of PPC-X, as reflected by a systematic increase in elongation at break. At a fixed PVA content of 20 wt%, the elongation at break increased from 3.04% to 51.42% as the PPC-PU content increased from 10 wt% to 40 wt%, while the tensile strength decreased from 25.25 MPa to 12.56 MPa. A similar linear enhancement in elongation was observed when the PVA content was held constant at 30 wt% or 40 wt% with increasing PPC-PU loadings. Furthermore, increasing the PVA content enhanced the toughening effect at any given PPC-PU content. For example, with 40 wt% PPC-PU, the elongation at break increased from 51.42% to 94.95% as the PVA content increased from 20 wt% to 40 wt%.
These results indicate that PPC-PU acts as an effective compatibilizer and toughening agent for PVA/PPC-X blends. This behavior is attributed to two primary factors: the presence of shared poly(propylene carbonate) segments in PPC-PU and PPC-X, which improves miscibility, and the hydrogen bonding and polar–polar interactions between the urethane and carbonate groups in PPC-PU and the hydroxyl groups in PVA. Additionally, carbonate groups in PPC-X contribute to polar interactions with PVA, further enhancing the interfacial adhesion between the phases (Fig. 2). The Young's modulus values of pure PPC-X, PPC-PU, and preplasticized PVA were 1789.17 MPa, 332.22 MPa, and 50.07 MPa, respectively. Upon forming composites, the Young's modulus of the composite films showed a decreasing trend. The PPC-PU elastomer contributed most significantly to this reduction. When the PVA content was held constant at 30 wt% and the PPC-PU content increased from 10 wt% to 40 wt%, the modulus of the ternary composite films dropped from 1381.73 MPa to 283.63 MPa. Similarly, with PPC-PU fixed at 30 wt% and PVA content rising from 20 wt% to 40 wt%, the modulus decreased from 737.10 MPa to 574.77 MPa. This decline in modulus is mainly due to the substantially lower modulus of PPC-PU and pre-plasticized PVA compared to PPC-X, resulting in an overall reduction in the modulus of the composite system after blending. The synergistic effect of these interactions results in significant toughening of the PPC-X matrix. However, a conventional trade-off between mechanical strength and toughness was observed, consistent with typical behavior in polymer blends.
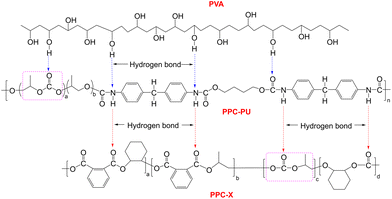 |
| | Fig. 2 Interface interaction in PVA/PPC-PU/PPC-X blends. | |
3.2 FTIR spectra of PVA/PPC-PU/PPC-X composites
As shown in Fig. 3, the FTIR spectra of PPC-X, PPC-PU, plasticized PVA, and their composite PVA/PPC-PU/PPC-X exhibit similar spectral features due to common functional groups. Three characteristic absorption bands were observed: (i) a broad peak at 3331 cm−1 attributed to hydrogen-bonded N–H and O–H stretching vibrations, (ii) a strong band at 1735 cm−1 associated with C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching of carbonyl groups, and (iii) a distinct peak at 1530 cm−1 resulting from N–H bending vibrations, collectively indicative of intermolecular hydrogen bonding within the composites. The signals at 2986 cm−1 are assigned to aromatic C–H stretching vibrations present in both PPC-X and PPC-PU, while adjacent peaks at 2934 cm−1 and 2881 cm−1 correspond to asymmetric and symmetric –CH2– stretching modes, respectively, common to all components. The spectral region between 1400 and 1600 cm−1 reveals fingerprint vibrations associated with aromatic ring skeletons, and absorptions between 1061 and 1230 cm−1 are characteristic of asymmetric C–O–C stretching from ester/ether linkages and terminal hydroxyl groups. These spectral features confirm the molecular structures and intermolecular interactions within the PVA/PPC-PU/PPC-X composite system, consistent with the mechanism proposed in Fig. 2.
O stretching of carbonyl groups, and (iii) a distinct peak at 1530 cm−1 resulting from N–H bending vibrations, collectively indicative of intermolecular hydrogen bonding within the composites. The signals at 2986 cm−1 are assigned to aromatic C–H stretching vibrations present in both PPC-X and PPC-PU, while adjacent peaks at 2934 cm−1 and 2881 cm−1 correspond to asymmetric and symmetric –CH2– stretching modes, respectively, common to all components. The spectral region between 1400 and 1600 cm−1 reveals fingerprint vibrations associated with aromatic ring skeletons, and absorptions between 1061 and 1230 cm−1 are characteristic of asymmetric C–O–C stretching from ester/ether linkages and terminal hydroxyl groups. These spectral features confirm the molecular structures and intermolecular interactions within the PVA/PPC-PU/PPC-X composite system, consistent with the mechanism proposed in Fig. 2.
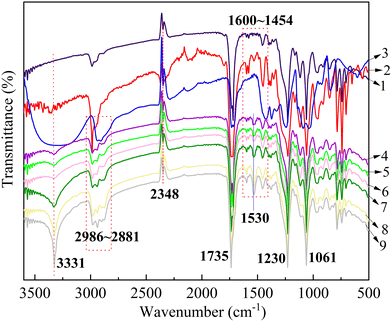 |
| | Fig. 3 FTIR spectra of PVA/PPC-PU/PPC-X composites (1: PPC-X; 2. PPC-PU; 3: preplasicized PVA. 4–9: xPVA/yPPC-PU/zPPC-X (wt%) = 20/30/50, 30/10/60, 30/20/50, 30/30/40, 30/40/30, 40/30/30). | |
3.3 Thermal properties
3.3.1 Analysis of thermal transition behavior.
The thermal transition behavior of PPC-X, PPC-PU, and their composites with PVA was presented in Fig. 4. Both amorphous PPC-PU and PPC-X showed single glass transition temperatures (Tg) near 13.2 °C and 47.7 °C, respectively, with no discernible melting endotherms below 160 °C; the subambient Tg of PPC-PU confirms its elastomeric character. Due to the onset of thermal degradation in PPC segments occurring below the melting point of PVA, the upper temperature limit was intentionally set below the PVA melting region. All composites except neat PPC-X and PPC-PU exhibited two separate glass transition endothermic peaks. With PVA content fixed at 30 wt% and PPC-PU content increasing from 10 wt% to 30 wt%, only slight reductions in Tg were observed for both phases, where the Tg of PPC-X decreased modestly to 44.6 °C. However, at 40 wt% PPC-PU, a sharp decrease in Tg was seen for both phases: the Tg of PPC-X dropped to 25.0 °C and that of PPC-PU fell to −13.2 °C. For the binary composite containing 30 wt% PPC-PU without PVA, the Tg values were 46.4 °C for PPC-X and 12.7 °C for PPC-PU. When PPC-PU content was held constant at 30 wt% and PVA was increased from 20 wt% to 30 wt%, the composite Tg values decreased slightly, with the PPC-X phase Tg reduced to 44.6 °C and the PPC-PU phase Tg lowered to 5.8 °C. Notably, at 40 wt% PVA, both phases again exhibited a pronounced drop in Tg, with the PPC-X phase transitioning at 23.2 °C and the PPC-PU phase at −15.0 °C. These results demonstrate that the incorporation of PVA into PPC-PU/PPC-X binary blends reduces the glass transition temperature of the ternary composites. It is further recommended that both PPC-PU and PVA contents be limited to 30 wt% in the ternary system, as higher concentrations significantly deteriorate the thermal transition behavior of PPC-X. The notable decrease in Tg observed beyond this threshold for PPC-PU is due to molecular encapsulation of PPC-X chains by PPC-PU, which expands interchain distance and enhances chain flexibility, thereby increasing segmental mobility. Similarly, when PVA loading exceeds 30 wt%, it tends to form the continuous phase, while the amorphous PPC-PU and PPC-X act as plasticizers and interfacial binders. This reorganization increases free volume around the PPC-X and PPC-PU chains compared to their pure state, facilitating enhanced chain motion and leading to a substantial reduction in the Tg of the PPC-X and PPC-PU phases.
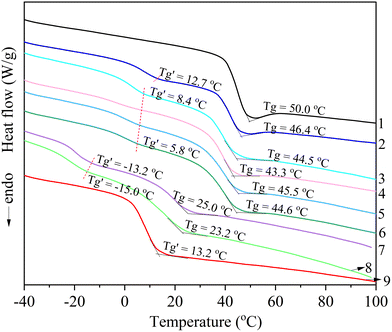 |
| | Fig. 4 DSC thermograms of representative PVA/PPC-PU/PPC-X specimens (1: PPC-X; 2–8: xPVA/yPPC-PU/zPPC-X (wt%) = 0/30/70, 20/30/50, 30/10/60, 30/20/50, 30/30/40, 30/40/30, 40/30/30. 9: PPC-PU). | |
3.3.2 Analysis of thermogravimetric properties.
The thermal degradation behavior of PPC-X, PPC-PU, preplasticized PVA and their composites was presented in Fig. 5(a)–(c) and Table 3. All materials except PPC-PU exhibited two-stage degradation profiles. Neat PPC-X began decomposing at 227.5 °C (Td,5%), with maximum degradation rates observed at 245.5 °C (Td,max1, polycarbonate segments) and 353.3 °C (Td,max2, polyphthalate-based segments). Neat PPC-PU and preplasticized PVA began decomposing at 236.3 °C and 107.5 °C (Td,5%), respectively (Fig. 5(a)). The initial weight loss in pre-plasticized PVA is mainly due to the evolution of water and glycerol. The addition of 30 wt% PPC-PU improved thermal stability, increasing Td,5% to 233.6 °C and influencing Td,max1 positively, though Td,max2 decreased, reflecting the higher initial stability of PPC-PU's aliphatic carbonate soft segments compared to PPC-X, yet lower stability of its hard segments during main-chain degradation. Introducing PVA further altered degradation behavior: with PPC-PU fixed at 30 wt% and PVA increased from 20 wt% to 40 wt%, the Td,5% decreased linearly from 233.6 °C to 166.5 °C mainly resulting from the evolution of water and glycerol. When PVA was held at 30 wt% and PPC-PU varied from 10 to 40 wt%, Td,5% initially rose then fell, yet remained below that of the binary blend. Theincorporation of pre-plasticized PVA, which contains 10 wt% glycerol, introduces strong hydrogen-bond interactions within the ternary composite system and further enhances the inherent hygroscopicity of the composite. As a result, both the first and second maximum decomposition temperatures (Td,max1 and Td,max2) of the ternary system are significantly lower compared to the binary composite (Fig. 5(b)). These effects are attributed to PVA's hygroscopicity and glycerol evolution, which introduce moisture during processing, storage, and testing, thereby depressing the initial decomposition temperatures. Simultaneously, thermal dehydration of PVA's hydroxyl groups above 200 °C facilitates decomposition of poly(propylene carbonate) segments in both PPC-X and PPC-PU.27 Both mechanisms collectively contribute to the reduction in Td,max1 and Td,max2. Despite the reduced thermal stability upon PVA incorporation, the actual degradation (excluding moisture evaporation) of all components occurs above 200 °C. This degradation temperature remains well above the typical processing temperatures for these ternary composites. As exhibited in Fig. 5(c), the isothermal thermogravimetric analysis (ITG) curve of the 30 wt%PVA/30 wt%PPC-PU/PPC-X ternary composite was performed at 200 °C. An initial mass loss of approximately 18 wt% is observed within the first 15 minutes, which is attributed to the volatilization of moisture in various hydrogen-bonded states and glycerol. After 20 minutes of isothermal holding, the baseline stabilizes, indicating the complete evolution of these small molecules. No further mass change is detected up to 35 minutes, demonstrating that no polymer chain degradation occurs in the ternary system at 200 °C. This confirms the thermal stability of the material during processing at or below this temperature.
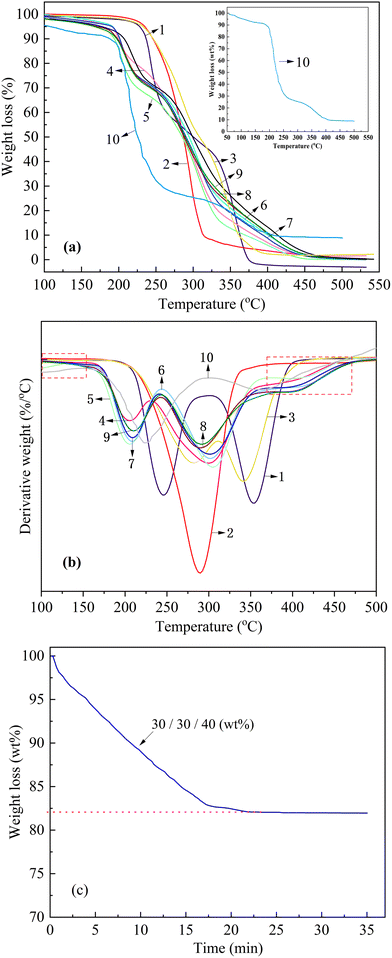 |
| | Fig. 5 Thermogravimetric analysis of PVA/PPC-PU/PPC-X composites: (a) TGA; (b) DTG; (c) ITG (isothermal TGA) (1: PPC-X; 2: PPC-PU; 3–9: xPVA/yPPC-PU/zPPC-X (wt%) = 0/30/70, 20/30/50, 30/10/60, 30/20/50, 30/30/40, 30/40/30, 40/30/30. 10: prepalsticized PVA). | |
Table 3 Thermogravimetric analysis data of xPVA/yPPC-PU/zPPC-X (wt%) composites
| Entry |
T
d,5%
/°C |
T
d,max1
/°C |
T
d,max2
/°C |
|
5 wt% weight loss temperature.
The first maximum degradation rate temperature.
The second maximum degradation rate temperature.
Not detected.
|
| PPC-X |
227.5 |
245.5 |
353.3 |
| PPC-PU |
236.3 |
289.6 |
NDd |
| Peplasticized PVA |
107.5 |
224.2 |
366.4 |
| 0/30/70 |
233.6 |
282.2 |
341.1 |
| 20/30/50 |
192.5 |
206.8 |
301.1 |
| 30/10/60 |
169.3 |
205.4 |
305.6 |
| 30/20/50 |
192.0 |
208.3 |
301.4 |
| 30/30/40 |
191.5 |
209.2 |
302.6 |
| 30/40/30 |
176.2 |
210.7 |
290.7 |
| 40/30/30 |
166.5 |
210.2 |
292.5 |
3.4 Microstructure analysis of samples
The microstructure of brittle fracture surfaces from PPC-X and its binary PPC-PU/PPC-X composites as well as ternary xPVA/yPPC-PU/zPPC-X composites was characterized by SEM with different magnification (Fig. 6(a1–h1) 2000× and (a2–h2) 5000×). As presented in Fig. 6, Pure PPC-X exhibits smooth, featureless fracture surfaces with cleavage planes characteristic of brittle fracture (Fig. 6a1/a2), establishing the morphological benchmark. Composites containing 30 wt% PPC-PU without PVA (Fig. 6b1/b2) uniformly dispersed ductile protrusions emerge, signaling the initiation of ductile fracture mechanisms without phase-separated boundaries between PPC-PU and PPC-X. The absence of interfacial features confirms exceptional PPC-PU/PPC-X compatibility, facilitating molecular-level blending that forms homogeneous co-continuous networks with indistinguishable components in fracture surfaces. The incorporation of 20 wt% PVA into the PPC-X composite system containing 30 wt% PPC-PU (Fig. 6c1/c2) resulted in the dispersion of PVA as micrometer-sized particles with relatively indistinct interfacial boundaries. When the PVA content was fixed at 30 wt% and the PPC-PU loading increased from 10 wt% to 40 wt% (Fig. 6(d1–h1) and (d2–h2)), the PVA initially formed well-defined spherical microparticles with clear interfaces. As the PPC-PU content increased, the interfacial boundaries of the PVA dispersed phase became progressively less distinct, accompanied by a reduction in particle size and a morphological transition from spherical to elliptical and eventually cylindrical shapes. This evolution indicates a significant compatibilizing effect of PPC-PU within the blend. Notably, at 40 wt% PPC-PU, the interfacial distinction was nearly eliminated and the PVA domain size was further reduced to the sub-micrometer level. When the PPC-PU content was fixed at 30 wt% and the PVA loading increased from 20 wt% to 40 wt% (Fig. 6c1/f1/h1 and c2/f2/h2), although the size and density of the spherical PVA aggregates increased, the interfacial regions remained diffuse, suggesting favorable interfacial adhesion and compatibility. This further confirms that a minimum of 30 wt% PPC-PU is optimal in this ternary system. These morphological observations align well with the mechanical trends presented in Fig. 6, where higher PPC-PU content led to improved compatibility, finer dispersion, and consequently enhanced toughness and greater elongation at break. These results collectively demonstrate that PPC-PU acts as an effective compatibilizer and toughening agent for PVA and PPC-X blends, while also helping to maintain the biodegradability of the composite system.
 |
| | Fig. 6 SEM images of cryogenically fractured PVA/PPC-PU/PPC-X blends with varying compositions (xPVA/yPPC-PU/zPPC-X, wt%): (a1–h1) 2000×; (a2–h2) 5000×. | |
3.5 Water contact angle and absorption behavior of specimens
As shown in Fig. 7(a), the water contact angles of pristine PPC-X, PPC-PU, and PVA were measured as 67.3°, 78.3°, and 34.1°, respectively. In contrast, the ternary melt-blended composites exhibited moderately hydrophilic surfaces, with contact angles consistently ranging from 71° to 75°, indicating that variations in PPC-PU (10–40 wt%) or PVA (20–40 wt%) content had minimal impact on surface wettability. An exception was observed for the blend with a composition of PVA/PPC-PU/PPC-X = 30/20/50 wt%, which showed a maximum contact angle of 81°. This mild hydrophilicity is advantageous for packaging applications as it helps maintain internal humidity, facilitates uniform surface wetting, reduces water loss from fresh produce, and preserves optical clarity during use. As illustrated in Fig. 7(b), the water absorption behavior after 24 h immersion in deionized water revealed a distinct trend. Although PPC-X and PPC-PU display slight surface hydrophilicity, their bulk hydrophobic nature resulted in low saturation water uptake values of 0.597 wt% and 0.465 wt%, respectively. Pure PVA, being water-soluble, was not tested separately; however, its introduction into the PPC-PU/PPC-X system markedly increased the water absorption of the ternary composites, with uptake escalating significantly at higher PVA loadings. When PPC-PU content was fixed at 30 wt% and PVA increased from 20 wt% to 30 wt% and 40 wt%, the water absorption rose from 25.55 wt% to 42.73 wt% and 67.24 wt%, respectively, with slight dimensional swelling observed at 40 wt% PVA. When PVA content was maintained at 30 wt% and PPC-PU was varied from 10 wt% to 40 wt%, water absorption initially increased, reaching a maximum of 45.8 wt% at 20 wt% PPC-PU, then decreased linearly to 39.04 wt% at 40 wt% PPC-PU. Given that ambient humidity conditions are substantially less severe than full immersion, it is recommended that the PVA content be limited to 30 wt% to ensure the dimensional stability and transparency of the composite films. Furthermore, it is anticipated that water uptake will be markedly reduced by incorporating a cross-linked structure within the ternary polymer system.
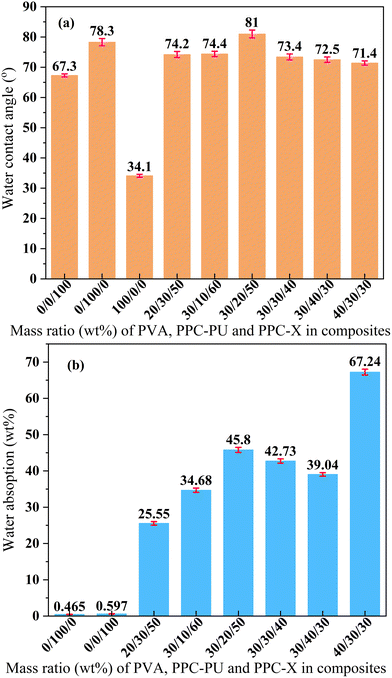 |
| | Fig. 7 Water contact angle (a) and absorption behavior (b) of the PVA/PPC-PU/PPC-X composites (xPVA/yPPC-PU/zPPC-X, wt%). | |
3.6 Gas barrier performance in PVA/PPC-PU/PPC-X composite films
The barrier performance of polymer films against oxygen (O2), carbon dioxide (CO2), and water vapor (H2O) is a critical metric in packaging science, fundamentally governed by the solution-diffusion mechanism where the permeability coefficient (P) is the product of the solubility (S) and diffusion (D) coefficients (P = S × D). The diffusion coefficient (D), a kinetic parameter, is primarily dictated by the polymer's internal structure, where increased free volume facilitates gas mobility, while greater chain rigidity, density, or the inclusion of fillers creates a more tortuous path, significantly reducing D. The solubility coefficient (S), a thermodynamic parameter, depends on the specific affinity between gas molecules and the polymer matrix; for instance, the high condensability and potential dipole interactions of CO2 with polar groups (e.g., carbonyl, hydroxyl) can lead to a higher S, whereas water vapor permeability is predominantly controlled by hydrophilic interactions. The overall permeation process involves adsorption, dissolution, diffusion, and desorption, with diffusion typically being the rate-determining step. Therefore, the efficacy of a material as a barrier is a function of its chemical composition, which governs solubility (S), and its structural morphology, which controls diffusion (D).38–42
3.6.1 Carbon dioxide permeability.
The thicknesses of all samples are listed in Table 4. As illustrated in Fig. 8, the carbon dioxide permeability coefficient (CO2P) of neat PPC-X was measured as 5180 cm3 µm (m2 day)−1. The introduction of PVA into the PPC-PU/PPC-X system markedly increased the CO2 permeability coefficient of the ternary composite films, with values rising consistently as the PVA content increased. Specifically, the highest permeability coefficient value of 6906 cm3 µm (m2 day)−1 was observed for the film containing 30 wt% PVA and 10 wt% PPC-PU. In contrast, when the PVA content was fixed at either 20 wt% or 30 wt% and the PPC-PU loading was raised from 10 wt% to 40 wt%, the CO2 permeability coefficient decreased linearly. For instance, at 40 wt% PPC-PU, the permeability coefficient values reached 5417 and 5360 cm3 µm (m2 day)−1 for the 20/40/40 and 30/40/30 (wt%) of PVA/PPC-PU/PPC-X blends, respectively. Furthermore, increasing the PVA content from 30 wt% to 40 wt% at a constant PPC-PU content of 30 wt% had only a minimal effect on CO2 permeability coefficient.
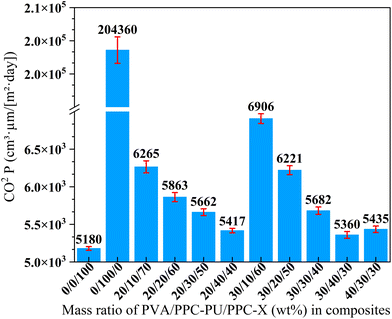 |
| | Fig. 8 Carbon dioxide permeability coefficient of PVA/PPC-PU/PPC-X composite films (xPVA/yPPC-PU/zPPC-X, wt%). | |
Table 4 Sample thickness of PVA/PPC-PU/PPC-X composite films
| Entry |
xPVA/yPPC-PU/zPPC-X (wt%) |
Thickness (µm) (mean ± SD) |
Entry |
xPVA/yPPC-PU/zPPC-X (wt%) |
Thickness (µm) (mean ± SD) |
| 1 |
0/0/100 |
260 ± 4.24 |
7 |
30/10/60 |
233 ± 2.37 |
| 2 |
0/100/0 |
216 ± 2.38 |
8 |
30/20/50 |
241 ± 2.82 |
| 3 |
20/10/70 |
237 ± 1.41 |
9 |
30/30/40 |
213 ± 2.13 |
| 4 |
20/20/60 |
212 ± 1.51 |
10 |
30/40/30 |
210 ± 2.19 |
| 5 |
20/30/50 |
231 ± 2.82 |
11 |
40/30/30 |
231 ± 2.42 |
| 6 |
20/40/40 |
227 ± 2.53 |
|
|
|
These variations are primarily governed by the compatibility of PVA within the composite. As indicated in Fig. 6, low PPC-PU content resulted in poor compatibility between PVA and PPC-X, yielding weak interfacial adhesion and ample interfacial free volume, which enhanced CO2 diffusion coefficient. Conversely, when the PPC-PU content reached 30 wt% or higher, the enhanced interfacial adhesion constrained gas transport along the interfaces, necessitating diffusion through the bulk matrix and thereby decreasing diffusion coefficient while reducing permeability. Moreover, with PPC-PU fixed at 30 wt%, only a marginal reduction in permeability coefficient was observed as the PVA content increased from 20 wt% to 40 wt%, further underscoring the critical role of interfacial compatibility in governing gas transport behavior. Even at 40 wt% PPC-PU, the permeability coefficient of the composite remained slightly higher than that of neat PPC-X. The result may be attributed to an overall increase in diffusion and solubility resulting from the incorporation of both preplasticized PVA and PPC-PU, combined with moisture-induced plasticization caused by the hydrophilic PVA, thereby collectively compromising the barrier resistance to CO2.
3.6.2 Oxygen permeability.
The thicknesses of all samples are listed in Table 5. Fig. 9 shows oxygen permeability coefficients of 1020 cm3 µm (m2 day)−1 for pristine PPC-X and 24740 cm3 µm (m2 day)−1 for neat PPC-PU, respectively. The oxygen permeability coefficient of the ternary blend films exhibited a decreasing trend with increasing content of both PVA and PPC-PU. When the PVA content was held constant, the oxygen permeability coefficient decreased linearly with higher PPC-PU loadings. Similarly, with PPC-PU fixed at 30 wt%, increasing the PVA content from 20 wt% to 40 wt% also resulted in a linear reduction in permeability coefficient. These findings clearly demonstrate that both components contribute to improved oxygen barrier performance. Notably, when PVA content was maintained at 30 wt% and PPC-PU exceeded 30 wt%, the oxygen permeability coefficient of certain composite films fell slightly below that of neat PPC-X. Specifically, compositions of 30/30/40 and 30/40/30 (PVA/PPC-PU/PPC-X, wt%) exhibited oxygen permeability coefficient values of 1015 cm3 µm (m2 day)−1 and 1002 cm3 µm (m2 day)−1, respectively. Moreover, with PPC-PU content fixed at 30 wt%, increasing PVA to 40 wt% further reduced the oxygen permeability coefficient to 982 cm3 µm (m2 day)−1. This systematic variation indicates that PVA content plays a primary and more sensitive role in controlling the oxygen barrier properties, primarily by significantly reducing the oxygen diffusion coefficient (D). This reduction is largely attributed to the increased crystallinity and strong hydrogen bonding within the PVA phase, which decreases the polymer's free volume and restricts chain mobility, thereby hindering the passage of gas molecules. To achieve high barrier performance, the PPC-PU content should be no less than 30 wt%, which aligns with its compatibilizing function within the blend to assure a dense interface and minimize defects. Above this threshold, the strengthened interfacial adhesion eliminates easy diffusion pathways along the interfaces, forcing oxygen molecules to traverse the bulk material, which correspondingly lowers the effective diffusion coefficient (D). Furthermore, when the PVA content exceeds 30 wt%, its inherent superior barrier properties, driven by its low intrinsic oxygen solubility (S) due to high polarity and strong interchain interactions, significantly enhance the overall oxygen resistance of the composite films.
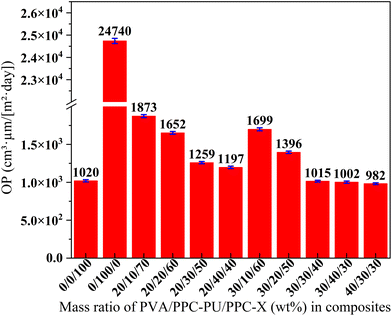 |
| | Fig. 9 Oxygen permeability coefficient of PVA/PPC-PU/PPC-X composite films (xPVA/yPPC-PU/zPPC-X, wt%). | |
Table 5 Sample thickness of PVA/PPC-PU/PPC-X composite films
| Entry |
xPVA/yPPC-PU/zPPC-X (wt%) |
Thickness (µm) (mean ± SD) |
Entry |
xPVA/yPPC-PU/zPPC-X (wt%) |
Thickness (µm) (mean ± SD) |
| 1 |
0/0/100 |
253 ± 2.52 |
7 |
30/10/60 |
226 ± 1.28 |
| 2 |
0/100/0 |
220 ± 1.23 |
8 |
30/20/50 |
235 ± 2.24 |
| 3 |
20/10/70 |
230 ± 2.01 |
9 |
30/30/40 |
221 ± 1.52 |
| 4 |
20/20/60 |
217 ± 1.21 |
10 |
30/40/30 |
220 ± 1.34 |
| 5 |
20/30/50 |
235 ± 1.67 |
11 |
40/30/30 |
235 ± 2.15 |
| 6 |
20/40/40 |
222 ± 1.23 |
|
|
|
3.6.3 Water vapor permeability.
The thicknesses of all samples are listed in Table 6. As shown in Fig. 10, the water vapor permeability coefficient (WVP) of the ternary composite films was strongly influenced by their composition. Specifically, the permeability coefficient increased progressively with higher PVA content but decreased linearly with increasing PPC-PU loading when the PVA content was fixed. For example, with PVA constant at 20 wt%, the WVP reached a minimum value of 1075 mg µm (m2 day)−1 at 40 wt% PPC-PU. Similarly, with 30 wt% PVA, the blend with a composition of 30/40/30 (PVA/PPC-PU/PPC-X, wt%) exhibited the lowest WVP value of 4052 mg µm (m2 day)−1. In contrast, when the PVA content was increased to 40 wt% in the 40/30/30 formulation, the WVP rose markedly to 7872 mg µm (m2 day)−1. These results demonstrate that the incorporation of PVA substantially impaired the water vapor barrier property, whereas PPC-PU contributed to its improvement, primarily by modulating the solubility and diffusion coefficients within the composite. Nevertheless, all ternary composites showed significantly higher water vapor permeability (WVP) than neat PPC-X, indicating that the overall permeability behavior is co-dominated by the hydrophilic nature of PVA and the hydrophobic character of PPC-PU, with PVA playing the more decisive role. The high hydrophilicity of PVA led to an elevated solubility coefficient (S) for water vapor due to strong polar interactions, while moisture absorption and subsequent plasticization under testing conditions (50% relative humidity) increased the free volume in PVA-rich regions, thereby raising the diffusion coefficient (D). In contrast, PPC-PU likely reduced the effective D by promoting a denser interface and morphology, tortuous pathways. Consequently, excessive PVA incorporation elevates both S and D, synergistically increasing WVP and degrading barrier performance. It is anticipated that WVP can be markedly reduced by introducing a chain extension/crosslinking structure, which enhances component cohesion and packing density to suppress D, and by incorporating nanomaterials such as nanocellulose or mica sheets, which further impede diffusion by increasing path tortuosity and potentially reducing solubility at interfaces.
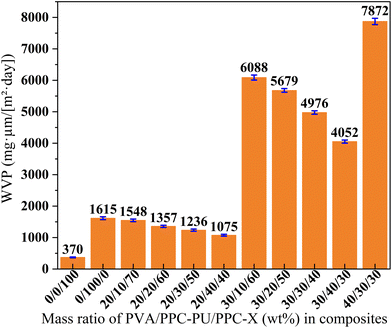 |
| | Fig. 10 Water vapor permeability coefficient of PVA/PPC-PU/PPC-X composite films (xPVA/yPPC-PU/zPPC-X, wt%). | |
Table 6 Sample thickness of PVA/PPC-PU/PPC-X composite films
| Entry |
xPVA/yPPC-PU/zPPC-X (wt%) |
Thickness (µm) (mean ± SD) |
Entry |
xPVA/yPPC-PU/zPPC-X (wt%) |
Thickness (µm) (mean ± SD) |
| 1 |
0/0/100 |
240 ± 2.52 |
7 |
30/10/60 |
235 ± 1.97 |
| 2 |
0/100/0 |
231 ± 2.13 |
8 |
30/20/50 |
226 ± 1.42 |
| 3 |
20/10/70 |
235 ± 2.32 |
9 |
30/30/40 |
232 ± 1.63 |
| 4 |
20/20/60 |
225 ± 1.67 |
10 |
30/40/30 |
218 ± 1.22 |
| 5 |
20/30/50 |
227 ± 1.44 |
11 |
40/30/30 |
233 ± 2.25 |
| 6 |
20/40/40 |
230 ± 2.05 |
|
|
|
Overall, the PVA/PPC-PU/PPC-X ternary composite films exhibits superior barrier performance against CO2, O2 and water vapor. Nevertheless, compared to commercially established biodegradable composites such as PBAT, PLA, and other reported analogous biodegradable film systems (Table 7),29,43–50 the PVA/PPC-PU/PPC-X ternary composite films maintains superior comprehensive barrier efficacy.
Table 7 Gas barrier comparison of selected biodegradable films
| Entry |
Carbon dioxidea |
Oxygena |
Water vapora |
| CO2P/cm3 µm [m2 day]−1 |
OP/cm3 µm [m2 day]−1 |
WVP/mg µm (m2 day)−1 |
|
The permeability coefficient of carbon dioxide (CO2P), oxygen (OP) and water vapor (WVP) were detected under the identical condition, respectively.
The units are converted from other units in the references.
ND denotes ‘not determined’.
|
| 30 wt%PVA/30 wt%PPC-PU/PPC-X |
5.68 × 103 |
1.02 × 103 |
5.00 × 103 |
| 75 wt%PPC-P/25 wt%PBAT/2%MDI@Gly29 |
NDc |
1.79 × 103 |
4.75 × 104 |
| 30%PPC-P/70%PBAT/1.0 wt%ADR50 |
ND |
1.31 × 104 |
1.63 × 107 |
| PBAT/30 wt%PLLA43b |
2.32 × 106 |
5.13 × 104 |
ND |
| PBSb |
ND |
5.28 × 103,7,44 |
4.39 × 104,24,46 |
| PPC47 |
ND |
2.25 × 103 |
1.25 × 106 |
| PLA24,48b |
ND |
3.35 × 103 |
8.57 × 104 |
| PET49b |
ND |
1.54 × 103 |
9.80 × 104 |
| PBAT50b |
ND |
4.97 × 104 |
2.73 × 107 |
4. Conclusions
A series of fully biodegradable films were fabricated via melt blending of pre-plasticized PVA1788 with PPC-PU and PPC-X at various ratios, followed by hot-pressing. Comprehensive analysis including mechanical testing, FTIR, SEM, DSC, TGA, gas barrier performance, water contact angle, and water absorption measurements revealed that the PPC-PU elastomer acts as both an effective compatibilizer and a toughener. This multifunctional role originates from hydrogen bonding and polar–polar interactions among functional groups in PPC-PU, PPC-X, and PVA. At 30 wt% PPC-PU loading, the blend with a composition of xPVA/yPPC-PU/zPPC-X = 30/30/40 (wt%) exhibited a tensile strength of 14.32 MPa, an elongation at break of 30.29% (compared to 3.76% for neat PPC-X), a Tg of 44.6 °C, and a Td,5% of 191 °C. Although reduced thermal stability of the composites due to the evolution of water and glycerol containing in PVA, these thermal properties still remain suitable for practical processing and application conditions. Owing to the pronounced effect of PVA on water absorption, its content should be limited to below 30 wt%. The incorporation of PVA and PPC-PU into PPC-X resulted in superior CO2 and oxygen barrier performance; the 30/30/40 (wt%) blend showed a CO2 permeability (CO2P) of 5.68 × 103 cm3 µm (m2 day)−1, close to that of neat PPC-X, and an O2 permeability (OP) of 1.02 × 103 cm3 µm (m2 day)−1, almost equivalent to that of pure PPC-X. However, the hydrophilic characteristic of PVA led to an increase in water vapor permeability, reaching 5.00 × 103 cm3 µm (m2 day)−1. Although the water vapor permeability (WVP) is significantly higher than that of pure PPC-X, the findings of this study still demonstrate certain advantages and reference value when compared to commercially available packaging films and reports in the literature. Overall, the ternary composite films exhibit promising gas barrier properties, significantly enhanced mechanical performance compared to neat PPC-X, and viable processability, making them attractive sustainable alternatives to conventional non-biodegradable packaging materials such as LDPE, PVC, PET, and PP. These materials demonstrate considerable potential for reducing petroleum dependence, promoting CO2 valorization, and supporting the transition toward more sustainable packaging technologies.
Author contributions
Yong Chen: investigation, methodology, writing original draft, conceptualization, data curation, formal analysis, and project administration. Yang Zhao: data curation, and formal analysis. Min Xiao: writing – review and editing. Yuezhong Meng: project administration, conceptualization, supervision, and writing – review and editing.
Conflicts of interest
The authors declare no competing financial interest or personal relationships that could have appeared to influence the work reported in this paper.
Data availability
The authors confirm that the data supporting the findings of this study are available within the article.
Acknowledgements
The authors would like to thank the Natural Science Foundation of Higher Educational Bureau of Guangdong Province (2022KTSCX244) and the National Natural Science Foundation of China (Grant No. 21376276, 21643002) for financial support of this work.
References
-
OECD, Global Plastics Outlook: Policy Scenarios to 2060, OECD Publishing, Paris, 2022. DOI:10.1787/aa1edf33-en.
-
Plastics Europe, Plastics–the fast Facts 2024, Plastics Europe Publishing, Brussels, 2024, https://plasticseurope.org/knowledge-hub/plastics-the-fast-facts-2024/ Search PubMed.
- S. S. Yue, T. W. Zhang and S. J. Wang,
et al., Recent Progress of Biodegradable Polymer Package Materials: Nanotechnology Improving Both Oxygen and Water Vapor Barrier Performance, Nanomaterials, 2024, 14, 338 Search PubMed.
- F. Jahangiri, A. K. Mohanty and M. Misra, Sustainable biodegradable coatings for food packaging: challenges and opportunities, Green Chem., 2024, 26, 4934 Search PubMed.
- M. Ansari, A. Sabzevari and N. Joupari,
et al., Eco-Friendly Poly(Lactic Acid)/Poly (Butylene Adipate-Co-Terephthalate) Films Loaded by Zinc-Aluminum Layered Double Hydroxide Nanoparticles for Packaging Applications, Polym. Compos., 2025, 1–12 Search PubMed.
- N. Bumbudsanpharoke, N. Harnkarnsujarit and B. Chongcharoenyanon,
et al., Enhanced properties of PBAT/TPS biopolymer blend with CuO nanoparticles for promising active packaging, Food Packag. Shelf Life, 2023, 37, 101072 CrossRef CAS.
- H. L. Liu, Y. Zhao and F. Y. Wang,
et al., Gas Permeability Changes in Biodegradable Films and Litchi Preservation Performance, Polym. Adv. Technol., 2025, 36, e70360 CrossRef CAS.
- J. Jacob, N. Linson and S. Kuriakose,
et al., Poly(lactic acid)-Based Films with Functionalized Mesoporous Silica from Rice Husk for Sustainable Food Packaging, ACS Sustainable Chem. Eng., 2024, 12, 3702–3714 Search PubMed.
- Y. T. Zhou, Z. L. Jiang and Y. S. Huang,
et al., Zinc oxide nanoparticles enhance barrier and antimicrobial properties of PBAT (polybutylene adipate-co-terephthalate) degradable food packaging film, Food Biosci., 2025, 71, 107119 CrossRef CAS.
- F. Benbelaid, B. S. Bouakaz and A. Aouaitia,
et al., Composite Nanoarchitectonics of Poly(butylene adipate-coterephthalate) Reinforced with Metal-Organic Framework (MOF-2) and Epoxy-Functionalized Graphene (EFG) for Sustainable Food Packaging, J. Inorg. Organomet. Polym. Mater., 2025, 35 DOI:10.1007/s10904-025-04036-4.
- K. L. F. Cardoso, P. M. S. Souza and R. M. V. Alves,
et al., Poly(butylene adipate-co-terephthalate)/Graphene Oxide Nanocomposite Films for Food Packaging Applications, ACS Appl. Polym. Mater., 2025, 7, 5229–5239 CrossRef CAS.
- S. J. Zhou, D. X. Zhang and S. J. Xiong,
et al., A High-Performance and Cost-Effective PBAT/Montmorillonite/Lignin Ternary Composite Film for Sustainable Production, ACS Sustainable Chem. Eng., 2024, 12, 14704–14715 CrossRef CAS.
- Z. Cheng, M. Ning and K. Y. Mao,
et al., Bamboo powder lignocellulose/polybutylene adipate terephthalate biodegradable bioplastic composite film for food packaging materials, Int. J. Biol. Macromol., 2025, 316, 144781 CrossRef CAS PubMed.
- S. Mavai, A. Bains and K. Sridhar, Formulation and application of poly lactic acid, gum, and cellulose-based ternary bioplastic for smart food packaging: A review, Int. J. Biol. Macromol., 2024, 268, 131687 CrossRef CAS PubMed.
- Y. Fang, J. H. Ding and H. R. Zhao, High-strength, high-barrier, and biodegradable polyester composites via nanoconfined assembly of polydopamine nanosheets, Chem. Eng. J., 2025, 519, 165390 CrossRef CAS.
- D. T. Tadele, B. M. Trinh and T. H. Mekonnen, PBAT/corn zein ester blends: Rheology, morphology, and physicochemical properties, Polymer, 2023, 283, 126258 CrossRef CAS.
- L. Wu, D. Y. Niu and B. Liu,
et al., Stiffness-ductility balanced, high-barrier and UV-resistant biaxially drawn PLA/PPC/lignin films through orientation of matrix nanocrystals and layered dispersed phases, Polymer, 2025, 323, 128220 CrossRef CAS.
- F. Ghisoni, A. Fiorati and L. D. Nardo, Design strategies for sustainable gas barrier coatings in food packaging via wet deposition technologies, Food Packag. Shelf Life, 2025, 52, 101635 CrossRef.
- G. G. D. Lima, I. C. B. Zakaluk and M. A. Artner,
et al., Enhancing Barrier and Antioxidant Properties of Nanocellulose Films for Coatings and Active Packaging, A Review, ACS Appl. Nano Mater., 2025, 8, 4397–4421 CrossRef.
- R. Capuano, R. Avolio and R. Castaldo,
et al., Up-cycling coffee silverskin into biobased functional coatings, J. Cleaner Prod., 2024, 469, 143063 CrossRef CAS.
- C. J. D. S. Pens, T. V. Klug and L. Stoll,
et al., Poly (lactic acid) and its improved properties by some modifications for food packaging applications: A review, Food Packag. Shelf Life, 2024, 41, 101230 CrossRef.
- D. J. Darensbourg, Chemistry of Carbon Dioxide Relevant to Its Utilization: A Personal Perspective, Inorg. Chem., 2010, 49(23), 10765–10780 CrossRef CAS PubMed.
- G. A. Luinstra, Poly(Propylene Carbonate), Old Copolymers of Propylene Oxide and Carbon Dioxide with New Interests: Catalysis and Material Properties, Polym. Rev., 2008, 48(1), 192–219 CrossRef CAS.
- F. Wu, M. Misraa and A. K. Mohanty, Challenges and new opportunities on barrier performance of biodegradable polymers for sustainable packaging, Prog. Polym. Sci., 2021, 117, 101395 CrossRef CAS.
- C. X. Fan, J. X. Liang and S. X. Ye,
et al., Study of the Synthesis of CO2/Propylene Epoxide/Phthalic Anhydride Terpolymers with Different Sequence Structures and Their Properties, Acta Polym. Sin., 2022, 53(5), 497–504 CAS.
- W. J. Wang, S. X. Ye and J. X. Liang,
et al., Architecting Branch Structure in Terpolymer of CO2, Propylene Oxide and Phthalic Anhydride: An Enhancement in Thermal and Mechanical Performances, Chin. J. Polym. Sci., 2022, 40, 462–468 CrossRef CAS.
- S. X. Ye, W. J. Wang and J. X. Liang,
et al., Metal-Free Approach for a One-Pot Construction of Biodegradable Block Copolymers from Epoxides, Phthalic Anhydride, and CO2, ACS Sustainable Chem. Eng., 2020, 8, 17860–17867 CrossRef CAS.
- T. W. Zhang, J. X. Liang and S. S. Yue,
et al., Preparation and Characterization of Biodegradable Poly(propylene carbonate-co-phthalate)/Poly(butylene adipate-co-terephthalate) Blends, Acta Polym. Sin., 2023, 54(8), 1144–1154 CAS.
- J. T. Deng, S. S. Yue and M. Xiao,
et al., High Gas Barrier and Biodegradable PPC-P/PBAT Composite Films Coated by Poly(vinyl alcohol)/borax Complexes, Surfaces, 2024, 7, 517–528 CrossRef CAS.
- X. Huang, T. T. Zhao and S. J. Wang,
et al., Self-Healable, Transparent, Biodegradable, and Shape Memorable Polyurethanes Derived from Carbon Dioxide-Based Diols, Molecules, 2024, 29, 4364, DOI:10.3390/molecules29184364.
- G. Mallamaci, A. A. Faysal and A. Guinault,
et al., Innovative poly(vinyl alcohol) (PVA)-based nanolayered Films:Balancing mechanical and gas barrier properties, ACS Appl. Mater. Interfaces, 2025, 17, 36182–36191 Search PubMed.
- A. Blanchard, F. Melis and F. Gouanvé,
et al., Influence of the PVOH molar mass on the morphology and functional properties of EVOH/PVOH films prepared by melt blending, J. Polym. Sci., 2021, 59, 70–83 CrossRef CAS.
- J. W. Wu, Y. Q. Huang and H. B. Li,
et al., Properties of polyamide 6,10/poly(vinyl alcohol) blends and impact on oxygen barrier performance, Polym. Int., 2018, 67, 453–462 CrossRef CAS.
- S. L. Zeng, L. Li and Q. Wang, Structure-property correlation of polyvinyl alcohol films fabricated by different processing methods, Polym. Test., 2023, 126, 108143 CrossRef CAS.
- X. Huang, K. Alferov and T. T. Zhao,
et al., Facile and direct synthesis of oligocarbonate diols from carbon dioxide and their application as sustainable feedstock for polyurethane, J. CO2 Util., 2023, 75, 102571 CrossRef CAS.
- T. T. Zhao, S. R. Yang and S. J. Wang,
et al., Bottom-Up approach to synthesize polycarbonate-type polyurethanes from CO2 for high-performance cathode binder in Li-ion batteries, Chem. Eng. J., 2025, 509, 161097 CrossRef CAS.
- S. Su, J. T. Wang and Q. Yan,
et al., Biodegradable, wear-resistant and resilient thermoplastic polycarbonate-based polyurethane with nanoscale microphase structure, Polymers, 2025, 17, 1665 CrossRef CAS PubMed.
- S. Y. Cui, P. F. Wei and L. Li, Thermal decomposition behavior of poly(propylene carbonate) in poly(propylene carbonate)/poly(vinyl alcohol) blend, J. Therm. Anal. Calorim., 2019, 135, 2437–2446 CrossRef CAS.
- V. T. W. Thuppahige and M. A. Karim, A comprehensive review on the properties and functionalities of biodegradable and semibiodegradable food packaging materials, Compr. Rev. Food Sci. Food Saf., 2022, 21, 689–718 CrossRef PubMed.
- J. S. Song, H. F. Zhou and X. D. Wang,
et al., Role of chain extension in the rheological properties, crystallization behaviors, and microcellular foaming performances of poly (butylene adipate-co-terephthalate), J. Appl. Polym. Sci., 2019, 136(14), 47322–47332 CrossRef.
-
M. Alfaifi, Master's theses, West Virginia University, 2024.
- K. Ito, Y. Saito and T. Yamamoto,
et al., Correlation study between oxygen permeability and free volume of ethylene–vinyl alcohol copolymer through positronium life time measurement, Macromolecules, 2001, 34(18), 6153–6155 Search PubMed.
- L. M. Wang, B. X. Tu and Y. F. Yu,
et al., Thermal, Mechanical and Barrier Properties of PBAT/PLLA Blend Films, China Plast., 2019, 33(9), 41–45 Search PubMed.
- K. Chen, X. Y. Zhang and Z. R. Wang,
et al., Effect of poly(propylene carbonate) on properties of polylactic acid based composite films, Int. J. Mol. Sci., 2024, 25(9), 4730, DOI:10.3390/ijms25094730.
- L. Xie, H. Xu and J. B. Chen,
et al., From nanofibrillar to nanolaminar poly(butylene succinate): paving the way to robust barrier and mechanical properties for full-biodegradable poly(lactic acid) films, ACS Appl. Mater. Interfaces, 2015, 7, 8023–8032 CrossRef CAS PubMed.
- S. Charlon, N. Follain and C. Chappey,
et al., Improvement of barrier properties of bio-based polyester nanocomposite membranes by water-assisted extrusion, J. Membr. Sci., 2015, 496, 185–198 CrossRef CAS.
- G. F. Li, W. H. Luo and M. Xiao,
et al., Biodegradable poly(propylene carbonate)/layered double hydroxide composite films with enhanced gas barrier and mechanical properties, Chin. J. Polym. Sci., 2016, 34(1), 13–22 CrossRef CAS.
- M. Drieskens, R. Peeters and J. Mullens,
et al., Structure versus properties relationship of poly(lactic acid). I. Effect of crystallinity on barrier properties, J. Polym. Sci., Part B: Polym. Phys., 2009, 47(22), 2247–2258 Search PubMed.
- S. S. Yue, T. W. Zhang and S. J. Wang,
et al., Recent progress of biodegradable polymer package materials: Nanotechnology improving both oxygen and water vapor barrier performance, Nanomaterials, 2024, 14, 338 CrossRef CAS PubMed.
- J. Z. Ai, S. Y. Li and Y. Zhao,
et al., Biodegradable PBAT/PPC-P blown film with enhanced mechanical and barrier performances via in situ reaction compatibilization, Composites, Part A, 2025, 198, 109147 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2026 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
Yang
Zhao
b,
Min
Xiao
c and
Yue-Zhong
Meng
*a,
Yang
Zhao
b,
Min
Xiao
c and
Yue-Zhong
Meng











