
Efficient solar-driven hydrogen peroxide production enabled by a perovskite electrochemical device integrated with a cobalt-based chiral catalyst
Young Sun
Park†
a,
Jaerim
Kim†
 bc,
Subin
Moon†
a,
Eunji
Ahn
bd,
Hyeonwoong
Hwang
b,
Sang-Hoon
You
b,
Juwon
Lee
e,
Chang-Seop
Jeong
a,
Wooyong
Jeong
a,
Hyoung-il
Kim
e,
Yong-Tae
Kim
b,
Kug-Seung
Lee
f,
Donghwa
Lee
bd,
Jong Kyu
Kim
*b and
Jooho
Moon
*a
bc,
Subin
Moon†
a,
Eunji
Ahn
bd,
Hyeonwoong
Hwang
b,
Sang-Hoon
You
b,
Juwon
Lee
e,
Chang-Seop
Jeong
a,
Wooyong
Jeong
a,
Hyoung-il
Kim
e,
Yong-Tae
Kim
b,
Kug-Seung
Lee
f,
Donghwa
Lee
bd,
Jong Kyu
Kim
*b and
Jooho
Moon
*a
aDepartment of Materials Science and Engineering, Yonsei University, 50 Yonsei-ro Seodaemun-gu, Seoul 03722, Republic of Korea. E-mail: jmoon@yonsei.ac.kr
bDepartment of Materials Science and Engineering, Pohang University of Science and Technology (POSTECH), 77 Cheongam-Ro, Nam-Gu, Pohang 37673, Republic of Korea. E-mail: kimjk@postech.ac.kr
cDepartment of Chemistry, Northwestern University, 2145 Sheridan Road, Evanston, IL 60208, USA
dDivision of Advanced Materials Science, Pohang University of Science and Technology (POSTECH), 77 Cheongam-Ro, Nam-Gu, Pohang 37673, Republic of Korea
eDepartment of Civil & Environmental Engineering, Yonsei University, 50 Yonsei-ro Seodaemun-gu, Seoul 03722, Republic of Korea
fPohang Accelerator Laboratory, Pohang University of Science and Technology, Pohang 37673, Republic of Korea
First published on 25th November 2025
Abstract
The solar-driven production of hydrogen peroxide (H2O2, a valuable energy carrier with excellent energy density and industrial applicability) using the two-electron oxygen reduction reaction (2e− ORR) represents a straightforward and eco-friendly technology. However, its efficiency is limited by the accumulation of oxygen intermediates (*OOH) adsorbed on the electrocatalyst, originating from spin-antiparallel electrons migrating through the latter, which hinders the favorable two-electron transfer to the triplet O2 possessing two spin-parallel electrons. Herein, we present Co metal-based nanohelices (Co NHs) with inherently chiral structures, synthesized via the oblique angle deposition method, as effective 2e− ORR catalysts based on their chirality-induced spin selectivity (CISS) effect. The chiral Co NH electrocatalyst, exhibiting a spin polarization efficiency of 78.1%, enhanced the 2e− ORR kinetics by facilitating the transfer of spin-parallel electrons and suppressing the accumulation of *OOH intermediates. Therefore, the Co NHs exhibited superior intrinsic 2e− ORR activity compared to achiral Co metal-based thin films, as confirmed by operando experiments and theoretical calculations. Subsequently, the Co NHs were integrated into a perovskite-based electrochemical device, and the resulting immersed perovskite-based cathode achieved a photocurrent density of −18.0 mA cm−2 at 1.2 V vs. reversible hydrogen electrode (VRHE), with a significant onset potential of 1.75 VRHE. Furthermore, we assembled an unbiased H2O2 production device using our immersed perovskite-based cathode paired with a carbon cloth for driving the iodide oxidation reaction; this device yielded a remarkable solar-to-chemical conversion efficiency of nearly 9.4%, with stable operation for 20 h.
Broader contextThe unassisted photosynthesis of hydrogen peroxide (H2O2) via the two-electron oxygen reduction reaction (2e− ORR) offers an eco-friendly route for on-site H2O2 production. However, 2e− ORR activity is hampered by the accumulation of oxygen-based intermediates caused by spin-antiparallel electrons passing through the catalyst, followed by suppression of the transport of two-electron transfer to the triplet O2 bearing two spin-parallel electrons. Here, we tackle this bottleneck by leveraging the chirality-induced spin selectivity effect in inherently chiral cobalt nanohelices (Co NHs). Their strong spin polarization induces efficient and selective 2e− ORR kinetics by promoting the migration of spin-parallel electrons and hindering the accumulation of intermediates. Therefore, the Co NHs yielded excellent intrinsic 2e− ORR activity with respect to achiral Co metal-based thin films, verified by in situ Raman and density functional theory analysis. Subsequently, an immersed perovskite-based electrochemical device integrated with Co NHs achieved a high photocurrent density of −18.0 mA cm−2 at 1.2 V vs. reversible hydrogen electrode (VRHE) and an onset potential of 1.75 VRHE. Additionally, we demonstrated an unassisted H2O2 generation system by employing our immersed perovskite-based electrochemical device combined with a carbon cloth-based electrode for the iodide oxidation reaction as an alternative oxidation reaction of the oxygen evolution reaction, showing a record-high solar-to-chemical conversion efficiency of approximately 9.4%, along with stable operation for 20 h. |
Introduction
Hydrogen peroxide (H2O2) is recognized as a valuable product, widely utilized in the medical and chemical industries.1,2 Additionally, aqueous H2O2 can be readily transported and employed as a key energy carrier, exhibiting an excellent energy density (60%; 3.0 MJ L−1), comparable to that of compressed hydrogen (2.8 MJ L−1 at 35 MPa).3,4 However, the traditional synthesis of H2O2 has been predominantly based on the complex anthraquinone oxidation process, which requires a substantial amount of energy due to the multistep reaction involved, and generates environmentally harmful organic waste.5,6 Thus, the development of a straightforward and environmentally friendly synthetic method for H2O2 is crucial to support sustainable energy production. In this context, the solar-assisted H2O2 synthesis driven by the two-electron oxygen reduction reaction (2e− ORR) represents an important approach, due to its ability to mitigate energy consumption and the generation of harmful byproducts.7–9Despite these advantages, the solar-driven production of H2O2 has been hampered by the lack of suitable 2e− ORR electrocatalysts with favorable catalytic activity and selectivity toward H2O2. Conventional noble metals and their alloys, such as Pt–Hg, Pd–Hg, and Au–Pd, are considered efficient catalysts for the 2e− ORR, but their large-scale application is hindered by their scarcity and high toxicity of Hg.10–12 Therefore, the exploration of earth-abundant and non-toxic electrocatalysts with efficient 2e− ORR catalytic activity is essential for realizing the cost-effective photosynthesis of H2O2. To address this need, non-noble transition metal-based catalysts (i.e., Mo, Fe, and Co) have been reported as prominent candidates because of their low cost, non-toxicity, and intrinsically high oxygen reduction activity.2,13–15 Among these materials, Co metal-based systems can be promising candidates as electrocatalysts, due to their resistance to demetallation of active sites as well as to free radical attack with respect to their counterparts.12 However, the efficiency of the 2e− ORR driven by Co metal-based catalysts is limited by the accumulation of oxygen intermediates (*OOH) adsorbed on the Co metal site during the 2e− ORR process (O2 + H2O + e− → *OOH + OH− + e− → HO2−).10,16 The accumulation of *OOH species can occur when the O2 molecule only reacts with the first electron, without accepting the second electron. This is likely due to the fact that natural O2 exists in the triplet state with two unpaired and spin-parallel electrons, and thus the transfer of the second electron to triplet O2 can be hindered when the spins of the first and second electrons are not parallel, based on the Pauli exclusion principle.17,18 Hence, the spin orientation of electrons in Co metal-based catalysts for the 2e− ORR should be regulated for competitive solar-assisted H2O2 synthesis.
Recently, the chirality-induced spin selectivity (CISS) effect has emerged as an effective tool to efficiently control the spin state of electrons migrating through electrocatalysts.19–21 The CISS effect involves a chiral structure acting as a spin polarizer that can align the spin states of electrons as they migrate through it.22,23 To date, the CISS effect has been easily induced by introducing chirality to an electrocatalyst through the simple intercalation of chiral organic molecules within inorganic catalytic materials.24,25 In fact, several Co metal-based electrocatalysts, obtained using chiral organic ligands, revealed chiral characteristics along with the CISS effect.26,27 However, these catalysts, which incorporated chiral organic molecules, exhibited inferior chirality, solely derived from small chiral ligands. Moreover, these organic ligands are likely to cause instability issues in the electrolyte, as well as low conductivity and blockage of the active sites of the catalyst.28,29 In contrast, inorganic chiral catalysts exhibiting intrinsic structural chirality at the mesoscale can be fabricated using a geometry-controlled deposition process, rather than relying on externally introduced chiral molecules; this represents a unique approach that ensures enhanced spin selectivity, a stable chiral structure, and full exposure of active catalytic sites under electrochemical conditions.
In this study, we successfully fabricated Co metal-based nanohelices (Co NHs) with inherently chiral structures for 2e− ORR catalysis via an oblique angle deposition (OAD) method. The obtained Co NHs exhibited excellent spin selectivity, yielding a CISS-based spin polarization efficiency of 78.1%, as shown by magnetic conductive probe–atomic force microscopy (mCP-AFM) analysis. The CISS effect of the Co NHs obviously induced faster catalytic kinetics for the 2e− ORR compared with an achiral-structured Co metal-based thin film (Co TF). Moreover, in-depth operando tests and theoretical calculations confirmed the improved catalytic activity induced by the spin-modulated electrochemistry, which mitigated the accumulation of oxygen intermediates. Subsequently, an immersed perovskite-based cathode decorated with the Co NHs delivered a saturated 2e− ORR photocurrent density of –18.0 mA cm−2 at 1.2 V vs. reversible hydrogen electrode (VRHE), with a remarkable onset potential of 1.75 VRHE. Furthermore, we fabricated an efficient unassisted H2O2 production device using our immersed perovskite-based cathode paired with a carbon cloth electrocatalyst to drive the iodide oxidation reaction (IOR); this device yielded an excellent unbiased photocurrent density of −16.0 mA cm−2, with stable operation for 20 h. Moreover, this bias-free device for solar fuel production exhibited a selectivity of nearly 98% toward H2O2 synthesis, resulting in a record-high solar-to-chemical conversion (SCC) efficiency of approximately 9.4%. These results confirm the highly efficient solar-driven H2O2 generation via the CISS effect enabled by the present Co metal-based chiral electrocatalyst.
Results and discussion
The OAD method was applied to fabricate a catalyst with an inherently chiral structure, consisting of an array of Co NHs on fluorine-doped tin oxide (FTO) (Fig. 1a). The OAD method, which involves a geometrical deposition process, is a cost-effective, scalable, and highly versatile nanofabrication technique.30–34 In this method, evaporation of a source element proceeds toward a tilted substrate, in such a way that the vapor flux has a glancing incident angle (θ) with respect to the direction of the substrate normal. Based on the geometric self-shadowing effect, various three-dimensional nanostructured catalysts, including nanohelices, nanorods, and nanozigzags, can be easily fabricated by adjusting the deposition conditions, including the oblique angle, the substrate rotation speed, and the deposition rates.35–38 Fig. S1, SI shows a cross-sectional scanning electron microscopy (SEM) image of a vertically aligned Co NHs array on FTO, with a thickness of 340 nm, prepared by rotating the substrate in a clockwise direction (Co NHs-CW). Fig. 1b shows the corresponding cross-sectional transmission electron microscopy (TEM) image, which confirms the successful fabrication of the nanohelical morphology of the Co NHs-CW catalyst on the FTO substrate. The high-resolution TEM (HR-TEM) image of Co NHs-CW, shown in Fig. 1c, reveals lattice distances of 0.19, 0.20, and 0.21 nm; these values are consistent with the interplanar spacings corresponding to the (101), (002), and (100) planes of the hexagonal close-packed crystal structure of Co metal, indicating the polycrystalline characteristics of the Co NHs.39,40 Moreover, the selected-area electron diffraction (SAED) and X-ray diffraction (XRD) patterns confirmed the polycrystalline nature of the fabricated Co NHs-CW catalyst (Fig. S2, SI). Subsequently, the chemical states of Co NHs-CW were identified by X-ray photoelectron spectroscopy (XPS), as shown in Fig. S3, SI. The peaks located at 780.0 and 795.5 eV are characteristic of Co3+ species in CoO6 octahedral coordination, while those at 781.8 and 797.3 eV can be assigned to Co2+ ions in CoO4 tetrahedral coordination (Fig. S3a, SI).41 These oxidation states are ascribed to the formation of native surface oxides (CoOx), originating from the intrinsic susceptibility of metallic Co to oxidation under ambient conditions. Moreover, the high-resolution XPS O 1s spectra of Co NHs-CW revealed three distinct peaks centered at 530.0, 531.4, and 532.4 eV, corresponding to lattice oxygen (OL), oxygen vacancies (VO), and adsorbed hydroxide (OH) species, respectively (Fig. S3b, SI).41 The Co NHs-CW catalyst exhibited a higher intensity for the VO than the OL peak, indicating that the CoOx phase at the surface of Co metal existed in an amorphous state. This observation is consistent with the XRD results, which showed no visible diffraction peaks corresponding to crystalline CoOx. The VO-enriched CoOx phase at the Co metal surface can provide critical active sites for the 2e− ORR, as confirmed by recent studies.41,42 | ||
| Fig. 1 (a) Schematic of the OAD process for fabrication of inherently chiral-structured Co NHs. (b) Cross-sectional TEM image showing helical morphology of Co NHs-CW on FTO substrate. (c) HR-TEM image of Co NHs-CW on FTO substrate. (d) CD spectra of Co NHs and Co TF. (e) Schematic illustration of mCP-AFM analysis. Average I–V curves for (f) Co NHs-CW and (g) Co TF. | ||
To assess the chiral properties of the Co NHs-CW catalyst, its chiroptical activity was elucidated using transmission circular dichroism (CD) spectroscopy (Fig. 1d). The chiroptical properties of a catalyst predominantly originate from its geometrically engineered helical crystal configuration. This chiral arrangement establishes an asymmetric electronic environment, resulting in distinct interactions with left- and right-circularly polarized light. These different interactions give rise to a measurable CD signal, which reflects the extent of the chiroptical activity of the chiral catalyst.43,44 In addition to the Co NHs-CW sample, we fabricated a chiral-structured Co NH array with a counterclockwise helical growth orientation (Co NHs-CCW), as well as an achiral-structured Co TF using a similar deposition process, with an oblique angle of zero. The cross-sectional SEM images of the two different samples are shown in Fig. S4, SI. The Co TF and Co NHs-CCW exhibited thicknesses of 125 and 337 nm, respectively. The CD spectra of Co NHs-CW and Co NHs-CCW exhibited inverse signal directions, because of the opposite helical growth orientations of Co NHs-CW and Co NHs-CCW (Fig. 1d). In contrast, the CD signal was absent in the spectrum of the achiral Co TF. Impressively, the CD signals of Co NHs-CW and Co NHs-CCW revealed a considerably high degree of ellipticity, with a value of almost 400 mdeg. This is a significantly larger value compared with those obtained for catalysts incorporating chiral organic molecules in previous studies,27,28 because of the highly asymmetric crystallographic distortion induced by the OAD process. Moreover, mCP-AFM measurements were conducted to evaluate the CISS effect-based spin-polarized current of the helical-structured Co metal catalysts (Fig. 1e). A cobalt–chrome-coated tip was pre-magnetized using a permanent magnet, aligned in either the north or south orientation with respect to the Co metal-based catalysts on the FTO substrate. Next, a potential was applied between the substrate and the pre-magnetized tip. To ensure the accuracy of the evaluation, I–V curves were measured at least 20 times on different points of the Co TF and Co NHs-CW catalysts. All the raw I–V measurement data are shown in Fig. S5, SI. Interestingly, the Co NHs-CW sample showed significantly distinct average current values for the two opposite magnetization orientations (Fig. 1f). The current value for the downward orientation was significantly larger than that corresponding to the upward orientation in the potential range of −0.3 to 0.3 V, whereas the Co TF sample exhibited no notable difference in current values between the two opposite magnetization directions (Fig. 1g). These results of the mCP-AFM measurements indicate a considerable spin-polarized current in the Co NHs-CW catalyst, derived from the CISS effect. The spin polarization (P) of this material was determined by the following equation:
 | (1) |
To evaluate the effect of the spin polarization on the 2e− ORR activity of the electrocatalysts, linear sweep voltammetry (LSV) curves of Co NHs-CW and TF were measured in O2-saturated 0.1 M KOH electrolyte (pH ∼ 13) using a conventional three-electrode system. As shown in Fig. 2a, the LSV curves of the two catalytic electrodes showed that Co NHs-CW exhibited an onset potential of 0.71 VRHE, which was positively shifted compared to that of Co TF (0.65 VRHE). This result demonstrates the key role of the chirality-induced spin polarization in mitigating the required potential for the 2e− ORR process, emphasizing its impact on the spin-polarized electrocatalysis. Moreover, Co NHs-CW achieved a superior oxygen reduction current density of −2.9 mA cm−2 at 0.2 VRHE, while Co TF exhibited a considerably lower current density of −0.7 mA cm−2 at the same potential. An achiral Co nanostructure was synthesized under similar deposition conditions to those used for the Co NHs catalyst (Fig. S9, SI), except that the substrate was continuously rotated at 1 rpm. As shown in Fig. S10, SI, the CD signal was absent in the spectrum of the achiral nanostructured Co catalyst, confirming its non-chiral nature. Moreover, the mCP-AFM analysis also demonstrated that the achiral Co nanostructure revealed the absence of a spin polarization effect (Fig. S11, SI). Impressively, the Co NHs-CW exhibited a superior 2e− ORR current density and a more positive onset potential compared to the achiral Co nanostructure (Fig. S12, SI). The 2e− ORR capability of Co NHs-CCW is nearly identical to that of Co NHs-CW (Fig. S13, SI), because Co NHs-CW and Co NHs-CCW exhibited comparable morphologies and spin polarization efficiencies. Furthermore, the Tafel slopes of the Co NHs-CW and TF catalysts in the 2e− ORR were determined to be 97 and 141 mV dec−1, respectively, confirming the faster 2e− ORR kinetics of Co NHs-CW compared to Co TF (Fig. S14, SI). To provide further evidence for the actual 2e− ORR catalytic activity of the two different catalysts, LSV measurements were also conducted in Ar-saturated 0.1 M KOH in the absence of O2 (Fig. S15, SI). Both Co NHs-CW and TF exhibited negligible current densities in Ar-saturated 0.1 M KOH without O2 purging, indicating that the current densities observed in Fig. 2a obviously originated from the reduction of O2 on the two catalysts.
 | ||
| Fig. 2 (a) LSV curves of Co NHs-CW and Co TF in O2-saturated 0.1 M KOH. (b) In situ Raman spectra for Co NHs-CW and (c) Co TF under varying bias potentials from 0.6 to 0.3 VRHE in O2-saturated NaClO4 (pH ≈ 13). Schematic illustrations of spin-dependent electron transfer mechanisms during the 2e− ORR enabled by (d) Co NHs-CW and (e) Co TF. (f) Surface Pourbaix diagram of the Co(002) surface as a function of applied bias (U). (g) Gibbs free energy profiles for 2e− ORR on the O-covered Co(002) surfaces in high-spin and low-spin states at 0.70 VRHE. (h) Long-term stability of Co NHs-CW at 0.2 VRHE. (i) Co K-edge XANES and (j) FT-EXAFS spectra of Co NHs-CW before and after 2e− ORR catalysis. | ||
To determine whether the enhanced 2e− ORR activity of Co NHs-CW originates from its larger surface area compared to Co TF, the electrochemical active surface areas (ECSAs) of the two samples were determined from cyclic voltammetry (CV) curves, obtained at scan rates ranging from 10 to 50 mV s−1 (Fig. S16a and b, SI). The double-layer capacitance (Cdl, corresponding to the ECSA) of Co NHs-CW (0.975 mF cm−2) was higher than that of Co TF (0.4 mF cm−2), due to the helical nanostructure of the former (Fig. S16c, SI). Moreover, to assess the intrinsic 2e− ORR catalytic performances, the specific catalytic activity (Js), representing the specific current density per ECSA of the two different catalysts, was determined via the following equations:
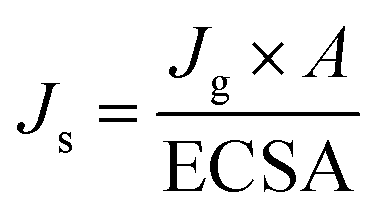 | (2) |
 | (3) |
| TOF = j × A/(n × F × Nactive) | (4) |
Moreover, the TOF surface normalized to the surface redox-active Co sites (TOFsurface) was also evaluated using the number of moles of active sites at the surface. The surface of Co NHs-CW and Co TF is mainly composed of a VO-enriched CoOx phase, as confirmed by XPS measurements (Fig. S3 and Fig. S17, SI). Based on previous results, the Co2+ of CoOx acts as an active site for 2e− ORR, whereas Co3+ only supplied the double-layer capacitance without obvious activity.41 The number of these Co2+ sites can be obtained by integrating the oxidation peak of Co2+ to Co3+ in LSV of our Co metal-based electrodes in Ar-saturated 0.1 M KOH.46 As shown in Fig. S18b, SI, the LSV curves of Co NHs-CW and Co TF exhibited two oxidation peaks at 1.25 and 1.47 VRHE, corresponding to oxidation peaks of Co2+ → Co3+ and Co3+ → Co4+.47 Accordingly, the oxidation peak at 1.25 VRHE was integrated under baseline, yielding areas under oxidation peaks of 0.2986 and 0.0817 V mA cm−2 for Co NHs-CW and Co TF, respectively, and the estimated charges were calculated to be 0.0037 and 0.0010 C by dividing these areas by voltage scan rate (80 mV s−1).46 Since Co2+ to Co3+ oxidation is a one-electron transfer reaction, the number of moles of Co2+ sites can be determined by dividing the calculated charge by the Faraday constant, and thus the Γactive of Co NHs-CW and Co TF are 0.038 and 0.01 µmol cm−2, respectively. Consequently, the TOFsurface of two different samples was obtained in Fig. S18c, SI, indicative of the superior TOFsurface of Co NHs-CW compared with that of Co TF. These results clearly show that the spin-polarized electrons originating from the CISS effect can significantly enhance the inherent electrocatalytic performance for the 2e− ORR.
In situ Raman spectroscopy measurements were performed to clarify the adsorption/desorption behaviors of reaction intermediates, which are intimately associated with the intrinsic 2e− ORR activities of catalytic electrodes. The Co metal-based catalysts were immersed in O2-saturated NaClO4 electrolyte (pH ∼ 13) while varying the applied voltage. The Raman spectra of the two different catalysts displayed clear peaks at 478, 519, and 685 cm−1,48–50 attributed to the vibrational modes of the CoOx phase (Fig. 2b and c). Furthermore, the peak at 933 cm−1, corresponding to the symmetric stretching mode of ClO4−, was observed in the Raman spectra of both Co NHs-CW and Co TF, and thus employed as the calibration signal.2 Interestingly, when the applied bias to both Co NHs-CW and Co TF was varied from 0.6 to 0.3 VRHE, no change was observed in the Raman spectra, except for the peaks located at 613 and 1074 cm−1, ascribed to *OOH adsorbed on Co metal sites and superoxide (O2−) species, representing oxygen intermediates of the 2e− ORR.51,52 O2− can be produced by deprotonation of *OOH when its desorption from the catalyst surface is delayed.53 The low intensities of the *OOH and O2− peaks detected at the Co NHs-CW surface clearly show that Co NHs-CW generated a small amount of oxygen-based adsorbates (Fig. 2b). On the other hand, the Raman spectrum of Co TF displayed a considerable increase of the peaks at 613 and 1074 cm−1 as the reduction potential was applied (Fig. 2c), indicating the accumulation of oxygen intermediates during the 2e− ORR. This observation confirms that the CISS effect of Co NHs-CW can reduce oxygen adsorbates because triplet O2, possessing two spin-parallel electrons, can readily accept another two spin-parallel electrons with opposite spin orientation based on the Pauli exclusion principle, resulting in a favorable 2e− ORR (Fig. 2d).17,18 In contrast, in the case of the Co TF catalyst, the suppressed transfer of the second electron induces accumulation of oxygen species at the surface of the catalyst, because the first and second electrons are likely to exhibit a spin-antiparallel configuration due to the absence of the CISS effect, leading to inferior 2e− ORR activity (Fig. 2e).
To further understand the difference in 2e− ORR activity between Co TF and NHs-CW, we performed density functional theory (DFT) calculations to evaluate the Gibbs free energy changes along the reaction pathway. Based on XRD and HR-TEM analyses, which showed that both catalysts predominantly expose the Co(002) facet, we constructed Co(002) surface slab models. To represent the experimentally observed differences in magnetic properties, we modeled the Co surfaces in high-spin and low-spin states, corresponding to Co NHs and Co TF, respectively. This modeling approach is consistent with recent literature. In a study of CISS-mediated electrocatalysis using chiral Au nanoparticles, Chae et al. modeled the surface in high- and low-spin states to represent chiral and achiral environments, respectively, and examined how spin polarization affects the electrochemical reaction.54
The density of states (DOS) for each electronic configuration is provided in Fig. S19, SI. Given the surface oxidation of Co NHs and TF, as confirmed by XPS, we first identified the thermodynamically favorable surface coverage by evaluating the adsorption energies of O, OH and H at 0.25, 0.5, 0.75, and 1 monolayer (ML) coverages.55–58 The atomic structures corresponding to each coverage are shown in Fig. S20, SI. Our free energy analysis relative to the clean surface revealed that the Co(002) surface with 0.5 ML O coverage is the most stable configuration under 2e− ORR-relevant conditions (0.2–0.7 VRHE), as shown in Fig. 2f. Using this surface model, we calculated the Gibbs free energy profiles of the 2e− ORR pathway at the equilibrium potential (0.7 VRHE) for both high-spin and low-spin Co surfaces. The relaxed atomic structures of the O-covered Co surface and the *OOH intermediate are shown in Fig. S21, SI. In both cases, *OOH adsorbs strongly onto the surface, and the subsequent H2O2 formation step was identified as the rate-determining step (Fig. 2g). The free energy change for the H2O2 formation step was 0.75 eV for the low-spin Co surface and 0.61 eV for the high-spin Co surface, indicating a lower reaction barrier for the high-spin Co state. The sluggish H2O2 formation for the low-spin Co surface is consistent with in situ Raman spectra showing the accumulation of *OOH species on Co TF. In contrast, the reduced barrier for the high-spin Co surface suggests that the high-spin state facilitates H2O2 formation. This enhancement can be attributed to the spin-selective nature of the surface: in a high-spin system, electrons can be more efficiently transferred to triplet O2, thereby promoting the overall 2e− ORR process.
Moreover, the long-term stability of the Co NHs-CW catalyst for the 2e− ORR was evaluated using a chronoamperometry test at a constant potential of 0.2 VRHE (Fig. 2h). During a 10-h period under 2e− ORR catalysis, Co NHs-CW exhibited excellent stability, maintaining a constant current density without degradation. Interestingly, the 2e− ORR current density of Co NHs-CW increased from −2.7 to −5.0 mA cm−2 after a 10-h chronoamperometric test, corresponding to a 1.85-fold increment. To elucidate the origin of the high current density observed for Co NHs-CW during electrolysis, SEM analysis of this catalyst was carried out after the stability test (Fig. S22, SI). The cross-sectional SEM image shows that Co NHs-CW was deposited with a nanoparticle-based layer. Synchrotron-based X-ray absorption spectroscopy (XAS) measurements were performed to further investigate the corresponding changes in the structural properties of Co NHs-CW during the 2e− ORR. The Co K-edge X-ray absorption near-edge structure (XANES) spectrum of Co NHs-CW before the stability test indicates the presence of oxidized Co rather than metallic Co states, due to the native oxide (CoOx) formed at the surface of the Co metal phase (Fig. 2i), in agreement with the XPS analysis (Fig. S3, SI). The Co NHs-CW catalyst exhibited an increased white-line intensity and a positive shift in the edge position after the stability test, indicating further oxidized surface states during the 2e− ORR process.59 The Fourier transform extended X-ray absorption fine structure (EXAFS) analysis revealed the local coordination structures in Co NHs-CW. As shown in Fig. 2j, the EXAFS spectra of Co NHs exhibited a main peak at 2.15 Å, corresponding to Co–Co coordination, with the peak at 1.35 Å assigned to the Co–O scattering shell. After electrolysis, the intensity of the Co–O coordination peak was significantly enhanced, while that of the Co–Co coordination peak decreased. These results imply the additional formation of the CoOx phase on the surface of Co NHs-CW during the 2e− ORR, because these oxides can be readily formed at the potential range of the 2e− ORR in alkaline electrolytes, according to the Pourbaix diagram (Fig. 2f) and previous work.60 Further insights were obtained from XPS analysis in the Co 2p and O 1s regions. After the stability test, only Co2+ and Co3+ states were detected in the spectra, with no evidence of metallic Co0 (Fig. S23a, SI). In addition, the lattice oxygen peak associated with crystalline CoOx exhibited a decreased intensity, while the signal corresponding to oxygen vacancies became more pronounced, indicating an increased proportion of amorphous CoOx phase during the 2e− ORR process (Fig. S23b, SI). Additional XRD analysis showed that that no crystalline peaks were generated after the stability test of Co NHs-CW (Fig. S24, SI), providing further evidence of the amorphous phase of CoOx. Therefore, the increase in current density observed during the chronoamperometric test can be attributed to the formation of an amorphous CoOx phase enriched with oxygen vacancies, which act as active sites for the 2e− ORR. The current density reached a steady state after 1 h, likely due to the complete deposition of the amorphous CoOx layer (Fig. 2g). In fact, the ECSA of Co NHs-CW after the stability test was evaluated by estimating the Cdl value from the CV curves (Fig. S25, SI), and the results revealed a Cdl of 1.85 mF cm−2, a 1.9-fold increase compared to the initial state. This improved ECSA was closely correlated with the enhanced current density of Co NHs-CW during the stability test. Moreover, as shown in Fig. S26a, SI, the CD signal in the spectrum of Co NHs-CW still exhibited a high value, exceeding nearly 150 mdeg; this was due to the Co NHs retaining the helical nanorod structure with an ∼18 nm CoOx overlayer, as confirmed by the cross-sectional TEM analysis of Co NHs-CW after the stability test (Fig. S26b, SI). Moreover, we conducted mCP-AFM analysis of Co NHs-CW after the stability test. The I–V curves for Co NHs-CW after stability measurement exhibited larger current values under downward magnetization compared with upward magnetization (Fig. S27, SI). The calculated P value of Co NHs-CW after stability at 0.3 V was 64%, which is lower than that of Co NHs-CW before the stability test (78.1%). This is because a CoOx layer was generated at the surface of Co NHs-CW during the 2e− ORR. Nevertheless, this value is considerably higher compared to chiral catalysts modified with chiral organic molecules in previous results,28,61 indicating that efficient spin transport is still maintained through the CoOx overlayer. Thus, the spin-dependent electrochemistry via the CISS effect persisted during long-term electrolysis.
To investigate the electrochemical H2O2 synthesis driven by Co metal-based catalysts, an ultraviolet-visible (UV-vis) spectroscopy-based colorimetric procedure was used for quantifying the H2O2 amount generated over the catalytic electrodes. The concentration of H2O2 was calculated using a calibration curve based on the absorption peak located at 551 nm (Fig. S28, SI).20 Fig. S29a, c, and e, SI show the absorption spectra of the electrolyte as a function of the reaction time during the chronoamperometry test at 0.2 VRHE with the Co metal-based catalysts. The time-dependent formation of H2O2 from Co NHs-CW is illustrated in Fig. S29b, SI; the catalyst exhibited excellent selectivity, with a faradaic efficiency (FE) of 98%. This result also demonstrated that the selectivity was unaffected by the formation of the CoOx overlayer during the electrolysis. The Co TF catalyst also showed a high selectivity with a FE of 97%, as shown in Fig. S29d, SI. The Co NHs-CCW exhibited a selectivity of 99% toward H2O2 synthesis (Fig. S29f, SI). These results indicate that Co NHs-CW and Co TF exhibited similar selectivity for H2O2, due to the CoOx phase (i.e., favorable active sites for the 2e− ORR) formed at the surface of the Co metal phase; however, the superior intrinsic 2e− ORR current density of the chiral Co NHs-CW catalyst promoted the generation of a higher amount of H2O2 during electrolysis, compared to the achiral Co TF counterpart.
Next, we fabricated an immersed perovskite-based electrochemical device, as reported in our previous studies,62,63 for the solar-driven production of H2O2. The configuration of the obtained immersed perovskite-based cathode, illustrated in Fig. 3a (see the experimental section for further details), shows that the underlying FTO region of the Co NHs-CW electrode was directly attached to the bottom electrode (FTO) of the immersed perovskite-based electrochemical device using a silver paste (with the obtained device denoted as Co NHs-CW PSK). To protect the hygroscopic perovskite layer from electrolyte penetration and prevent degradation, the obtained device was encapsulated in an epoxy matrix. The solar-driven oxygen reduction activity of Co NHs-CW PSK was evaluated by LSV measurements under 1-sun illumination in an O2-saturated 0.1 M KOH electrolyte. The Co NHs-CW PSK device was back-illuminated to avoid light blocking induced by the epoxy-based encapsulation, and achieved a superior saturation photocurrent density of −18.0 mA cm−2 at 1.2 VRHE, with a remarkable onset potential of 1.75 VRHE. On the other hand, the immersed perovskite-based cathode based on Co TF (denoted as Co TF PSK) delivered a lower photocurrent density of −4.0 mA cm−2 at 1.2 VRHE, with an inferior onset potential of 1.55 VRHE (Fig. 3b). To determine the intrinsic 2e− ORR photocurrent density, the ECSAs of Co NHs-CW PSK and Co TF PSK were calculated by evaluating the Cdl value from the CV curves measured under 1-sun illumination (Fig. S30a and b, SI). The results showed that the Cdl values of Co NHs-CW and TF were 1.33 and 0.68 mF cm−2, respectively (Fig. S30d, SI). Then, the Js of Co NHs-CW PSK was compared with that of Co TF PSK, confirming that the CISS effect effectively improved the intrinsic 2e− ORR activity, even in the photoelectrolysis system (Fig. S30c, SI). The incident photon-to-current conversion efficiency (IPCE) of the two devices was estimated at 1.0 VRHE (Fig. 3c). Co NHs-CW PSK achieved a higher IPCE of over 75% in a wide wavelength range from 350 to 780 nm, while Co TF PSK yielded a significantly lower IPCE of nearly 20% in the same wavelength range, reflecting the superior light utilization ability associated with Co NHs-CW PSK. Furthermore, the integrated photocurrent densities of Co NHs-CW PSK and Co TF PSK obtained from the IPCE results were close to –17.5 and –4.9 mA cm−2, respectively, in good agreement with the LSV measurements.
 | ||
| Fig. 3 (a) Schematic illustrations of top and side views of PSK electrochemical device integrated with Co NHs-CW. (b) LSV curves of Co NHs-CW PSK and Co TF PSK under 1-sun illumination in O2-saturated 0.1 M KOH. (c) IPCE spectra of Co NHs-CW PSK and Co TF PSK at 1.0 VRHE. Nyquist plots for (d) Co NHs-CW PSK and (e) Co TF PSK, obtained from EIS measurements performed at various potentials under 1-sun illumination. (f) Charge transfer rates of Co NHs-CW PSK and Co TF PSK, calculated from IMPS analysis at 1.2 VRHE. (g) Long-term stability test of Co NHs-CW PSK at 1.2 VRHE in O2-saturated 0.1 M KOH under 1-sun illumination. | ||
To elucidate the transfer behavior of photoinduced charge carriers in the two samples, electrochemical impedance spectroscopy (EIS) measurements were carried out at 1.0, 1.1, 1.2, and 1.3 VRHE under 1-sun illumination (Fig. 3d and e). A Randles–Ershler circuit model consisting of a series resistance (Rs), a charge transfer resistance (Rct), and a constant phase element was used to fit the results of the EIS analysis,64 as illustrated in the inset of Fig. 3d and e. The Nyquist plots of the two samples displayed a single semicircle at low frequencies, attributed to the Rct corresponding to the 2e− ORR kinetics. As the reduction potential decreased from 1.3 to 1.0 VRHE, the Rct values of two devices were reduced, due to the fast reaction kinetics at higher reduction potential (Table S1, SI). The Co NHs-CW PSK device exhibited smaller Rct values at all the applied biases compared with the Co TF PSK counterpart, due to the larger density of active sites, as well as to the CISS effect improving the intrinsic 2e− ORR reaction kinetics. Moreover, intensity-modulated photocurrent spectroscopy (IMPS) measurements were performed at a potential of 1.2 VRHE to examine the surface kinetics; the Nyquist plots of the IMPS spectra for the Co NHs-CW PSK and Co TF PSK devices are displayed in Fig. S31, SI. The charge transfer rate (τd) could be estimated as 1/2πfmin, where fmin represents the frequency of the lowest point in the IMPS spectra.65 The Co NHs-CW PSK device exhibited a higher charge transfer rate (1.25 µs) compared to the Co TF PSK counterpart (2.60 µs), reflecting the enhanced reaction kinetics via spin-dependent electrocatalysis (Fig. 3f). Moreover, the long-term stability of Co NHs-CW PSK for the 2e− ORR was evaluated in an O2-saturated 0.1 M KOH electrolyte (Fig. 3g). The device achieved excellent durability at 1.2 VRHE, without a decrease in photocurrent density for 25 h, while the photocurrent density was enhanced from –17.9 to –22.1 mA cm−2, due to the generation of amorphous CoOx at the surface of Co NHs-CW. The selectivity of Co NHs-CW PSK for H2O2 production was also measured using a UV-vis spectroscopy-based colorimetric method during the chronoamperometric test at 1.2 VRHE, which revealed a FE of nearly 98% for H2O2 generation (Fig. S32, SI). Moreover, the selectivity for H2O2 formation of Co NHs-CW PSK was estimated by an additional UV-vis spectroscopy-based colorimetric method at various applied potentials (1.4–1.7 VRHE) for 15 min. Notably, our Co NHs-CW PSK revealed excellent H2O2 selectivity of 99% at all the applied potentials (Fig. S33, SI), evidencing the efficient solar-driven 2e− ORR capability.
Finally, we investigated an unbiased solar-driven H2O2 production device consisting of Co NHs-CW PSK for driving the 2e− ORR and a carbon cloth electrode to catalyze the IOR (Fig. 4a). Light irradiation on the back side of Co NHs-CW PSK yielded a photovoltage that enabled the Co NHs and carbon electrode catalysts to drive the 2e− ORR and IOR, respectively. We used a H-type cell comprising O2-purged 0.1 M KOH as the catholyte for the 2e− ORR and 0.1 M KOH + 0.5 M KI as the anolyte for the IOR, with the two electrolytes being physically separated by a Nafion 117 membrane. The IOR, characterized by fast reaction kinetics involving a two-electron transfer, was employed as an alternative oxidation process instead of the oxygen evolution reaction (OER), which exhibits sluggish kinetics due to the four-electron-transfer step.66–69 The carbon cloth served as the catalytic electrode for the IOR, due to its excellent iodide oxidation capability, derived from the sp2 bond of graphite, as well as its superior electrochemical stability.66,70 This electrode exhibited an IOR current density of 10 mA cm−2 at a small anodic potential of 1.4 VRHE in 0.1 M KOH + 0.5 M KI electrolyte (Fig. S34, SI). Increasing the concentration of KI in the anolyte from 0.1 to 0.7 M resulted in an enhanced IOR activity due to the increased ionic conductivity, and the optimal concentration of KI was 0.5 M KI. Moreover, the evaluation of the OER performance of the carbon cloth revealed a superior IOR activity compared to the OER one, because of the faster reaction kinetics of the IOR process. The operational point of the unbiased H2O2 production device was 16.2 mA cm−2, as determined from the intersection of the LSV curves of Co NHs-CW PSK for the 2e− ORR and the carbon cloth electrode for the IOR (Fig. 4b). Next, using the two-electrode configuration, we carried out a chronoamperometric test on the unassisted H2O2 production device at 0 V under 1-sun illumination, as shown in Fig. 4c. The bias-free unassisted H2O2 production device exhibited an initial photocurrent density of –16.0 mA cm−2 at 0 V. This value is approximately consistent with the expected value based on the operational point of the LSV curves (Fig. 4b). Our unassisted solar-driven H2O2 production device exhibited not only remarkable stability for 20 h, but also an increased photocurrent density from −16.0 to −19.5 mA cm−2, due to the generation of additional amorphous CoOx during the photoelectrolysis. Notably, the device enabled the unbiased solar-to-H2O2 conversion with a superior FE of nearly 98% (Fig. S35, SI). The FE of carbon cloth for the IOR was also estimated using rotating ring-disk electrode (RRDE) measurements (Fig. S36a, SI). Because the carbon cloth was primarily composed of the graphite phase, graphite powders were cast on the disk electrode. The current delivered by the disk electrode (ID) was attributed to the two-electron iodide oxidation of the graphite powders, while the current on the ring electrode (IR) originated from the two-electron iodine (I2) reduction reaction (IRR) enabled by the Pt ring electrode. The FE for the IOR driven by the graphite powders was obtained by the following equation:
 | (5) |
| IO3− + 5I− + 6H+ → 3I2 + 3H2O | (6) |
 | ||
| Fig. 4 (a) Schematic illustrations of the unassisted H2O2 production system comprising Co NHs-CW PSK (driving the 2e− ORR) and carbon cloth (catalyzing the IOR) electrodes. (b) LSV curves for Co NHs-CW PSK and carbon cloth electrodes driving the 2e− ORR and IOR processes, respectively. (c) Durability test of the unassisted H2O2 production system under 1-sun illumination at 0 V. (d) Comparison of SCC efficiencies for unassisted H2O2 production devices based on the present ORR-IOR system and conventional OER-based systems. | ||
Subsequently, I2 can be extracted from the solution by adding granulated activated carbon, on which I2 molecules are favorably adsorbed, enabling stable storage and transportation of I2 molecules as an I2-adsorbed carbon composite (i.e., I2/C).74 The I2/C composite can be treated with ethanol to dissolve and separate the adsorbed I2, or added to an alkaline aqueous solution to convert I2 into IO3−. Then, the SCC efficiency of our unassisted H2O2 generation device was calculated using the following equation:
 | (7) |
Conclusions
In summary, we successfully demonstrated a high-performance unbiased system for H2O2 photosynthesis based on the spin-modulated electrochemistry of an immersed perovskite-based cathode integrated with a chiral Co NHs electrocatalyst. The chiral-structured Co NH catalytic electrode induced a pronounced CISS effect, favoring the spin-parallel configuration of the electrons. Spin polarization effects promoted an efficient 2e− ORR, suppressing the formation of oxygen intermediates because triplet O2 can readily accept two spin-parallel electrons with opposite spin orientation to that of its electrons. This spin-polarized electrocatalysis of the Co NHs was thoroughly investigated by employing in situ Raman analysis as well as theoretical calculations, which evidenced the superior intrinsic 2e− ORR activity of the chiral Co NHs with respect to the achiral-structured Co TF. Afterward, the Co NH catalyst was attached to an immersed perovskite-based electrochemical device, which exhibited an impressive photocurrent density of –18.0 mA cm−2 at 1.2 VRHE, accompanied by a significant onset potential of 1.75 VRHE. Moreover, we assembled an unbiased H2O2 production system combining our immersed perovskite-based cathode with a carbon cloth electrode for the IOR, achieving an outstanding SCC efficiency of 9.4%, along with stable operation for 20 h. Therefore, our results demonstrate the excellent feasibility of a competitive unassisted device for solar-to-H2O2 conversion, exploiting the CISS effect of a chiral-structured Co metal-based catalyst obtained by a geometry-controlled OAD process.Experimental section
Synthesis of Co metal-based catalysts on FTO
FTO glass was first cleaned by sonication in acetone and isopropyl alcohol for 5 min, followed by rinsing with deionized (DI) water for 5 min; the precleaned FTO glass was used as the substrate. An array of Co NHs was directly grown on the FTO glass through the OAD method with a custom-designed electron-beam evaporator. The base pressure of the vacuum chamber was maintained at 1 × 10−6 Torr during the deposition. Co evaporation was carried out with a deposition rate of 2.0 Å s−1, monitored by a quartz crystal microbalance (QCM). The incidence angle of the vapor flux relative to the substrate normal was maintained at 80°, and the substrate was repeatedly rotated at 1 rpm for 2 s followed by a pause of 12 s to fabricate an array of Co NHs. For comparison, an achiral Co nanostructure was synthesized under similar deposition conditions to those used for the Co NHs catalyst, except that the substrate was continuously rotated at 1 rpm. The achiral Co TF sample was fabricated on the FTO glass using the electron-beam evaporator with a vapor flux incidence angle of zero. The deposition rate and substrate rotation speed were maintained at 1.0 Å s−1 and 25 rpm, respectively.Fabrication of the immersed perovskite-based cathode
The FTO substrate was laser-etched using a Korthem Science (Korea) system and cleaned sequentially with DI water, acetone (Duksan, Korea), and ethyl alcohol (Duksan, Korea) for 20 min each, followed by O2 plasma treatment for 10 min. To prepare the electron transport layer, 275 mg of SnCl2 2H2O (9.99%, trace metal basis), 1.25 g of urea, 1.25 mL of HCl (Duksan, Korea), and 25 mL of thioglycolic acid (TGA, >99%) were dissolved in DI water, and the FTO substrates were immersed in the chemical bath for 2 h. After reaction, the substrates were rinsed with 2-propanol (IPA, anhydrous, 99.5%) and DI water for 10 min and annealed at 170 °C for 1 h. The perovskite precursor solution, consisting of (FAPbI3)0.95(MAPbBr3)0.05, was prepared by dissolving 1.4 M formamidinium iodide (FIA, >99.99%), 1.4 M PbI2(99.99%), 0.023 M methylammonium bromide (MABr, >99.99%), 0.023 M lead bromide (PbBr2, 99.99%), 0.023 M cesium iodide (CsI, 99.99%, trace metals basis), and 0.5 M MACl with 3.8 mol.% methylenediamine dihydrochloride (MDACl2, >98.0%) in a 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of dimethylformamide (DMF, anhydrous, 99.8%) and dimethyl sulfoxide (DMSO, anhydrous > 99.9%) solvents. For substrate doping with 10 µmol potassium iodide (KI, 99.0%), 0.1 mL of a KI stock solution (166 mg KI dissolved in 10 mL DMF) was added to the precursor solution. The precursor solutions were spin-coated onto glass/FTO/SnO2 substrates using a two-step process (1000 rpm for 12 s, 5800 rpm for 20 s). Ethyl acetate (Duksan, Korea) was dripped onto the film into the spin-coating process at 22 s, followed by annealing at 100 °C for 60 min. A 25 µL cyclohexylammonium iodide (CHAI, > 99.99%) solution (6 mg CHAI in 1 mL IPA) was spin-coated onto the perovskite surface at 6000 rpm, followed by annealing at 70 °C for 10 min. The hole transport layer (HTL) precursor was prepared by mixing 72 mg of spiro-OMeTAD, 17.5 µL of lithium bis(fluorosulfonyl)imide (Li-FSI, 98%) solution (260 mg Li-TFSI in 0.5 mL acetonitrile), 6.85 µL of tris(2-(1H-pyrazol-1-yl)-4-tert-butylpyridine)cobalt(III) tri[bis(trifluoromethane)sulfonimide] (KF209 Co TFSI) solution (375 mg KF209 Co TFSI in 1 mL acetonitrile), and 28.8 µL of 4-tert-butylpyridine (TBP, 96%) in 1.2 mL of chlorobenzene. After cooling the substrate, the HTL precursor was spin-coated at 3000 rpm for 30 s. Finally, an 80 nm-thick Au electrode was deposited using a thermal evaporator. Cu tape was attached to the Au layer and a Cu wire was connected using silver paste. To encapsulate the perovskite, the whole components except the FTO bottom electrode were passivated with an insulating resin (HYSOL 9642, Henkel). Subsequently, the Co NHs-CW and/or Co TF catalytic electrodes were partially scraped using a sharp blade to expose a portion of the underlying FTO substrate. Then, the exposed FTO substrate of the Co NHs-CW and/or Co TF electrodes, which exhibited a geometrical active area of 0.6 cm2, was directly attached to the exposed FTO bottom electrode of the perovskite-based electrochemical device using silver paste (Elcoat). Afterward, the obtained device was fully encapsulated in an epoxy matrix, except for the active areas (0.6 cm2) of the Co metal-based electrodes and the back side FTO substrate of the perovskite-based cathode, which was exposed to light illumination. The illuminated area on the back side of the perovskite-based cathode was set to 0.1 cm2 using masking tape.
:1 mixture of dimethylformamide (DMF, anhydrous, 99.8%) and dimethyl sulfoxide (DMSO, anhydrous > 99.9%) solvents. For substrate doping with 10 µmol potassium iodide (KI, 99.0%), 0.1 mL of a KI stock solution (166 mg KI dissolved in 10 mL DMF) was added to the precursor solution. The precursor solutions were spin-coated onto glass/FTO/SnO2 substrates using a two-step process (1000 rpm for 12 s, 5800 rpm for 20 s). Ethyl acetate (Duksan, Korea) was dripped onto the film into the spin-coating process at 22 s, followed by annealing at 100 °C for 60 min. A 25 µL cyclohexylammonium iodide (CHAI, > 99.99%) solution (6 mg CHAI in 1 mL IPA) was spin-coated onto the perovskite surface at 6000 rpm, followed by annealing at 70 °C for 10 min. The hole transport layer (HTL) precursor was prepared by mixing 72 mg of spiro-OMeTAD, 17.5 µL of lithium bis(fluorosulfonyl)imide (Li-FSI, 98%) solution (260 mg Li-TFSI in 0.5 mL acetonitrile), 6.85 µL of tris(2-(1H-pyrazol-1-yl)-4-tert-butylpyridine)cobalt(III) tri[bis(trifluoromethane)sulfonimide] (KF209 Co TFSI) solution (375 mg KF209 Co TFSI in 1 mL acetonitrile), and 28.8 µL of 4-tert-butylpyridine (TBP, 96%) in 1.2 mL of chlorobenzene. After cooling the substrate, the HTL precursor was spin-coated at 3000 rpm for 30 s. Finally, an 80 nm-thick Au electrode was deposited using a thermal evaporator. Cu tape was attached to the Au layer and a Cu wire was connected using silver paste. To encapsulate the perovskite, the whole components except the FTO bottom electrode were passivated with an insulating resin (HYSOL 9642, Henkel). Subsequently, the Co NHs-CW and/or Co TF catalytic electrodes were partially scraped using a sharp blade to expose a portion of the underlying FTO substrate. Then, the exposed FTO substrate of the Co NHs-CW and/or Co TF electrodes, which exhibited a geometrical active area of 0.6 cm2, was directly attached to the exposed FTO bottom electrode of the perovskite-based electrochemical device using silver paste (Elcoat). Afterward, the obtained device was fully encapsulated in an epoxy matrix, except for the active areas (0.6 cm2) of the Co metal-based electrodes and the back side FTO substrate of the perovskite-based cathode, which was exposed to light illumination. The illuminated area on the back side of the perovskite-based cathode was set to 0.1 cm2 using masking tape.
Characterization
The phase composition of the Co metal-based catalysts was investigated by XRD analysis (MinFlex 600, Rigaku, Japan) with Cu Kα radiation (λ = 0.15406 nm). The morphologies of the Co metal-based catalysts were examined using field-emission SEM (JSM-7001F, JEOL, Japan). TEM (JEM-ARM200F, JEOL, Japan) analysis at an acceleration voltage of 200 kV was performed to examine the morphology of the Co NHs-CW catalyst. A focused ion beam lift-out procedure was employed for sample preparation. Furthermore, XPS measurements were performed using a K-Alpha system (Thermo Scientific, UK) with a monochromatic Al Kα radiation source; all XPS spectra were calibrated based on the binding energy of the C 1s peak (284.5 eV). Raman spectra were obtained using a confocal microscope (Alpha 300 Apyron, WITec, Germany). CD spectra of Co metal-based catalysts were measured using a J-815 spectrometer (JASCO Corporation, Tokyo, Japan). mCP-AFM measurements (NX10, Park Systems, South Korea) were conducted using a Co–Cr-coated cantilever to evaluate the spin-polarized current of the Co metal-based catalysts. The RIE (reactive ion etcher, Young Hi-Tech, Korea) process was conducted at a power of 200 W and an Ar flow rate of 100 sccm to reduce the thickness of Co NHs-CW.XAS measurements were conducted at the 8C nanoprobe XAFS beamline (BL8C) of the Pohang Light Source (PLS-II), in a 3.0 GeV storage ring with a ring current of 250 mA. The X-ray beam was monochromated by a Si(111) double crystal, where the beam intensity was reduced by 30% to eliminate higher-order harmonics. The X-ray beam was then delivered to a secondary source aperture with the beam size adjusted to 0.5 mm (v) × 1 mm (h). For the Co K-edge measurements, the monochromator energies were calibrated using Co foil. All measurements were performed at room temperature in transmission mode, and the detectors were based on ionization chambers. The obtained spectra were processed using the Demeter package, which provides an interface to the IFEFFIT library.
Electrochemical measurements
All electrochemical experiments were conducted on an electrochemical workstation (SI 1287, Solartron, UK). The LSV and chronoamperometry measurements of the Co metal-based electrodes and PSK-based electrochemical devices were conducted under dark and 1-sun illumination (AM 1.5G) conditions, using a three-electrode system comprising Ag/AgCl/KCl (saturated) as the reference electrode and Pt coil as the counter electrode. For the LSV measurements of Co metal-based electrodes, all potentials were iR-corrected. Moreover, in all electrochemical measurements, the applied potential values were converted to the RHE scale using the following equation:| ERHE = EAg/AgCl + 0.05916 × pH + 0.197 | (8) |
The Cdl values obtained from the CV curves were used to determine the ECSA according to the following equation:
 | (9) |
Quantitative H2O2 detection
The N,N-diethyl-1,4-phenylenediamine sulfate (DPD)–peroxidase (POD) method was used to determine the concentration of H2O2 at 551 nm. Briefly, DPD (50 mg, 98.0%, Sigma) and POD (5 mg, horseradish, Sigma) were dissolved in 0.1 N H2SO4 (5 mL) and DI water (5 mL), respectively. Finally, 0.1 M sodium phosphate buffer (2.7 mL), aliquots of the DPD (0.05 mL) and POD (0.05 mL) solutions, and the catholyte (0.3 mL) were mixed together. For quantification, a standard calibration curve was obtained by plotting the absorbance against the concentration, and the H2O2 concentration was determined as a function of the reaction time.H2O2 detection
Density functional theory (DFT) calculations were performed using the Vienna Ab initio Simulation Package (VASP) with the projector augmented wave (PAW) method.78,79 The exchange–correlation interactions were treated within the generalized gradient approximation (GGA) using the Perdew–Burke–Ernzerhof (PBE) functional.80 Spin-polarization was included in all calculations, and van der Waals interactions were accounted for using the Grimme's DFT-D3 method.81 A plane-wave cutoff energy of 520 eV was used. For bulk hcp Co, the Brillouin zone was sampled with a 16 × 16 × 8 Monkhorst–Pack k-point grid. Surface calculations were performed to optimize the surface coverage using a p(2 × 2) Co(002) slab consisting of five atomic layers, where the bottom two layers were fixed and the top three layers were relaxed. A vacuum region of 15 Å was introduced along the z-direction to avoid interactions between periodic images. A 8 × 8 × 1 k-point grid was used for the surface models. For calculation of ORR energetics, a larger p(4 × 4) surface supercell was used with a 4 × 4 × 1 k-point mesh. Electronic convergence was set to 10−6 eV, and geometry optimization was performed until all atomic forces were below 0.01 eV Å−1. To investigate the surface coverage and 2e-ORR pathway, we employed the computational hydrogen electrode (CHE) method developed by Nørskov and co-workers, which allows for the calculation of electrochemical reaction energies without explicit modeling solvated protons or electrons.82–84 Gibbs free energy changes (ΔG) for each elementary step were computed as:| ΔG = ΔE + ΔZPE − TΔS − neU | (10) |
The free energy of the *OOH intermediate was computed using:
 | (11) |
The asterisk (*) is the active site of the catalyst.
This expression avoids the direct use of O2 (g), whose energy is poorly described in DFT, by referencing to the experimental reaction energy of  .
.
Conflicts of interest
There are no conflicts to declareData availability
The data supporting this article have been included as part of the supplementary information (SI). Supplementary information is available. See DOI: https://doi.org/10.1039/d5ee04313a.Acknowledgements
This research was supported by the National Research Foundation (NRF) of Korea (grant no. 2021R1A3B1068920, 2021M3H4A1A03049662, RS-2024-00344690, and RS-2025-02633327), funded by the Ministry of Science and ICT. Additional support was provided by the Yonsei Signature Research Cluster Program of 2021 (2021-22-0002) and the Yonsei Fellowship, funded by Lee Youn Jae.References
- Y. Y. Jiang, P. J. Ni, C. X. Chen, Y. Z. Lu, P. Yang, B. Kong, A. Fisher and X. Wang, Adv. Energy Mater., 2018, 8, 1801909 CrossRef.
- S. Moon, Y. S. Park, H. Lee, W. Jeong, E. Kwon, J. Lee, J. Yun, S. Lee, J. H. Kim, S. Yu and J. Moon, Energy Environ. Sci., 2024, 17, 5588–5600 RSC.
- Y. Y. Sun, L. Han and P. Strasser, Chem. Soc. Rev., 2020, 49, 6605–6631 RSC.
- J. Y. Tang, T. S. Zhao, D. Solanki, X. B. Miao, W. G. Zhou and S. Hu, Joule, 2021, 5, 1432–1461 CrossRef CAS.
- J. M. Campos-Martin, G. Blanco-Brieva and J. L. G. Fierro, Angew. Chem., Int. Ed., 2006, 45, 6962–6984 CrossRef CAS PubMed.
- M. Ko, Y. Kim, J. Woo, B. Lee, R. Mehrotra, P. Sharma, J. Kim, S. W. Hwang, H. Y. Jeong, H. Lim, S. H. Joo, J. W. Jang and J. H. Kwak, Nat. Catal., 2022, 5, 37–44 CrossRef CAS.
- R. Mehrotra, D. Oh and J. W. Jang, Nat. Commun., 2021, 12, 6644 CrossRef CAS PubMed.
- Z. M. Zhang, X. Chen, R. Hao, Q. L. Feng and E. Q. Xie, Adv. Funct. Mater., 2023, 33, 2303391 CrossRef CAS.
- H. Xu, M. Jin, S. B. Zhang, X. Y. Zhang, M. Xu, Y. X. Zhang, G. Z. Wang and H. M. Zhang, Adv. Sci., 2025, 12, 6644 Search PubMed.
- Y. P. Zhu, C. L. Yang, T. C. Lu, Y. Z. Gan, H. Zhao, Y. B. Chen, Q. Q. Cheng and H. Yang, Nano Energy, 2025, 141, 111142 CrossRef CAS.
- S. Siahrostami, A. Verdaguer-Casadevall, M. Karamad, D. Deiana, P. Malacrida, B. Wickman, M. Escudero-Escribano, E. A. Paoli, R. Frydendal, T. W. Hansen, I. Chorkendorff, I. E. L. Stephens and J. Rossmeisl, Nat. Mater., 2013, 12, 1137–1143 CrossRef CAS PubMed.
- R. X. Zheng, Q. L. Meng, L. Zhang, J. J. Ge, C. P. Liu, W. Xing and M. L. Xiao, Chem - Eur. J., 2023, 29, e202203180 CrossRef CAS PubMed.
- Y. A. Shi, L. L. Zhang, C. Y. Wang and S. H. Sun, Adv. Sci., 2025, 12, 2502388 CrossRef CAS.
- Z. T. Zhang, W. B. Chen, H. K. Chu, F. Xiong, K. X. Zhang, H. C. Yan, F. Q. Meng, S. Gao, B. Ma, X. Hai and R. Q. Zou, Angew. Chem., Int. Ed., 2024, 63, e202410123 CrossRef CAS PubMed.
- J. Wu, A. Mehmood, G. H. Zhang, S. Wu, G. Ali and A. Kucernak, ACS Catal., 2021, 11, 5035–5046 CrossRef CAS.
- Y. H. Chen, C. Zhen, Y. B. Chen, H. Zhao, Y. D. Wang, Z. Y. Yue, Q. S. Wang, J. Li, M. D. Gu, Q. Q. Cheng and H. Yang, Angew. Chem., Int. Ed., 2024, 63, e202407163 CrossRef CAS PubMed.
- Z. J. Yang, Y. Gao, L. L. Zuo, C. Long, C. Y. Yang and X. F. Zhang, ACS Catal., 2023, 13, 4790–4798 CrossRef CAS.
- J. S. Li, J. J. Ma, Z. Ma, E. L. Zhao, K. Du, J. X. Guo and T. Ling, Adv. Energy Sust. Res., 2021, 2, 2100034 CrossRef CAS.
- T. L. Feng, W. H. Chen, J. Xue, F. F. Cao, Z. W. Chen, J. C. Ye, C. X. Xiao and H. P. Lu, Adv. Funct. Mater., 2023, 33, 2215051 CrossRef CAS.
- Y. S. Park, J. Lee, H. Lee, J. B. Park, J. Yun, C. U. Lee, S. Moon, S. Lee, S. Kim, J. H. Kim, D. Kim, J. Han, D. W. Kim and J. Moon, ACS Appl. Mater. Interfaces, 2025, 17, 18228–18242 CrossRef CAS PubMed.
- Y. R. Jin, W. L. Fu, Z. H. Wen, L. L. Tan, Z. Chen, H. Wu and P. P. Wang, J. Am. Chem. Soc., 2023, 146, 2798–2804 CrossRef PubMed.
- T. Bai, J. Ai, L. Y. Liao, J. W. Luo, C. Song, Y. Y. Duan, L. Han and S. A. Che, Angew. Chem., Int. Ed., 2021, 60, 9421–9426 CrossRef CAS PubMed.
- T. Bai, J. Ai, J. Ma, Y. Y. Duan, L. Han, J. G. Jiang and S. N. Che, Angew. Chem., Int. Ed., 2021, 60, 20036–20041 CrossRef CAS PubMed.
- Z. Y. Bian, K. Kato, T. Ogoshi, Z. Cui, B. S. Sa, Y. Tsutsui, S. Seki and M. Suda, Adv. Sci., 2022, 9, 2201063 CrossRef CAS PubMed.
- Z. Y. Bian, Y. Nakano, K. Miyata, I. Oya, M. Nobuoka, Y. Tsutsui, S. Seki and M. Suda, Adv. Mater., 2023, 35, 2306061 CrossRef CAS PubMed.
- A. Vadakkayil, C. Clever, K. N. Kunzler, S. S. Tan, B. P. Bloom and D. H. Waldeck, Nat. Commun., 2023, 14, 1067 CrossRef CAS PubMed.
- S. Ghosh, B. P. Bloom, Y. Y. Lu, D. Lamont and D. H. Waldeck, J. Phys. Chem. C, 2020, 124, 22610–22618 CrossRef CAS.
- H. Im, S. Ma, H. Lee, J. Park, Y. S. Park, J. Yun, J. Lee, S. Moon and J. Moon, Energy Environ. Sci., 2023, 16, 1797 RSC.
- W. L. Fu, L. L. Tan and P. P. Wang, ACS Nano, 2023, 17, 16326–16347 CrossRef CAS PubMed.
- J. Kim, H. Jung, S. M. Jung, J. Hwang, D. Y. Kim, N. Lee, K. S. Kim, H. Kwon, Y. T. Kim, J. W. Han and J. K. Kim, J. Am. Chem. Soc., 2021, 143, 1399–1408 CrossRef CAS PubMed.
- M. P. Garcia, D. Gibson, K. L. McAughey, D. A. Hughes and C. G. Núñez, Adv. Phys. Res., 2024, 3, 240020 Search PubMed.
- J. Kim, S. M. Jung, N. Lee, K. S. Kim, Y. T. Kim and J. K. Kim, Adv. Mater., 2023, 35, 2305844 CrossRef CAS PubMed.
- J. Kim, S. M. Jung, H. Im, H. Hwang, D. S. Kim, G. W. Jeon, Y. T. Kim, J. W. Han and J. K. Kim, Chem. Eng. J., 2024, 501, 157385 CrossRef CAS.
- H. Hwang, H. Bae, E. Ahn, D. Lee, H. H. Cho, S. Cheong, T. Y. Kim, J. Kim, Y. Yun, D. Lee, S. K. Hahn and J. K. Kim, Small, 2025, 2504976 CrossRef CAS PubMed.
- G. Lee, Y. Han, H. K. Lee, H. Yoo and Y. J. Kim, Adv. Opt. Mater., 2024, 12, 2303050 CrossRef CAS.
- S. Singh, A. S. Dev, M. Priyadarsini, P. Gupta, S. K. Vayalil, B. Sochor, V. Srihari and D. Kumar, Appl. Surf. Sci., 2025, 695, 162857 CrossRef CAS.
- J. A. Delgado-Notario, D. López-Díaz, D. McCloskey and J. M. Caridad, Adv. Funct. Mater., 2024, 34, 2401599 CrossRef CAS.
- S. Ju, N. Lee, H. Sung, S. Son, N. Kim, J. Kim, J. K. Kim and H. Lee, J. Mater. Chem. A, 2023, 11, 17644–17650 RSC.
- M. Avila-Gutierrez, A. Moisset, A. T. Ngo, S. Costanzo, G. Simon, P. Colomban, M. Petit, C. Petit and I. Lisiecki, Colloids Surf., A, 2023, 676, 132281 CrossRef CAS.
- D. H. Lee, M. Kang, S. M. Paek and H. Jung, Electrochim. Acta, 2016, 217, 132–138 CrossRef CAS.
- L. N. Yan, X. Cheng, Y. S. Wang, Z. Z. Wang, L. R. Zheng, Y. Yan, Y. Lu, S. R. Sun, W. G. Qiu and G. Chen, Mater. Today Energy, 2022, 24, 100931 CrossRef CAS.
- J. X. Hou, K. W. Wang, X. Zhang, Y. Wang, H. Su, C. Y. Yang, X. Y. Zhou, W. N. Liu, H. W. Hu, J. X. Wang, C. Li, P. J. Ma, R. Zhang, Z. Wei, Z. C. Sun, Q. H. Liu and K. Zheng, ACS Catal., 2024, 14, 10893–10903 CrossRef CAS.
- J. Ahn, S. Ma, J. Y. Kim, J. Kyhm, W. Yang, J. A. Lim, N. A. Kotov and J. Moon, J. Am. Chem. Soc., 2020, 142, 4206–4212 CrossRef CAS PubMed.
- S. S. Andrews and J. Tretton, J. Chem. Educ., 2020, 97, 4370–4376 CrossRef CAS.
- M. Yang, Y. F. Li, Y. Yu, X. C. Liu, Z. Shi and Y. Xing, Chem – Eur. J., 2018, 24, 13002–13008 CrossRef CAS PubMed.
- S. Anantharaj, H. Sugime, B. Chen, N. Akagi and S. Noda, J. Phys. Chem. C, 2020, 124, 9673–9684 CrossRef CAS.
- Z. H. Xiao, Y. C. Huang, C. L. Dong, C. Xie, Z. J. Liu, S. Q. Du, W. Chen, D. F. Yan, L. Tao, Z. W. Shu, G. H. Zhang, H. G. Duan, Y. Y. Wang, Y. Q. Zou, R. Chen and S. Y. Wang, J. Am. Chem. Soc., 2020, 142, 12087–12095 CrossRef CAS PubMed.
- X. Y. Quek, Q. H. Tang, S. Q. Hu and Y. H. Yang, Appl. Catal., A, 2009, 361, 130–136 CrossRef CAS.
- T. Priyadharshini, B. Saravanakumar, G. Ravi, A. Sakunthala and R. Yuvakkumar, J. Nanosci. Nanotechnol., 2018, 18, 4093–4099 CrossRef CAS PubMed.
- L. L. Zhang, X. J. Yao, Y. Y. Lu, C. Z. Sun, C. J. Tang, F. Gao and L. Dong, J. Colloid Interface Sci., 2018, 509, 334–345 CrossRef CAS PubMed.
- C. Yang, J. Chen, L. Yan, Y. J. Gao, J. Q. Ning and Y. Hu, Appl. Catal., B, 2024, 352, 124060 CrossRef CAS.
- Y. B. Shi, Z. P. Yang, L. J. Shi, H. Li, X. P. Liu, X. Zhang, J. D. Cheng, C. Liang, S. Y. Cao, F. R. Guo, X. Liu, Z. H. Ai and L. Z. Zhang, Environ. Sci. Technol., 2022, 56, 14478–14486 CrossRef CAS PubMed.
- Y. Luo, B. P. Zhang, C. C. Liu, D. H. Xia, X. W. Ou, Y. P. Cai, Y. Zhou, J. Jiang and B. Han, Angew. Chem., Int. Ed., 2023, 62, e202305355 CrossRef CAS PubMed.
- K. Chae, H. Lee, W. T. Huang, J. Son, B. Pavageau, T. H. Kim, S. E. Lee, J. Kim, J. Moon, R. S. Liu, J. Bang and D. H. Kim, Adv. Mater., 2025, 37, 2507658 CrossRef CAS PubMed.
- H. A. Hansen, J. Rossmeisl and J. K. Norskov, Phys. Chem. Chem. Phys., 2008, 10, 3722–3730 RSC.
- S. H. Ma, Z. Y. Jiao, T. X. Wang and X. Q. Dai, Surf. Sci., 2014, 619, 90–97 CrossRef CAS.
- Q. W. Chang, P. Zhang, A. H. B. Mostaghimi, X. R. Zhao, S. R. Denny, J. H. Lee, H. P. Gao, Y. Zhang, H. L. L. Xin, S. Siahrostami, J. G. G. Chen and Z. Chen, Nat. Commun., 2020, 11, 2178 CrossRef CAS PubMed.
- T. Kim, T. H. Lee, S. Y. Park, T. H. Eom, I. Cho, Y. Kim, C. Kim, S. A. Lee, M. J. Choi, J. M. Suh, I. S. Hwang, D. Lee, I. Park and H. W. Jang, ACS Nano, 2023, 17, 4404–4413 CrossRef CAS PubMed.
- J. Kim, S. H. Her, D. S. Kim, B. Kim, S. M. Jung, J. Kim, H. Hwang, K. S. Kim, Y. T. Kim, S. Y. Choi and J. K. Kim, Small, 2025, 2505383 CrossRef CAS PubMed.
- B. B. Sheng, D. F. Cao, H. W. Shou, O. A. Moses, W. J. Xu, Y. J. Xia, Y. Z. Zhou, H. J. Wang, P. Wan, S. Zhu, W. S. Chu, X. J. Wu, S. M. Chen and L. Song, Nano Res., 2022, 15, 1831–1837 CrossRef CAS.
- V. Kiran, S. P. Mathew, S. R. Cohen, I. H. Delgado, J. Lacour and R. Naaman, Adv. Mater., 2016, 28, 1957 CrossRef CAS PubMed.
- Y. S. Park, J. W. Lee, H. Lee, J. Yun, G. Jang, J. Lee, S. Son, C. U. Lee, C. S. Jeong, S. Moon, S. Lee, H. I. Kim and J. Moon, Adv. Energy Mater., 2023, 13, 2301166 CrossRef CAS.
- J. Yun, H. Lee, Y. S. Park, W. Jeong, C. S. Jeong, J. Lee, J. Lee, J. Tan, S. Ma, G. Jang, C. U. Lee, S. Moon, H. Im, S. Lee, D. Y. Yee, J. H. Kim and J. Moon, Adv. Energy Mater., 2023, 13, 2301693 CrossRef CAS.
- H. Y. Sun, R. Tang, X. M. Zhang, S. B. Zou, Y. H. Liang, N. Namuerkesaihan, Q. F. Gu, A. Baiker and J. Huang, J. Mater. Chem. A, 2025, 13, 14205–14215 RSC.
- Z. L. Tian, P. F. Zhang, P. Qin, D. Sun, S. N. Zhang, X. W. Guo, W. Zhao, D. Y. Zhao and F. Q. Huang, Adv. Energy Mater., 2019, 9, 1901287 CrossRef.
- Y. S. Park, G. Jang, I. Sohn, H. Y. S. Lee, J. Tan, J. Yun, S. Ma, J. Lee, C. U. Lee, S. Moon, H. Im, S. M. Chung, S. H. Yu, H. Kim and J. Moon, Carbon Energy, 2023, 5, e366 CrossRef CAS.
- D. B. Adam, M. C. Tsai, Y. A. Awoke, W. H. Huang, Y. W. Yang, C. W. Pao, W. N. Su and B. J. Hwang, ACS Sustain. Chem. Eng., 2021, 9, 8803–8812 CrossRef CAS.
- J. Yun, Y. S. Park, H. Lee, W. Jeong, C. S. Jeong, C. U. Lee, J. Lee, S. Moon, E. Kwon, S. Lee, S. Kim, J. Kim, S. Yu and J. Moon, Adv. Energy Mater., 2024, 14, 202401055 Search PubMed.
- L. Yao, Y. P. Liu, H. H. Cho, M. Xia, A. Sekar, B. P. Darwich, R. A. Wells, J. H. Yum, D. Ren, M. Grätzel, N. Guijarro and K. Sivula, Energ. Environ. Sci., 2021, 14, 3141–3151 RSC.
- S. Ito, M. Sugimasa, Y. Toshimitsu, A. Orita, M. Kitagawa and M. Sakai, Electrochim. Acta, 2021, 379, 138181 CrossRef CAS.
- Y. C. Wen, T. Zhang, J. Y. Wang, Z. L. Pan, T. F. Wang, H. Yamashita, X. F. Qian and Y. X. Zhao, Angew. Chem., Int. Ed., 2022, 134, e202205972 CrossRef.
- H. Tan, Z. P. Yu, A. P. LaGrow, S. Y. Ma, J. W. Wang, H. Li, D. H. Xiong and L. F. Liu, J. Mater. Chem. A, 2023, 11, 26152–26163 RSC.
- E. L. Hu, Y. Yao, Y. Chen, Y. J. Cui, Z. Y. Wang and G. D. Qian, Nanoscale Adv., 2021, 3, 604–610 RSC.
- T. A. Dessie, W. H. Huang, D. B. Adam, Y. A. Awoke, C. H. Wang, J. L. Chen, C. W. Pao, N. G. Habtu, M. C. Tsai, W. N. Su and B. J. Hwang, Nano Lett., 2022, 22, 7311–7317 CrossRef CAS PubMed.
- Y. Choi, R. Mehrotra, S. H. Lee, T. V. T. Nguyen, I. Lee, J. Kim, H. Y. Yang, H. Oh, H. Kim, J. W. Lee, Y. H. Kim, S. Y. Jang, J. W. Jang and J. Ryu, Nat. Commun., 2022, 13, 5709 CrossRef CAS PubMed.
- C. R. Raj, A. Samanta, S. H. Noh, S. Mondal, T. Okajima and T. Ohsaka, J. Mater. Chem. A, 2016, 4, 11156–11178 RSC.
- Y. S. Park, X. Y. Jin, J. W. Tan, H. Lee, J. Yun, S. Ma, G. M. Jang, T. Kim, S. G. Shim, K. Kim, J. Lee, C. U. Lee, S. J. Hwang and J. Moon, Energy Environ. Sci., 2022, 15, 4725–4737 RSC.
- G. Kresse and J. Furthmuller, Phys. Rev. B:Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed.
- G. Kresse and D. Joubert, Phys. Rev. B:Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS.
- M. Unzog, A. Tal and G. Kresse, Phys. Rev. B, 2022, 106, 155133 CrossRef CAS.
- S. Grimme, J. Comput. Chem., 2006, 27, 1787–1799 CrossRef CAS PubMed.
- J. K. Norskov, J. Rossmeisl, A. Logadottir, L. Lindqvist, J. R. Kitchin, T. Bligaard and H. Jónsson, J. Phys. Chem. B, 2004, 108, 17886–17892 CrossRef CAS PubMed.
- V. Viswanathan, H. A. Hansen, J. Rossmeisl and J. K. Norskov, ACS Catal., 2012, 2, 1654–1660 CrossRef CAS.
- Z. Y. Sang, Y. Q. Qiao, R. Chen, L. C. Yin, F. Hou and J. Liang, Nat. Commun., 2025, 16, 4050 CrossRef CAS PubMed.
Footnote |
| † These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2026 |