
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Explainable active learning framework for ligand binding affinity prediction
Satya Pratik
Srivastava
a,
Rohan
Gorantla
 bc,
Sharath Krishna
Chundru
a,
Claire J. R.
Winkelman
c,
Antonia S. J. S.
Mey
*c and
Rajeev Kumar
Singh
*a
bc,
Sharath Krishna
Chundru
a,
Claire J. R.
Winkelman
c,
Antonia S. J. S.
Mey
*c and
Rajeev Kumar
Singh
*a
aShiv Nadar University, Delhi-NCR, India. E-mail: rajeev.kumar@snu.edu.in
bSchool of Informatics, University of Edinburgh, Edinburgh EH8 9AB, UK
cEaStCHEM School of Chemistry, University of Edinburgh, Edinburgh EH9 3FJ, UK. E-mail: Antonia.mey@ed.ac.uk
First published on 23rd December 2025
Abstract
Active learning (AL) prioritises which compounds to measure next for protein–ligand affinity when assay or simulation budgets are limited. We present an explainable AL framework built on Gaussian process regression and assess how molecular representations, covariance kernels, and acquisition policies affect enrichment across four drug-relevant targets. Using recall of the top active compound, we find that dataset identity which is a target's chemical landscape sets the performance ceiling and method choices modulate outcomes rather than overturn them. Fingerprints with simple Gaussian process kernels provide robust, low-variance enrichment, whereas learned embeddings with non-linear kernels can reach higher peaks but with greater variability. Uncertainty-guided acquisition consistently outperforms random selection, yet no single policy is universally optimal; the best choice follows structure–activity relationship (SAR) complexity. To enhance interpretability beyond black-box selection, we integrate SHapley Additive exPlanations (SHAP) to link high-impact fingerprint bits to chemically meaningful fragments across AL cycles, illustrating how the model's attention progressively concentrates on SAR-relevant motifs. We additionally provide an interactive active learning analysis platform featuring SHAP traces to support reproducibility and target-specific decision-making.
1 Introduction
Drug discovery is the process of identifying new molecules that can target a disease state with novel chemical compounds ranging from small molecules1–3 to anti-bodies.3,4 One way of approaching small molecule drug discovery is by identifying a biological target, e.g., a protein or other relevant biomolecule to alter their functional state by inhibition. One key property that can help identify novel inhibitors for a protein target is optimisation of the protein-ligand binding affinity. As such, accurate in silico and experimental estimation of protein–ligand binding affinities are essential properties to measure and predict during hit identification across vast chemical libraries and systematic optimization of congeneric series during hit–to–lead campaigns.5–7 High–throughput screening remains a cornerstone of small-molecule discovery, but rising assay complexity and cost increasingly preclude exhaustive use.8 As discovery shifts toward medium-throughput, biophysics-rich assays supported by structure-guided optimisation with alchemical free-energy methods (AFEs),2,6,9–11 the goal centers around exploring chemical space effectively under a budget. This budget can determine both the number of evaluations measurements or computational predictions by only assessing a few hundred compounds per cycle.5,6,12 Biophysical assays such as surface plasmon resonance (SPR) provide kinetics-resolved confirmation of binding and are widely used when functional assays are noisy, non-specific, or fail to identify tractable series.13,14 Yet their medium-to-low throughput constrains campaign scale. Similarly, AFE calculations can prospectively prioritise substitutions but remain computationally intensive. In both cases, identifying the most promising set of compounds with the fewest computational or experimental evaluations and minimising the overall budget is desirable.Active learning (AL), a subset of machine learning, has emerged as a framework to address this challenge.2,9,15 By training a surrogate model, quantifying predictive uncertainty, and iteratively prioritising the next most informative compounds, AL maximises information gain from a limited number of experimental assay measurements or physics-based computations, enabling efficient enrichment without relying on brute-force screening.9,15–18 In practice, AL balances exploitation that is refining known high-activity scaffolds, against exploration that probes novel chemotypes that may unlock new structure–activity relationships (SARs). This trade-off is controlled by the acquisition strategy.19,20 As a result, AL has been deployed for ligand binding affinity prediction and multi-property lead optimisation under assay- or simulation-constrained budgets.2,9,15,18,20 Notwithstanding its potential, AL is not a “one-size-fits-all” solution.15,21 Its performance is significantly dependent on a complex interplay of methodological choices, including the underlying machine learning model, the molecular representation, the kernel function, and the acquisition protocol.19,21 Outcomes vary with the chemical landscape of the library, the molecular representation, the surrogate model choice, and the acquisition protocol.15,19,21 Moreover, surrogate models and representations from deep learning models can behave as “black boxes,” limiting chemical intuition and trust in recommendations.22–24 AL has been applied successfully on individual targets2,25,26 and specific workflows,10,27,28 and recent efforts have begun to systematically explore different strategies and parameters.15,21 Open questions remain around clarifying when different AL designs are most effective, why performance varies across chemical spaces, and finding ways to incorporate explainability into the selection process of the AL cycles to help with guiding design choices that can be experimentally verified.
In this work we combine explainability while exploring seven acquisition protocols with five Gaussian-process kernels and three molecular representations (ECFP4, MACCS, and ChemBERTa) in a fixed budget-setting for pharmaceutically relevant targets taken from literature (TYK2, USP7, D2R, and MPro). We show that the inherent chemical landscape of each target substantially dictates achievable enrichment, and that the choice of representation–kernel combination presents a trade-off between robustness (e.g., fixed fingerprints with simple kernels) and peak performance (e.g., learned embeddings with non-linear kernels). To move beyond black-box selection, we integrate SHapley Additive exPlanations29 (SHAP) to map high-impact fingerprint bits to chemically interpretable fragments over AL cycles, revealing how model focus sharpens onto SAR-relevant motifs. To allow easy visualisation and analysis of various AL strategies in combination with the SHAP analysis, we provide an active learning analysis platform. This platform provides a way to visualise this comprehensive analysis across all diverse settings and targets and integrates SHAP traces to support reproducibility and target-specific decision-making. It can easily be adapted to different protocols and targets to provide a comprehensive and interactive understanding of different AL strategies and their impact on chemical space. Our code is available at https://github.com/meyresearch/explainable_AL.
 | ||
| Fig. 1 Screenshots of the interactive active learning analysis platform. (A) The main landing page offers two primary ways to engage with the tool: loading a complete, pre-analyzed demo dataset or uploading custom data files via the sidebar. (B) A view of the ‘Feature Evolution Analysis’ tab, which visualizes how the importance of top molecular features, as measured based on their mean SHAP values, changes dynamically across the different phases (random, explore, and exploit) of the active learning cycles. (C) The “How to Explore Our Results” section, providing clear, step-by-step instructions for users to either explore the platform's built-in findings or analyze their own data for comparison. | ||
2 Methods
2.1 Active learning setup
Central to our active learning framework is a methodology that employs principles of Bayesian Optimization (BO).30 BO is an iterative strategy for optimizing black-box functions that are expensive to evaluate. It operates by building a probabilistic surrogate model of the objective function, which is then used to intelligently select the most promising points to evaluate next. In our framework, the surrogate model approximates the relationship between the molecular structure and binding affinity across the chemical space of ligands. An acquisition function (AF) uses the model's estimates and uncertainties to select the next batch of compounds for evaluation. The model is then updated with the new data, and the process is repeated. The ultimate goal of this iterative process is to find the compound with the highest affinity, as summarised by the objective in eqn (1). | (1) |
2.2 Gaussian process as the surrogate model
The most common and effective class of surrogate models for Bayesian optimization are Gaussian Processes (GPs).31 A GP is a non-parametric model, defined by its mean function m(x) and covariance, i.e., the kernel function k(x, x′) which measures the similarity between two points. The GP is defined by using the following eqn (2)| f(x) ∼ GP(m(x),k(x, x′)) | (2) |
Gaussian functions can model the unknown affinity function f(x) on a distribution of functions, and they are incredibly adaptable at approximating nonlinear functions, which are needed to traverse the vast chemical space.
2.3 Acquisition strategies in AL cycles
Compound selection within the active learning loop is guided by an acquisition strategy; here we use the generalized Upper Confidence Bound (UCB) acquisition function.20,32 This function balances exploring new molecules with exploiting known good binders by linearly weighting the model's estimated mean affinity and its associated uncertainty. The acquisition score for a compound x is determined as follows,| sacq(x) = α µ(x) + β σ(x) | (3) |
The seven distinct active learning acquisition protocols in this study were designed to systematically probe the trade-off between exploration and exploitation. Each protocol began with an initial random batch of 60 compounds to seed the model, followed by 10 acquisition cycles of 30 compounds each. The exploration-exploitation balance was controlled by dynamically varying the α and β parameters in the generalized Upper Confidence Bound (UCB) acquisition function: sacq(x) = α µ(x) + β σ(x).
This framework allows for three primary modes: pure exploration (α = 0, β = 1), which prioritizes molecules with the highest uncertainty (σ(x)); pure exploitation (α = 1, β = 0), which selects the most promising estimated affinity (µ(x)); and a balanced strategy (α = 0.5, β = 0.5). The specific schedules for each protocol are summarised in Table 1.
| Protocol name | Acquisition schedule (10 cycles of 30 compounds) |
|---|---|
| Random baseline | [R(30)] × 10 |
| UCB-balanced | [B(30)] × 10 |
| UCB-alternate | [E(30), X(30)] × 5 |
| UCB-sandwich | [E(30)] × 2 + [X(30)] × 6 + [E(30)] × 2 |
| UCB-explore-heavy | [E(30)] × 7 + [X(30)] × 3 |
| UCB-exploit-heavy | [X(30)] × 7 + [E(30)] × 3 |
| UCB-gradual | [E(30)] × 3 + [B(30)] × 4 + [X(30)] × 3 |
Beyond simple baselines like the random and UCB-balanced protocols, we designed several dynamic strategies to model different discovery campaign philosophies:
2.4 Model validation and hyperparameter handling
To ensure the robustness of our models and the validity of their uncertainty estimates, we incorporated several validation and regularization techniques.2.5 Molecular representations and kernel choices
When using GPs, we need to convert chemical SMILES strings with numerical feature vectors. An efficient molecular representation can reduce the complexity of the problem by capturing only relevant information. Capturing all the relevant structure and chemical information, maintaining low dimensionality and providing chemical intuition are the challenges that any representation method has to deal with. By using three different molecular representations, we explore different aspects each with their unique tradeoffs. We use ECFP fingerprints, i.e., Extended-Connectivity Fingerprints with radius 4, consisting of 4,096 binary features.33 MACCS Keys, with 166-bit binary fingerprints representing predefined molecular fragments,34 and, ChemBERTa Embeddings, generated using the pre-trained ChemBERTa-77M-MTR model.35The choice of kernel function is fundamental to the GP's ability to model correlations between data points based on their similarity. We explore five distinct covariance kernel functions viz., Tanimoto, linear, Radial Basis Function (RBF), Rational Quadratic (RQ), and Matérn (ν = 1.5). Please refer to the SI for further details. For all kernels that include hyperparameters (i.e., linear, RBF, RQ, and Matérn), these parameters (e.g., lengthscale ℓ, shape parameter α, outputscale s, and noise variance σ2n) were optimized by maximizing the marginal log-likelihood during model training.36,37 For further information please refer to the SI.
2.6 Model explainability with SHAP
We incorporated SHapley Additive exPlanations (SHAP)29,38 to quantify the contribution of individual molecular features to GP model predictions across active learning cycles. For a molecule x, the prediction f(x) is decomposed into a baseline ϕ0 and additive contributions from M features,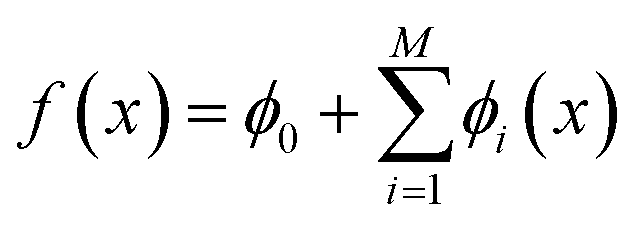 | (4) |
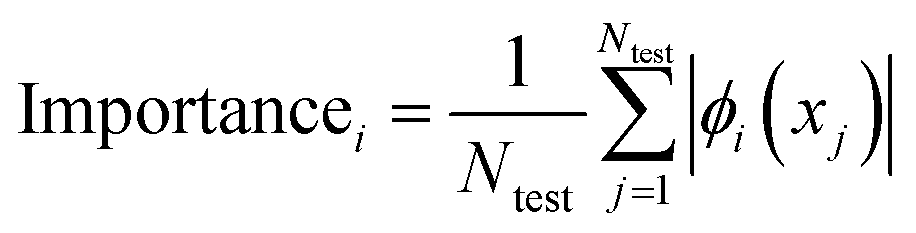 | (5) |
For each AL cycle, SHAP values were evaluated on 100 test molecules randomly sampled from the unqueried pool, using a background of 50 randomly sampled compounds from the training set to initialise the shap.KernelExplainer. The top ten features ranked based on the mean absolute SHAP value were retained for detailed analysis. The stability and robustness of these feature attributions were validated through quantitative analysis across different acquisition protocols.
For models trained on ECFP fingerprints, selected features were mapped back to molecular fragments using RDKit. To address the ambiguity of mapping ECFP bits (due to bit collisions or multiple environments), we implemented an affinity-prioritized algorithm. Atom environments corresponding to top-ranked fingerprint bits were first identified in all molecules containing the bit. These molecules were then sorted by descending affinity. The environment from the highest-affinity compound was extracted using Chem.FindAtomEnvironmentOfRadiusN, canonicalised to a SMILES string, and used as the representative fragment. These fragments were then ranked by a combined score of frequency and SHAP magnitude. This procedure ensures that the identified chemical substructures are those most strongly associated with the high-potency predictive signal and allows for a mechanistic interpretation of how AL reshapes the model's representation of structure–activity relationships.
2.7 Experimental setup and evaluation
In order to evaluate the AL setup, we follow the fixed cost approach by Gorantla et al.15 in acquiring a total of 360 compounds for each individual experiment. Each experiment starts with 60 randomly selected compounds, followed by 10 cycles of selecting 30 new compounds per cycle, using different exploration/exploitation strategies.The cycle is then repeated for each experiment, and parameter combinations undergo repeated cycles. Suitable steps for updating and acquisition are undertaken to allow for unbiased comparison across datasets.
In this work, a single “experiment” refers to one complete, 10-cycle active learning simulation for a specific combination of datasets, molecular representations, kernels, acquisition protocols, and random seeds.
For each dataset–representation–kernel combination, all seven acquisition strategies were evaluated, resulting in a total of 4 × 3 × 5 × 7 = 420 distinct experiments. The vast scope of the experiments poses a challenge to visualise and evaluate these results.
The entire computational study, including the training of all GP models, required approximately 4 hours of wall-clock time on a single NVIDIA RTX 4090 GPU. This demonstrates the practical feasibility of applying our comprehensive benchmarking framework.
The recall of top compounds (Rk) metric quantifies the fraction of truly high-affinity compounds (top k%) that are successfully identified by the active learning process, relative to the total number of such compounds present in the entire dataset. It is calculated using the following eqn (6),
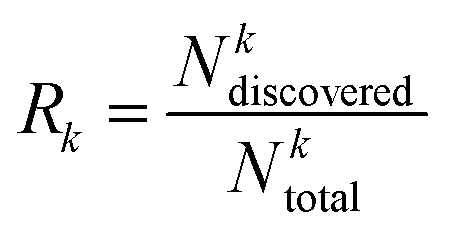 | (6) |
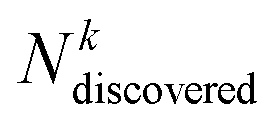 is the number of compounds found in the acquired set that belong to the top k% class, and
is the number of compounds found in the acquired set that belong to the top k% class, and  is the total number of compounds that actually belong in the top k% most active ones based on the observed activity in the entire dataset. Recall was computed for the top 2% (R2) and 5% (R5) of compounds.
is the total number of compounds that actually belong in the top k% most active ones based on the observed activity in the entire dataset. Recall was computed for the top 2% (R2) and 5% (R5) of compounds.
To provide a more comprehensive and robust assessment of early enrichment performance, we also report two additional standard metrics. The Enrichment Factor (EFk) measures how many times more frequently active compounds are found within the top k% of a ranked list compared to a random selection. It is defined as give in eqn (7):
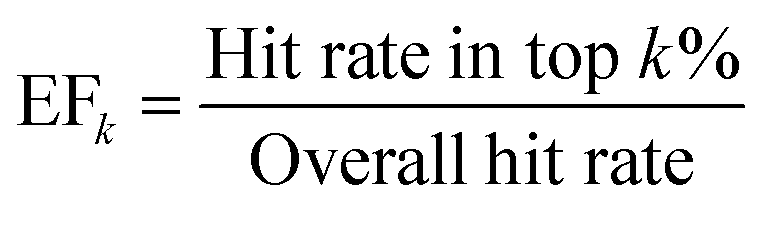 | (7) |
An EFk of 1.0 corresponds to random performance. In this study, we report the EF at 1%, 2%, and 5%.
To mitigate the sensitivity to a fixed cutoff k, we also report the Boltzmann-Enhanced Discrimination of ROC (BEDROC) score.39 BEDROC is a metric that preferentially rewards the identification of active compounds at the top of a ranked list without requiring an arbitrary cutoff. It applies an exponential weight to each compound based on its rank, such that hits at the beginning of the list contribute much more to the final score than those found later. Following common practice for virtual screening, we use an α parameter of 20.0, which heavily focuses the evaluation on the top portion of the ranked list. The score ranges from 0 (no enrichment over random) to 1 (perfect ranking).
2.8 Data for the study
The active learning framework has been evaluated using four diverse protein target datasets viz., TYK2 (Tyrosine Kinase 2),9 USP7 (Ubiquitin Specific Peptidase 7),40 D2R (Dopamine D2 Receptor),41 and MPRO (SARS-CoV-2 Main Protease).26 It is important to note that while Thompson et al.9 describes TYK2 as a congeneric series derived from a single synthetic scaffold, our analysis using RDKit's Murcko decomposition identified 104 distinct Murcko scaffolds, reflecting minor structural variations within the series. In contrast, USP7, D2R, and MPRO demonstrate substantially higher diversity (N/M ≈ 0.41 − 0.45), reflecting more structurally varied compound collections. Details of datasets are provided in the SI. Table 2 summarises some relevant details across the four datasets used.| Property | TYK2 | USP7 | D2R | MPRO |
|---|---|---|---|---|
| Binding measure | pKi | pIC50 | pKi | pIC50 |
| Ligands (total) | 9997 | 1799 | 2502 | 2062 |
| Scaffolds (unique) | 104 | 770 | 1034 | 934 |
| Std dev (p-value) | 1.36 | 1.31 | 1.44 | 0.91 |
| N/M ratio | 0.0104 | 0.428 | 0.413 | 0.452 |
We note that the datasets employ different affinity measures (pKi for TYK2 and D2R; pIC50 for USP7 and MPRO), as shown in Table 2. As these units are derived from different assay types and are not directly comparable, our study does not make direct, quantitative comparisons of the absolute affinity values across targets. Instead, our primary performance metric, recall of top compounds (Rk), is based on a relative, percentile-based threshold. For each dataset, the “top k%” active compounds are determined by internally ranking the molecules based on their specific affinity measure. This approach allows for a valid comparison of the enrichment efficiency of the AL strategies across the different chemical landscapes, without relying on a comparison of the raw activity scales.
3 Results and discussion
While the conceptual idea of an active learning cycle is quite straightforward, the myriad of choices that one can make around surrogate models, acquisition functions, kernel choices, and molecular representations poses a challenge. Finding an optimal combination of choices may not be practical and evaluating the increasingly large number of combinations is difficult to assess and visualise. Lastly, active learning cycles are often black box systems allowing for little explainability of what the models are learning. Combining active learning with a SHapley Additive exPlanations (SHAP)29,38 analysis can provide some indications of model learning. With our results, we highlight that optimal AL strategies are highly context-dependent, underscoring the critical influence of inherent dataset characteristics and the complex interactions among methodological choices. In the following section, we present a comprehensive study of dataset characteristics followed by an analysis of how different methodological choices influence active learning performance. Furthermore, we explore how a versatile web-based tool aids in understanding complex results. Lastly, we use SHAP to understand if the AL cycles pick up patterns that lead to explainable properties that could be harnessed by medicinal chemists in designing effective AL strategies.3.1 Chemical landscape sets difficulty – scaffold diversity patterns anticipate AL headroom
We evaluated four therapeutically relevant targets with distinct chemistry—TYK2, USP7, D2R, and MPRO—to probe how dataset composition shapes active learning (AL) outcomes.Scaffold diversity, as determined by the ratio of unique scaffolds to total molecules (N/M) is the main differentiator between the datasets. TYK2 exhibits exceptionally low diversity (N/M ≈ 0.01), indicating a highly constrained chemical space dominated by few structural motifs. On the other hand, USP7, D2R, and MPRO exhibit significantly greater diversity (N/M ≈ 0.41 − 0.45), which is indicative of more structurally diverse compound collections.
Scaffold diversity directly impacts molecular similarity patterns within each dataset as evident in Fig. 2. For instance, TYK2's constrained chemical space is particularly evident with ECFP fingerprints, which show highly skewed similarity distributions with the majority of compound pairs exhibiting low Tanimoto similarities as evident from Fig. 2A. ChemBERTa embeddings and MACCS, on the other hand, display broader distributions centered at higher similarity values as evident from Fig. 2B and C demonstrating how different representations highlight structural homogeneity differently. In contrast, USP7, D2R, and MPRO show wider and more diverse internal similarity distributions across all three molecular representations—ECFP, MACCS, and ChemBERTa (Fig. 2A–C). ECFP fingerprints produce sharp peaks at low similarity values, whereas MACCS keys and ChemBERTa embeddings give more spread-out distributions because they capture the molecular structure in different ways.
 | ||
| Fig. 2 Molecular similarity distributions across datasets and representations. Kernel Density Estimate (KDE) plots illustrate the distribution of pairwise Tanimoto similarity scores for compounds within the TYK2, D2R, MPRO and USP7 datasets, as perceived by different molecular representations. For each dataset, the similarity profiles generated by ECFP4, MACCS, and ChemBERTa are compared. The ECFP fingerprints consistently show distributions which are heavily skewed towards low similarity across all datasets, particularly for TYK2, D2R, and MPRO. In contrast, both MACCS keys and ChemBERTa embeddings provide broader similarity distributions, often centered at higher values, indicating their capacity to capture more diverse structural relationships than ECFP. (A–D) Demonstrates kernel density estimate with respect to similarity scores across TYK2, USP7, D2R and MPRO targets respectively. | ||
Dataset diversity patterns have direct implications for active learning performance. While the more expansive chemical landscape of USP7, D2R, and MPRO offers more chance of strategic compound selection, constrained chemical space like TYK2 restricts the opportunity for diversified exploration. Further dataset diagnostics are provided in the SI.
3.2 AL analysis platform
With a 420 experiment run, the complexity of our results, which include four datasets, three molecular representations, five kernels, and seven protocols, necessitates a new way of presenting the results beyond graphs. To address this and promote transparency and reproducibility, we have developed the active learning analysis platform, an interactive web tool which is freely accessible. As shown in Fig. 1, this platform offers access to all the comprehensive experiments and analysis we did on each target dataset, enabling researchers to explore our findings interactively.3.3 Dataset characteristics drive performance variation
The chemical space properties have a significant effect on active learning performance. Recall of top compounds Rk values ranging from 0.5052 for the constrained TYK2 dataset to 0.9942 for the more diverse MPRO dataset indicate that performance varies significantly across datasets as shown in Fig. 4. Visual summary of the distribution of all experimental outcomes with every dataset is available at https://shapanalysis.streamlit.app/.Statistical analysis demonstrates that the intrinsic properties of the target dataset are the most dominant factor in determining achievable performance. To quantify the relative contributions of our methodological choices, we conducted a four-factor ANOVA (Type II Sums of Squares) on the final recall (Rk) values from all non-random protocols. The full model explained a substantial proportion of the variance in performance (R2 = 0.84; Adjusted R2 = 0.82).
To properly assess effect sizes, we computed omega-squared (ω2), an unbiased estimator of the population effect size, along with 95% bootstrap confidence intervals (1,000 iterations). Dataset identity exhibited the largest effect (ω2 = 0.31, 95% CI [0.28, 0.35]; F(3, 994) = 640.37, p < 0.001), confirming that the chemical landscape sets fundamental performance constraints. Notably, the interaction between datasets and kernel interaction showed a similarly large effect (ω2 = 0.31; F(12, 994) = 160.22, p < 0.001), demonstrating that kernel effectiveness is highly context-dependent.
Other factors made smaller but significant contributions: kernel choice (ω2 = 0.09, 95% CI [0.07, 0.11]; F(4, 994) = 135.27, p < 0.001), molecular representation (ω2 = 0.03, 95% CI [0.02, 0.04]; F(2, 994) = 91.47, p < 0.001), and the kernel × fingerprint interaction (ω2 = 0.04; F(8, 994) = 29.08, p < 0.001). The acquisition protocol, while statistically significant (F(5, 994) = 12.44, p < 0.001), had the smallest main effect (ω2 = 0.01, 95% CI [0.004, 0.019]), suggesting that its role is to modulate outcomes within the constraints imposed by the dataset and model architecture. This statistical evidence reinforces that optimal active learning strategies are highly context-dependent, with dataset characteristics and their interactions with methodological choices playing the dominant role.
The Post-hoc Tukey HSD analysis showed that all UCB-based protocols performed significantly better than random selection in terms of mean recall of top compounds Rk with all adjusted p-values less than 0.05, indicating strong statistical significance. However, there is no significant difference between the UCB protocols themselves, as all adjusted p-values were greater than 0.05. The practical impact of these improvements is measured using Cohen's d effect sizes, which were larger, ranging from 0.934 ucb-balanced vs. random to 1.308 ucb-explore-heavy vs. random, revealing that UCB strategies had a strong advantage over random selection.
No one set of Kernel function, acquisition technique or molecular representation worked optimally in every circumstance. The best configuration for each dataset highlights the range of possible Rk values from 0.5052 for TYK2 to 0.9942 for MPRO, indicating that different datasets require different optimal setups as shown in Fig. 3.
 | ||
| Fig. 3 Overall active learning performance across datasets. This composite figure summarizes key active learning performance metrics for each dataset, aggregating results across all kernel, molecular representation, and acquisition strategy combinations. (A) Performance distribution across datasets: violin plots illustrating the distribution of final 2% recall of top compounds (Rk) values for each dataset (TYK2,USP7 and MPRO,D2R). The horizontal lines within each violin indicate the mean (µ) and median (red) Rk values, while the shape reflects the density of results. (B) Learning curves by dataset: average 2% recall of top compounds (Rk) over the 10 active learning cycles, demonstrating performance evolution for each dataset. All plots aggregate data across all method combinations and replicates unless otherwise specified. | ||
3.4 Impact of molecular representation and kernel functions
Performance is significantly impacted by the kernel function and selected molecular representation. Our findings demonstrate no universally optimal combination, consistent with significant partial values for Dataset:Kernel interaction (65.92%) and Kernel:Fingerprint interaction (18.97%) in the ANOVA analysis.
values for Dataset:Kernel interaction (65.92%) and Kernel:Fingerprint interaction (18.97%) in the ANOVA analysis.
ChemBERTa embeddings exhibited a high-variance performance profile characterized by exceptional peaks and notable failures. When optimally paired with non-linear kernels i.e. Matérn and RBF on USP7 and MPRO, ChemBERTa achieved the highest individual Rk of 0.99 on MPRO. This representation proved susceptible to significant performance loss under suboptimal conditions. On challenging datasets viz. D2R and TYK2, identical kernel combinations yielded dramatically lower mean Rk values, with some as low as 0.02 ± 0.01 and a mean BEDROC of 0.003 ± 0.01 for the Matérn kernel on TYK2, highlighting ChemBERTa's context-dependency and unpredictable efficacy.
MACCS fingerprints demonstrated the most consistent performance profile despite achieving the lowest overall mean Rk of 0.27 ± 0.18. This representation exhibited remarkably stable performance across different datasets, with substantially lower inter-dataset variance compared to ECFP or ChemBERTa. Even while MACCS rarely reached peak performance, its consistency makes it a reliable baseline when predictable results are prioritized over maximum performance. Notably, MACCS achieved competitive performance on D2R with Rk = 0.61 when paired with the Tanimoto kernel, demonstrating its potential for specific dataset-kernel synergies.
The linear and Tanimoto kernels delivered consistent, moderate performance across all tested conditions. Linear kernel achieved a mean Rk of 0.35 ± 0.14 on D2R and 0.29 ± 0.13 on TYK2, and a mean enrichment factor at 2% (EF2) of 17.1 ± 8.2. This EF2 value, indicating that the top 2% of compounds were identified at over 17 times the rate of random selection, stands in stark contrast to the near-random performance of the non-linear kernels on the same dataset (EF2 ≈ 1.1), while the Tanimoto kernel yielded 0.30 ± 0.12 and 0.26 ± 0.12 on the same datasets, respectively. These kernels maintained stable performance regardless of dataset difficulty or molecular representation. The Rational Quadratic (RQ) kernel consistently underperformed across all conditions, achieving a Rk as low as 0.12 ± 0.07, and EF2 of only 7.6 ± 4.0, on TYK2 and reaching only 0.26 ± 0.13 on MPRO. This demonstrates a trade-off wherein the non-linear kernels can offer high rewards but with high variability, while linear kernels offer reliable, moderate performance suitable for risk-averse applications as evident in Fig. 4.
 | ||
| Fig. 4 Mean 2% recall of top compounds (Rk) across protocols, kernels, and representations. A heatmap illustrating the average final Rk for each combination of active learning protocol (rows), Gaussian process kernel (main columns), and molecular representation (sub-columns) at cycle 10. Each cell represents the mean Rk across 3 replicate runs. The colour scale indicates performance, from low (dark purple/blue) to high (yellow). (A) TYK2 dataset: performance landscape for the challenging TYK2 dataset; highlights the relatively lower overall Rk and the best-performing combinations. (B) USP7 dataset: performance landscape for the receptive USP7 dataset; illustrates the generally higher Rk values and identifies highly effective combinations. | ||
Exploit-heavy strategies such as UCB-exploit-heavy, often designed for rapid prioritization, demonstrated effectiveness on USP7 and MPRO datasets, leading to rapid initial gains. Temporal SHAP analysis, which demonstrated top features for USP7 exploit-heavy strategies consistently peaking early in Cycles 2 or 3, indicates rapid initial SAR identification. In contrast, exploit-heavy strategies exhibited a noticeable ‘late spike’ in feature importance on datasets such as TYK2, suggesting that important SAR features are not immediately apparent, but are rather revealed after focused, persistent sampling in specific, high-reward regions of the chemical space. This ‘late spike’ reflects the model's attempt to progressively prioritize subtle features within a highly constrained or challenging SAR landscape as shown in Fig. 5.
 | ||
| Fig. 5 SHAP analysis reveals dynamic feature importance in TYK2 active learning. (A) Mean SHAP importance evolution for top-ranking ECFP features (699, 491, and 561) across active learning cycles using the UCB-exploit-heavy protocol. Feature importance shifts from exploration (cycles 1–3) to exploitation phases (cycles 4–10). (B) Binding affinity distributions for compounds containing key features. Dashed lines show mean pKi values: feature 699 (7.75 ± 1.35), feature 491 (7.76 ± 1.44), and feature 561 (7.77 ± 1.34), confirming association with high-affinity binders. (C) Representative TYK2 inhibitors with SHAP-identified substructure highlighted regions. Molecular structures demonstrate concrete chemical patterns underlying abstract feature importance scores. (D) Protein–ligand binding showing interaction modes for selected compounds in the TYK2 active site, with key residues Val100 and Asp107 labeled. | ||
On the other hand, explore-heavy strategies such as UCB-explore-heavy typical showed slower initial progress but could achieve higher long-term Rk on complex datasets like D2R, showing more consistent improvement patterns. This reflects a broader sampling approach and a more distributed learning of features across the chemical space, as evident by less pronounced temporal shifts in SHAP feature importance. This approach is advantageous where targets have more diffused SAR or where novel active regions need to be discovered beyond narrow, pre-defined areas. Balanced and adaptive protocols (e.g.,UCB-balanced and UCB-gradual) frequently achieved competitive performance and demonstrated robustness across varied complexities, providing reliable options when optimal configurations are not immediately apparent.
The importance of protocol choice varied significantly depending on the dataset selected. High-performing combinations such as Matérn + ChemBERTa achieved high Rk across most protocols with rapid convergence on datasets such as MPRO and USP7. On the other hand, protocol selection was more crucial for difficult datasets such as TYK2 and D2R which had significant Rk variation and demonstrated slow improvement beyond 300 compounds. This emphasizes how AL strategy effectiveness is highly dependent on dataset characteristics and the chosen kernel–fingerprint combinations, influencing the initial trajectory and overall performance outcome.
3.5 Mechanistic insights from feature importance analysis
To obtain deeper mechanistic insights into how Gaussian process models predict compound activity and how active learning influences the understanding of the SAR we perform explainability studies. The application of SHAP analysis on ECFP fingerprint models is a well-established method38 for understanding explainability. This analysis, focusing on TYK2 and USP7 targets uses exploit-heavy and explore-heavy AL protocols, to uncover distinct aspects of the model's learning and the underlying chemical determinants of activity.SHAP analysis consistently identified specific, chemically interpretable molecular fragments that were highly predictive of binding affinity, validating the model's ability to learn genuine SARs.42,43 Importantly, compounds containing these top-ranked features consistently exhibited high binding affinities (Fig. 5).
Our analysis demonstrates that the model learns stable and genuine SAR drivers. For the USP7 target, the set of the top 5, most important features was identical between the ucb-exploit-heavy and ucb-explore-heavy protocols, yielding a Jaccard index of 1.00. This perfect stability indicates that the model rapidly and consistently identified the core SAR. For the more challenging, low-diversity TYK2 dataset, the analysis still showed good stability with a Jaccard index of 0.43. While different protocols explored different nuances of the constrained chemical space, a core set of features (e.g., bits corresponding to cF and cNc fragments) were consistently ranked as the most important. This provides strong evidence that our model is learning genuine SARs rather than stochastic noise.
For USP7, prominent features were consistently associated with carbonyl groups such as Feature ID 2362, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and nitrogen-rich heterocycles such as Feature ID 3500, cnc for both protocols. These features with mean affinity for USP7 9.33–9.66 pIC50 are chemically relevant for deubiquitinase active sites, often involved in hydrogen bonding and electrostatic interactions.45,46 The identification of a complex fragment ID 875, i.e., nc1cncn(CC2(O)CCNCC2)c1=O suggests the model's capability to prioritize intricate patterns. USP7's top fragments were 100% aromatic, 24% nitrogen-containing, and 0% halogen-containing, aligning with DUB modulator characteristics.
O and nitrogen-rich heterocycles such as Feature ID 3500, cnc for both protocols. These features with mean affinity for USP7 9.33–9.66 pIC50 are chemically relevant for deubiquitinase active sites, often involved in hydrogen bonding and electrostatic interactions.45,46 The identification of a complex fragment ID 875, i.e., nc1cncn(CC2(O)CCNCC2)c1=O suggests the model's capability to prioritize intricate patterns. USP7's top fragments were 100% aromatic, 24% nitrogen-containing, and 0% halogen-containing, aligning with DUB modulator characteristics.
O) maintained high ranks and consistent affinities across protocols. This robustness suggests that the identification of core binding motifs is stable, even if the sampling strategy influences the diversity of compounds explored around them.44 This consistency provides further confidence in the model's generalizability and its robust mechanistic understanding of binding, even when the underlying sampling strategies might aim for different balances of exploration and exploitation within the chemical space.
4 Conclusion and outlook
In this work, we evaluated active learning (AL) strategies for ligand binding affinity prediction, investigating the interplay between molecular representations, kernel functions, and acquisition protocols across various chemical datasets. Our main conclusion is that AL's effectiveness varies significantly based on the dataset's chemical properties. Statistical analysis demonstrated that the dataset, and its interaction with techniques like kernel functions, is the primary factor influencing performance, establishing the limits for AL success.Our analysis revealed important trade-offs between different methodological choices. We discovered that simpler, explicit representations like ECFP fingerprints, paired with robust linear kernels, offer consistent and reliable performance across a wide range of dataset complexities. On the other hand, advance, pre-trained embeddings like ChemBERTa, when combined with flexible non-linear kernels such as Matérna and RBF, can achieve state-of-the-art peak performance; however, they are prone to catastrophic failures on difficult or mismatched chemical landscapes. Similar to this it was demonstrated the AL protocol selection is context-dependent. Exploit-heavy methods are better suited for rapid lead optimization within well-defined SARs, whereas explore-heavy strategies are beneficial for novel chemotype discovery in more diverse chemical spaces. Mechanistic insights from our SHAP analysis offer a framework for understanding why these choices matter, linking them to the model's dynamic learning of SARs throughout the AL cycles.
According to these results, there is no “one-size-fits-all” AL strategy that works in all circumstances. We proposed a context-aware framework for AL in drug discovery demonstrating promising results in terms of ease of their analysis. Practitioners should first analyze their dataset's chemical space, i.e., scaffold diversity and similarity to set reasonable expectations and select AL components accordingly. Challenging or unknown spaces may benefit from stable combinations such as ECFP with a linear kernel, while well-behaved SARs might justify using risky, high-reward methods like ChemBERTa with non-linear kernels.
While this study provides a robust framework, it has limitations, including its retrospective nature and the focus of SHAP analysis on ECFP models. Future work can focus on the prospective validation of these findings in real-world drug discovery campaigns. The most promising future direction, however, lies in the development of adaptive active learning frameworks. These systems could learn the characteristics of the chemical space in real-time and automatically select or adjust the molecular representation, kernel, and acquisition strategy during the campaign, moving beyond the static protocol choices. We can fully utilize active learning to speed up the development of novel medicine by balancing the performance improvement in ligand binding affinity prediction with explainability built in the model from the start. Further improvements could also be achieved by exploring more advanced surrogate models, such as warped Gaussian processes, which could allow the model to explicitly learn the non-Gaussian distribution of affinity data.
Conflicts of interest
There are no conflicts to declare.Data availability
All data sets curated for this study and results are publicly available on Zenodo here – https://doi.org/10.5281/zenodo.17935028. The code is available on Github here – https://github.com/meyresearch/explainable_AL.Supplementary information (SI) is available. See DOI: https://doi.org/10.1039/d5dd00436e.
Acknowledgements
Satya Pratik Srivastava was supported by the Department of Computer Science and Engineering, Shiv Nadar University, Delhi-NCR. Rohan Gorantla was funded by the United Kingdom Research and Innovation (grant EP/S02431X/1), UKRI Centre for Doctoral Training in Biomedical AI at the University of Edinburgh, School of Informatics, and Exscientia Plc, Oxford.Notes and references
- J. A. Weller and R. Rohs, J. Chem. Inf. Model., 2024, 64, 6450–6463 CrossRef CAS PubMed.
- Y. Khalak, G. Tresadern, D. F. Hahn, B. L. d. Groot and V. Gapsys, J. Chem. Theory Comput., 2022, 18, 6259–6270 CrossRef CAS PubMed.
- Y. Zhu, S. Alqahtani and X. Hu, Molecules, 2021, 26, 1776 CrossRef CAS PubMed.
- J. L. Medina-Franco, Front. Drug Discovery, 2021, 1, 728551 CrossRef.
- M. R. Shirts and V. S. Pande, Annu. Rev. Phys. Chem., 2007, 58, 219–246 Search PubMed.
- A. S. J. S. Mey, B. K. Allen, H. E. B. Macdonald, J. D. Chodera, D. F. Hahn, M. Kuhn, J. Michel, D. L. Mobley, L. N. Naden, S. Prasad, A. Rizzi, J. Scheen, M. R. Shirts, G. Tresadern and H. Xu, Living J. Mol. Sci., 2020, 2, 18378 Search PubMed.
- R. Gorantla, A. P. Gema, I. X. Yang, Á. Serrano-Morrás, B. Suutari, J. J. Jiménez and A. S. J. S. Mey, J. Chem. Inf. and Model., 2025, 65 Search PubMed.
- F. Stanzione, I. Giangreco and J. C. Cole, Prog. Med. Chem., 2021, 60, 273–343 Search PubMed.
- J. Thompson, W. P. Walters, J. A. Feng, N. A. Pabon, H. Xu, B. B. Goldman, D. Moustakas, M. Schmidt and F. York, Artif. Intell. Life Sci., 2022, 2, 100050 Search PubMed.
- K. Konze, P. H. Bos, M. K. Dahlgren, K. Leswing, I. T. Brohman, A. Bortolato, B. Robbason, R. Abel and S. Bhat, J. Chem. Inf. Model., 2019, 59, 3782–3793 CrossRef CAS PubMed.
- P. Ghanakota, P. H. Bos, K. D. Konze, J. Staker, G. Marques, K. Marshall, K. Leswing, R. Abel and S. Bhat, J. Chem. Inf. Model., 2020, 60, 4311–4325 CrossRef CAS PubMed.
- D. Boldini, L. Friedrich, D. Kuhn and S. A. Sieber, ACS Cent. Sci., 2024, 10, 823–832 CAS.
- W. Meschendoerfer, C. Gassner, F. Lipsmeier, J. T. Regula and J. Moelleken, J. Pharm. Biomed. Anal., 2017, 132, 141–147 CrossRef CAS PubMed.
- L. Tan, S. Hirte, V. Palmacci, C. Stork and J. Kirchmair, Nat. Rev. Chem., 2024, 8, 319–339 CrossRef CAS PubMed.
- R. Gorantla, A. Kubincova, B. Suutari, B. P. Cossins and A. S. Mey, J. Chem. Inf. Model., 2024, 65(22), 12279–12291 CrossRef PubMed.
- J. Yu, X. Li and M. Zheng, Artif. Intell. Life Sci., 2021, 1, 100023 CAS.
- D. E. Graff, E. I. Shakhnovich and C. W. Coley, Drug Discov. Today, 2015, 20, 458–465 CrossRef PubMed.
- A. T. Müller, M. Hierl, D. Heer, P. Westwood, P. Hartz, B. Wörsdörfer, C. Kramer, W. Haap, D. Roth and M. Reutlinger, J. Med. Chem., 2025, 68, 14806–14817 CrossRef PubMed.
- D. Reker, Drug Discovery Today: Technol., 2019, 32–33, 73–79 CrossRef PubMed.
- A. Krause and J. Hübotter, Probabilistic Artificial Intelligence, arXiv, 2025, preprint, arXiv:2502.05244, DOI:10.48550/arXiv.2502.05244.
- D. van Tilborg and F. Grisoni, Nat. Comput. Sci., 2024, 4, 786–796 CrossRef CAS PubMed.
- M. Gallegos, V. Vassilev-Galindo, I. Poltavsky, Á. Martín Pendás and A. Tkatchenko, Nat. Commun., 2024, 15, 4345 Search PubMed.
- R. K. Singh, R. Pandey and R. N. Babu, Neural Comput. Appl., 2021, 33, 8871–8892 CrossRef PubMed.
- R. K. Singh, R. Gorantla, S. G. R. Allada and P. Narra, PLoS One, 2022, 17, e0276836 CrossRef CAS PubMed.
- G. J. Correy, M. M. Rachman, T. Togo, S. Gahbauer, Y. U. Doruk, M. G. V. Stevens, P. Jaishankar, B. Kelley, B. Goldman, M. Schmidt, T. Kramer, D. S. Radchenko, Y. S. Moroz, A. Ashworth, P. Riley, B. K. Shoichet, A. R. Renslo, W. P. Walters and J. S. Fraser, Sci. Adv., 2025, 11, eadi5196 CrossRef PubMed.
- H. Achdout, BioRxiv, 2020, preprint, DOI:10.1101/2020.10.29.339317.
- F. Gusev, E. Gutkin, M. G. Kurnikova and O. Isayev, J. Chem. Info. and Model., 2023, 63, 583–594 CrossRef CAS PubMed.
- M. Ahmadi, M. Vogt, P. Iyer, J. Bajorath and H. Fröhlich, J. Chem. Inf. Model., 2013, 53, 553–559 CrossRef CAS PubMed.
- L. S. Shapley, Contributions to the Theory of Games, Princeton University Press, 1953, 2, pp. 307–317 Search PubMed.
- R. Garnett, Bayesian Optimization, Cambridge University Press, 2023 Search PubMed.
- C. E. Rasmussen and C. K. I. Williams, Gaussian Processes for Machine Learning, MIT Press, 2006 Search PubMed.
- A. Slivkins, Found. Trends Mach. Learn., 2019, 12, 1–286 CrossRef.
- D. Rogers and M. Hahn, J. Chem. Inf. Model., 2010, 50, 742–754 CrossRef CAS PubMed.
- J. L. Durant, B. A. Leland, D. R. Henry and J. G. Nourse, J. Chem. Inf. Comput. Sci., 2002, 42, 1273–1280 Search PubMed.
- W. Ahmad, E. Simon, S. Chithrananda, G. Grand and B. Ramsundar, Chemberta-2: Towards chemical foundation models, arXiv, 2022, preprint, arXiv:2209.01712, DOI:10.48550/arXiv.2209.01712..
- D. J. C. MacKay, in Bayesian Interpolation, ed. C. R. Smith, G. J. Erickson and P. O. Neudorfer, Springer Netherlands, Dordrecht, 1992, pp. 39–66 Search PubMed.
- S. Sundararajan and S. S. Keerthi, Neural Comput., 2001, 13, 1103–1118 CrossRef CAS PubMed.
- S. M. Lundberg and S.-I. Lee, Proceedings of the 31st International Conference on Neural Information Processing Systems, Red Hook, NY, USA, 2017, pp. 4768–4777 Search PubMed.
- J.-F. Truchon and C. I. Bayly, J. Chem. Inf. and Model., 2007, 47, 488–508 CrossRef CAS PubMed.
- W.-F. Shen, H.-w. Tang, J.-b. Li, X. Li and S. Chen, J. Cheminform., 2023, 15, 1–16 Search PubMed.
- D. Mendez, A. Gaulton, A. P. Bento, J. Chambers, M. De Veij, E. Félix, M. Magariños, J. Mosquera, P. Mutowo, M. Nowotka, M. Gordillo-Marañón, F. Hunter, L. Junco, G. Mugumbate, M. Rodriguez-Lopez, F. Atkinson, N. Bosc, C. Radoux, A. Segura-Cabrera, A. Hersey and A. Leach, Nucleic Acids Res., 2018, 47, D930–D940 CrossRef PubMed.
- R. Rodríguez-Pérez and J. Bajorath, J. Med. Chem., 2020, 63, 8761–8777 CrossRef PubMed.
- R. Rodríguez-Pérez and J. Bajorath, J. Comput. Aided Mol. Des., 2020, 34, 1013–1026 CrossRef PubMed.
- G. Li, J. Li, Y. Tian, Y. Zhao, X. Pang and A. Yan, Mol. Diversity, 2023, 28(4), 2429–2447 CrossRef PubMed.
- C. Kennedy, K. McPhie and K. Rittinger, Front. Mol. Biosci., 2022, 9, 1019636, DOI:10.3389/fmolb.2022.1019636.
- N. J. Schauer, R. S. Magin, X. Liu, L. M. Doherty and S. J. Buhrlage, J. Med. Chem., 2020, 63, 2731–2750 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2026 |