
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAdvancing metal organic framework and covalent organic framework design via the digital-intelligent paradigm
Bing
Ma
,
Na
Qin
,
Qianqian
Yan
,
Wei
Zhou
 *,
Sheng
Zhang
,
Xiao
Wang
*,
Lipiao
Bao
and
Xing
Lu
*
*,
Sheng
Zhang
,
Xiao
Wang
*,
Lipiao
Bao
and
Xing
Lu
*
School of Chemistry and Chemical Engineering, Hainan University, 58 Renmin Avenue, Haikou, 570228, P. R. China. E-mail: weichou2023@hainanu.edu.cn; xiaowang2025@hainanu.edu.cn; lux@hainanu.edu.cn
First published on 18th November 2025
Abstract
Porous framework materials—including metal–organic frameworks (MOFs) and covalent organic frameworks (COFs)—have attracted widespread attention due to their high surface areas, tunable pore structures, and diverse functionalities, enabling promising applications in gas separation, catalysis, and energy storage. However, the vast chemical configuration space and the complexity of multi-parameter synthesis conditions pose significant challenges to the rational design and controlled synthesis of materials with targeted properties. In recent years, artificial intelligence (AI), particularly machine learning (ML) and deep learning (DL), in combination with multiscale molecular simulation methods such as density functional theory (DFT), grand canonical Monte Carlo (GCMC), and molecular dynamics (MD), has emerged as a powerful tool for accelerating the screening and optimization of framework materials. This review systematically summarizes AI-assisted strategies for framework material design, focusing on data-driven prediction of synthetic routes, optimization of reaction conditions, and inverse design targeting specific functionalities. We evaluate key AI models, including interpretable tree-based algorithms and neural networks capable of modeling complex structure–property relationships, and highlight their integration with atomistic simulations to enhance predictive accuracy. Furthermore, the synergy between AI and automated experimental platforms is advancing the development of high-throughput experimentation and self-optimizing workflows, often referred to as self-driving laboratories. Several case studies illustrate the effectiveness of AI methods in identifying high-performance framework materials and achieving morphology control, particularly when leveraging the integration of experimental and simulation data. The review also discusses key challenges in AI-assisted materials design, including inconsistent data quality, limited model interpretability, and the gap between prediction and practical synthesis. Looking ahead, the continued expansion of materials databases, advances in AI algorithms, and deeper integration of domain knowledge are expected to play an increasingly vital role in framework material development, driving a paradigm shift in materials research from empirical trial-and-error to more efficient, predictive, and intelligent design.
 Bing Ma | Bing Ma received her master's degree from Hainan University, where she focused on computer networks and machine learning. In 2023, she began pursuing her PhD at Hainan University, engaging in research on the synthesis of covalent organic framework (COF) materials. Her current research primarily focuses on integrating large language models with machine learning to predict COF synthesis processes and to evaluate the adsorption and classification performance of COF materials. |
 Na Qin | Na Qin received her master's degree from Yili Normal University, where she focused on natural language processing (NLP) and electrocatalytic hydrogen evolution. In 2025, she began her PhD studies at Hainan University. She is currently dedicated to integrating her expertise in artificial intelligence with experimental materials science, employing digital-intelligent approaches to guide the design, performance prediction, and synthesis of hydrogen-bonded organic frameworks (HOFs) for CO2 capture and conversion, with the aim of developing efficient photocatalytic CO2 reduction systems. |
 Wei Zhou | Wei Zhou received his PhD in Materials Physics and Chemistry from the Institute of Frontier and Interdisciplinary Science, Shandong University, in 2023. He is currently an Associate Professor at the School of Chemistry and Chemical Engineering, Hainan University. His research focuses on the digital-intelligent design of porous framework materials and their applications in heterogeneous catalysis. |
 Sheng Zhang | Sheng Zhang received his PhD from Northwest University, where his research focused on low-dimensional magnetic coordination polymers and energetic coordination polymers. In 2021, he was appointed as an Associate Professor at Hainan University. His research group currently focuses on the design and synthesis of single-molecule magnets (SMMs) and metal–organic frameworks (MOFs). |
 Xiao Wang | Xiao Wang earned her PhD from Shandong University, where her research was dedicated to the development of metal–organic frameworks (MOFs) and covalent organic frameworks (COFs) for electrocatalytic applications. She was appointed as an Associate Researcher at Hainan University in 2025. Her current research focuses on the design and synthesis of MOFs/COFs and the investigation of their dynamic evolution under electrochemical conditions. |
 Xing Lu | Xing Lu is a full professor at Hainan University and Adjunct Professor at Huazhong University of Science and Technology. He received his BS, MS, and PhD from Xiamen University (1998), Changchun Institute of Applied Chemistry, Chinese Academy of Sciences (2001) and Peking University (2004), respectively. Then, he worked as a COE Postdoctoral researcher at Nagoya University, Japan. From 2006 to 2013, he was a Senior Scientist at The University of Tsukuba, Japan. He is the recipient of The Ambassador Award from the Chinese Embassy in Japan (2009), The Osawa Award from the Fullerenes and Nanotubes Research Society of Japan (2011) and The National Science Fund for Distinguished Young Scholars (2019). His research interests lie in the rational design and facile generation of novel hybrid carbon materials with applications in energy storage conversion and biology. He has published more than 180 peer-reviewed papers in international journals with >60 at J. Am. Chem. Soc., Angew. Chem. Int. Ed. and Chem. Sci. |
1 Introduction
Porous materials refer to solid materials with highly developed pore structures, with pore sizes spanning micropores (<2 nm), mesopores (2–50 nm), and macropores (>50 nm).1 These pores endow the materials with extremely high specific surface areas, significantly enhancing the exposure of active sites as well as the transport of matter and energy. Therefore, porous materials are widely applied in adsorption separation,2,3 catalysis,4 and energy storage5 and conversion.6 Representative materials include inorganic porous zeolites, metal–organic frameworks (MOFs) and covalent organic frameworks (COFs). Thanks to their tunable pore structures and surface chemical properties, these materials show great potential in gas adsorption and separation, catalysis, energy storage, and biomedicine. For example, zeolites are widely used in petrochemical catalysis; MOFs perform excellently in hydrogen and methane storage and in separating complex gas mixtures such as CO2/CH4 and C3H6/C3H8;7–9 COFs, with their ordered organic skeletons and functionalized pore walls, have promising prospects in optoelectronic devices and catalysis.10–12Traditional inorganic porous materials such as zeolites have long held an important position in catalysis and separation due to their stable pore structures and excellent adsorption performance.13 However, as demands for material performance and structural control increase, MOFs and COFs, as emerging porous material systems, have become research hotspots owing to their highly tunable pore structures, diverse chemical compositions, and excellent functionalization potential. Compared to traditional zeolites, MOFs and COFs can achieve precise pore size design from the nanoscale to mesoscale and provide a broader chemical tunability, meeting more complex requirements in gas storage, separation, and catalysis.14,15
Despite their broad prospects, rational design and synthesis of novel porous structures still face significant challenges. On one hand, the abundance of metal nodes, organic ligands, and monomer units offers nearly infinite combinatorial possibilities. Based on reticular chemistry principles, theoretically, thousands of MOF structures with different topologies and compositions can be constructed;16 COFs, through dynamic covalent chemistry, can be used to build ordered porous networks with flexible and diverse building block choices.17 This enormous “chemical space” far exceeds the capacity of traditional empirical or intuitive exploration. On the other hand, the synthesis and crystallization of porous materials are influenced by multiple interacting factors such as temperature, solvent systems, precursor concentration, additive types and dosages (e.g., modulators), pH, and reaction time. Especially for COFs, which require reversible covalent bonds to achieve long-range order, optimization is complex, involving both kinetic and thermodynamic considerations. The underlying mechanisms are complicated and lack a comprehensive theoretical framework, causing new material discovery to rely heavily on repeated experiments, with long cycles and often serendipitous success.
In recent years, the rapid development of artificial intelligence (AI) has brought new opportunities to materials science.18 Through machine learning (ML), data mining, molecular simulations, and intelligent optimization, researchers can uncover intrinsic relationships between material structures and properties from vast experimental and computational data, greatly accelerating materials design and performance prediction.19,20 Molecular simulations reveal structural evolution and property mechanisms at the atomic and molecular levels, providing critical support for theoretical studies and experimental design.21–23
In the design and synthesis of porous materials, AI, molecular simulations, and related computational methods jointly play important roles.24,25 By integrating literature, experimental data, and molecular simulations, researchers can predict the thermodynamic stability and synthetic feasibility of target structures.26 First-principles methods such as density functional theory (DFT) and ab initio molecular dynamics (AIMD) based on DFT elucidate electronic structures and reaction mechanisms; molecular dynamics (MD) and grand canonical Monte Carlo (GCMC) simulations study macroscopic behaviours such as gas adsorption and molecular diffusion.27–29 AI integrates simulation and experimental data to optimize experimental conditions, intelligently search high-dimensional parameter spaces, and reduce blind trial-and-error. Target-performance-based inverse design can recommend potential high-performance structures.30–32 Combining experimental automation and real-time data analysis, AI can dynamically adjust synthesis strategies, promoting the formation of autonomous “machine scientist” workflows.33–35 The fusion of data-driven methods and multiscale simulations significantly accelerates the discovery, prediction, and mechanistic understanding of porous framework materials, advancing efficient design and synthesis. The integration of AI and molecular simulations not only improves research efficiency but also shortens the cycle from design to material realization, fostering the intelligent development of materials science.36,37
Herein, this review systematically summarizes recent advances in the rational design of MOFs and COFs enabled by the integration of AI and molecular simulations, with a particular emphasis on their synergistic applications in synthesis pathway design and performance prediction. It provides an overview of widely used molecular simulation techniques and AI methods in metal–organic framework and covalent organic framework design, with an in-depth analysis of their integration mechanisms and collaborative effects in revealing structure–property relationships and guiding materials design.38,39 Finally, the review outlines the key technical challenges currently facing the field and discusses prospects for the deep integration of multiscale simulations and data-driven methodologies, aiming to accelerate the digitally intelligent design and autonomous discovery of novel porous materials.40
2 From trial-and-error to rational design: the role of AI and molecular simulations in optimizing MOF and COF design
The design of MOFs and COFs has undergone four distinct phases, reflecting advancements in computational and data-driven methods. As shown in Fig. 1, initially, material design relied on traditional trial-and-error approaches, where researchers manually adjusted synthesis conditions to explore structure–performance relationships and tested different experimental combinations to find potential optimal materials.41 In the second phase, methods such as DFT, MD, and GCMC simulations were integrated, complementing experimental research and enabling preliminary screening of feasible structures, reducing the reliance on extensive physical experiments.42 The third phase introduced ML algorithms, advancing data-driven modeling and strengthening the correlation between simulation results and experimental data, thus improving performance prediction efficiency.43–45 The fourth phase established a closed-loop system combining AI, standardized databases, and multi-scale simulations, facilitating autonomous iterative design based on big data and accelerating the process of material discovery and optimization.46 | ||
| Fig. 1 The evolution of MOF/COF design. | ||
The synthesis of framework materials, especially COFs, faces numerous challenges. These challenges arise from the diversity of ligands and metal centers (in metal-COFs), complex structural topologies, and the precise control required over synthesis conditions such as solvent type, dosage, temperature, and reaction time balance. Even slight variations in synthesis conditions, such as whether oxygen molecules are removed from the reaction solvent, can lead to different structures or properties, increasing experimental difficulty and uncertainty.47,48 Traditional framework material design relies on trial-and-error methods, where repeated experiments are conducted to synthesize samples with potentially superior properties. While effective in some cases, the high complexity of MOF and COF structures and properties makes predicting material performance during synthesis challenging, often resulting in inefficiency.49 Furthermore, the vast design space for organic ligands and the large number of potential metal nodes and organic linkers lead to an overwhelming number of possible structures. Relying solely on manual exploration delays the development process and wastes considerable human and material resources.50,51
In the design and optimization of MOFs and COFs, various computational methods, such as DFT, MD simulations, GCMC simulations, and other advanced techniques like quantum dynamics, coarse-grained, and ReaxFF simulations, play a crucial role in providing deep insights and enhancing material performance. DFT has been invaluable in accelerating the design of MOFs and COFs by calculating key parameters like reaction free energy, activation energy, and adsorption energy.52 This helps predict catalytic performance and material stability under various conditions. The applications of DFT calculations extend beyond catalysis, aiding in the design of materials for gas storage, separation, sensing, and electronic properties by calculating adsorption energies, evaluating gas selectivity, and assessing electronic structures.
It also helps in predicting and optimizing the behavior of MOFs and COFs for applications like CO2 capture, hydrogen storage, and also electrocatalysts.53 In our previous work, we reported a methodology for designing high-performance COF-based electrocatalysts by integrating DFT calculations, ML, and experimental validation. As shown in Fig. 2, first, 100 virtual M–NxOy (M = 3d transition metal) model catalysts were screened using DFT.54Fig. 2a illustrates the structural model of virtual M–NxOy catalysts (M = 3d transition metal) for DFT screening. It clearly defines three coordination sites (1,2-coordination, 1,3-coordination, and 1,4-coordination) and marks the metal centre (M) with distinct labelling, which lays the structural foundation for subsequent calculations of catalytic activity and the analysis of coordination environment effects on reaction performance. Fig. 2b presents the linear correlation between the Gibbs free energy change (ΔG) in the rate-determining step obtained from DFT calculations and the applied potential (U), with a high correlation coefficient (R2) of 0.933. This strong linear relationship fully validates the thermodynamic rationality of using ΔG as a key descriptor for evaluating OER activity, providing a reliable theoretical basis for further identifying high-activity catalyst structures. Fig. 2c visualizes the distribution of ΔG values for various 3d transition metal-based M–NxOy catalysts through a color-gradient scatter plot. The data points show that Ni-based catalysts are concentrated in the region with low ΔG values (typically <1.8 eV), which clearly implies their potential as high-performance OER candidate materials. This result effectively guides subsequent research to focus on the design and optimization of Ni-cantered catalysts, avoiding the inefficiency of blind screening across all metal centres. Then, intrinsic descriptors of OER activity were extracted and analysed through ML, enabling the prediction of the most promising structures. Finally, a COF-based electrocatalyst with excellent performance was successfully synthesized and experimentally verified. The electrocatalytic studies demonstrated that Ni-COF exhibits activity comparable to the best COF-based OER catalysts reported to date. Then, intrinsic descriptors of OER activity were extracted and analysed through ML, enabling the prediction of the most promising structures. Fig. 2d defines three key intrinsic descriptors regulating OER activity, namely the d–p bond length between the metal centre and coordinating atoms, the Mulliken charge (qmetal) of the metal centre, and the electron affinity (Eaff). These descriptors are all derived from structural and electronic property data obtained via DFT calculations, enabling precise capture of the core features that influence catalytic activity. Fig. 2e quantifies and ranks the importance of the descriptors using a machine learning model. The results show that the d–p bond length of Ni-based catalysts accounts for the highest contribution ratio (42%), followed by the Mulliken charge (28%) and electron affinity (20%). This clarifies the core targets for structural optimization and provides a basis for the targeted design of catalysts. Fig. 2f presents the consistency verification between OER activity predicted by machine learning and results calculated via DFT, with a correlation coefficient (R2) of 0.994. The embedded probability distribution plot further demonstrates that the model's prediction error is less than 0.08 eV, fully validating the value of machine learning as a reliable tool for rapidly screening many catalyst structures. Finally, a COF-based electrocatalyst with excellent performance was successfully synthesized and experimentally verified. Fig. 2g outlines the experimental validation workflow for the optimal Ni-COF electrocatalyst, encompassing three core steps: (1) solvothermal synthesis: the catalyst was prepared using Ni(NO3)2·6H2O as the metal precursor and Salen-type organic linkers as the organic building blocks, with a mixed solvent of N,N-dimethylformamide (DMF) and ethanol (volume ratio 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) at 120°C for 72 hours; (2) structural characterization: X-ray diffraction (XRD) was employed to confirm the crystalline structure of the Ni-COF, while X-ray photoelectron spectroscopy (XPS) verified the formation of the Ni-N2O2 coordination motif (the key active site predicted by DFT and ML); (3) electrochemical performance testing: OER activity was evaluated in a three-electrode electrochemical system with 1.0 M KOH as the electrolyte, including measurements of linear sweep voltammetry (LSV) and cyclic voltammetry (CV) for stability. The schematic in Fig. 2g clearly connects the theoretical design process (DFT screening of virtual catalysts and ML prediction of active descriptors) with experimental implementation, directly verifying the success of the “DFT screening-ML prediction-experimental validation” integrated workflow for COF-based electrocatalyst design. Specifically, the Ni-COF showed an overpotential of 260 mV at 10 mA cm−2 and a Tafel slope of 65 mV dec−1 in 1.0 M KOH electrolyte, with no obvious performance decay after 2000 cycles. The electrocatalytic studies demonstrated that Ni-COF exhibits activity comparable to the best COF-based OER catalysts reported to date. To further assess the generalizability of the ML model across diverse M–NxOy structures (beyond Ni-based catalysts), we extended the model's predictions to additional candidates and compared their ΔG(O–OH) (Gibbs free energy change for O–OH bond cleavage) with DFT results (Fig. 2h). As shown in Fig. 2h, the scatter plot contrasts ML-predicted ΔG(O–OH) and DFT-calculated ΔG(O–OH) for over 100 M–NxOy catalysts (M = 3d transition metals, including Fe, Co, Cu, Zn, etc.). Data points for all metal centres cluster closely around the parity line (ΔGml = ΔG_DFT), confirming the model's consistent performance across diverse single-metal structural configurations. The embedded inset further quantifies the prediction error, showing that over 92% of data points have an absolute error <0.1 eV—validating the model's ability to reliably screen high-performance M–NxOy catalysts beyond Ni-based systems. This result confirms the ML model's robustness in screening a broad range of M–NxOy structures, rather than being limited to Ni-based catalysts, and validates its ability to accurately prioritize high-performance candidates for experimental synthesis—laying a foundation for scalable catalyst design.
:1) at 120°C for 72 hours; (2) structural characterization: X-ray diffraction (XRD) was employed to confirm the crystalline structure of the Ni-COF, while X-ray photoelectron spectroscopy (XPS) verified the formation of the Ni-N2O2 coordination motif (the key active site predicted by DFT and ML); (3) electrochemical performance testing: OER activity was evaluated in a three-electrode electrochemical system with 1.0 M KOH as the electrolyte, including measurements of linear sweep voltammetry (LSV) and cyclic voltammetry (CV) for stability. The schematic in Fig. 2g clearly connects the theoretical design process (DFT screening of virtual catalysts and ML prediction of active descriptors) with experimental implementation, directly verifying the success of the “DFT screening-ML prediction-experimental validation” integrated workflow for COF-based electrocatalyst design. Specifically, the Ni-COF showed an overpotential of 260 mV at 10 mA cm−2 and a Tafel slope of 65 mV dec−1 in 1.0 M KOH electrolyte, with no obvious performance decay after 2000 cycles. The electrocatalytic studies demonstrated that Ni-COF exhibits activity comparable to the best COF-based OER catalysts reported to date. To further assess the generalizability of the ML model across diverse M–NxOy structures (beyond Ni-based catalysts), we extended the model's predictions to additional candidates and compared their ΔG(O–OH) (Gibbs free energy change for O–OH bond cleavage) with DFT results (Fig. 2h). As shown in Fig. 2h, the scatter plot contrasts ML-predicted ΔG(O–OH) and DFT-calculated ΔG(O–OH) for over 100 M–NxOy catalysts (M = 3d transition metals, including Fe, Co, Cu, Zn, etc.). Data points for all metal centres cluster closely around the parity line (ΔGml = ΔG_DFT), confirming the model's consistent performance across diverse single-metal structural configurations. The embedded inset further quantifies the prediction error, showing that over 92% of data points have an absolute error <0.1 eV—validating the model's ability to reliably screen high-performance M–NxOy catalysts beyond Ni-based systems. This result confirms the ML model's robustness in screening a broad range of M–NxOy structures, rather than being limited to Ni-based catalysts, and validates its ability to accurately prioritize high-performance candidates for experimental synthesis—laying a foundation for scalable catalyst design.
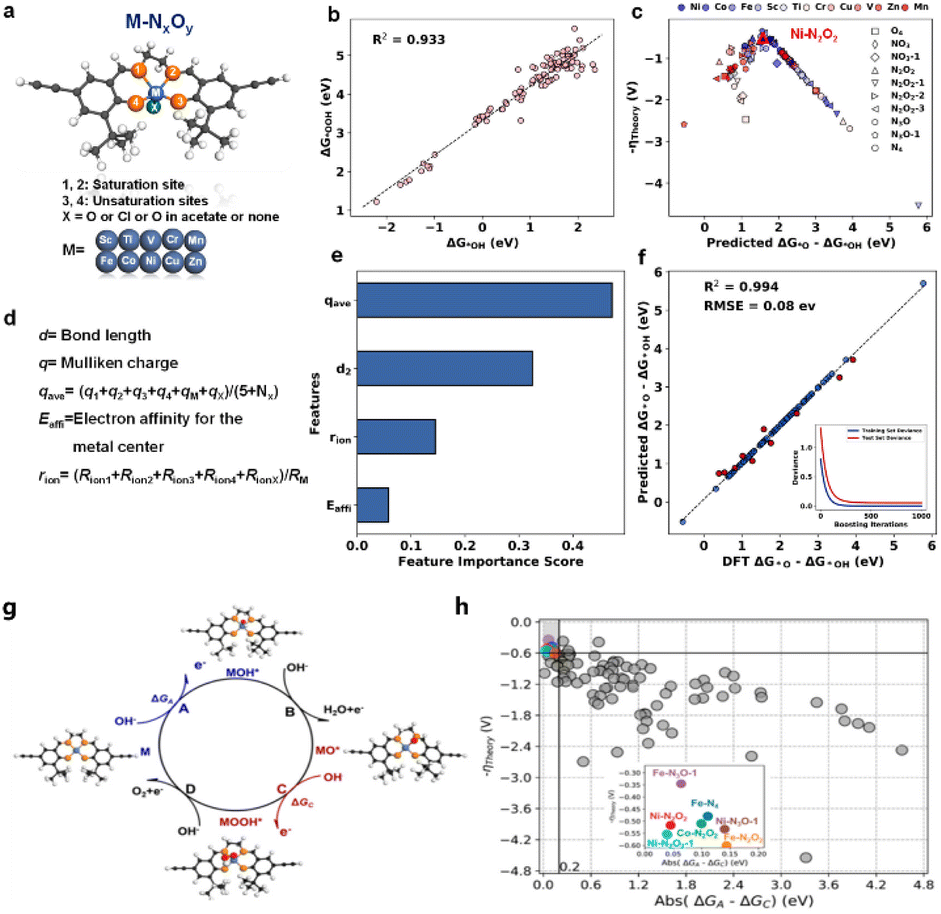 | ||
| Fig. 2 Workflow for designing high-performance COF-based electrocatalysts by integrating DFT calculations, ML, and experimental validation (reproduced with permission from American Chemical Society, Copyright© 2021).55 (a) Structural schematic of M–NxOy catalytic sites, labeling saturated sites (1 and 2) and unsaturated sites (3 and 4), with X being O or Cl (acetate or non-acetate ligand) and M representing transition metals (e.g., Fe, Co, Ni, etc.). (b) Scatter plot showing the correlation between ΔG(OOH) and ΔG(O–OH), with a goodness-of-fit R2 of 0.933, reflecting their energy relationship in the catalytic process. (c) Line graph depicting the relationship between adsorption energy of the NH3O2 intermediate in a NH3/O2 system for different metals (e.g., Mn, Fe, Co, etc.) and predicted ΔG(O–OH), distinguishing performance differences among metal sites. (d) List of structural and electronic characteristic parameters for analysis, including the d-band center (d(d)), Mulliken charge (q(metal)), electron affinity of the metal center (E(af)), and average bond length (r(av)). (e) Bar graph of feature importance, where d(d) has the highest impact on catalytic performance, followed by r(av) and E(aff). (f) Scatter plot with a fitted line illustrating the agreement between machine learning-predicted ΔG(O–OH) and DFT-calculated values (R2 = 0.994; RMSE = 0.08 eV), with an inset showing the model's learning curve to demonstrate generalization ability. (g) Cyclic workflow for COF-based electrocatalyst design, integrating DFT calculations, ML screening, and experimental validation, with labels for key reaction intermediates (e.g., MOH+) and steps (e.g., OH− adsorption and electron transfer). (h) Scatter plot of ΔG(O–OH) (Gibbs free energy change for O–OH bond cleavage) predicted by the ML model vs. that calculated via DFT for over 100 M–NxOy catalysts (M = 3d transition metals, e.g., Fe, Co, Ni, Cu, and Zn). Data points cluster around the parity line (ΔGml = ΔG_DFT), with an inset quantifying prediction errors (over 92% of points have absolute error <0.1 eV), validating the model's robustness across diverse single-metal catalytic sites. | ||
By integrating computational screening, ML, and experimental validation, this work demonstrates the potential of digital-intelligent approaches for the design of porous crystalline electrocatalysts. Nevertheless, further improvements are needed, including the incorporation of larger and more diverse catalyst datasets, the use of more sophisticated ML algorithms for higher predictive accuracy, and the integration of dynamic catalytic environments to better capture realistic reaction conditions.56
While DFT provides precise insights into thermodynamic and electronic properties, MD simulations complement this by offering a dynamic understanding. MD simulations are especially effective in studying the behavior of MOFs and COFs under varying environmental conditions such as temperature, pressure, or solvent presence. For example, MD simulations of ZIF-8 revealed temperature-induced cubic-to-orthorhombic phase transitions, with atomic displacement trajectories quantifying framework flexibility during phase switching; this observation was further corroborated by in situ X-ray diffraction (XRD) experiments, confirming the phase transition pathway and critical temperature range.31 For molecular diffusion, MD studies on UiO-66-NH2—calibrated against quasielastic neutron scattering data—demonstrated how amino-functional groups regulate CO2 diffusivity within pores (1.2–2.5 × 10−9 m2 s−1 at 298 K), with intra-cage jump events identified as the dominant transport mechanism; this mechanistic insight directly guides the design of high-efficiency CO2 separation materials.57 In terms of structural stability, MD simulations of imine-based COFs clarified the solvent-induced framework deformation mechanisms: ethanol environments were shown to reduce interlayer stacking order by 15% without collapsing the porous structure, a phenomenon attributed to reversible imine bond rotation that preserves long-range framework integrity—consistent with experimental powder XRD (PXRD) results of TpPa-1 after ethanol treatment.58 These simulations allow researchers to predict phase transitions, degradation, or structural changes during synthesis and application. Additionally, MD simulations help optimize gas adsorption and separation performance by providing data on molecular diffusion and transport properties, which are critical for enhancing gas storage capacity and separation efficiency.57 By studying interactions between reactant molecules and catalyst surfaces, MD simulations also play a significant role in optimizing catalytic performance.
Building on these insights, GCMC simulations play a crucial role in optimizing the pore structure of MOFs and COFs, especially in gas adsorption and thermodynamic behavior.59 By simulating gas molecule interactions within the pores, GCMC simulations help calculate adsorption isotherms and predict gas storage and separation performance under various temperature and pressure conditions. These simulations also provide valuable insights into the long-term stability of MOFs and COFs, identifying potential issues like pore collapse or structural changes that may arise under extreme operating conditions. Furthermore, GCMC simulations can guide the design of materials with tailored pore structures, improving adsorption properties and enabling the efficient storage and separation of specific gases. In addition, GCMC simulations can be combined with high-throughput computational screening methods to deeply analyze the competitive adsorption mechanisms of CO2 and H2O, providing valuable insights for the design of CO2 capture materials.60 For example, Snurr et al.3 proposed a high-throughput computational strategy to identify MOFs that can effectively adsorb CO2 under high humidity conditions. One of the key innovations of this study is the large-scale screening of MOFs to find materials with high CO2 selectivity over H2O at 80% relative humidity (Fig. 3). As illustrated in Fig. 3, each subfigure details the computational screening workflow and its outcomes:
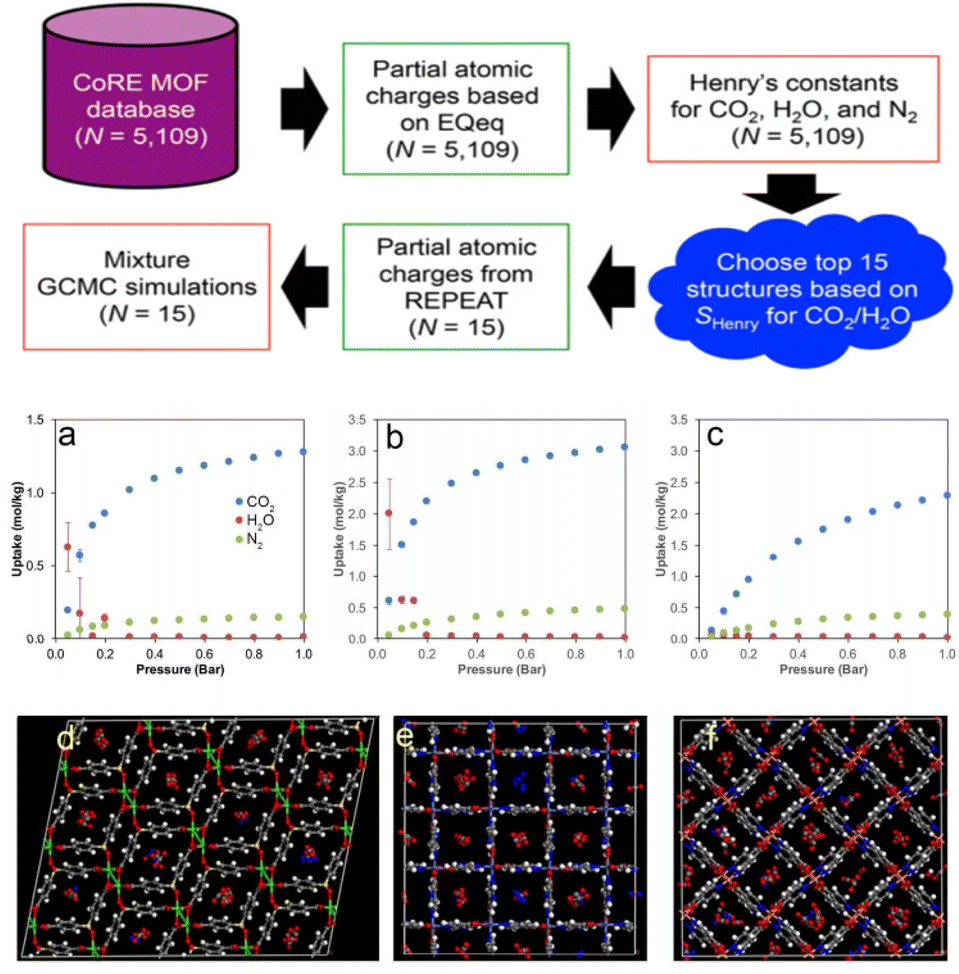 | ||
| Fig. 3 Workflow of the computational screening strategy employed in this study. N is the number of MOF structures involved in each step (reproduced with permission from American Chemical Society, Copyright© 2016).3 Workflow: initiates with the CoRE MOF database (N = 5109), computes partial atomic charges based on EQeq (N = 5109) to obtain Henry's constants for CO2, H2O, and N2 (N = 5109), selects the top 15 structures according to selectivity for CO2/H2O, repeats partial atomic charge calculation for these 15 structures, and conducts mixture GCMC simulations (N = 15). (a) Adsorption isotherms of CO2, H2O, and N2 as a function of pressure (bar), exhibiting their uptake disparities. (b) Adsorption isotherms of CO2, H2O, and N2 as a function of pressure (bar), demonstrating their uptake behaviours under different pressure conditions. (c) Adsorption isotherms of CO2, H2O, and N2 as a function of pressure (bar), depicting their adsorption features. (d) Structural visualization of a MOF, with various atoms and moieties distinguished by different colours (e.g., metal nodes and organic linkers). (e) Structural visualization of a MOF, showcasing its framework architecture and atomic arrangements. (f) Structural visualization of a MOF, illustrating its porous structure and component distribution. | ||
Notably, the reliability of such GCMC simulations—and thus the validity of CO2/H2O adsorption predictions—hinges critically on the accuracy of partial atomic charge models, which quantify the electrostatic interactions between adsorbates (CO2 and H2O) and MOF frameworks. Recent advances in this field have seen the adoption of models like DDEC6, CM5, EQeq, and PACMAN, each impacting screening reliability distinctively:
DDEC6 and CM5 offer high precision by accounting for local electron density variations, making them ideal for simulating CO2–H2O competitive adsorption (e.g., reducing errors in H2O cluster formation within MOF pores, a key factor in Snurr et al.'s high-humidity screening).
EQeq excels in speed, enabling large-scale preliminary screening but may slightly underpredict electrostatic interactions in polar frameworks.
PACMAN, optimized for porous materials, better matches experimental CO2 adsorption enthalpies, narrowing the gap between simulated and real-world performance.
These models directly shape the quality of data driving Snurr et al.'s screening, underscoring their role in ensuring the accuracy of high-humidity CO2 capture predictions.
Fig. 3a depicts the adsorption isotherms of CO2, H2O, and N2 in top-ranked MOF structures at 298 K. The x-axis represents pressure (Bar), and the y-axis denotes the amount adsorbed (mmol g−1). CO2 shows a significantly higher adsorption capacity than H2O and N2 across all pressures, highlighting the preferential affinity of MOFs for CO2—critical for CO2/H2O/N2 separation applications.
Fig. 3b focuses on H2O adsorption isotherms in the selected MOFs. The sharp increase in H2O adsorption at low pressures (≤0.2 Bar) emphasizes their strong hydrophilicity, essential for evaluating performance under humid conditions. Comparing this with Fig. 3a reveals differential adsorption behaviors that enable tailored selectivity design for CO2/H2O separation.
Fig. 3c illustrates N2 adsorption isotherms in the same MOFs. N2 adsorption remains low even at high pressures, confirming weak interactions with N2. Combined with Fig. 3a's high CO2 capacity, this underscores potential for efficient CO2/N2 separation in industrial flue gas purification.
Fig. 3d–f present crystal structures of the three top-performing MOFs. Fig. 3d features a complex 3D network with interconnected pores and metal clusters (coloured spheres) providing multiple adsorption sites. Fig. 3e displays a more open framework with larger pores to facilitate gas diffusion. Fig. 3f shows a distinct topological arrangement, highlighting structural diversity and how variations (e.g., pore size and metal coordination) correlate with adsorption behaviours in Fig. 3a–c. These structures link computational screening results to material features governing gas adsorption. The authors used a fast-screening method based on the ratio of Henry's law constants for CO2 and H2O, followed by more detailed GCMC simulations using more accurate framework charge calculations.61
The study revealed that electrostatic interactions play a critical role in the adsorption behavior of water molecules in MOFs, emphasizing the importance of accurate charge calculation methods for simulating water adsorption.62 Snurr et al.3 also highlighted the challenge of developing efficient MOFs for industrial CO2 capture at high humidity. They found that MOFs with smaller pore sizes provided stronger CO2 binding and limited water uptake by preventing water clusters from forming inside the pores. However, these small-pore MOFs might have lower working capacity compared to other adsorbents with larger pore volumes, posing a significant challenge in the MOF research field.
Additionally, Zhong et al. systematically explored and designed efficient COF materials by combining molecular simulations with ML techniques to achieve industrial applications, such as the reverse separation of ethane/ethylene (Fig. 4).30 To accelerate material screening, the authors used the CoRE COF database and evaluated C2H6/C2H4 uptakes at 1 bar and 0.1 bar; these values, together with derived selectivity/capacity trade-offs, were used to identify promising materials with excellent separation performance, with special attention given to balancing adsorption selectivity and capacity. Special attention was given to balancing adsorption selectivity and capacity. This approach not only greatly improved the screening efficiency but also provided new insights into understanding the interactions between gas molecules and the pore structures of materials.
 | ||
| Fig. 4 The workflow for designing ethane/ethylene separation COF materials as reported in Zhong's work (reproduced with permission from American Chemical Society, Copyright© 2022).30 | ||
Building upon molecular simulations, they further introduced ML algorithms, particularly the Random Forest (RF) model, to predict the separation performance of numerous COF materials. Experimental results showed that the RF model exhibited a high predictive accuracy (R2 = 0.97) and was able to effectively identify material density (ρ) as the key structural parameter influencing selectivity. By ranking the importance of structural parameters, the study revealed critical factors for optimizing COF material performance, providing important theoretical guidance for future material design.63,64 In addition to model training and performance prediction, Zhong et al.30 also leveraged the model results to screen several potential high-performance “hypothetical COFs”. These materials demonstrated exceptional selectivity and have great potential for achieving efficient gas separation in practical applications. Through this data-driven approach, the research significantly enhanced the speed and efficiency of material screening, laying a solid scientific foundation for the development of industrial-scale reverse gas separation technologies.
Despite the remarkable innovative potential demonstrated by this research in combining ML and molecular simulations, there are still areas for further optimization. Future studies could consider incorporating additional factors related to real-world applications, such as material synthesis difficulty, operational conditions, and long-term performance stability, to improve the adaptability of the model.65 Moreover, strengthening experimental validation and translating simulation results into experimental outcomes will further enhance the practical relevance and guiding significance of the research.
Building upon these insights, similar methodologies have been employed by Van Speybroeck et al., who also utilized molecular simulations in combination with ML techniques to design and optimize COF materials, with a specific focus on CO2 capture applications. In their approach, they leveraged a large database and advanced simulations to enhance the screening process for COF materials aimed at post-combustion CO2 capture.66
As shown in Fig. 5 Van Speybroeck et al.‘s research introduces an innovative high-throughput computational screening method designed to optimize COFs for post-combustion CO2 capture. The study uses the ReDD-COFFEE database, which contains over 268000 hypothetical COFs, and integrates ML algorithms to accelerate the material screening process. Through a multi-step screening strategy, the authors conduct idealized single-component GCMC simulations, followed by ML models for prediction, and then apply more accurate mixed GCMC simulations to identify the most promising COF materials for CO2 separation. This approach effectively narrows down the vast COF material space, significantly improving screening efficiency, and enabling faster identification of candidate materials with excellent performance.
 | ||
| Fig. 5 The workflow for designing CO2/N2 separation COF materials as reported in Van Speybroeck's work (reproduced with permission from American Chemical Society, Copyright© 2024).66 | ||
A key innovation of the study is the integration of ML algorithms with molecular simulations to predict CO2 working capacity and ideal CO2/N2 selectivity. By training models on a representative subset of COFs, the authors revealed that certain bonding types, such as amide and (acyl)hydrazone linkages, as well as functionalized aromatic rings, are particularly beneficial for CO2 adsorption. Additionally, they found that three-dimensional COFs with pore sizes of about 1.0 nm, especially those with a larger distance between aromatic rings, provide the strongest CO2 adsorption sites, thereby significantly optimizing the material's performance. This finding highlights the importance of the pore structure and functionalization in optimizing COF performance.
In this study, the authors also propose several design rules to guide experimental researchers in synthesizing high-performance COF materials, bridging the gap between computational predictions and real-world applications. The specific design rules are as follows: first, optimization of pore size and shape—research shows that three-dimensional COFs with a pore size of around 1.0 nm, especially those with a larger distance between aromatic rings, provide the strongest CO2 adsorption sites. Thus, optimizing pore size and shape is crucial for enhancing CO2 adsorption performance; second, functionalization design—certain bonding types, such as amide and (acyl) hydrazone linkages, along with functionalized aromatic rings, can significantly enhance CO2 adsorption. This suggests that incorporating specific functional groups, particularly those that strengthen gas–pore interactions, can improve adsorption performance in COF materials; third, the synergy of the pore structure and functionalization—the combined effect of the pore structure and functionalization design is critical for optimizing material performance. Designing COFs with appropriate pore sizes and functionalization can better achieve efficient CO2 capture; Finally, the advantages of multidimensional structures—three-dimensional COFs are typically more favorable for CO2 adsorption than two-dimensional COFs because the former offers more adsorption sites and higher surface area. Therefore, designing COFs with three-dimensional topologies may be more suitable for efficient CO2 capture.
Although this study provides valuable insights, there are some limitations and areas for improvement. First, the practical feasibility of synthesizing the top-performing COF materials identified in the computational models remains a major challenge. While the study has filtered out materials with lower synthesis difficulty, many of the proposed materials may still be difficult or impractical to synthesize. This is a common issue in high-throughput screening of hypothetical materials, and future research should focus on aligning computational predictions with synthetic feasibility to better enable experimental validation.67 Second, the study could benefit from further evaluation of the long-term stability and performance of the identified materials under real-world conditions. Specifically, experimental validation of these materials in dynamic industrial environments will help confirm their effectiveness in actual CO2 capture processes, thereby facilitating their successful industrial application.
ML can assist in the stability analysis of framework materials to a certain extent.68 Jiang et al. proposed a hierarchical high-throughput computational screening (HTCS) strategy that integrates ML-assisted stability analysis with molecular simulations, aiming to identify ultrastable MOFs capable of efficiently capturing CO2 under wet flue gas conditions. This study highlights the importance of both performance and stability in the practical application of MOFs for CO2 capture, making it highly significant for environmental protection and sustainable development.
A major strength of this study is its comprehensive approach. As is shown in Fig. 6 by utilizing large-scale screening from the “ab initio REPEAT charge MOF” database and ML-driven stability predictions, the authors significantly improve screening efficiency. By predicting the stability of MOFs in water, under thermal conditions, and in activation processes, the study narrows down approximately 280000 candidates to 9,755, and then further evaluates these materials' performance using molecular simulations. This hierarchical method offers an innovative and practical solution to the challenge of identifying stable MOFs, reducing the time and computational resources typically needed for experimental validation.
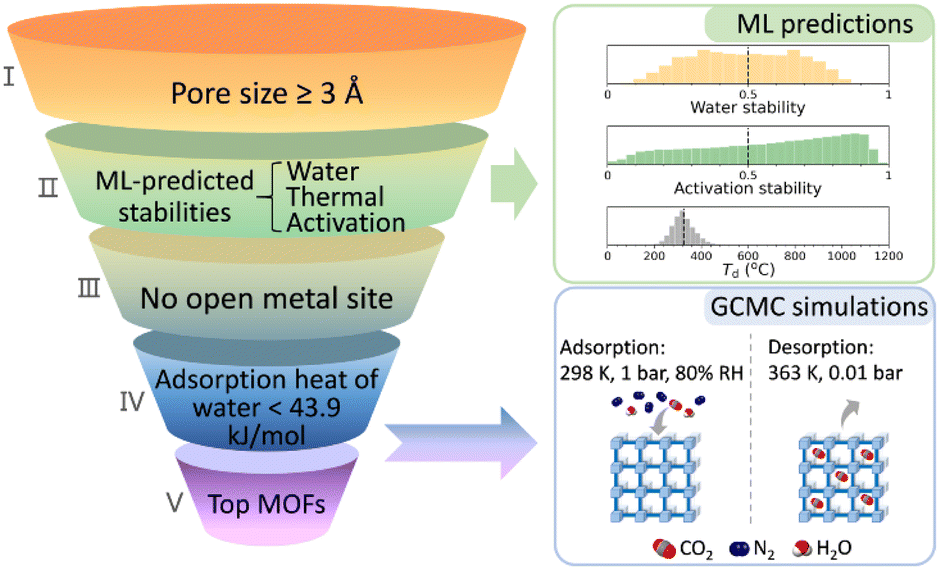 | ||
| Fig. 6 Workflow for CO2 capture from wet flue gas using MOFs, as presented in Jiang's work (reproduced with permission from American Chemical Society, Copyright© 2025).60 | ||
In the development of ML models, the authors derive crucial design principles for MOFs that can overcome performance trade-offs. By identifying key geometric features that affect CO2 capture and selectivity, such as void surface area (VSA), pore limiting diameter (PLD), and high void fraction of the pore openings (VF_PO), they provide vital guidance for future MOF design. Additionally, the authors developed an ML classifier to assess the impact of water on CO2 capture performance, providing a more rigorous perspective and helping to better understand the behavior of these materials under real-world conditions, especially in high-humidity environments. The work also discovered several vanadium-based MOFs, which feature conjugated aromatic linkers and RNA-like topologies, showing exceptional stability and CO2 capture ability under wet flue gas conditions. These findings, along with the identification of geometric and elemental features that influence MOF performance, provide valuable design guidelines for future research and development. The identified MOFs exhibit CO2 capture capacities ranging from 3 to 7 mmol g−1, with CO2/N2 selectivity as high as 401, demonstrating the potential of vanadium-based MOFs in real-world CO2 capture applications.
While GCMC and MDs simulations primarily focus on molecular behavior related to the structure and gas interactions, quantum dynamics simulations provide a more precise understanding of the electronic properties of materials. This study addresses a critical gap in many CO2 capture studies, which often lack stability analysis. While performance metrics such as CO2 uptake and selectivity are essential, their relevance is diminished without a thorough understanding of the material's long-term stability, especially under the high-temperature and humid conditions typical of flue gas streams. By considering both stability and performance, the authors ensure that their findings are directly applicable to practical scenarios, paving the way for the real-world deployment of MOFs in CO2 capture. Traditional high-throughput computational screening (HTCS) methods often overlook material stability, yet stability is a crucial factor determining whether MOFs can perform successfully in practical applications. Based on this fact, Jiang's work provides important theoretical foundations and methodological innovations for the screening and application of MOFs in CO2 capture. By integrating stability assessments with high-throughput computational screening, they offer a new perspective and tool for MOF screening and optimization. One of the key contributions of their research is the incorporation of four stability metrics—thermodynamic stability, mechanical stability, thermal stability, and activation stability—into the MOF screening process, proposing a more comprehensive screening approach. Specifically, the study evaluates thermodynamic and mechanical stability through MD simulations, while activation and thermal stabilities are predicted using ML models (Fig. 7). This approach identifies MOFs that not only efficiently capture CO2 but also possess high stability. The research underscores the central importance of stability in MOF screening, particularly for CO2 capture in environments with high humidity, corrosive gases, or organic solvents, providing strong theoretical support for such applications.69
 | ||
| Fig. 7 Workflow for high-throughput computational screening (HTCS) of hMOFs for CO2 capture as reported in Jiang's work (reproduced with permission from Springer Nature, Copyright© 2023).70 | ||
2.1 MOFs and COFs
The GCMC and MDs simulations allow researchers to model the electronic structures and dynamic behaviors of materials with high accuracy, offering crucial insights into reaction mechanisms, charge transport, and adsorption processes. Quantum dynamics are especially important for applications in catalysis and electronic devices, where precise material–molecule interactions play a key role in functionality.58 Additionally, these simulations are vital for studying catalytic reactions, providing in-depth analysis of energy band structures, electron densities, and reaction pathways, thereby guiding the optimization of MOFs and COFs for catalytic applications.Lang et al. proposed a universal screening model for the rapid prediction and selection of efficient helium separation materials.71 The innovation of this model lies in its transferability across different types of porous materials (such as COFs and MOFs), based on helium's chemical inertness and the appropriate choice of descriptors. As shown in Fig. 8, this transferability enables the model to be applied broadly. Fig. 8a shows the helium adsorption isotherm of 3D-5p-COF-1 at 298 K, with pressure ranging from 0 to 10 bar. The isotherm exhibits a steep increase in helium uptake at low pressures, indicating strong affinity, and reaches a saturation capacity of 0.872 mol kg−1—consistent with the MD simulation results. This high-capacity positions 3D-5p-COF-1 as a top candidate for helium capture applications. Fig. 8b compares the membrane selectivity of 3D-5p-COF-1 for He/CH4 and He/N2 against other tested materials. The selectivity ratios (3.37 for He/CH4 and 4.48 for He/N2) are plotted, demonstrating that 3D-5p-COF-1 outperforms conventional adsorbents. This superior selectivity is attributed to its tailored pore size and surface properties that favour helium over methane and nitrogen. Fig. 8c visualizes the crystal structure of 3D-5p-COF-1, highlighting its interconnected 3D pore network and the arrangement of organic building blocks. The large pore volume and high porosity (evident from the structural visualization) directly correlate with the enhanced helium adsorption observed in Fig. 8a, while the specific surface chemistry contributes to the selective interactions that drive the high selectivity in Fig. 8b. Through MD simulations, the researchers further validated the model's effectiveness and found that 3D-Sp-COF-1 outperformed all other tested materials, showing the highest helium adsorption capacity (0.872 mol kg−1), and membrane selectivity ratios of 3.37 for He/CH4 and 4.48 for He/N2. Additionally, SHAP analysis revealed that larger pore volume and higher porosity contribute to enhanced helium adsorption, while a higher adsorption heat ratio and specific surface area favour increased helium selectivity.
 | ||
| Fig. 8 Workflow for screening high-performance COFs/MOFs for helium separation as reported in Lang's work (reproduced with permission from Elsevier Ltd, Copyright© 2025).71 (a) Workflow of machine learning (ML) models built on the COF database, including data partitioning (training/test sets), feature engineering (e.g., Top2D-ERA and HKUST-1 COF features), model training (e.g., LGBM and XGBoost), and feature importance analysis. (b) Scatter plot showing the correlation between ML-predicted and actual values for a performance metric, demonstrating model accuracy. (c) Scatter plot presenting the relationship between ML-predicted and true values, validating model performance. (d) Scatter plot illustrating the correlation between predicted and actual values for another metric, further verifying model reliability. (e) Schematic of COF/MOF pore or structural features, highlighting topological or pore size characteristics. (f) Schematic of COF/MOF pore channels or nodes, emphasizing structural details. (g) Visualization of the COF/MOF framework structure, showcasing its porous architecture as the basis for helium separation. | ||
Despite the significant innovation and practical value of this study, there are aspects that warrant further exploration. First, while the model demonstrates significant findings regarding the impact of pore volume and specific surface area on helium separation performance, other factors such as the heterogeneity of pore structures and the chemical stability of materials may also influence helium separation efficiency, and these factors may not have been fully accounted for in the current model. Secondly, although MD simulations provided effective validation of material performance, the accuracy of the simulation results could be limited by computational methods and simulation time, suggesting that the real-world performance of materials may slightly differ from the simulated results. Therefore, further experimental validation and real-world testing are crucial. Finally, although the study screened many MOF materials from the CoRE MOF database, many other COFs and MOF materials were not included in the screening process. Future research could expand the material database to explore a wider variety of porous materials, further improving the model's applicability and accuracy.
Quantum dynamics simulations can more precisely describe the electronic properties of materials such as MOFs and COFs, especially in scenarios where material–molecule interactions may involve electronic-level behaviors (e.g., catalytic reaction mechanisms). These simulations are capable of providing insights into energy band structures, electron density distributions, and reaction pathways, though their application in framework materials research is still evolving.72 These simulations not only provide crucial information on energy band structures, electron density distributions, and reaction pathways, but also accurately simulate the dynamic changes in materials in different environments, including electron transfer and excited-state transitions. For helium separation—where the focus is on molecular diffusion and adsorption (rather than electronic processes)—classical MD simulations remain effective for studying such macro-scale behaviours. In contrast, MD simulations focus on atomic-level mechanical interactions, which are well-suited for analysing helium adsorption capacity and membrane selectivity, as demonstrated in subsequent studies. Thus, quantum dynamics simulations can complement traditional molecular dynamics simulations in specific scenarios requiring electronic-level analysis, while MD is more directly applicable to helium separation-related research that prioritizes atomic motion and adsorption behavior. By selecting the appropriate simulation method based on research objectives, researchers can achieve a targeted assessment of material performance, further optimizing the selective adsorption and separation processes for helium.
Meanwhile, coarse-grained simulations offer a different approach by simplifying molecular models, reducing computational complexity, and enabling large-scale systems to be studied. This technique is particularly useful for screening various pore structures quickly, making it easier to identify materials with superior gas storage and separation properties.73 Coarse-grained simulations also provide insights into diffusion and transport properties of molecules within MOF and COF structures, enhancing their efficiency in applications like gas storage and separation. Furthermore, these simulations can be used to study the effects of solvents on material behavior, offering insights into how MOFs and COFs perform under different environmental conditions, such as in liquid-phase reactions.
Finally, ReaxFF simulations, a type of reactive force field, provide a powerful tool for studying chemical reactions and the dynamic nature of MOF and COF materials during catalytic processes.74 These simulations are ideal for exploring reactions where bonds are continuously formed and broken, making them particularly useful for studying catalytic reactions and associated changes in the pore structure of MOFs and COFs. ReaxFF simulations can help researchers understand reaction mechanisms, activation energies, and reaction pathways, optimizing catalytic efficiency. They also provide insights into the long-term stability and performance of materials in catalytic applications.
In conclusion, the integration of DFT, MD, GCMC, quantum dynamics, coarse-grained, and ReaxFF simulations significantly enhances the design and functionality of MOFs and COFs. Each simulation method offers unique advantages: DFT provides precision in the electronic structure and thermodynamics, MD offers dynamic insights into material stability and transport properties, GCMC excels in gas adsorption and separation studies, quantum dynamics delivers high-accuracy insights into catalytic behavior, coarse-grained simulations enable large-scale pore structure screening, and ReaxFF is used to model dynamic chemical reactions and material stability during catalysis. Together, these advanced techniques form a comprehensive toolkit for accelerating the development of high-performance MOF and COF materials, enabling their application in energy storage, gas separation, catalysis, electronics, and beyond. As computational resources continue to improve, these simulations will play an increasingly crucial role in advancing the design and optimization of these materials.
High-throughput screening, combined with advanced simulation techniques, has revolutionized the design and optimization of MOF and COF materials. By integrating methods such as DFT, MD, and GCMC simulations, researchers can efficiently calculate key material properties, such as adsorption energies, stability, and performance under various conditions.75 This enables rapid identification of materials with superior gas storage capabilities or specific gas selectivity, thus enhancing the material discovery process. For example, Yang's work combined molecular simulation and ML methods to establish a large database of COF properties and calculate their methane adsorption capacity through GCMC simulations. Subsequently, as shown in Fig. 9, feature selection and traditional ML models, such as multiple linear regression (MLR), support vector machine (SVM), decision trees (DT), and random forests (RF), were used for performance prediction. At the same time, the AutoML tool TPOT was introduced to automate feature engineering, model selection, and hyperparameter tuning to improve prediction accuracy and work efficiency.
 | ||
| Fig. 9 The workflow combining GCMC simulation and automated machine learning as reported in Yang's work (reproduced with permission from American Chemical Society, Copyright© 2021).70 | ||
The research results showed that the model automatically generated by Tree-based Pipeline Optimization Tool (TPOT) significantly outperformed traditional ML models in predicting COF performance, with a coefficient of determination (R2) as high as 0.992, greatly improving prediction accuracy (error reduction) and enabling rapid and efficient performance screening. On the other hand, AutoML not only alleviated the burden of manual parameter tuning but also lowered the entry barrier for non-expert researchers in material screening, thus promoting the development of data-driven material design. Despite the significant advantages demonstrated by the study in introducing AutoML technology for COF performance prediction, there are still some shortcomings that need attention. Firstly, the model was trained based on simulation data, and although the prediction performance is excellent, its generalization ability in real-world applications still needs to be confirmed through extensive experimental validation, especially regarding stability and reliability under different experimental conditions. Secondly, while the features used by the model cover many structural and chemical properties, some key factors, such as defects, impurities, or changes in the microstructure, might have been overlooked, leading to potential biases in the prediction results. Furthermore, this study primarily focused on predicting methane storage capacity and did not analyse other performance metrics, such as adsorption selectivity or kinetics, thus limiting the model's application scope. Moreover, although AutoML tools provide automated modelling, their “black-box” nature might affect the interpretability of the model, which is still essential for understanding the factors influencing performance in scientific research. Finally, while model training and prediction are efficient, in large-scale industrial applications, practical processes such as data collection, model maintenance, and updates still face challenges and need further optimization and validation.
High-throughput workflows allow for the simultaneous exploration of how various pore structures influence gas adsorption and separation performance.76 By simulating thousands of configurations, researchers can rapidly assess how temperature, pressure, and solvent conditions affect material behavior, enabling the quick optimization of properties such as gas storage, separation, and catalysis, without the need for extensive experimental validation. This approach significantly reduces computational costs and accelerates material design.
It should be noted that in Fig. 3 (where Snurr et al. conducted studies on screening MOFs for CO2 adsorption at high humidity) and Fig. 8 (where Lang et al. screened materials for efficient helium separation) earlier in the manuscript, the CoRE MOF database has been briefly mentioned as a core data source. However, the core solution logic of this database for addressing the difficulty in high-throughput screening caused by experimental MOF crystal structures (including solvent molecules, partially occupied sites, etc.) and its supporting value for the reliability of subsequent molecular simulations have not been elaborated. Considering that this database is a key foundation for breaking through the technical bottleneck of MOF high-throughput computational screening, and its structural pretreatment methods and property parameters directly affect the scientificity of simulation results, it is therefore elaborated in detail here in conjunction with Fig. 10 to provide data-level rational support for subsequent studies based on this database.
 | ||
| Fig. 10 Workflow for the CoRE MOF database construction as reported in Snurr's work (reproduced with permission from American Chemical Society, Copyright© 2014).44 | ||
However, in the context of MOFs, experimental refinement of crystal structures often includes solvent molecules, partially occupied sites, or disordered atoms, which presents a significant challenge for high-throughput computational screening. To address this, Snurr et al. developed the computation-ready MOF (CoRE MOF) database, which contains over 4700 experimentally derived porous structures that are immediately suitable for molecular simulations.77 The database includes the atomic coordinates of these structures, along with essential physical and chemical properties such as surface area and pore dimensions, making it a valuable tool for MOF material screening and performance evaluation.
As shown in Fig. 10, to demonstrate the utility of the CoRE MOF database, they performed GCMC simulations of methane adsorption on all the structures in the database. They analysed the structural features influencing methane storage capacity and found that these relationships aligned well with those derived from a large database of hypothetical MOFs. This not only highlights the effectiveness of the database but also showcases that computational simulations can provide physical and chemical property data consistent with experimental results. Furthermore, the database was expanded to include over 5000 computation-ready MOF structures, derived directly from experimental crystal data. By applying efficient algorithms, the database removes solvent molecules and retains charge-balancing ionic species, ensuring that the structures are suitable for atomistic simulations. It is noteworthy that while the initial data for these structures came from the Cambridge structural database (CSD), they were significantly modified to ensure that they were compatible with atomic-level simulations.
Despite the usefulness of the CoRE MOF database, there are still areas for improvement. First, experimental MOF materials may not correspond exactly to the fully desolvated crystal structures reported in the database. Incomplete activation and material defects can significantly reduce the porosity of real materials, impacting their adsorption properties. This discrepancy means that some of the high-performance structures in the CoRE MOF database may not have experimentally reported BET or Langmuir surface areas that match the theoretical values. This limitation indicates that relying solely on experimental data for simulations may not always yield accurate predictions of real material performance.
Second, although the CoRE MOF database provides substantial support for high-throughput screening, its structural diversity is still less than that of hypothetical MOF databases. The MOF structures in the CoRE MOF database are limited in terms of topological variety, which may constrain the predictive capabilities for certain applications. Future research should further investigate the impact of MOF topology on structure–property relationships, particularly whether certain combinations of textural properties are only accessible with specific topologies. These unresolved questions will provide crucial insights for the design of optimal materials and open new directions for future research.78
To address these limitations of CoRE MOF and strengthen the data infrastructure for AI-driven framework material discovery, several recent open-access datasets and tools serve as valuable complements. The ReDD-COFFEE database—featured in Van Speybroeck et al.'s work on CO2 capture-oriented COF screening—contains over 268000 hypothetical COF structures, expanding the exploration of chemical space beyond CoRE MOF's focus on experimentally synthesized MOFs and enabling high-throughput AI modeling for targeted applications. Additionally, the Quantum MOF (QMOF) database offers comprehensive DFT-calculated quantum-chemical data (e.g., electronic structures and stability metrics) for thousands of MOFs, providing atomic-level precision to enhance AI model training accuracy. The MOF Classifier, an open-access machine learning tool, further streamlines AI-driven screening by rapidly predicting key MOF properties (e.g., water stability and gas adsorption selectivity), bridging the gap between structural data and functional performance evaluation.
Moreover, the integration of quantum dynamics, coarse-grained simulations, and ReaxFF simulations within high-throughput approaches further accelerates the optimization process. These simulations provide valuable insights into reaction mechanisms, catalytic performance, and molecular diffusion, enhancing material stability and optimizing performance under real-world conditions. By simplifying molecular models for large-scale systems, coarse-grained simulations facilitate the screening of pore structures, while ReaxFF is used to model dynamic chemical reactions and material stability under catalytic conditions.
The true advantage of high-throughput screening lies in its ability to integrate these advanced techniques into a cohesive framework. This approach allows researchers to systematically screen large material libraries, calculate reaction energetics with DFT, assess material stability with MD, and predict adsorption properties with GCMC—all in a single workflow. As computational power continues to improve, the integration of these techniques will play an increasingly important role in accelerating the design of MOF and COF materials, particularly for energy, environmental, and electronic applications.
Undoubtedly, the combination of high-throughput screening with advanced simulation techniques is transforming the design and optimization of MOFs and COFs. These methods enable fast prediction, screening, and optimization, dramatically accelerating material discovery and reducing the reliance on traditional experimental approaches. As computational capabilities grow, high-throughput simulations will continue to drive faster, more efficient material development in a wide range of applications.
One of the main ways ML is integrated with high-throughput screening is through data-driven performance prediction. Once large datasets of material properties—such as adsorption energy, gas selectivity, or stability—are generated through DFT or MD simulations, ML models can be trained on this data to predict the performance of new materials. This approach allows researchers to quickly identify materials with excellent properties, such as high gas storage capacity or specific gas selectivity, without incurring additional high computational costs.79 ML models can generalize across a wide range of material combinations, thereby accelerating the screening and discovery of potential candidate materials.
Another important application of ML is in material design optimization. ML can identify correlations between material structures—such as pore size, shape, and connectivity—and the desired properties, such as gas storage capacity or catalytic efficiency.64 Once trained, ML models can predict how changes in material composition or structure might improve performance, helping researchers design new MOFs and COFs. With this predictive capability, researchers can rapidly conduct multiple iterations in the design process, reducing the time and cost associated with traditional trial-and-error methods and accelerating material discovery. A typical example is Yıldırım's work, which applied ML to optimize MOFs for methane (CH4) storage and delivery. As shown in Fig. 11, the team constructed a database containing 2224 data points and used decision tree analysis and artificial neural networks (ANNs), along with other data mining tools, to successfully identify underlying patterns. For model validation, the researchers compared their findings with experimental results from the literature, confirming the reliability of the ML models based on structural properties. The study highlighted that structural features, such as pore volume and pore diameter, are crucial factors in predicting MOF performance in CH4 storage and delivery. These factors fall into two categories: user-defined descriptors and structural properties.
 | ||
| Fig. 11 The procedures for decision-tree analysis and artificial neural networks as reported in Yıldırım's work (reproduced with permission from American Chemical Society, Copyright© 2019).64 | ||
Among the user-defined descriptors, the crystal structure (especially tetragonal and cubic structures) was found to be associated with higher CH4 delivery capacity, while the total unsaturation degree was considered an effective indicator of storage capacity. In terms of structural properties, pore volume was identified as one of the most important factors for achieving high CH4 delivery capacity, with larger pore volumes generally corresponding to better storage ability. Maximum pore diameter was also an important parameter affecting CH4 storage capacity.
Additionally, the study showed that high pore volume is a necessary condition for improving performance, and both crystal structure type (especially tetragonal and cubic) and structural properties like pore volume and maximum pore diameter significantly influence CH4 storage and delivery performance. These findings provide guidance for the future design and screening of MOFs, particularly in optimizing pore structures and crystal structures to enhance gas storage performance. This work not only demonstrates the potential of ML in materials science but also provides new insights into the performance prediction of MOF materials. However, despite the significant progress made in this study, optimizing MOF materials still faces several challenges. For example, factors such as the crystal structure and total unsaturation degree, while helpful for performance prediction, require further in-depth exploration to fully understand their comprehensive impact on MOF performance. Future research can continue to expand the application range of ML models and combine more experimental data, to promote the development and practical application of MOF materials in broader fields such as energy storage and gas separation.
Moreover, ML plays a significant role in feature selection and data dimensionality reduction. High-throughput screening often generates large amounts of data, which can be overwhelming and difficult to handle manually. ML techniques can automatically identify the most important features affecting material performance and reduce the data's dimensionality, allowing researchers to focus on key design parameters. This enables more efficient prioritization of the most promising materials and further streamlines the material design process.
ML can also help create surrogate models that approximate the results of expensive simulations, such as DFT or MD.80 Surrogate models run much faster and can help optimize material performance in real-time, providing quick predictions. This approach enables rapid evaluation of material potential without conducting full-scale simulations, greatly enhancing efficiency.
When combined with high-throughput screening, ML also enables an automated material discovery process. Reinforcement learning and active learning techniques can use prior simulation results to select the next materials to simulate, continually improving the selection process. This makes the material design process iterative and dynamic, with ML “learning” from previous results and continuously refining the choice of candidate materials.
In MOF and COF design, ML employs a variety of methods, including supervised learning, unsupervised learning, reinforcement learning, and deep learning (DL). Supervised learning trains models using labelled data (such as known material performance) to predict new material properties. Unsupervised learning helps identify patterns or relationships in the data without predefined outcomes, which aids in uncovering complex correlations between the material structure and performance. Reinforcement learning accelerates material discovery by optimizing design through simulated feedback, while DL, particularly neural networks, can identify subtle relationships between material features and performance when analyzing complex datasets.
The integration of ML with high-throughput screening and molecular simulation techniques brings numerous advantages. It makes material performance prediction faster, reduces reliance on computationally expensive simulations, and helps researchers efficiently handle large and complex datasets.81 Additionally, ML can reveal hidden correlations that traditional modeling methods might miss, driving the discovery of new materials. By continuously optimizing models, ML improves the accuracy of material predictions, further accelerating the design and optimization process.
When combining ML with high-throughput screening, certain challenges do arise, one of the key issues being the quality of the data used for training. If the data are poor or sparse, ML models may make inaccurate predictions, leading to design process errors. While ML models can make predictions quickly, they are often difficult to interpret, especially with complex models like deep neural networks.82 This “black box” issue can limit our understanding of the model results, particularly when trying to explain why certain materials perform well, as there is a lack of transparency.
To address this, advanced tools like SHAP (SHapley Additive exPlanations) and LIME (Local Interpretable Model-agnostic Explanations) can be used to “open the black box.” SHAP and LIME provide both local and global explanations of ML model predictions, helping reveal which features were critical in making a specific prediction.83 These tools allow researchers to better understand why certain MOF or COF materials were selected, enabling more targeted adjustments to the model or material design strategy. Not only does this improve the transparency of the model, but it also provides a deeper understanding of the design process, enhancing the model's usability and credibility. Additionally, overfitting is another challenge in ML, where models perform well on training data but fail to generalize to new materials, potentially impacting prediction accuracy.
Despite these challenges, integrating ML with high-throughput screening and advanced molecular simulation techniques can significantly accelerate the discovery and optimization of MOF and COF materials. By combining simulations and ML, researchers can design materials with specific functions in a much shorter time, speeding up their application in energy, environmental, and electronic fields. As computational power continues to grow and data quality improves, ML will play an increasingly important role in the accelerated design of MOF and COF materials.
With the continuous development of AI, the design methods for MOFs and COFs are evolving toward more efficient, rational, and intelligent approaches.84,85 AI accelerates the material design and discovery process through three core functionalities: automated feature extraction, generative design, and multi-task predictive modeling.
First, AI leverages advanced data processing tools (such as web crawlers, Python data pipelines, etc.) to automatically extract structural features from heterogeneous datasets, converting complex information like metal cluster identities, organic linker geometries, and topological connectivity into high-dimensional feature vectors suitable for ML. This process not only eliminates the reliance on manual descriptor engineering but also systematically captures chemical patterns, providing a solid foundation for subsequent model training and prediction.
Second, AI explores novel chemical spaces through generative design. By learning from existing frameworks, AI generates new chemically plausible candidates. Advanced architectures such as Generative Adversarial Networks (GANs) and variational autoencoders (VAEs) manipulate molecular building blocks within learned chemical spaces, proposing innovative combinations of metal nodes, linkers, and topologies that extend beyond traditional chemical databases and human intuition.86
Finally, AI constructs multi-task predictive models using deep learning techniques, correlating structural descriptors with material properties like gas adsorption capacity, thermal stability, and catalytic activity. These models efficiently identify complex nonlinear relationships between the structure and performance, enabling rapid virtual screening and providing reliable guidance for material synthesis.87
Together, these three functionalities create an integrated workflow: automated feature extraction grounds models in empirical knowledge, generative design explores untapped chemical spaces, and predictive modeling validates functional potential. This streamlined process accelerates material discovery, reducing the timeline from years to weeks, while systematically navigating the vast combinatorial design space of MOFs and COFs.
The synergy between AI and molecular simulations has transformed the design and optimization of MOFs and COFs.46,88,89 This collaboration allows the two methods to complement each other, enhancing both the efficiency and accuracy of material design. AI accelerates the development process by rapidly screening candidate materials, optimizing design workflows, and generating new material candidates. Molecular simulations, on the other hand, provide the essential physical foundation that ensures more accurate and reliable AI predictions, validating the performance of new designs.
Molecular simulation techniques play an indispensable role by offering atomistic-level insights and rigorous quantitative evaluations of material properties, which are crucial for rational design. For instance, DFT calculations provide precise analysis of key characteristics, such as metal–ligand bonding energies, electronic band structures, charge transfer mechanisms, and reaction activation barriers, which are critical for assessing the stability and chemical reactivity of materials under operational conditions.90 MD simulations uncover intricate dynamic behaviors, including framework flexibility, guest-induced structural transitions, and conformational changes during adsorption or catalytic processes, capturing the temporal evolution of systems at femtosecond resolution.
Meanwhile, GCMC simulations utilize statistical mechanics to predict gas adsorption isotherms across different pressures, identify preferential binding sites, quantify adsorption enthalpies, and calculate mixture selectivity. These simulations generate comprehensive thermodynamic metrics essential for optimizing material performance. Together, these integrated computational methods bridge the gap between abstract structural design concepts and tangible functional performance. They establish direct connections between atomic configurations and macroscopic properties through physics-based modeling.
Moreover, these simulations provide critical validation benchmarks for data-driven approaches, offering mechanistic insights that would be difficult to obtain through experimental characterization alone. This collaborative effort creates a foundational knowledge base that supports the development of predictive models, enhancing the accuracy and efficiency of MOF and COF material discovery.
In practical applications, AI can expedite the preliminary screening phase of molecular simulations. Traditional molecular simulations, such as DFT or MD, require substantial computational resources and time. AI, however, can swiftly predict key material properties—such as adsorption energy, stability, and pore structure—based on existing simulation data, screening promising candidates for further detailed simulations. This reduces computational costs and improves screening efficiency.
Molecular simulations provide critical training data for AI, especially in the design of MOFs and COFs.91 These simulations generate vast amounts of data, including molecular structures, adsorption properties, and molecular diffusion behavior. AI models can leverage these data to learn the relationships between the material structure and performance. For instance, stability data from MD simulations allows AI to quickly assess material stability, accelerating the screening process.
AI's role extends beyond prediction and screening—it also optimizes the design process. By employing reinforcement learning (RL) or genetic algorithms (GA), AI can autonomously generate new MOF or COF designs, adjusting them based on feedback from molecular simulations. This “design-test-optimize” cycle enhances the efficiency of material design, eliminating the need for traditional trial-and-error methods.92
Moosavi's research demonstrates the innovative combination of multimodal AI technologies with molecular simulation methods for predicting the properties and applications of MOFs.25 As shown in Fig. 12, the study introduces two AI techniques—transformer models and Convolutional Neural Networks (CNNs)—to process chemical precursor SMILES strings and PXRD spectral data, capturing both the chemical properties and geometric structural information of the materials. This fusion of modalities improves the model's accuracy and adaptability, particularly when handling different types of data. Furthermore, the research employs a self-supervised learning strategy, utilizing large-scale unlabeled data in conjunction with Crystal Graph Convolutional Neural Networks (CGCNNs). This allows the model to learn local chemical environments, significantly improving performance on smaller labeled datasets. This strategy not only reduces reliance on large labeled datasets but also enhances the model's ability to predict MOF properties.
 | ||
| Fig. 12 Workflow for linking MOF synthesis to applications using multimodal ML, as presented in Moosavi's work (reproduced with permission from Springer Nature, Copyright© 2025).82 (a) Schematic of the multimodal machine learning workflow, integrating crystal structure (via OC-CNN), PXRD pattern (via CNN), and precursor (via transformer/processor) modules. It involves pre-training, concatenated embedding, and downstream tasks for property prediction (e.g., gas adsorption, mechanical stability, and band gap) and recommendations (e.g., retrosynthesis and defect engineering). (b) Scatter plot comparing CH4 uptake at 65 bar (mol kg−1) between CSD and CoRE-2019 databases, with SRCC (CSD–CoRE) = 0.73 and relative error ≤ 19%, illustrating the consistency of uptake data. (c) Structural visualizations of MOFs from the CoRE-2019 database, showcasing their framework architectures. (d) Structural visualizations of MOFs from the CSD database, depicting their distinct topological and atomic arrangements. (e) Structure of a solvent-containing MOF. | ||
A groundbreaking innovation in Moosavi's work is the model's ability to predict material properties accurately without relying on complete crystal structure data. Traditional MOF property predictions typically depend on detailed crystal structure information, but Moosavi's model demonstrates that predictions can be made using only PXRD data and chemical precursor information. This accelerates the evaluation of potential applications for new materials. Moreover, the study introduces a “time-travel” validation method, using historical data to predict the future applications of MOF materials. This provides a new perspective on cross-domain applications, such as identifying MOF materials initially designed for other purposes that may show excellent performance in CO2 capture. This approach facilitates the exploration of diverse material applications.
However, despite the strong predictive capabilities, several challenges remain. First, the “black-box” nature of the model limits its interpretability, which is crucial for the design and optimization of MOF materials. Without a clear physical explanation for the model's predictions, researchers may struggle to fully understand the rationale behind the outputs, which can hinder model optimization. Second, the quality and coverage of the crystal structure databases and PXRD spectral data used in the study may affect the model's generalizability. Given the structural complexity and diversity of MOFs, existing databases may not fully represent all types of MOFs, especially those not yet synthesized. Therefore, improving data quality and expanding the database's coverage is essential for enhancing the model's performance. While self-supervised learning and pretraining strategies reduce the need for labeled data, the model still relies on substantial amounts of training data. Its performance may be suboptimal for novel or atypical MOF structures, posing a challenge for future work in adapting the model to accommodate a broader range of structures.
In addition to predictive models, generative AI techniques, such as generative adversarial networks (GANs), are advancing the discovery of new materials. These models generate new material candidates, which can be validated through molecular simulations. The AI-generated models are then optimized based on the simulation results, accelerating material discovery and revealing structural features and performance advantages that traditional methods may overlook.
For example, MOFs and COFs are widely used for gas storage applications, such as hydrogen, methane, and carbon dioxide storage, due to their tunable pore structures. MD simulations provide detailed information on the diffusion, adsorption, and desorption of gas molecules within the material's pores. However, running full MD simulations for each candidate material can be computationally expensive. By training ML models on large MD datasets, AI learns the relationship between the material structure and gas adsorption performance, enabling the rapid prediction of gas adsorption capabilities. This allows for efficient screening of potential candidates for further validation via MD simulations.
The combination of AI and molecular simulations not only accelerates the design of MOFs and COFs but also ensures the physical plausibility and accuracy of predictions.93,94 By rapidly screening potential materials, optimizing designs, and validating simulations, the synergy between AI and molecular simulations significantly accelerates the discovery of novel materials, unlocking vast opportunities in energy, environmental, and electronic applications.
The synergistic mechanism between AI and molecular simulations operates as a sophisticated closed-loop framework that integrates database construction, computational screening, predictive modeling, and experimental validation into a continuous design cycle. Initially, a comprehensive multi-source database is established by consolidating heterogeneous experimental literature, high-throughput characterization data, and curated simulation outcomes, ensuring broad coverage of structural and property spaces. This database serves as the foundation for subsequent computational screening, which employs hierarchical simulations. Techniques such as coarse-grained DFT, GCMC, and MD are used to rapidly narrow down candidate materials by evaluating their fundamental stability and baseline performance metrics.95
AI then processes these high-dimensional datasets through methods like graph neural networks and multi-task learning architectures, uncovering complex patterns and establishing quantitative structure–property relationships across multiple performance objectives. Promising candidates identified through ML models undergo further refinement using fine-grained DFT validation, ensuring that predicted properties are thermodynamically feasible and accurate at an electronic structure level before moving to experimental synthesis. Following synthesis, the materials undergo comprehensive characterization, and the resulting performance data are systematically fed back into the central database and predictive models.
This iterative feedback mechanism enhances model accuracy and gradually expands the design space. The closed-loop system continually incorporates lessons from previous iterations, systematically reducing empirical uncertainty and significantly accelerating discovery timelines—by orders of magnitude compared to traditional methods. The integration ensures that each cycle of the design process benefits from prior knowledge, leading to increasingly precise navigation of the complex MOF and COF design landscape, which ultimately accelerates material discovery and optimization.
AI multi-task networks and other advanced architectures significantly streamline the material screening process. By simultaneously predicting multiple properties—such as gas storage capacity, catalytic efficiency, thermal stability, and water stability—AI captures the interdependencies between these properties, thereby enhancing the accuracy and efficiency of material design. This approach shifts material design from a traditional trial-and-error method to an active, prediction-driven process, enabling researchers to systematically explore the complex design space of MOFs and COFs, quickly identify novel materials, and move material research toward rational design.
The ML method proposed by Jiang et al. to predict and accelerate the discovery of water-stable MOFs is closely related to the application of AI multi-task networks. As presented in Fig. 13, the authors first constructed the largest water stability database to date, which includes 1133 synthesized MOFs, categorized according to their experimental stability in aqueous solutions and water vapor, forming the foundation for the ML model.96 By combining structural and chemical descriptors, Jiang et al. developed a random forest classifier specifically for predicting MOF water stability under different water exposure conditions. This method ensures the robustness and adaptability of the model, which performs with high prediction accuracy and excellent transferability in out-of-sample validation.
 | ||
| Fig. 13 Workflow to predict water stability of MOFs as reported in Jiang's work (reproduced with permission from Wiley-VCH GmbH, Copyright© 2024).87 (a) Data collection, distinguishing water-stable (S) and unstable (U) MOFs with their structural schematics. (b) Featurization, including global descriptors (pore size, surface area, void fraction, etc.) and building unit-based descriptors (metal node and organic linker characteristics). (c) Data processing, categorizing S and U MOFs, followed by oversampling for the U group to balance the dataset. (d) Prediction, presenting the percentage of MOF water stability prediction results (e.g., bar chart for different MOF categories and heatmap for prediction distribution). (e) Model training, employing an ensemble learning approach with majority voting to train the predictive model for MOF water stability. | ||
The identification of key factors further validates the application of AI in material design. The study reveals that the water stability of MOFs is closely related to multiple factors, such as high surface area, narrow pore sizes, metals with larger atomic mass and radius, and the connectivity and electronegativity variation of organic linkers. These factors not only enhance the understanding of material behavior but also provide a framework for designing MOFs with stronger water stability. Jiang et al.‘s work aligns closely with the core concept of AI multi-task networks. Both approaches aim to capture the interrelationships between various material properties, thereby improving the accuracy and efficiency of material design. Through ML, Jiang et al. accelerated the discovery of MOFs, further demonstrating the powerful potential of AI in material design and offering new tools and insights for material research, driving the shift from traditional trial-and-error methods to intelligent, prediction-driven design approaches.
In conclusion, the integration of AI with molecular simulations creates a synergistic ecosystem where molecular simulations provide high-fidelity atomic-level data essential for training and validating AI models. Conversely, AI accelerates the pre-screening phase, improving design efficiency and reducing computational costs. The use of AI generative models further supports the design of new materials. As computational capabilities advance, the integration of AI and molecular simulations will become increasingly crucial in MOF and COF design, expediting the discovery of new materials and meeting the demands of more complex applications.
3 Typical case of digital and intelligent design in framework materials
Building on the previous framework, the concept of the digital-intelligent design framework emerges. This new research paradigm transforms the design process of MOFs and COFs from traditional, experience-based trial-and-error methods to a more systematic and intelligent approach that integrates molecular simulations, experimental data, and advanced AI technologies.97 In this framework, “digital” refers to the vast amounts of data derived from molecular simulations, topological information, experimental measurements, and materials performance data extracted from the literature. These diverse data sources are integrated into a structural database for MOFs and COFs, built using high-throughput simulations, providing comprehensive support and a solid data foundation for material design. For instance, MD simulations offer data on material stability and diffusion behaviours, while DFT provides precise electronic structure information. These datasets serve as rich sources for training and validating AI models.“Intelligence” refers to the wisdom extracted through AI and ML algorithms, based on the trial-and-error experience accumulated by researchers in the early stages of the design process. In this framework, AI algorithms not only automatically identify the potential and performance of materials but also optimize the design process through DL, reinforcement learning, and other advanced techniques. The AI-driven optimization process dynamically adjusts design strategies based on different application needs, quickly identifying and selecting high-performance MOF and COF candidates. Through this approach, digital-intelligent design accelerates both the discovery and optimization of MOFs and COFs, enabling intelligent control over the material design process through continuous iterative learning. This dramatically enhances the accuracy and efficiency of the entire design process.
This digital-intelligent design paradigm opens a new model for the development of MOF and COF materials, driving rapid progress in applications such as energy storage, environmental protection, catalytic reactions, and industrial processes. By leveraging efficient data-driven design and AI-powered optimization, digital-intelligent design accelerates material development, making it both faster and more efficient while better addressing diverse and complex application demands. With the ongoing advancements in data science and computational capabilities, the digital-intelligent design framework will continue to lead the development of MOFs and COFs into an era of greater intelligence and precision.
A prime example of digital-intelligent design in the development of MOFs is Berend Smit's groundbreaking work. Limiting the increase in CO2 in the atmosphere is one of the greatest challenges of our generation, directly contributing to global warming and climate change. As a result, the development of effective CO2 capture and storage technologies is critical for mitigating current emissions. Among the promising materials for CO2 capture, MOFs have emerged as a key candidate due to their tunable structures, high surface areas, and large pore volumes.98
While many MOFs exhibit significant performance in CO2/N2 separation under ideal conditions, their efficiency drops dramatically when exposed to real-world flue gases, particularly those containing water vapor. Water competes with CO2 for the same adsorption sites, causing MOFs to lose selectivity and reducing their overall separation performance. Thus, the task of screening MOFs that can maintain high CO2 capture efficiency in the presence of water remains labour-intensive and resource-consuming. Smit's work addresses this challenge by employing a novel approach that combines computational screening and data mining techniques, utilizing a database of over 300000 MOFs (Fig. 14). Through data mining, the researchers identified a variety of strong CO2-binding sites, which they termed “adsorbaphores.” These adsorbaphores endow MOFs with CO2/N2 selectivity in wet flue gases, effectively solving the problem of water interference.
 | ||
| Fig. 14 Computational screening of MOFs for strong CO2 adsorption and selectivity as reported in Smit's work (reproduced with permission from Springer Nature, Copyright© 2019).53 (a) Scatter plot illustrating the distribution of MOFs regarding CO2/N2 selectivity and related performance metrics, with a color gradient representing a specific parameter (e.g., CO2 adsorption capacity). (b) Scatter plot of MOF performance parameters (e.g., CO2 adsorption capacity vs. another descriptor), with data points categorized by different groups (e.g., metal nodes or linker types). (c) Schematic of a MOF's crystal structure, highlighting its pore architecture and potential CO2 adsorption sites. (d) Gas adsorption isotherms of CO2 and H2O as a function of relative humidity, accompanied by molecular structure illustrations, demonstrating the adsorption behavior of a MOF. (e) Gas adsorption isotherms of CO2 and H2O as a function of relative humidity for another MOF, depicting its uptake features under different humidity conditions. (f) Structural visualization of a MOF in the dry state, showcasing its framework architecture. (g) Structural visualization of a MOF in a humid environment, illustrating the incorporation of adsorbed water molecules and their impact on the framework. | ||
The research not only successfully identified these adsorbaphores but also synthesized two water-stable MOFs incorporating the most hydrophobic adsorbaphores. These MOFs exhibited outstanding CO2 capture performance, maintaining efficiency even in the presence of water, and outperforming several commercial materials.
By combining computational screening with data mining, Smit's work significantly accelerated the discovery and optimization of MOFs with ideal performance. Moreover, this study showcases the potential of the digital-intelligent design framework, which integrates molecular simulations, experimental data, and AI technologies, to expedite material discovery and optimization. This innovative approach has made MOF material development more efficient and capable of addressing increasingly complex application needs, laying a solid foundation for the continued advancement of CO2 capture and storage technologies.
Additionally, as shown in Fig. 15, Jiang et al. presented an innovative multiscale computational screening approach to identify fluorinated metal–organic frameworks (FMOFs) with high CO2 capture performance from wet flue gas. The research team systematically screened 5061 FMOFs and shortlisted 19 top candidates, demonstrating the potential of fluorinated MOFs for CO2 capture in humid environments. By calculating the geometric properties, pore size, and water adsorption heat, the study first identified FMOFs with suitable pore sizes and weak water affinity, laying the foundation for subsequent performance screening.
 | ||
| Fig. 15 Workflow for multiscale computational screening of hydrostable fluorinated MOFs for CO2 capture from a wet flue gas as reported in Jiang's work (reproduced with permission from American Chemical Society, Copyright© 2024).69 (a) Multiscale screening workflow schematic, featuring layers of fluorinated MOFs (FMOFs) with stepwise reduction in quantity: starting from 16441 FMOFs (screened by |ΔG| < 5.0 Å3), 7138 FMOFs (with a CO2/N2 separation metric), 6782 FMOFs (with Q_CO2 < 42 wt%), 5061 FMOFs (via GCMC simulation), top 19 FMOFs (via FPMD simulation), and finally hydratable FMOFs. (b) Scatter plot illustrating the correlation between two performance metrics of FMOFs (e.g., CO2/N2 separation ability and another descriptor), with a color gradient representing a specific parameter. (c) Scatter plot depicting the distribution of FMOFs based on CO2 adsorption capacity and related performance indicators, distinguished by color or category. (d) Schematic illustration of the hydrostable fluorinated MOF's structure and mechanism for CO2 capture from wet flue gas, showing steps like hydrolysis, CO2 adsorption, and framework interactions. | ||
A key contribution of this work is the use of GCMC simulations to evaluate the adsorption of CO2/N2/H2O mixtures (at 60% relative humidity) across the FMOF library. Among the 19 top candidates, Cu-based FMOFs were found to have the highest adsorption performance. The study also revealed the importance of the position, rather than the amount, of F atoms in CO2 adsorption. Moreover, FMOFs with nitrogen-containing pillar groups demonstrated enhanced selectivity for CO2 adsorption in the presence of humidity. Furthermore, Jiang et al. confirmed the hydrostability of these top FMOFs through first-principles molecular dynamics (FPMD) simulations. The results showed that these materials maintain excellent structural stability even in the presence of co-adsorbed CO2 and H2O. One of the notable findings of this study is the strong interaction between CO2 and the F atom, which effectively traps CO2 within the framework, particularly in FMOFs with nitrogen-decorated pillars. Additionally, as observed in VOFFIVE-3_Fe with pyrazine, the pore size of the framework is adjusted via the rotation of pyrazine, allowing it to preferentially accommodate CO2 over H2O.
Jiang et al.69 provided important theoretical insights into CO2 capture by fluorinated MOFs in humid environments. By revealing the impact of the position, rather than the quantity, of F atoms on CO2 adsorption performance, and highlighting the superior selective adsorption ability of FMOFs with nitrogen-containing pillars under humid conditions, this study offers valuable guidance for the future development of hydrostable CO2 capture materials suitable for real-world industrial conditions. However, it is worth noting that while the study primarily focuses on material properties such as adsorption capacity and selectivity, the authors emphasize that future research should integrate process and system-level optimization for a more holistic evaluation of these materials' practical application potential.
The present study demonstrates that large-scale, database-driven simulations can reveal adsorption and catalytic mechanisms that are difficult to uncover through conventional small-scale experiments or single-case studies. In our previous work, we constructed a dataset comprising 10994 M-Salen-COF structures, which were optimized using MD methods. Subsequently, the excess adsorption amounts of CO2 under ambient conditions were simulated for these structures using GCMC simulations. Analysis of the simulation results revealed significant differences in the adsorption performance among the COFs, providing a basis for further investigation of the pore enrichment effect. As shown in Fig. 16, COFs with high adsorption capacity exhibit pore architectures and functional groups that interact strongly with CO2 molecules, leading to local concentration and pronounced pore enrichment, which enhances both adsorption performance and catalytic activity.
 | ||
| Fig. 16 Workflow of GCMC simulations for screening COF candidates for CO2 fixation based on a dataset of 10994 M-Salen-COF structures (reproduced with permission from Springer Nature, Copyright© 2020).99 | ||
In contrast, low-adsorption COFs show a relatively uniform distribution of CO2, with weak molecular interactions that fail to effectively enrich the gas; additionally, limited pore accessibility or reduced interlayer spacing further restricts CO2 accumulation. These observations indicate that the pore enrichment effect is a key factor in improving CO2 adsorption and storage in porous materials.
Based on these findings, we defined the concept of the pore enrichment effect, whereby the pore structure and functional groups within the framework exert characteristic adsorption interactions with gas molecules, significantly increasing the local concentration of CO2 within the pores and promoting catalytic reactions in confined environments. Guided by this insight, we synthesized a COF predicted to exhibit the strongest pore enrichment effect, denoted as Zn-Salen-COF-SDU113, and evaluated its catalytic performance in the coupling reaction between CO2 and terminal epoxides.
Remarkably, under ambient conditions, Zn-Salen-COF-SDU113 achieved a CO2 conversion yield of 98.2% with a turnover frequency (TOF) of 3068.9, comparable to the best-performing catalysts reported globally for this type of reaction. Furthermore, Zn-Salen-COF-SDU113 represents the first porous material to catalyse the reaction between CO2 and 2, 3-epoxybutane at room temperature and atmospheric pressure. These experimental results not only validate the role of the pore enrichment effect in promoting catalytic reactions within porous materials but also establish a closed-loop approach from theoretical prediction to experimental verification, in which large-scale, database-driven simulations directly guide the rational design and synthesis of high-performance COF catalysts for CO2 fixation.
Building upon the fourth phase of autonomous AI-driven design, recent advances in prompt engineering have further expanded the capabilities of MOF and COF research. By carefully crafting prompts to guide large language models (LLMs) and generative AI, researchers can extract design strategies, predict structural properties, and even propose synthetic routes, complementing traditional computational and experimental approaches while bridging the gap between human expertise, data-driven simulations, and experimental validation.100 The integration of prompt engineering provides an additional layer of acceleration, enabling more efficient generation of candidate structures, proposal of functional modifications, and informed decision-making in material discovery. The design and synthesis of MOFs traditionally rely heavily on experimental literature and existing data. However, with the exponential growth of publications, manually extracting and organizing key information has become time-consuming, labor-intensive, and inefficient. This approach limits researchers' ability to rapidly access synthesis conditions, performance data, and structural information, thereby hindering high-throughput design and systematic optimization. To address this issue, conventional natural language processing (NLP) methods have been employed for literature data extraction. Nevertheless, these approaches typically require solid programming skills, as well as expertise in computer science and data science, and often need to be redesigned or reprogrammed for different research objectives, limiting their generalizability and scalability. Collectively, these challenges make the rapid and accurate extraction of MOF synthesis and performance information from vast literature a critical bottleneck for materials discovery and intelligent design.
A representative example is the work reported by Yaghi and co-workers, who developed an efficient workflow for MOF design by integrating text mining, ML, and AI tools.101 As shown in Fig. 17, they employed prompt engineering to guide ChatGPT in automatically extracting MOF synthesis conditions from the literature, capable of handling diverse formats and styles of scientific articles. This workflow enabled parsing, searching, screening, classifying, summarizing, and structuring the information. Using this approach, the team successfully extracted 26257 unique MOF synthesis parameters covering approximately 800 MOFs, and rigorously evaluated the text-mining results using precision, recall, and F1-score metrics, all achieving 90–99% accuracy.
 | ||
| Fig. 17 The ChatGPT chemistry assistant workflow reported in Yaghi's work (reproduced with permission from American Chemical Society, Copyright© 2023).102 | ||
The systematically organized data were compiled into a structured synthesis database, laying the foundation for subsequent data-driven simulations and ML models, which exemplifies the “data” aspect of digital-intelligent design. Traditional MOF design relies heavily on human experience and trial-and-error, which is time-consuming and may overlook latent patterns. In contrast, this approach enables a closed loop from design to experimental validation through systematic data and intelligent modelling. Based on these data-driven models, it is possible not only to predict crystallization outcomes but also to identify key factors influencing synthesis, thereby guiding the rational design of new MOFs and optimization of synthesis conditions. This method significantly improves design efficiency and provides a feasible paradigm for the digital-intelligent design of MOFs and other framework materials, accelerating the transformation from data to knowledge to experimental verification, and marking a shift in MOF research from experience-driven to data and intelligence-driven design. Prompt engineering can facilitate the discovery of unique physicochemical properties of framework materials, particularly in terms of crystallinity and the realization of high performance. The Yaghi group first constructed a dataset of metal–organic framework (MOF) linkers and employed a fine-tuned GPT assistant to mutate and modify existing linker structures, thereby proposing new MOF linker designs.103 This strategy enabled the discovery of high-performance water-harvesting MOFs: the resulting 10 Long-Arm MOFs (LAMOFs) set new benchmarks in water uptake (up to 0.64 g g−1) and operational humidity range (13–53%).
Furthermore, the Yaghi group further developed a multi-AI-driven laboratory system using ChatGPT and Bayesian optimization.96,103 The system, comprising seven large language model-based assistants and ML algorithms, can coordinate multiple tasks in a chemistry laboratory. It accelerated the optimization of microwave synthesis conditions for MOF-321, MOF-322, and COF-323, enhancing crystallinity while achieving desired porosity and water uptake. Within the workflow, different AI assistants handle strategy planning, literature search, coding, robotic operation, labware design, safety inspection, and data analysis, providing comprehensive support to human researchers. By reducing human biases in experimental screening and balancing exploration and exploitation of synthesis parameters, the Bayesian search efficiently identified optimal conditions from a vast pool, enabling a single researcher collaborating with AI to reach productivity comparable to a full traditional research team. This work demonstrates the potential of AI in MOF/COF synthesis and provides a compelling example of laboratory automation and intelligent research.
Another representative work is from the Li group, who developed a method for constructing a knowledge graph of framework materials based on large language models (LLMs).84 Framework materials exhibit significant potential in structural diversity, precise pore regulation, and functional modification. However, as research progresses, the volume of related literature has rapidly increased, and the information has become highly fragmented, posing challenges for researchers in systematically accessing and utilizing the vast data. Li's group analysed over 100000 publications on various types of framework materials to build a knowledge network containing 2.53 million nodes and 4.01 million relationships. LLMs were employed to automatically extract key information, perform semantic analysis, and conduct logical reasoning, converting scattered and unstructured data into a structured knowledge graph. This approach greatly enhanced information integration efficiency, reduced manual curation efforts, and provided researchers with a clear and navigable knowledge network.104
Furthermore, they combined the knowledge graph with LLMs to develop a question-answering system called Qwen2-KG (Fig. 18). In the field of framework materials, the system achieved an accuracy of 91.67%, far exceeding other models such as GPT-4 (33.33%). Importantly, as is shown in Fig. 1, Qwen2-KG provides precise information sources, ensuring reliability and traceability of answers, and offers an efficient and trustworthy tool for knowledge retrieval and decision-making in research.105
 | ||
| Fig. 18 A comparison of the use of a knowledge graph reported in Li's work (reproduced with permission from Springer Nature, Copyright© 2025).84 | ||
This work holds significant implications for the digital-intelligent design of framework materials. By constructing a large-scale, structured, and searchable knowledge network, researchers can systematically access dispersed information on synthesis conditions, structural features, and performance data, providing a solid foundation for data-driven design. Coupled with the LLM-based question-answering system, it not only enhances the efficiency of information utilization but also ensures data reliability, supporting rapid decision-making and high-throughput screening. This approach integrates human expertise, experimental data, and intelligent algorithms, exemplifying the convergence of “data” and “intelligence” and establishing a new paradigm and set of tools for the digital and intelligent design of framework materials.106–109
4 Key challenges in digital-intelligent design of MOFs and COFs
Despite the remarkable progress in integrating AI, molecular simulations, and high-throughput screening for MOF/COF design, several critical challenges persist, hindering the translation of computational predictions to practical material development. These challenges, previously mentioned in scattered sections, are systematically discussed herein.4.1. Inconsistent data quality and limited data scalability
The reliability of data-driven models (e.g., ML/DL for performance prediction) heavily depends on the quality, consistency, and coverage of training data. However, existing datasets for MOFs/COFs often suffer from three issues: (1) structural inconsistency—experimental crystal structures may contain residual solvents, partial occupancy, or disordered atoms (e.g., in CoRE MOF database preprocessing), while hypothetical structures lack experimental validation; (2) property measurement variability—gas adsorption capacity, catalytic activity, and stability are often measured under different experimental conditions (e.g., temperature, pressure, and humidity), leading to non-comparable data; (3) data sparsity for novel systems—high-throughput simulations (e.g., DFT and GCMC) are computationally expensive, resulting in limited data for emerging functionalized MOFs/COFs (e.g., fluorinated MOFs and M-Salen-COFs). Recent studies have highlighted that such data inconsistencies can reduce ML model accuracy by up to 20–30%.87 Notably, efforts to address this challenge include the development of unified data standards (e.g., including experimental metadata in databases) and hybrid data generation methods (combining DFT with semi-empirical simulations to expand datasets). For example, a study on MOF water stability prediction demonstrated that integrating curated experimental data (from the ARC-MOF database) with DFT-calculated structural descriptors reduced prediction error to <13%, underscoring the value of high-quality data integration.87,1104.2. Limited model interpretability (the “black-box” problem)
Advanced AI models (e.g., deep neural networks and GANs) exhibit high predictive accuracy for MOF/COF properties (e.g., gas selectivity and thermal stability), but their “black-box” nature limits mechanistic understanding—critical for rational material design. For instance, while RF models achieved R2 = 0.97 for COF-based ethane/ethylene separation prediction, identifying the specific role of pore size vs. functional groups required additional SHAP/LIME analysis, increasing research complexity.30,83 Similarly, multimodal AI models for MOF property prediction (e.g., using PXRD and precursor data) can predict adsorption performance without crystal structures, but the lack of clear structure–property relationship interpretation hinders the optimization of synthesis strategies.82Recent advances in explainable AI (XAI, also referred to as interpretable AI) have shown promise. For example, Wang et al. applied random forest (RF) models to screen propane-selective MOFs from the CoRE MOF database, and their analysis revealed two key advances for improving model interpretability: (1) quantifying the relative importance of descriptors (Henry coefficient ratio S0 accounted for 36.89% and adsorption heat difference ΔQst accounted for 13.61%), directly linking structural/energy parameters to C3H8/C3H6 separation performance; (2) establishing clear descriptor ranges (e.g., pore limiting diameter 3.5–6.5 Å; largest cavity diameter 4.8–8.0 Å) for pre-screening, avoiding the “black-box” issue of unguided model predictions. This work proved that ML models can balance prediction accuracy (R2 = 0.83) and mechanistic interpretability, providing a reference for solving the disconnection between models and physics in MOF design.111
4.3. Gap between computational simulations and practical synthesis
Molecular simulations (e.g., DFT, MD, and GCMC) provide valuable insights into MOF/COF stability and performance, but their predictions often deviate from experimental results due to simplified assumptions: (1) simulation condition mismatch—DFT calculations typically use idealized structures (e.g., defect-free lattices) and vacuum environments, while experiments involve impurities, solvents, and dynamic conditions; (2) kinetic vs. thermodynamic bias—GCMC simulations focus on thermodynamic adsorption equilibrium, but practical synthesis and application depend on kinetic factors (e.g., nucleation rate and guest diffusion); (3) scalability limitations—quantum dynamics simulations, though critical for electronic processes (e.g., catalytic electron transfer), are restricted to small systems (<1000 atoms), making them incompatible with high-throughput screening of large MOF/COF libraries.58,112This gap is particularly evident in hypothetical material design: for example, 268000 hypothetical COFs in the ReDD-COFFEE database were screened for post-combustion CO2 capture, but only ∼5% were experimentally synthesizable, with most failing due to poor hydrothermal stability under industrial flue gas conditions.66
5 Conclusions
In summary, the rational design of MOFs and COFs is rapidly moving toward a digital-intelligent paradigm, integrating data-driven analysis with AI to shift from empirical trial-and-error to predictive and intelligent design. The vast chemical configuration space and complex multi-parameter synthesis conditions of porous framework materials pose challenges for traditional approaches, whereas digital-intelligent strategies combine high-throughput experiments, molecular simulations (e.g., DFT, GCMC, and MD), and ML/DL to enable predictive synthesis, performance optimization, and inverse design, significantly accelerating materials discovery and property control. Case studies demonstrate that workflows integrating experimental and simulation data within a digital-intelligent framework allow rapid screening of high-performance materials and morphology control while enhancing model interpretability and reliability. Despite the remaining challenges, such as data quality, model generalization, and the gap between computational predictions and practical synthesis, future efforts in expanding high-quality databases, developing interpretable AI algorithms, and establishing autonomous experimental platforms will position digital-intelligent design as a central methodology in framework material research. Looking ahead, deep digital-intelligent integration will foster the convergence of AI, multiscale simulations, and automated experimentation, enabling autonomous materials design and discovery, and providing efficient, intelligent solutions for energy, catalysis, environmental applications, and beyond—heralding a new era in porous materials research.6 Outlook
In future work, we plan to address two critical technical bottlenecks with actionable research directions. First, for database expansion, we will construct a heterogeneous MOF/COF database that integrates experimental data (e.g., in situ structural changes under gas adsorption and long-term stability tests) and multi-scale simulation results (DFT and MD). This database will specifically include edge cases like defective structures and performance under extreme conditions (high pressure and humidity), solving the current scarcity of diverse, validated data for robust ML model training.Second, to enhance algorithm interpretability and tackle black-box limitations, we will develop physics-informed interpretable frameworks by fusing quantum chemical descriptors (e.g., d-band centre and adsorption energy decomposition) with graph neural networks. Specifically, we will integrate SHAP analysis with first-principles calculations to decode structure–property relationships, enabling rational material design. Additionally, we will focus on cross-scale validation between theory and experiments for targeted applications (e.g., helium capture in industrial flue gas and CO2 reduction in alkaline electrolytes), bridging the gap between computational predictions and real-world performance.
Author contributions
Bing Ma: conceptualization, writing – original draft, visualization. Na Qin: writing – original draft, investigation, data curation. Qianqian Yan: writing – original draft, investigation, formal analysis. Wei Zhou: writing – review & editing, supervision, project administration, funding acquisition. Sheng Zhang: writing – review & editing, validation. Xiao Wang: writing – review & editing, supervision, resources. Lipiao Bao: writing – review & editing, methodology. Xing Lu: writing – review & editing, supervision, conceptualization, funding acquisition. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
There are no conflicts to declare.Data availability
No primary research results, software, or code have been included, and no new data were generated or analyzed as part of this review.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (Grant No. 22401062, 92261204 and 22431005), the Innovational Fund for Scientific and Technological Personnel of Hainan Province (KJRC2023C10), and the Hainan Provincial Natural Science Foundation of China (Grant No. 225QN212).References
- X. Y. Yang, L. H. Chen, Y. Li, J. C. Rooke, C. Sanchez and B. L. Su, Chem. Soc. Rev., 2017, 46, 481–558, 10.1039/C6CS00829A.
- R. L. Siegelman, E. J. Kim and J. R. Long, Nat. Mater., 2021, 20, 1060–1072, DOI:10.1038/s41563-021-01054-8.
- S. Li, Y. G. Chung and R. Q. Snurr, Langmuir, 2016, 32, 10368–10376, DOI:10.1021/acs.langmuir.6b02803.
- J. K. Wychowaniec, H. Saini, B. Scheibe, D. P. Dubal, A. Schneemann and K. Jayaramulu, Chem. Soc. Rev., 2022, 51, 9068–9126, 10.1039/D2CS00585A.
- Z. A. Sandhu, M. A. Raza, N. S. Awwad, H. A. Ibrahium, U. Farwa, S. Ashraf, A. Dildar, E. Fatima, S. Ashraf and F. Ali, Mater. Adv., 2024, 5, 30–50, 10.1039/D3MA00822C.
- G. Singh, J. Lee, A. Karakoti, R. Bahadur, J. Yi, D. Zhao, K. AlBahily and A. Vinu, Chem. Soc. Rev., 2020, 49, 4360–4404, 10.1039/D0CS00075B.
- J. Letwaba, U. O. Uyor, M. L. Mavhungu, N. O. Achuka and P. A. Popoola, RSC Adv., 2024, 14, 14233–14253, 10.1039/D4RA00865K.
- B. Li, H. M. Wen, W. Zhou, J. Q. Xu and B. Chen, Chem, 2016, 1, 557–580, DOI:10.1016/j.chempr.2016.09.009.
- Z. Duma, P. R. Makgwane, M. Masukume, A. Swartbooi, K. Rambau, T. Mehlo and T. Mavhungu, Mater. Today Sustain., 2024, 27, 100812, DOI:10.1016/j.mtsust.2024.100812.
- N. Keller and T. Bein, Chem. Soc. Rev., 2021, 50, 1813–1845, 10.1039/D0CS00793E.
- S. S. A. Shah, M. S. Javed, T. Najam, M. A. Nazir, A. ur Rehman, A. Rauf, M. Sohail, F. Verpoort and S. J. Bao, Mater. Today, 2023, 67, 229–255, DOI:10.1016/j.mattod.2023.05.023.
- H. Wang, H. Wang, Z. Wang, L. Tang, G. Zeng, P. Xu, M. Chen, T. Xiong, C. Zhou and X. Li, Chem. Soc. Rev., 2020, 49, 4135–4165, 10.1039/D0CS00278J.
- E. Pérez Botella, S. Valencia and F. Rey, Chem. Rev., 2022, 122, 17647–17695, DOI:10.1021/acs.chemrev.2c00140.
- H. Furukawa, K. E. Cordova, M. O'Keeffe and O. M. Yaghi, Science, 2013, 341, 1230444, DOI:10.1126/science.1230444.
- K. Li, J. Yang and J. Gu, Acc. Chem. Res., 2022, 55, 2235–2247, DOI:10.1021/acs.accounts.2c00262.
- S. Wuttke, Angew. Chem., 2019, 131, 14162, DOI:10.1002/ange.201906230.
- Y. Liu, X. Liu, A. Su, C. Gong, S. Chen, L. Xia, C. Zhang, X. Tao, Y. Li, Y. Li, T. Sun, M. Bu, W. Shao, J. Zhao, X. Li, Y. Peng, P. Guo, Y. Han and Y. Zhu, Chem. Soc. Rev., 2024, 53, 502–544, 10.1039/D3CS00287J.
- D. Morgan and R. Jacobs, Annu. Rev. Mater. Res., 2020, 50, 71–103, DOI:10.1146/annurev-matsci-070218-010015.
- S. Amini and S. Mohaghegh, Fluids, 2019, 4, 126, DOI:10.3390/fluids4030126.
- Y. Hong, B. Hou, H. Jiang and J. Zhang, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2020, 10, e1450, DOI:10.1002/wcms.1450.
- F. Noé, A. Tkatchenko, K. R. Müller and C. Clementi, Annu. Rev. Phys. Chem., 2020, 71, 361–390, DOI:10.1146/annurev-physchem-042018-052331.
- S. Axelrod, D. Schwalbe Koda, S. Mohapatra, J. Damewood, K. P. Greenman and R. Gómez Bombarelli, Acc. Mater. Res., 2022, 3, 343–357, DOI:10.1021/accountsmr.1c00238.
- E. Prašnikar, M. Ljubič, A. Perdih and J. Borišek, Artif. Intell. Rev., 2024, 57, 102, DOI:10.1007/s10462-024-10731-4.
- A. Vishnyakov, Materials, 2025, 18, 534, DOI:10.3390/ma18030534.
- S. M. Moosavi, F. Daeyaert, M. W. Deem and G. Sastre, AI-guided Des. Prop. Predict. Zeolites Nanoporous Mater., 2023, pp. 411–424, DOI:10.1002/9781119819783.ch15.
- Z. Han, Y. Yang, J. Rushlow, J. Huo, Z. Liu, Y. C. Hsu, R. Yin, M. Wang, R. Liang and K. Y. Wang, Chem. Soc. Rev., 2025, 54, 367–395, 10.1039/D4CS00432A.
- E. Xie, X. Wang, J. I. Siepmann, H. Chen and R. Q. Snurr, Digital Discovery, 2025, 4, 2336–2363, 10.1039/D5DD00221D.
- K. T. Butler, D. W. Davies, H. Cartwright, O. Isayev and A. Walsh, Nature, 2018, 559, 547–555, DOI:10.1038/s41586-018-0337-2.
- H. Lyu, Z. Ji, S. Wuttke and O. M. Yaghi, Chem, 2020, 6, 2219–2241, DOI:10.1016/j.chempr.2020.08.008.
- X. Cao, Z. Zhang, Y. He, W. Xue, H. Huang and C. Zhong, Ind. Eng. Chem. Res., 2022, 61, 11116–11123, DOI:10.1021/acs.iecr.2c01385.
- B. Kim, S. Lee and J. Kim, Sci. Adv., 2020, 6, eaax9324, DOI:10.1126/sciadv.aax9324.
- P. C. St John, C. Phillips, T. W. Kemper, A. N. Wilson, Y. Guan, M. F. Crowley, M. R. Nimlos and R. E. Larsen, J. Chem. Phys., 2019, 150, 234111, DOI:10.1063/1.5099132.
- S. M. Moosavi, K. M. Jablonka and B. Smit, J. Am. Chem. Soc., 2020, 142, 20273–20287, DOI:10.1021/jacs.0c09105.
- P. S. Gromski, A. B. Henson, J. M. Granda and L. Cronin, Nat. Rev. Chem., 2019, 3, 119–128, DOI:10.1038/s41570-018-0066-y.
- X. Bai and X. Zhang, Nano Micro Lett., 2025, 17, 135, DOI:10.1007/s40820-024-01634-8.
- H. C. Gulbalkan, G. O. Aksu, G. Ercakir and S. Keskin, Ind. Eng. Chem. Res., 2023, 63, 37–48, DOI:10.1021/acs.iecr.3c03817.
- H. Daglar, H. C. Gulbalkan, N. Habib, O. Durak, A. Uzun and S. Keskin, ACS Appl. Mater. Interfaces, 2023, 15, 17421–17431, DOI:10.1021/acsami.3c02130.
- Z. Zheng, O. Zhang, C. Borgs, J. T. Chayes and O. M. Yaghi, J. Am. Chem. Soc., 2023, 145, 18048–18062, DOI:10.1021/jacs.3c05819.
- Z. Zheng, O. Zhang, H. L. Nguyen, N. Rampal, A. H. Alawadhi, Z. Rong, T. Head Gordon, C. Borgs, J. T. Chayes and O. M. Yaghi, ACS Cent. Sci., 2023, 9, 2161–2170, DOI:10.1021/acscentsci.3c01087.
- C. Altintas and S. Keskin, Mater. Today Energy, 2023, 38, 101426, DOI:10.1016/j.mtener.2023.101426.
- G. Férey, Chem. Soc. Rev., 2008, 37, 191–214, 10.1039/B618320B.
- A. Sturluson, M. T. Huynh, A. R. Kaija, C. Laird, S. Yoon, F. Hou, Z. Feng, C. E. Wilmer, Y. J. Colón and Y. G. Chung, Mol. Simul., 2019, 45, 1082–1121, DOI:10.1080/08927022.2019.1648809.
- Z. Shi, W. Yang, X. Deng, C. Cai, Y. Yan, H. Liang, Z. Liu and Z. Qiao, Mol. Syst. Des. Eng., 2020, 5, 725–742, 10.1039/D0ME00005A.
- Y. G. Chung, J. Camp, M. Haranczyk, B. J. Sikora, W. Bury, V. Krungleviciute, T. Yildirim, O. K. Farha, D. S. Sholl and R. Q. Snurr, Chem. Mater., 2014, 26, 6185–6192, DOI:10.1021/cm502594j.
- X. Bai, Z. Shi, H. Xia, S. Li, Z. Liu, H. Liang, Z. Liu, B. Wang and Z. Qiao, Chem. Eng. J., 2022, 446, 136783, DOI:10.1016/j.cej.2022.136783.
- K. Neikha and A. Puzari, Langmuir, 2024, 40, 21957–21975, DOI:10.1021/acs.langmuir.4c03126.
- E. Issaka, M. Adams, J. Baffoe, E. Danso Boateng, L. Melville and A. Fazal, Clean Technol. Environ. Policy, 2025, 27, 833–860, DOI:10.1007/s10098-024-03102-8.
- D. Wu, N. Gu, J. Yao, Y. Cao, L. Wang, I. Shakir, Y. Sun and Y. Xu, Chem. Sci., 2025, 16, 5447, 10.1039/d5sc00109a.
- H. Vardhan, G. Rummer, A. Deng and S. Ma, Membranes, 2023, 13, 696, DOI:10.3390/membranes13080696.
- Z. Cao, R. Magar, Y. Wang and A. Barati Farimani, J. Am. Chem. Soc., 2023, 145, 2958–2967, DOI:10.1021/jacs.2c11420.
- V. Bernales, M. A. Ortuño, D. G. Truhlar, C. J. Cramer and L. Gagliardi, ACS Cent. Sci., 2018, 4, 5–19, DOI:10.1021/acscentsci.7b00500.
- R. Jose, G. Bangar, S. Pal and G. Rajaraman, J. Chem. Sci., 2023, 135, 19, DOI:10.1007/s12039-022-02130-5.
- P. G. Boyd, A. Chidambaram, E. García Díez, C. P. Ireland, T. D. Daff, R. Bounds, A. Gładysiak, P. Schouwink, S. M. Moosavi and M. M. Maroto Valer, Nature, 2019, 576, 253–256, DOI:10.1038/s41586-019-1798-7.
- R. Goeminne, L. Vanduyfhuys, V. Van Speybroeck and T. Verstraelen, J. Chem. Theory Comput., 2023, 19, 6313–6325, DOI:10.1021/acs.jctc.3c00495.
- W. Zhou, L. Yang, X. Wang, W. Zhao, J. Yang, D. Zhai, L. Sun and W. Deng, JACS Au, 2021, 1, 1497–1505, DOI:10.1021/jacsau.1c00258.
- S. Xu, Z. Chen, M. Qin, B. Cai, W. Li, R. Zhu, C. Xu and X. D. Xiang, npj Comput. Mater., 2024, 10, 194, DOI:10.1038/s41524-024-01386-4.
- Y. Liu, N. Li, X. Cui, W. Yan, J. Su and L. Jin, Membranes, 2022, 12, 1274, DOI:10.3390/membranes12121274.
- I. Choudhuri, J. Ye and D. G. Truhlar, Chem. Phys. Rev., 2023, 4, 031304, DOI:10.1063/5.0153656.
- O. F. Altundal, Z. P. Haslak and S. Keskin, Ind. Eng. Chem. Res., 2021, 60, 12999–13012, DOI:10.1021/acs.iecr.1c01742.
- Z. Zhang, A. S. Palakkal, X. Wu, J. Jiang and Z. Jiang, Environ. Sci. Technol., 2025, 59, 9123–9133, DOI:10.1021/acs.est.5c00768.
- S. Kancharlapalli and R. Q. Snurr, ACS Appl. Mater. Interfaces, 2023, 15, 28084–28092, DOI:10.1021/acsami.3c04079.
- M. Xu, Z. Liu, X. Huai, L. Lou and J. Guo, RSC Adv., 2020, 10, 34621–34631, 10.1039/D0RA06363K.
- G. O. Aksu and S. Keskin, J. Mater. Chem., 2023, 11, 14788–14799, 10.1039/D3TA02433D.
- Z. Gulsoy, K. B. Sezginel, A. Uzun, S. Keskin and R. Yıldırım, ACS Comb. Sci., 2019, 21, 257–268, DOI:10.1021/acscombsci.8b00150.
- S. Han and J. Kim, ACS Omega, 2023, 8, 4278–4284, DOI:10.1021/acsomega.2c07517.
- J. S. De Vos, S. Ravichandran, S. Borgmans, L. Vanduyfhuys, P. Van Der Voort, S. M. J. Rogge and V. Van Speybroeck, Chem. Mater., 2024, 36, 4315–4330, DOI:10.1021/acs.chemmater.3c03230.
- W. Temmerman, R. Goeminne, K. S. Rawat and V. Van Speybroeck, Adv. Mater., 2024, 2412005, DOI:10.1002/adma.202412005.
- R. Batra, C. Chen, T. G. Evans, K. S. Walton and R. Ramprasad, Nat. Mach. Intell., 2020, 2, 704–710, DOI:10.1038/s42256-020-00249-z.
- A. S. Palakkal, S. A. Mohamed and J. Jiang, Chem Bio Eng., 2024, 1, 970–978, DOI:10.1021/cbe.4c00111.
- S. A. Mohamed, D. Zhao and J. Jiang, Commun. Mater., 2023, 4, 79, DOI:10.1038/s43246-023-00409-9.
- Y. Lang, L. Sun, Y. Pan, M. Xu, D. Zhai, J. Liu, W. Deng and L. Yang, Chem. Eng. Sci., 2025, 305, 121175, DOI:10.1016/j.ces.2024.121175.
- G. Greene Diniz, D. Z. Manrique, W. Sennane, Y. Magnin, E. Shishenina, P. Cordier, P. Llewellyn, M. Krompiec, M. J. Rančić and D. Muñoz Ramo, EPJ Quantum Technol., 2022, 9, 37, DOI:10.1140/epjqt/s40507-022-00155-w.
- C. M. S. Alvares and R. Semino, J. Chem. Theory Comput., 2025, 21, 3142–3155, DOI:10.1021/acs.jctc.4c01341.
- Y. Yang, Y. K. Shin, H. Ooe, X. Yin, X. Zhang, A. C. T. van Duin, Y. Murase and J. C. Mauro, J. Phys. Chem. B, 2023, 127, 6374–6384, DOI:10.1021/acs.jpcb.3c00563.
- H. C. Gulbalkan, A. Uzun and S. Keskin, Appl. Phys. Lett., 2024, 124, 200501, DOI:10.1063/5.0202175.
- E. Ren, P. Guilbaud and F. X. Coudert, Digital Discovery, 2022, 1, 355–374, 10.1039/D2DD00018K.
- Y. G. Chung, E. Haldoupis, B. J. Bucior, M. Haranczyk, S. Lee, H. Zhang, K. D. Vogiatzis, M. Milisavljevic, S. Ling and J. S. Camp, J. Chem. Eng. Data, 2019, 64, 5985–5998, DOI:10.1021/acs.jced.9b00835.
- S. Chibani and F.-X. Coudert, Machine Learning Approaches for the Prediction of Materials Properties, APL Mater., 2020, 8, 080701, DOI:10.1063/5.0018384.
- H. Tang, L. Duan and J. Jiang, Langmuir, 2023, 39, 15849–15863, DOI:10.1021/acs.langmuir.3c01964.
- P. Yang, H. Zhang, X. Lai, K. Wang, Q. Yang and D. Yu, ACS Omega, 2021, 6, 17149–17161, DOI:10.1021/acsomega.0c05990.
- N. S. Bobbitt and R. Q. Snurr, Mol. Simul., 2019, 45, 1069–1081, DOI:10.1080/08927022.2019.1597271.
- S. T. Khan and S. M. Moosavi, Nat. Commun., 2025, 16, 5642, DOI:10.1038/s41467-025-60796-0.
- M. M. Hasan, J. Sci. Technol. Res., 2023, 5, 17–26, DOI:10.59738/jstr.v5i1.23(17-26).eaqr5800.
- X. Bai, S. He, Y. Li, Y. Xie, X. Zhang, W. Du and J. R. Li, npj Comput. Mater., 2025, 11, 51, DOI:10.1038/s41524-025-01540-6.
- Z. Zheng, A. H. Alawadhi, S. Chheda, S. E. Neumann, N. Rampal, S. Liu, H. L. Nguyen, Y. h. Lin, Z. Rong, J. I. Siepmann, L. Gagliardi, A. Anandkumar, C. Borgs, J. T. Chayes and O. M. Yaghi, J. Am. Chem. Soc., 2023, 145, 28284–28295, DOI:10.1021/jacs.3c12086.
- H. Wang, Y. Li, X. Xuan, K. Wang, Y. f. Yao and L. Pan, Environ. Sci. Technol., 2025, 59, 6361–6378, DOI:10.1021/acs.est.5c00390.
- Z. Zhang, F. Pan, S. A. Mohamed, C. Ji, K. Zhang, J. Jiang and Z. Jiang, Small, 2024, 20, 2405087, DOI:10.1002/smll.202405087.
- N. Rabiee, Clin. Transl. Discovery, 2023, 3, e207, DOI:10.1002/ctd2.207.
- J. Cao, L. Zhou, F. Gan and Z. You, JOM, 2025, 77, 1452–1465, DOI:10.1007/s11837-024-07065-5.
- S. D. Harding, T. Liu, L. Chen, S. Yang, I. Borne, T. Fellowes, A. W. Peters, S. C. Weston, J. W. Ward and A. I. Cooper, J. Am. Chem. Soc., 2025, 147, 23160–23169, DOI:10.1021/jacs.5c06861.
- C. Ji and K. Zhang, J. Mater. Sci., 2024, 59, 9371–9383, DOI:10.1007/s10853-024-09744-9.
- M. Wu, S. Yan and J. Ren, npj Comput. Mater., 2025, 11, 106, DOI:10.1038/s41524-025-01583-9.
- M. Chafiq, A. Chaouiki and Y. G. Ko, Adv. Compos. Hybrid Mater., 2024, 7, 222, DOI:10.1007/s42114-024-01044-9.
- W. Chen, M. Li, T. Yao, J. Liu, S. Jia, Z. Gao and J. Gong, Ind. Eng. Chem. Res., 2024, 63, 18241–18262, DOI:10.1021/acs.iecr.4c03224.
- A. K. Mishra and Y. Kan, Int. J. Artif. Intell. Sci., 2025, 1, DOI:10.63619/ijais.v1i1.005.
- Y. Zhao, Y. Zhao, J. Wang and Z. Wang, Ind. Eng. Chem. Res., 2025, 64, 4637–4668, DOI:10.1021/acs.iecr.4c04636.
- A. S. Rosen, J. M. Notestein and R. Q. Snurr, Curr. Opin. Chem. Eng., 2022, 35, 100760, DOI:10.1016/j.coche.2021.100760.
- Z. Zhang, X. Cao, C. Geng, Y. Sun, Y. He, Z. Qiao and C. Zhong, J. Membr. Sci., 2022, 650, 120399, DOI:10.1016/j.memsci.2022.120399.
- W. Zhou, Q. W. Deng, G. Q. Ren, L. Sun, L. Yang, Y. M. Li, D. Zhai, Y. H. Zhou and W. Q. Deng, Nat. Commun., 2020, 11, 4481, DOI:10.1038/s41467-020-18154-9.
- W. Yuan, G. Chen, Z. Wang and F. You, Adv. Mater., 2025, 2502771, DOI:10.1002/adma.70084.
- L. Gagliardi and O. M. Yaghi, Chem. Mater., 2023, 35, 5711–5712, DOI:10.1021/acs.chemmater.3c01706.
- Y. Su, X. Wang, Y. Ye, Y. Xie, Y. Xu, Y. Jiang and C. Wang, Chem. Sci., 2024, 15, 12200–12233, 10.1039/D3SC07012C.
- Z. Zheng, Z. Rong, N. Rampal, C. Borgs, J. T. Chayes and O. M. Yaghi, Angew. Chem., Int. Ed., 2023, 62, e202311983 CrossRef CAS PubMed.
- Y. Ye, J. Ren, S. Wang, Y. Wan, I. Razzak, B. Hoex, H. Wang, T. Xie and W. Zhang, Adv. Neural Inf. Process. Syst., 2024, 37, 56878–56897 Search PubMed.
- G. Zhao, L. M. Brabson, S. Chheda, J. Huang, H. Kim, K. Liu, K. Mochida, T. D. Pham, Prerna, G. G. Terrones, S. Yoon, L. Zoubritzky, F.-X. Coudert, M. Haranczyk, H. J. Kulik, S. M. Moosavi, D. S. Sholl, J. I. Siepmann, R. Q. Snurr and Y. G. Chung, Matter, 2025, 8, 102140, DOI:10.1016/j.matt.2025.102140.
- M. Gibaldi, A. Kapeliukha, A. White, J. Luo, R. A. Mayo, J. Burner and T. K. Woo, Chem. Sci., 2025, 16, 4085–4100, 10.1039/d4sc07438.
- X. Chen and R. L. Martin, Charge Model 5: An Extension of Hirshfeld Population Analysis for the Accurate Description of Molecular Interactions in Gaseous and Condensed Phases, J. Chem. Theory Comput., 2015, 11(12), 5275–5291, DOI:10.1021/ct600487c.
- D. Ongari, L. Talirz and B. Smit, Too Many Materials and Too Many Applications: An Experimental Problem Waiting for a Computational Solution, ACS Cent. Sci., 2020, 6, 1890–1900, DOI:10.1021/acscentsci.0c00988.
- X. Jin, K. M. Jablonka, E. Moubarak, Y. Li and B. Smit, MOFChecker: An Algorithm for Validating and Correcting Metal-Organic Framework (MOF) Structures, ChemRxiv, 2025, preprint, DOI:10.26434/chemrxiv-2025-bh607Chibani.
- P. Krokidas, S. Karozis, G. Giannakopoulos, S. Moncho, M. E. Kainourgiakis, I. G. Economou and T. A. Steriotis, J. Mater. Chem. A, 2022, 10, 13697–13703, 10.1039/d2ta02624d.
- Y. Wang, Z.-J. Jiang, D.-R. Wang, W.-G. Lu and D. Li, Machine Learning-Assisted Discovery of Propane-Selective Metal−Organic Frameworks, J. Am. Chem. Soc., 2024, 146, 6955–6961, DOI:10.1021/jacs.3c14610.
- Y. Wang, Z.-J. Jiang, W. Lu and D. Li, Angew. Chem., Int. Ed., 2025, 64, e202500783, DOI:10.1002/anie.202500783.
| This journal is © The Royal Society of Chemistry 2026 |