
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Predicting polyacrylate–microplastic interactions with atomistic simulation
Timothy M. E.
Jugovic
a,
Henry E.
Thurber
 b,
Michael T.
Robo
a,
Woojung
Ji
a,
Madeline E.
Clough
a,
Anne J.
McNeil
ab and
Paul M.
Zimmerman
*a
b,
Michael T.
Robo
a,
Woojung
Ji
a,
Madeline E.
Clough
a,
Anne J.
McNeil
ab and
Paul M.
Zimmerman
*a
aDepartment of Chemistry, University of Michigan, 930 North University Avenue, Ann Arbor, Michigan 48109-1055, USA. E-mail: paulzim@umich.edu
bMacromolecular Science and Engineering Program, University of Michigan, 2800 Plymouth Road, Ann Arbor, Michigan 48109-2800, USA
First published on 9th December 2025
Abstract
Adhesive-coated substrates have been used to non-selectively capture microplastics (MPs) in water. This work explores whether selective MP capture is plausible by modifying the adhesive structure. To find the degree of selectivity, adhesive strength might be estimated by examining surface interactions between specific adhesives and MPs. Herein, we describe the use of atomistic simulations to predict the aqueous adhesive strength via the aqueous work-of-adhesion (WoA(aq)), a thermodynamic measure of adhesion. Simulation of four common MPs and five different polyacrylate adhesives were conducted. The simulations show that fluorinated sidechains, due to their low interfacial energies and higher surface energies, exhibit increased selectivity toward polystyrene over other plastics. To provide experimental support for these predictions, probe-tack studies of aqueous adhesion were performed and found to agree with the simulations. Simulations also revealed non-intuitive interactions that govern WoA(aq), arising from the complex intra- and intermolecular interactions that occur when polyacrylates interface with water and MPs. This expanded understanding of the microscopic features of adhesion can be used to design next-generation adhesives and MP remediation technology.
Introduction
Acrylate polymers are versatile materials used in industrial and commercial applications.1 Products include Plexiglas (poly(methyl methacrylate)), water-soluble thickeners (sodium polyacrylate) and adhesives (poly(cyanoacrylate) and poly(2-ethylhexyl acrylate)).2,3 These applications are possible due to the acrylate functionality, which contains an ester-linked side-chain, permitting modular tuning of the polymer properties, including polarity, mass, and volume. As an example, polymers with mixed side-chains like poly(methyl/t-butyl acrylate) have higher work of adhesion (WoA), a measure of the adhesive strength, than either homopolymer.4 Adhesive applications range from mild pressure-sensitive adhesives (PSAs) such as poly(2-ethylhexyl acrylate) with a WoA ∼20–30 mN m−1, to strong cyanoacrylate adhesives with a WoA >100 mN m−1.1,5 Similarly, the aqueous work of adhesion (WoA(aq)), or the strength of an adhesive in water, is important for applications in water and has previously been determined for an epoxy adhesive/carbon reinforced fiber composite interface at ∼22 mN m−1.6Microplastics (MPs) are a pervasive form of plastic pollution and are difficult to remediate due to their small size and heterogenous composition.7–14 Moreover, environmental degradation of MPs into even smaller nanoplastics is a growing concern.15 Capturing microplastics before they are released into the environment is the best option. Once captured, those MP could be recycled into new plastic products. However, the physical separation of different types of microplastics would be challenging given their small size.16–18 Slight differences in MP properties might allow for adhesives to selectively bind them, providing a means to simultaneously capture and separate MPs by type. Acrylate polymers are potential materials for this purpose because their adhesive strength can be tailored with structural modifications. A common way to predict adhesion is to compare surface tensions. The surface tension represents the energy of a given surface when in contact with another material (i.e., liquid). Adhesion occurs between two materials when the surface tension is reduced. For plastics, surface tension (γ) spans from non-polar, at ∼15 mN m−1, to polar, ∼40 mN m−1,19,20 where common MP materials (polyethylene, polystyrene, polyamides, and polyesters21) are in the range of 15–30 mN m−1.22 Unfortunately, the small range in γ mean plastic surfaces are more alike in adhesive properties than they are different.23 For context, the γ of water is 72 mN m−1,24 and common metal surfaces have γ of 500–1000 mN m−1.25 Selectivity in adhering to less distinct surfaces therefore requires exploiting slight differences in composition that might bias intermolecular interactions, such as π-stacking or H-bonding, between the adhesive and MPs. These intermolecular interactions can also lead to deformation of the adhesive–MP interface, leading to changes in polymer conformation and further contributing to changes in adhesive properties. Examining the precise polyacrylate compositions necessary to achieve such selectivity is a significant undertaking. By developing models that are better able to predict adhesive behavior, this process could be accelerated.
The relationship that determines adhesion between surfaces26 can be expressed via the difference between their individual surface tensions and their combined interfacial tension, which gives the following equation:
| WoA(aq) = γadh-H2O + γplas-H2O − γadh-plas | (1) |
We hypothesized that polyacrylate compositions can be tailored to enable selectivity in binding specific MPs. Acrylate polymers can contain a range of side chains, from non-polar alkyl and fluorocarbon groups to highly polar carboxylate groups. This range provides a large design space. Achieving selectivity would require tuning side chain characteristics, such as polarity, volume, and mass, all of which play a role in determining polymer properties.34 In addition, the polymer length can also affect the γ, as longer polymers have different cohesive behavior.35 Similarly, emergent properties are more difficult to predict. Nevertheless, these interactions may become predictable through computational modeling efforts, as described below.
Computational methods, such as atomistic molecular dynamics (MD) simulations, can be used to build predictive models of polymer structure, behavior, and properties. The most common use of MD simulation is analysis of structure and function of proteins and peptides.36–38 This focus has led to most MD forcefields being optimized for amino acids, and other biologically relevant species. Commonly modeled protein interactions, such as quaternary structures, are analogous to adhesion between polymers and microplastics.39 Also, MD forcefields have been constructed to simulate interfacial adhesion40,41 as well as behavior and properties of plastic polymers.42–49 Recent work on polyacrylate adhesives highlights the power of MD simulation to elucidate properties that are normally difficult to determine using experimental methods.5 These previous works inform how interactions between polymers result in unexpected behaviors. Conceptual understanding of how these behaviors arise will enable better predictive models of novel polyacrylate adhesives. To develop design principles for polyacrylates with selective plastic adhesive properties, we examine structure–property relationships that connect polymer composition with WoA to MPs. The models are comprised of pairs of polymer chains that interact via surface contacts, allowing prediction of the microscopic interactions that lead to adhesion at the bulk level. To ensure the reliability of these atomistic models, tests against known literature quantities were conducted and then applied to determine behaviors that govern adhesion. These models provide qualitatively useful concepts for tailoring interactions between acrylates and common MP types, including non-obvious behaviors for fluorine-containing acrylates.
Methods
Polymer generation
Novel polyacrylate compositions were generated using an in-house polymer generation code (see SI for further details). Each polyacrylate strand was constructed from a racemic mixture of 2-ethylhexyl acrylate side chains and a different side chain (R*) with a composition indicated by its % R* value (see Chart 1). Each generated polymer strand was relaxed in vacuo to a final simulation structure in 1 ns CHARMM MD simulations. For MP surfaces, CHARMM-GUI was used to generate polymer strands of PA-6, HDPE, and PS of sufficient size to approximate macroscopic properties, which were annealed into MP particles of around 10–100 nm3 in size in 1 ns CHARMM-MD simulations (see SI, Section 2 for more details).50 These surfaces are representative of pristine MP models, lacking in situ changes or modifications that may occur in the environment.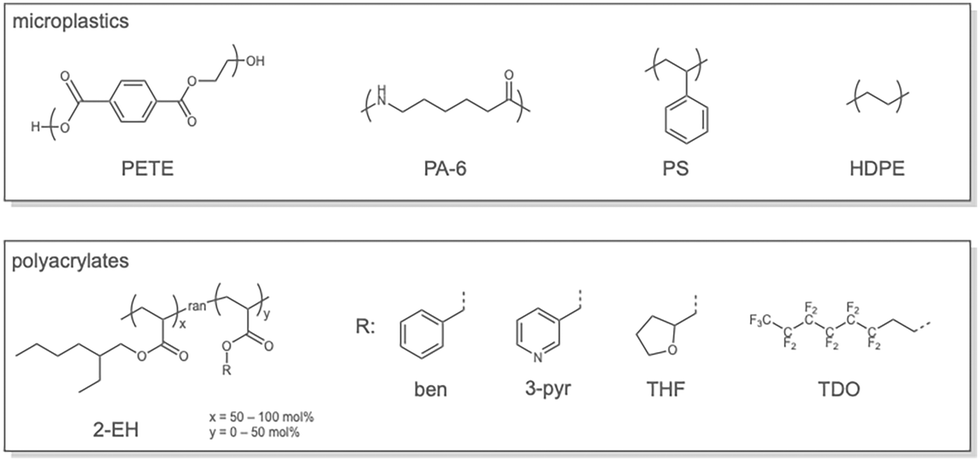 | ||
| Chart 1 Structures of tested MPs and polyacrylates. | ||
Force field validation
The CHARMM general force field version 4.0 (cgenff) was used to model the interactions, as prior work had parameterized force fields for similar atom types.42–49 The forcefields were tested by comparison to known literature values, in particular the density and aqueous interfacial surface tension (γSL). Table S2 shows the computed and literature aqueous interfacial surface tension (γSL with SL for solid–liquid) between water and surfaces. Literature values are derived from contact angle (θSL) and surface energy (γSG with SG for solid-gas) using the Young-Dupré equation.28 For all examined MPs, γSL are within 1–2 standard errors of literature or experimental values.51–55 The values of γSL are not available for the novel polyacrylates, so the estimated density is used as an alternative comparison (Table S1). Literature values for density of R:R* mixed polyacrylates were estimated using the weighted average of the known pure values. Similar agreement is found between the computed and estimated experimental density. Because density is intrinsically tied to the degree of crystallinity, this agreement (Table S1) suggests the MP models are reasonable. The agreement between these values and experiment, combined with the initial parameter validation done for cgenff force field for these bond types, give a measure of confidence in the reliability of our computational results.Statistical validation
An important concern when analyzing mixed polyacrylates is to sample multiple polymers of the same composition to arrive at meaningful averages for their properties. Another possible statistical issue is clustering of R* groups within the polymer, resulting in structures that resemble block copolymers more closely than random copolymers. To avoid these issues in simulations, six versions of each % R* composition were independently generated using an algorithm that randomly distributed the desired % R* sidechains throughout the structure. These structures are kept at a fixed molecular mass, and polymeric length was varied as composition varied, around 150–300 monomeric units. Each of these six chains was then independently equilibrated for 250 ns for the aqueous and MP boxes. After equilibration, trajectories were simulated for 5 ns with a frame taken every 200 ps to determine an average γ value for each case. This resulted in 60 simulations for each % composition that were then averaged to determine the WoA(aq). Error was quantified as the standard deviation of the combined simulation data for each data point. Outliers were identified using the Q-test and excluded from the reported results. Further description of methods can be found in the SI.Results and discussion
Our discussion is delineated in four general areas. First, the choice of specific polyacrylates and microplastics to represent realizable copolymers and environmentally relevant plastic types is outlined. Next, the WoA is determined from MD simulations for each microplastic across the span of copolymer compositions. Then, the same atomistic models are examined to determine the acrylate properties that affect adhesive ability for varying polymeric composition. Finally, adhesive simulations are analyzed for behaviors or trends that could be used to separate MPs by plastic type using adhesion.Chemical space: MPs and acrylates
Four types of plastic were chosen to represent some of the most abundant microplastics in the environment (Chart 1).8 In particular, high-density polyethylene (HDPE), polystyrene (PS), nylon-6 (PA-6), and polyethylene terephthalate (PETE). These plastics also span the polarity space of common plastics from purely nonpolar (HDPE) to moderately polar (PA-6, PS). Here, polarity refers to the fraction (in %) of surface energy attributed to polar components (e.g., dipoles) compared to the total energy, which also includes dispersive components.34 The reported polarity values for the four plastics are shown in Table 1.34 Beyond the polarity criterion, crystallinity was another property of general interest. In our atomistic models, HDPE and PETE are crystalline at standard temperature and pressure, PA-6 is amorphous, and PS is atactic and therefore amorphous. These microstructures are characteristic of MP waste.8| Polymer | γ d (mN m−1) | γ p (mN m−1) | % Polarity |
|---|---|---|---|
| a Estimated as an average of literature values for pure compositions. | |||
| Adhesives | |||
| 2-EH | 27.1 | 2.2 | 7.5 |
| 50% bena | 28 | 2.2 | 7.3 |
| 50% 3-pyra | 28 | 6.75 | 19.4 |
| 50% THFa | 23.05 | 5.9 | 20.5 |
| 50% TDOa | 20.5 | 1.8 | 8.1 |
| Microplastics | |||
| PA-652 | 30.3 | 5.5 | 15.4 |
| PS51 | 34.5 | 6.1 | 15.0 |
| HDPE53 | 25.9 | 0 | 0.0 |
| PETE53 | 39.3 | 4.2 | 9.7 |
A range of adhesive compositions was created to bind MPs, with polyacrylates containing 2-EH side chains as the baseline case (Chart 1). This polymer was chosen because it was shown to bind MPs under aqueous conditions.50 Derivatization by side-chain substitution should lead to different adhesive properties, which may enable selective MP binding. From Table 1 the percent polarities of acrylates go from 7.3% for poly(benzyl acrylate) (ben), which less than 2-EH at 7.5%, to 20.5% for THF. If polarity correlates with adhesive preference, polar adhesives should bind more strongly to polar MPs and vice versa. Both 3-pyridyl (3-pyr) and THF sidechains were chosen to form hydrogen bonds (H-bonds), an important consideration for aqueous adhesion. As plastics like PA-6 form H-bonds between amide groups, H-bonding acrylates could substitute for intra- and interchain H-bonds and enhance surface adhesion by more than would be expected from polarity considerations alone. Similarly, 3-pyr and ben have aromatic rings, which could boost adhesion to similar MPs (e.g., PS) via pi-stacking. Finally, the C–F bonds of poly(tridecafluorooctyl acrylate) (TDO) is more hydrophobic,56 which could enhance binding to the non-polar MPs (e.g., HDPE) compared to polar ones. Together with the four distinct MPs, and the five side-chains under consideration, we anticipated differences in adhesion. Snapshots of the equilibrated MP-adhesive structures are in Fig. 1. Because the MP and adhesive atomistic models are of similar size, the surface contacts are mostly geometrically flat. Further analysis reveals that side-chain clustering should not be a concern in these surface contacts, as the side-chain composition sampling was performed randomly (SI, Section 2).
 | ||
| Fig. 1 (Top) 100% 2-EH adhesive interacting with microplastics. (Bottom) Contact area, average interfacial surface tension, and aqueous work of adhesion for the same surfaces. Averages were determined by periodic sampling of MD simulations with error from standard deviation. | ||
Aqueous work-of-adhesion
Aqueous WoA was determined through MD simulations for all four MP types combined with the acrylate copolymers. Fig. 1 shows the mean WoA(aq) for a given adhesive composition interacting with each MP, whereas Fig. 2 provides the WoA as a function of degree of substitution. For copolymers containing ben, THF, and 3-pyr side chains, the WoA increases with degree of substitution compared to 100% 2-EH (Fig. 2A, C and D), meaning that these side chains bind more strongly than the 100% 2-EH baseline.19 In contrast, the trend in WoA reverses with TDO substitution, showing a decreasing trend (Fig. 2B). Of the four side chains, THF resulted in the largest WoA(aq) for the four MP types. While all four tested polyacrylates exhibit a preference for PS over other MP types, only TDO shows increasing selectivity with TDO composition. This preference can be seen in Fig. 2B, where at 50% TDO composition the WoA(aq) gap between PS and HDPE has increased from ∼10 mN m−1 to ∼20 mN m−1. | ||
| Fig. 2 WoA(aq) results by R* percent composition compared to microplastic surfaces. (A) ben, (B) 3-pyr, (C) THF, (D) TDO. | ||
Overall, the results can be used to check the hypotheses from the previous section. First, the degree of interaction between MP and adhesive is uncorrelated to adhesive side-chain aromaticity, as the slopes for PS and PETE are similar to HDPE and PA-6 for the ben-substituted acrylate. Similarly, the hypothesized H-bonding between THF and 3-pyr with PA-6 was not observed, as evidenced by PA-6 remaining the lowest WoA(aq) regardless of adhesive composition. Similarly, no correlation between percent polarity and WoA(aq) was found (nor between the individual polar and dispersive surface energy values that determine percent polarity). For example, PS and PA-6 have similar % polarity values, yet the WoA(aq) values are quite different regardless of the polyacrylate tested. Similarly, the more polar THF-based polymer exhibits the highest WoA(aq) for all tested MPs irrespective of their polarity. Quantitative comparisons of % polarity versus WoA(aq) can be found in the SI (Fig. S6–S9).
Structural changes of adhesives in aqueous conditions
To better understand how adhesives perform in water, several qualitative and quantitative metrics related to adhesion were examined. The equilibrated polyacrylate structures with 50% R* side chain compositions in the gas phase are compacted to maximize internal dispersive and polar interactions and minimize surface exposure (Fig. S10). Such behavior is generally expected under vacuum as there are no stabilizing surface interactions between vacuum and polymer. As seen in Fig. 3A, the polyacrylates undergo significant changes in the aqueous phase. For example, the TDO-substituted polyacrylate is still relatively compact whereas the THF-substituted polyacrylate is highly disrupted in aqueous simulations (e.g., see hole formation). Similar deformations can be observed for 3-pyr, though to a lesser degree (Fig. S10). The TDO and ben-substituted polyacrylates distort in shape but still maintain somewhat compacted geometries (Fig. S6). These qualitative morphological changes suggest that there may be substantial differences in surface-area-to-volume ratios (SA![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :V) among different adhesives. Indeed, there was a quite significant increase in surface area for all adhesives relative to their initial conformations, as polymers containing 50% THF, TDO, 3-pyr and ben increased by ∼27%, ∼15%, 24%, and 16%, respectively, under aqueous conditions (Fig. S6).
:V) among different adhesives. Indeed, there was a quite significant increase in surface area for all adhesives relative to their initial conformations, as polymers containing 50% THF, TDO, 3-pyr and ben increased by ∼27%, ∼15%, 24%, and 16%, respectively, under aqueous conditions (Fig. S6).
 | ||
| Fig. 3 (A) Representative structures of in aqueous simulation structures for 50% TDO (top) and THF (bottom) polyacrylates after simulation. (B). Interaction area versus γ(adh-H2O) for representative polyacrylate adhesives, intercept is set to zero as non-interacting surfaces have no interaction energy. | ||
Further examining the relationship between SA:V and γ(adh-H2O) is worthwhile to explain changes to the adhesives occurring in the aqueous phase. We found that the γ(adh-H2O) increases as SA:V increases, as highlighted in Fig. 3B. While all the materials follow roughly the same relationship between SA:V and γ(adh-H2O) (R2 = 0.997), there are nuances worth noting. For instance, H-bond acceptor acrylates have significant changes in SA:V and γ(adh-H2O) compared to non-H-bond acceptor acrylates. The H-bond acceptor 3-pyr acrylate at 50% R* composition shows 25% increases in SA:V and γ(adh-H2O) over the 100% 2-EH case. Conversely, for ben, which does not have an H-bond acceptor, the trend is somewhat steeper, as SA:V increases only ∼2% at 50% R*, with a 10% increase in γ(adh-H2O). This result indicates that H-bond accepting acrylates undergo greater morphological changes in the aqueous phase than non-acceptors. In contrast to the other three acrylate types, polymers with TDO sidechains decrease in SA:V and in γ(adh-H2O) with increasing % R*. This trend can be attributed to the highly hydrophobic fluorocarbons, which resist contact with water. Overall, there is a notable SA:V correlation to γ(adh-H2O), and H-bonding appears to play a significant role in this relationship.
As hypothesized, H-bonds substantially influence polyacrylate behavior under aqueous conditions. Shown in Fig. 4A is the H-bond formation percentage (see SI, Section 10 for details on how H-bonds were assigned) for THF and 3-pyr side chains, which describes the ratio of H-bonds present compared to the total possible number of H-bonds for a given % R* composition. This metric is not a measure of H-bonding strength, but rather the quantity. In the model, THF H-bonds were longer and weaker than their 3-pyr counterparts (Fig. 4A), which is expected given the weaker H-bond accepting strength of an ether. This finding is consistent with trends in Fig. 3B, since H-bonding between water and polyacrylate side chains is generally more favorable than water interacting with the non-polar polymer backbone. This energetic driving force also leads to increasing disruption of the compact, non-aqueous morphology of polyacrylates as more H-bonds form, resulting in higher SA:V. The somewhat fewer number of H-bonds for 3-pyr-relative to THF-substituted polymers is also consistent with this phenomenon, where the 3-pyr polyacrylates have lower γ(adh-H2O) than THF polymers of the same % R* composition. Near non- (or weakly) polar surfaces, water exists in an ordered structure without strong interactions to the surface.57 The water significantly rearranges when in contact with THF- or 3-pyr-containing polymers through formation of intermolecular H-bonds. The kernel density estimate (KDE) plots—similar to the pairwise distribution function but non-parametric to not bias the statistical analysis58—shown in Fig. 4B quantify that water molecules form H-bonds at a median distance of 1.95 Å of THF and 3-pyr side chains and range in length from 1.5–2.5 Å.
 | ||
| Fig. 4 (A) Normalized H-bond formation for representative polyacrylate adhesives (B). KDE of water molecules distance from sidechains (sampled within 4.0 Å of sidechains) for aqueous simulations of 50% polyacrylate composition compared against 100% 2-EH. Top is polar sidechains; bottom is non-polar sidechains. | ||
There is also an increase in density of water molecules in the 2.5–3.0 Å range for the non-polar carbon and hydrogen atoms in THF and 3-pyr. In contrast, few water molecules are found for 2-EH, ben, and TDO in the same range. As the water molecules form H-bonds to the acrylate, interfacial surface tension increases, rather than decreases, as non-polar regions comprise the majority of polyacrylates. Ultimately, the driving force for increasing WoA(aq) with H-bond accepting comonomer substitution is partly accounted for by disruption of the organized water shells, which increases interactions between water and non-polar regions. This disruption forces water molecules closer to the nonpolar regions of the same adhesive, which increases the surface tension and WoA(aq). These relationships are contained in just one variable of the WoA(aq) equation, so interactions between MPs and polyacrylate adhesives need to be examined next.
Properties of adhesive-plastic interactions
The determined γ(adh-plas) are approximately one-half the magnitude of γ(adh-H2O), due to the similarity in surface polarity between MPs and polyacrylates, resulting in low interfacial tension (Fig. S13). For acrylates with ben, 3-pyr and THF sidechains, γ(adh-plas) to the four MP types increases by 4–5 mN m−1 at 50% R* composition compared to the 100% 2-EH case. These values compare to changes in γ(adh-H2O) of 10–20 mN m−1 for the same acrylates. For TDO-containing polymers, HDPE increases by ∼7 mN m−1 across % R* compared to the reference case (Fig. S13). At 50% TDO, γ(adh-plas) for HDPE, PA-6, and PETE are all around ∼25 mN m−1 while PS remains ∼19 mN m−1 regardless of % R*. These results show that γ(adh-H2O) is a strong factor for predicting trends in WoA(aq) for TDO-containing polymers, while γ(adh-plas) is more important for differentiating PS. This substantiates that the qualitatively different WoA(aq) trend for PS originates from the interaction of PS and TDO where the γ(adh-plas) remains low across % TDO, rather than from γ(adh-H2O) interactions.In all, while γ(adh-H2O) is quantitively more important to WoA(aq), selectivity in MP adhesion is not tuned by adhesive interactions with water. Therefore γ(adh-plas) remains a key consideration, despite its observed lower variance with adhesive composition.
Analysis of emergent behaviors in adhesion
The unique trends in γ(adh-plas) for TDO warrant further examination to reveal their physical origin. Recall that while the contact area varies for each MP (Fig. 1), nearly all contacts are “flat” meaning that there is no obvious interweaving. This behavior is observed across almost all tested polyacrylate compositions. (Further images of flat surface contacts can be found in SI, Fig. S15–S26). The important exception to the observed flat contact occurs for TDO and PS (Fig. 5), where interweaving of 50% TDO is observed with PS but not HDPE. The degree of interweaving can be quantified by comparing the distance between the MP “centerline” and the “center” of TDO side chains. The KDE plots for 50% R* repeat units interacting with PS show that most repeat units only approach ∼10 Å of the centerline, whereas TDO has density near 3.5 Å from the PS centerline. This partial interweaving occurs because γ(adh-plas) is lower than γadh and γplas, which creates a driving force for increasing interaction between them. The effect can be traced to increased surface tension of the acrylate due to TDO substitution. | ||
| Fig. 5 (Top) Surface contacts between 50% TDO (white) and microplastics HDPE and PS (yellow). (Bottom) KDE plot of adhesive side chain center distance (defined as the average cartesian coordinates for all atoms in a side chain) from PS centerline (defined as a line parallel to the contact between MP and adhesive that includes the volumetric center of the MP). | ||
Analysis of interweaving for TDO sidechain versus % composition (Fig. S14) indicates that increasing TDO from 10% to 50% increases the acrylate surface tension to the point of interweaving. This trend also provides evidence that TDO side chains are increasing the surface tension of the polyacrylate with increasing % TDO. Given that PA-6, HDPE, and PETE all have higher γ(adh-plas) than PS, the surface tension of 50% TDO must be between 19–25 mN m−1. To summarize, when γ(adh-plas) is less than the surface tension of both the adhesive and MP, increased adhesion and interweaving can occur. These results show promise in achieving the goal of potentially separating PS from a mixture of the four tested plastics using TDO (or a similarly behaved) side chain.
Summary of properties that control adhesion ability
The above analysis revealed key properties that govern the overall behavior of polyacrylate adhesives. The H-bond accepting ability of the acrylate substituents was found to improve aqueous adhesion for THF and 3-pyr to all tested MPs. This effect was driven by the rearrangement of the ordered shells of water molecules surrounding non-polar surfaces. This disruption brings water molecules closer to non-polar regions in addition to forming H-bonds. As a result, the aqueous interfacial tension for H-bond accepting polyacrylates increases which, per eqn (1), increases WoA(aq).While ben, 3-pyr, and THF sidechains improved adhesive ability, the fluorocarbon TDO was found to reduce it. The overall decrease in adhesive strength for TDO polyacrylates is a result of their lower interfacial tension with water, where acrylate-water contacts are minimized. TDO polyacrylates also showed a change in binding selectivity to PS compared to the other MPs. This effect is caused by TDO's WoA(aq) towards PS staying higher than for other MPs across changing side chain composition (% R*). The unusual trend in WoA(aq) for PS and HDPE originates from the interweaving between adhesive and MP; this was traced to the γ(adh-plas) for 50% TDO binding to PS being less than γadh and γplas. This interweaving was a unique effect for TDO among the adhesive types tested in this work and may be a useful property for design of new selective acrylates.
Experimental comparison
An important consideration in developing novel adhesives is the difference between thermodynamic (from simulations) and practical adhesion (from experiments).56 Practical adhesion is often orders of magnitude larger than thermodynamic adhesion.56 The relationship between the two is typically assumed to follow:| GC = WoA(aq)·A(1 + ψ(a,T,…)) | (2) |
Fig. 6 shows GC from probe-tack measurements of 100% 2-EH and 42% ben with probes of the four MP compositions in aqueous conditions. Ben substitution was chosen as it would have a strong increase in WoA(aq), relative to 100% 2-EH while having a smaller effect on ψ(a,T,…), as they are more similar to 2-EH than the polar acrylates, THF and 3-pyr, as well as the fluorinated TDO. Overall, the probe-tack measure of practical adhesion replicates the general trends of the computed WoA(aq). Uncertainties in the measurements remain, however, stemming from the measurement being taken in water with plastic probes. For example, plastic probes will have differences in surface roughness that affect the practical WoA(aq) through changes in probe contact area. Keeping these uncertainties in mind, comparisons can still be made between GC and computed WoA. The lowest GC values come with PA-6, which matches the computational results. PS, PETE, and HDPE have similar values of GC and are also the highest in computational WoA(aq). While comparisons between ben-substituted adhesives with 100% 2-EH adhesives could further test the computed trends, these differences could not be resolved within the error bars of the experiments.
 | ||
| Fig. 6 Experimental Probe-tack comparison for select polyacrylate adhesives. Experiments were performed under aqueous conditions. | ||
Nevertheless, this work demonstrates that computationally determined values of thermodynamic adhesion are substantially more predictive of practical adhesion than comparing percent polar compositions (see SI, Fig. S6–S9), which represents a significant advance in available tools for developing adhesives.
Conclusions
While H-bonding produced a strong shift in aqueous interaction energy, the hypothesized H-bonding between nylon and H-bond-accepting acrylates (3-pyr, THF) was not observed. Along the same lines, aromatic acrylates, with ben and 3-pyr side chains, did not show π-stacking with the aromatic polystyrene. Instead, the interfacial energies between the four MPs and adhesives with ben, THF, and 3-pyr sidechains remained relatively consistent in both value and trend (Fig. 2). This consistency limited the viability of using these three adhesives for selective capture of MPs. Adding perfluoro sidechains, however, was found to increase selectivity for PS. This behavior arose from an increase in aqueous/side-chain surface energies with increasing substitution, which eventually surpassed the interfacial energy between TDO and PS. When this occurs, the interface becomes more favorable than wetting both surfaces, driving interweaving between TDO and PS. The result was an emergent phase not seen in the other adhesive-MP interactions. This phenomenon may warrant further attention in future studies.The concepts formulated in this work may be useful to precisely tailor the next generation of polyacrylate adhesives for MP binding. In particular, the microscopic features, such as H-bond accepting in aqueous adhesion, intra- and inter-polymeric interactions, and interweaving, should all be considered when selecting an adhesive polymer design. Highly selective adhesives might be achieved by further reducing interfacial energies to a specific MP and increasing overall surface energy of polyacrylates. The computational tools presented here are well-suited to further refining mechanistic understanding in search of this behavior. Besides these concepts, atomistic modeling can likely be used to predict WoA for novel acrylate compositions, further expanding the scope of knowledge in this area. The inability to fully model all components of practical adhesion remains a limitation, preventing a complete determination of experimental adhesive utility. In all, this understanding and these tools can be used to design improved polyacrylate adhesives.
Author contributions
T. M. E. J.: methodology, formal analysis, writing – original draft. H. E. T.: formal analysis, investigation, writing – review & editing. M. T. R.: conceptualization. W. J.: methodology, investigation. M. E. C.: investigation. A. J. M.: resources, writing – review & editing, supervision, project administration, Funding acquisition. P. M. Z.: conceptualization, resources, writing – review & editing, supervision, project administration, funding acquisition.Conflicts of interest
There are no conflicts to declare.Data availability
Additional data is available from the corresponding author upon reasonable request.Supplementary information (SI): further computational details, polyacrylate monomer sequences, experimental methods, tables, and figures can be found in the supplemental information. See DOI: https://doi.org/10.1039/d5cp03631c.
Acknowledgements
The authors acknowledge NSF EFRI E3P Grant 2029251 through the National Science Foundation–Emerging Frontiers in Research and Innovation 2020 program for support of this work.References
- E. Penzel, Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2000 Search PubMed.
- T. Ohara, T. Sato, N. Shimizu, G. Prescher, H. Schwind, O. Weiberg, K. Marten, H. Greim, T. D. Shaffer and P. Nandi, Ullmann's Encyclopedia of Industrial Chemistry, Wiley, 2020 Search PubMed.
- N. Xu, J. Cao and X. Liu, J. Macromol. Sci., Part B: Phys., 2015, 54, 1153–1168 CrossRef CAS.
- Y. Peykova, O. v Lebedeva, A. Diethert, P. Müller-Buschbaum and N. Willenbacher, Int. J. Adhes. Adhes., 2012, 34, 107–116 CrossRef CAS.
- P. T. Chazovachii, J. M. Rieland, V. V. Sheffey, T. M. E. Jugovic, P. M. Zimmerman, O. Eniola-Adefeso, B. J. Love and A. J. McNeil, ACS ES&T Eng., 2021, 1, 1698–1704 Search PubMed.
- D. H. Kaelble, J. Appl. Polym. Sci., 1974, 18, 1869–1889 CrossRef CAS.
- J. P. G. L. Frias and R. Nash, Mar. Pollut. Bull., 2019, 138, 145–147 CrossRef CAS PubMed.
- J. Barrett, Z. Chase, J. Zhang, M. M. B. Holl, K. Willis, A. Williams, B. D. Hardesty and C. Wilcox, Front. Mater. Sci., 2020, 7, 576170 Search PubMed.
- M. Padervand, E. Lichtfouse, D. Robert and C. Wang, Environ. Chem. Lett., 2020, 18, 807–828 CrossRef CAS.
- J. Boucher and D. Friot, Primary microplastics in the oceans: A global evaluation of sources, IUCN International Union for Conservation of Nature, 2017.
- M. Bergmann, S. Mützel, S. Primpke, M. B. Tekman, J. Trachsel and G. Gerdts, Sci. Adv., 2019, 5, 1157 CrossRef PubMed.
- M. R. Michielssen, E. R. Michielssen, J. Ni and M. B. Duhaime, Environ. Sci., 2016, 2, 1064–1073 CAS.
- M. Wang, P. Zhou, S. DuBay, S. Zhang, Z. Yang, Y. Wang, J. Zhang, Y. Cao, Z. Hu, X. He, S. Wang, M. Li, C. Fan, B. Zou, C. Zhou and Y. Wu, J. Hazard. Mater., 2025, 487, 137274 CrossRef CAS PubMed.
- Y. Xiang, Z. Wang, Y. Zhao, J. Liu, J. Wang, Q. Lu and L. Xie, J. Colloid Interface Sci., 2025, 683, 347–356 CrossRef CAS PubMed.
- A. Chamas, H. Moon, J. Zheng, Y. Qiu, T. Tabassum, J. H. Jang, M. Abu-Omar, S. L. Scott and S. Suh, ACS Sustain. Chem. Eng., 2020, 8, 3494–3511 CrossRef CAS.
- E. Schmaltz, E. C. Melvin, Z. Diana, E. F. Gunady, D. Rittschof, J. A. Somarelli, J. Virdin and M. M. Dunphy-Daly, Environ. Int., 2020, 144, 106067 CrossRef CAS PubMed.
- G. Casoli and S. Ramkumar, Mare Plasticum – The Plastic Sea, Springer International Publishing, Cham, 2020 Search PubMed.
- J. Hopewell, R. Dvorak and E. Kosior, Philos. Trans. R. Soc., B, 2009, 364, 2115–2126 CrossRef CAS PubMed.
- J. R. Dann, J. Colloid Interface Sci., 1970, 32, 302–320 CrossRef CAS.
- C. A. Finch, Polymer Handbook, 3rd edn, Br. Polym. J., 1990, 23, 277–280 Search PubMed.
- W. J. Shim, S. H. Hong and S. Eo, Microplastic Contamination in Aquatic Environments, Elsevier, 2018 Search PubMed.
- A. Kowalski, Z. Czech and Ł. Byczyński, J. Coat. Technol. Res., 2013, 10, 879–885 CrossRef CAS.
- L. W. McKeen, Film Properties of Plastics and Elastomers, Elsevier, 2012 Search PubMed.
- A. J. Kinloch, Adhesion and Adhesives, Springer, Netherlands, Dordrecht, 1987 Search PubMed.
- D. Briggs, D. G. Rance and B. J. Briscoe, Comprehensive Polymer Science and Supplements, Elsevier, 1989 Search PubMed.
- J. J. Bikerman, Surface Chemistry: Theory and Applications, Elsevier, Burlington, 2nd edn, 2013 Search PubMed.
- M. K. Abdelrahman, H. Kim, J. Maeng, P. Ondrusek and T. H. Ware, Macromolecules, 2020, 53, 2388–2395 CrossRef CAS.
- M. A. Firestone, S. C. Hayden and D. L. Huber, MRS Bull., 2015, 40, 760–767 CrossRef CAS.
- C. W. Abney, S. Das, R. T. Mayes, L.-J. Kuo, J. Wood, G. Gill, M. Piechowicz, Z. Lin, W. Lin and S. Dai, Phys. Chem. Chem. Phys., 2016, 18, 23462–23468 RSC.
- T. Khudiyev, E. Huseyinoglu and M. Bayindir, Sci. Rep., 2014, 4, 4607 CrossRef PubMed.
- R. G. Winkler, J. Elgeti and G. Gompper, J. Phys. Soc. Japan, 2017, 86, 101014 CrossRef.
- L. Xie, F. Liu, J. Liu and H. Zeng, ACS Appl. Mater. Interfaces, 2020, 12, 58360–58368 CrossRef CAS PubMed.
- L. Xie, X. Cui, J. Liu, Q. Lu, J. Huang, X. Mao, D. Yang, J. Tan, H. Zhang and H. Zeng, ACS Appl. Mater. Interfaces, 2021, 13, 6941–6950 CrossRef CAS PubMed.
- S. Ebnesajjad, Surface Treatment of Materials for Adhesive Bonding, Elsevier, 2014 Search PubMed.
- K. O’Driscoll and R. A. Sanayei, Macromolecules, 1991, 24, 4479–4480 CrossRef.
- J. A. McCammon, B. R. Gelin and M. Karplus, Nature, 1977, 267, 585–590 CrossRef CAS PubMed.
- M. Levitt and S. Lifson, J. Mol. Biol., 1969, 46, 269–279 CrossRef CAS PubMed.
- S. A. Hollingsworth and R. O. Dror, Neuron, 2018, 99, 1129–1143 CrossRef CAS PubMed.
- N. V. Bhagavan, Medical Biochemistry, Elsevier, 2002 Search PubMed.
- B. Park, M. Chandross, M. J. Stevens and G. S. Grest, Langmuir, 2003, 19, 9239–9245 CrossRef CAS.
- K. F. Mansfield and D. N. Theodorou, Macromolecules, 1991, 24, 4295–4309 CrossRef CAS.
- D. L. Cheung, D. P. McMahon and A. Troisi, J. Phys. Chem. B, 2009, 113, 9393–9401 CrossRef CAS PubMed.
- D. M. Huang, R. Faller, K. Do and A. J. Moulé, J. Chem. Theory Comput., 2010, 6, 526–537 CrossRef CAS PubMed.
- K. N. Schwarz, T. W. Kee and D. M. Huang, Nanoscale, 2013, 5, 2017–2027 RSC.
- D. Alberga, G. F. Mangiatordi, L. Torsi and G. Lattanzi, J. Phys. Chem. C, 2014, 118, 8641–8655 CrossRef CAS.
- M. Moreno, M. Casalegno, G. Raos, S. V. Meille and R. Po, J. Phys. Chem. B, 2010, 114, 1591–1602 CrossRef CAS PubMed.
- C. Poelking and D. Andrienko, Macromolecules, 2013, 46, 8941–8956 CrossRef CAS.
- O. Alexiadis and V. G. Mavrantzas, Macromolecules, 2013, 46, 2450–2467 CrossRef CAS.
- S. L. Mayo, B. D. Olafson and W. A. Goddard, J. Phys. Chem., 1990, 94, 8897–8909 CrossRef CAS.
- S. Jo, T. Kim, V. G. Iyer and W. Im, J. Comput. Chem., 2008, 29, 1859–1865 CrossRef CAS PubMed.
- Y. Li, J. Q. Pham, K. P. Johnston and P. F. Green, Langmuir, 2007, 23, 9785–9793 CrossRef CAS PubMed.
- S. N. Omenyi, A. W. Neumann and C. J. van Oss, J. Appl. Phys., 1981, 52, 789–795 CrossRef CAS.
- D. K. Owens and R. C. Wendt, J. Appl. Polym. Sci., 1969, 13, 1741–1747 CrossRef CAS.
- M. S. Hanqi Wen, Johns Hopkins University, 2019.
- CRC Handbook of Chemistry and Physics, ed. W. M. Haynes, CRC Press, 2014 Search PubMed.
- P. Kirsch, Modern Fluoroorganic Chemistry, Wiley-VCH, Weinheim, 1st edn, 2004, vol. 1 Search PubMed.
- D. A. Dillard, Advances in Structural Adhesive Bonding, Elsevier, 2010, pp. 350–388 Search PubMed.
- M. Kooi and A. A. Koelmans, Environ. Sci. Technol. Lett., 2019, 6, 551–557 CrossRef CAS.
| This journal is © the Owner Societies 2026 |