
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDefect engineering in β-ketoenamine-linked covalent organic frameworks for high-efficiency uranium extraction†
Douchao
Mei
and
Bing
Yan
 *
*
Shanghai Key Lab of Chemical Assessment and Sustainability, School of Chemical Science and Engineering, Tongji University, Siping Road 1239, Shanghai 200092, China. E-mail: byan@tongji.edu.cn
First published on 31st March 2025
Abstract
In view of energy and the environment, the development of suitable adsorbents for high-efficiency uranium extraction is highly desired. However, most reported adsorbents showcase poor stability, terrible anti-biofouling activity and dissatisfactory adsorption capacity. Herein, we designed and prepared three β-ketoenamine-linked COFs (Tp-DAAQ, Tp-AD and Tp-DAQ) with varying anthraquinone conjugated units for uranium extraction. Compared to Tp-DAQ, Tp-DAAQ and Tp-AD exhibited higher uranium adsorption capacities of 738.4 and 834.4 mg g−1 at pH = 6, which may be ascribed to their high specific surface areas and abundant uranium-binding sites. In addition, defect engineering was creatively introduced into the DAAQ-COFs and AD-COFs to enhance the uranium adsorption performance by exposing more active sites. Notably, the experimental results revealed that the defective COFs possessed higher U-uptake capacities in the same conditions, demonstrating the paramount role of defective sites for uranium extraction. More importantly, good anti-biofouling activity was observed in all these COFs under visible light irradiation conditions, indicating that they can serve as proper candidates for uranium extraction from complex water environments. This work paves a new way for uranium extraction application by rationally designing the bridging units and introducing defect engineering.
1. Introduction
On the one hand, nuclear energy with a low-carbon footprint plays a pivotal role in the global energy system, and uranium is considered the essential material for nuclear power plants.1–5 The rapid development of nuclear energy has led to low abundance of the known geological reserved uranium resource. On the other hand, the large-scale development of nuclear energy and the improper discharge of nuclear wastewater have caused serious environmental pollutant problems due to the high toxicity and radioactivity of soluble UO22+.6–8 Hence, the recovery and removal of uranium from complex water environments such as seawater and river water show extreme significance in view of energy and the environment. To date, several porous adsorbents, including metal organic frameworks (MOFs), covalent organic frameworks (COFs), hydrogen-bonded organic frameworks (HOFs) and porous organic polymers (POPs), have been developed for the extraction of uranium.9–18 Disappointingly, most of the above-mentioned materials exhibit poor stability, low selectivity and low anti-biofouling activity, resulting in unsatisfactory uranium extraction performances.In particular, COFs represent a class of porous adsorption platforms wherein functional groups with relatively high affinity toward uranium can be finely incorporated into the frameworks by postsynthetic modification. For example, amidoxime groups were grafted onto a 2D COF with a cyano group by Ma and coworkers,19 and the obtained COF material (named COF-TpAb-AO) exhibited good uranium adsorption capacity up to 408 mg g−1. Liu et al. reported a carboxyl-functionalized COF (JUC-505-COOH)20 that displayed a satisfactory uranium adsorption performance of 464 mg g−1 due to the existence of effective chelating sites. Our previous work also synthesized β-ketoenamine- and benzoxazole-linked COFs anchored with phosphonic groups (TpPa-2OH-Phos and TpDBD-Phos),21 which showed high selectivity and admirable application value in contaminated water systems. However, the complicated postsynthetic modification reactions lead to the decrease in surface area and pore volume, as well as increased synthesis cost, which hinder the practical applications of the COF adsorbents.22–24 More importantly, anti-biofouling activity is rarely mentioned in most reported COFs U-adsorbents, which may be attributed to the poor releasing performance of reactive oxygen species (ROS) as a consequence of the weak charge separation and migration properties. Taking the aforementioned dilemma into account, there remains great necessity and significance to explore novel COFs adsorbents for efficient uranium capture.
Defect engineering serves as an effective strategy to improve the chemical property of COFs and has aroused considerable attention in recent years.25–28 This strategy modulates the atomic environment by disrupting the strong crystal symmetry of original COFs, which reduces the crystallinity but induces the generation of active defect sites.29,30 To our knowledge, the active sites are of great importance for chelation with uranium. Moreover, the construction of COFs with donor–acceptor (D–A) type structures is conducive to the generation of ROS, which may be ascribed to the formation of effective electron transport channels.31–34 In particular, electrons can be easily transferred from electron-rich moieties (donor) to electron-poor moieties (acceptor) in D–A COFs under the irradiation of visible light.35,36 The generation of ROS in COFs can impede the covering of active adsorption sites by microorganisms, thereby significantly improving the uranium extraction capability. As reported, the β-ketoenamine-linked COFs are favorable for π-electron delocalization,37,38 which is preferential for the construction of D–A COFs.
Inspired by these, we designed and synthesized three β-ketoenamine-linked COFs (Tp-DAAQ, Tp-AD and Tp-DAQ), featuring similar building blocks, via a solvothermal method, aimed at efficient uranium extraction (Scheme 1a). These COFs exhibit high crystallinity, good thermal and chemical stability as well as regular pore structures. Strikingly, Tp-AD shows the highest uranium adsorption capacity of 834.4 mg g−1, which is higher than those of most previously reported COFs U-adsorbents. In addition, the 1,3,5-triformylphloroglucinol (Tp) parts display distinct electron-donating characterizations, while the 2,6-diaminoanthraquinone (DAAQ) and 1,5-diaminoanthraquinone (DAQ) units exhibit electron-accepting properties. The pronounced D–A structural characteristics in Tp-DAAQ and Tp-DAQ endow the outstanding property of reduction of U(VI) to U(IV) and excellent anti-biofouling activity. Due to the better U-uptake performances of Tp-DAAQ and Tp-AD, eight defective COFs (Tp-DAAQ-1, Tp-DAAQ-2, Tp-DAAQ-3, Tp-DAAQ-4, Tp-AD-1, Tp-AD-2, Tp-AD-3 and Tp-AD-4) were constructed by regulating the ratio of building blocks (Scheme 1b and c). Compared to the original COFs, the defective DAAQ-COFs and AD-COFs show high uranium adsorption capacities at the same conditions, demonstrating the crucial role of active defective sites. Furthermore, the defective COFs (Tp-DAAQ-4 and Tp-AD-4) show satisfactory extraction efficiencies in actual U-spiked environments (tap water, river water and seawater). More importantly, all the COFs exhibit excellent antibacterial activity, which may indicate that these COFs materials are ideal candidates for uranium extraction from complex water environments under harsh conditions.
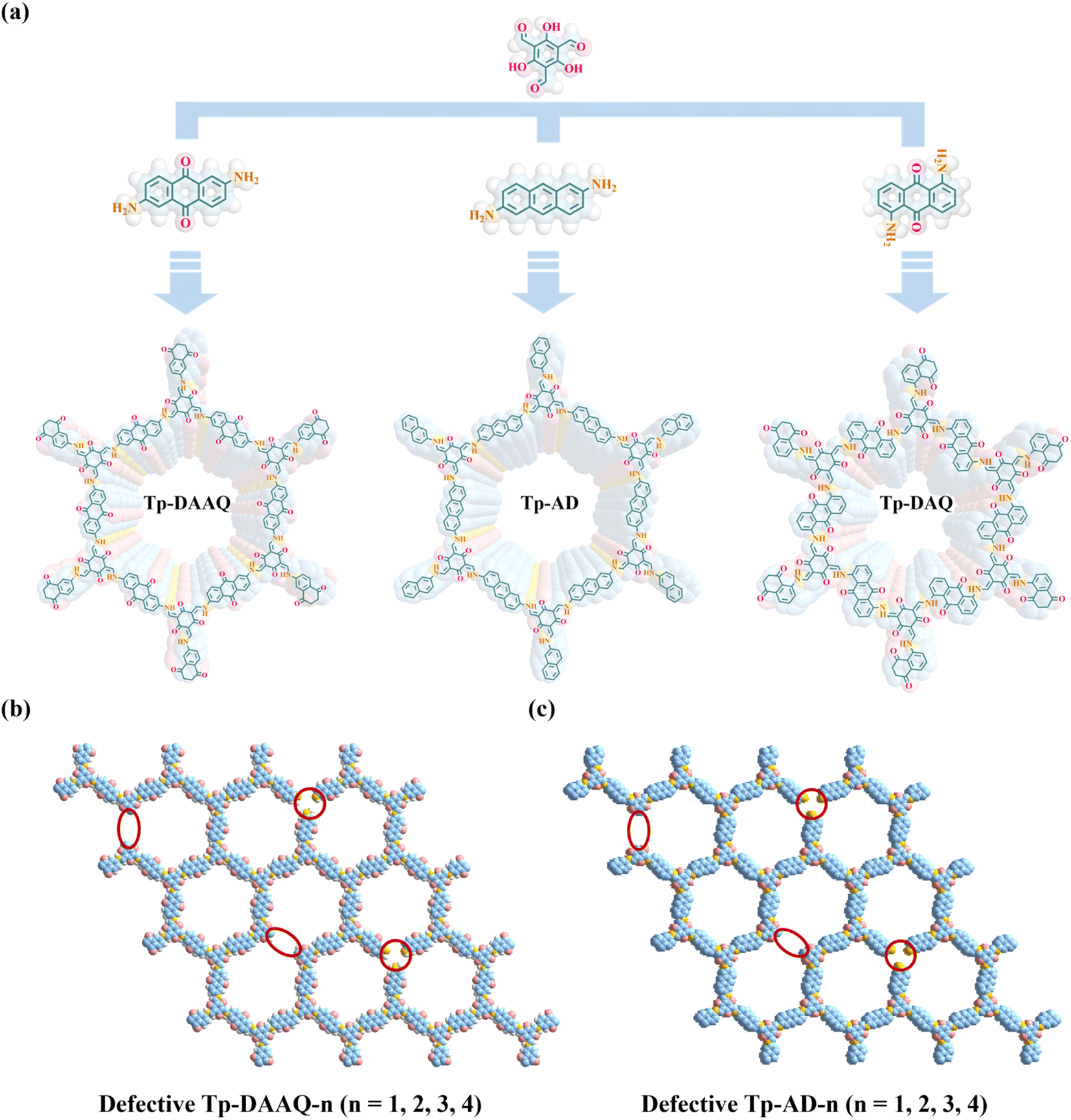 | ||
| Scheme 1 (a) Designed synthesis and crystal structures of Tp-DAAQ, Tp-AD and Tp-DAQ. Illustration of defects introduced in (b) Tp-DAAQ and (c) Tp-AD. | ||
2. Experimental section
2.1 Synthesis of Tp-DAAQ, Tp-DAAQ-1, Tp-DAAQ-2, Tp-DAAQ-3 and Tp-DAAQ-4
Tp-DAAQ was prepared similarly to the previously reported method with mild optimization.38 Tp (0.095 mmol, 20 mg) and DAAQ (0.142 mmol, 34 mg) were added into a 10 mL Schlenk tube. After adding N,N-dimethylacetamide (DMAc) (0.9 mL), 1,3,5-trimethylbenzene (0.3 mL) and aqueous acetic acid (6 M, 0.05 mL), the mixture was treated by ultrasonication for 20 min. Then, it was heated at 120 °C for 72 h after three freeze–pump–thaw cycles. After the reaction, the collected precipitate was washed with N,N-dimethylformamide (DMF) (3 × 30 mL) and acetone (3 × 30 mL). Finally, the obtained powder was dried at 120 °C under vacuum for 12 h. A similar synthesis procedure was followed for the synthesis of defective DAAQ-COFs except for slightly changing the amount of Tp. Specifically, the corresponding amounts of Tp were 0.048 mmol, 0.071 mmol, 0.12 mmol and 0.142 mmol for Tp-DAAQ-1, Tp-DAAQ-2, Tp-DAAQ-3 and Tp-DAAQ-4, respectively.2.2 Synthesis of Tp-AD, Tp-AD-1, Tp-AD-2, Tp-AD-3 and Tp-AD-4
Tp-AD was synthesized based on the previously reported method with slight modification.39 In the typical synthesis, a Schlenk tube was charged with Tp (0.095 mmol, 20 mg), anthracene-2,6-diamine (AD) (0.142 mmol, 31.2 mg), 1,4-dioxane (1 mL) and aqueous acetic acid (6 M, 0.2 mL). To obtain a homogeneous dispersion, the mixture was sonicated for 20 min. After three freeze–pump–thaw cycles, the tube was heated at 120 °C for 72 h. After that, the brown-colored precipitate was collected and washed with tetrahydrofuran (THF) (3 × 30 mL) and acetone (3 × 30 mL). Finally, the obtained material was evacuated at 120 °C under vacuum overnight. A similar preparation procedure was followed for the preparation of defective AD-COFs except for slightly changing the amount of Tp. To be specific, the corresponding amounts of Tp were 10 mg, 15 mg, 25 mg and 30 mg for Tp-AD-1, Tp-AD-2, Tp-AD-3 and Tp-AD-4, respectively.2.3 Synthesis of Tp-DAQ
A mixture of Tp (0.06 mmol, 12.4 mg) and DAQ (0.09 mmol, 21.5 mg) was added into a mixture of 1,4-dioxane (0.25 mL) and 1,3,5-trimethylbenzene (0.75 mL) in a Schlenk tube. The resulting mixture was sonicated for 20 min, followed by heating at 120 °C for 72 h. Afterward, the obtained solid was washed with THF (3 × 30 mL) and acetone (3 × 30 mL) and dried at 120 °C under vacuum for 12 h, and Tp-DAQ was obtained as a black powder.2.4 Uranium adsorption tests
The detailed adsorption experiments can be found in Section 3 (ESI†).2.5 Antibacterial property tests
The detailed antibacterial experiments are provided in Section 4 (ESI†).3. Results and discussion
3.1 Synthesis and characterizations
The crystalline structures of all the COFs were analyzed by powder X-ray diffraction (PXRD). As depicted in Fig. 1a–c, all three COFs show an intense and narrow reflection peak at about 3.6° and a broad peak at 26.8° (2θ), which correspond to the (100) and (001) crystal facets,39–41 implying the high crystallinity of Tp-DAAQ, Tp-AD and Tp-DAQ. The experimental diffraction patterns match well with the simulated peaks of the eclipsed AA stacking model, corroborating the successful synthesis of the three COFs. Pawley refinement was conducted on the experimental PXRD patterns, giving the unit cell parameters for Tp-DAAQ (a = b = 29.7470 Å, c = 3.6255 Å, α = β = 90°, γ = 120°, Rp = 6.32% and Rwp = 9.37%), Tp-AD (a = b = 31.2270 Å, c = 3.8087 Å, α = β = 90°, γ = 120°, Rp = 4.65% and Rwp = 6.48%) and Tp-DAQ (a = b = 25.2370 Å, c = 3.5282 Å, α = β = 90°, γ = 120°, Rp = 6.46% and Rwp = 8.50%), further indicating that the actual structures are consistent with the simulated structures (Fig. S1–S3†). The detailed unit cell parameters and atomic coordinates of the three COFs are presented in Tables S1–S3.† Comparing Tp-DAAQ and Tp-AD, the peak positions have no obvious variation, while the peak intensities show varying degrees of decrease for Tp-DAAQ-1, Tp-DAAQ-2, Tp-DAAQ-3, Tp-DAAQ-4, Tp-AD-1, Tp-AD-2, Tp-AD-3 and Tp-AD-4 (Fig. 2a, d, S4 and S16†), which suggests that the defective COFs were successfully synthesized with relatively low crystallinity. The distortion of the crystal lattice and decrease of crystallinity may lead to the generation of active defect sites.29,30 | ||
| Fig. 1 Experimental and Pawley refined PXRD of (a) Tp-DAAQ, (b) Tp-AD and (c) Tp-DAQ. Solid-state 13C NMR spectra of (d) Tp-DAAQ, (e) Tp-AD and (f) Tp-DAQ. N2 adsorption–desorption isotherms and the corresponding pore size distributions (inset) of (g) Tp-DAAQ, (h) Tp-AD and (i) Tp-DAQ. | ||
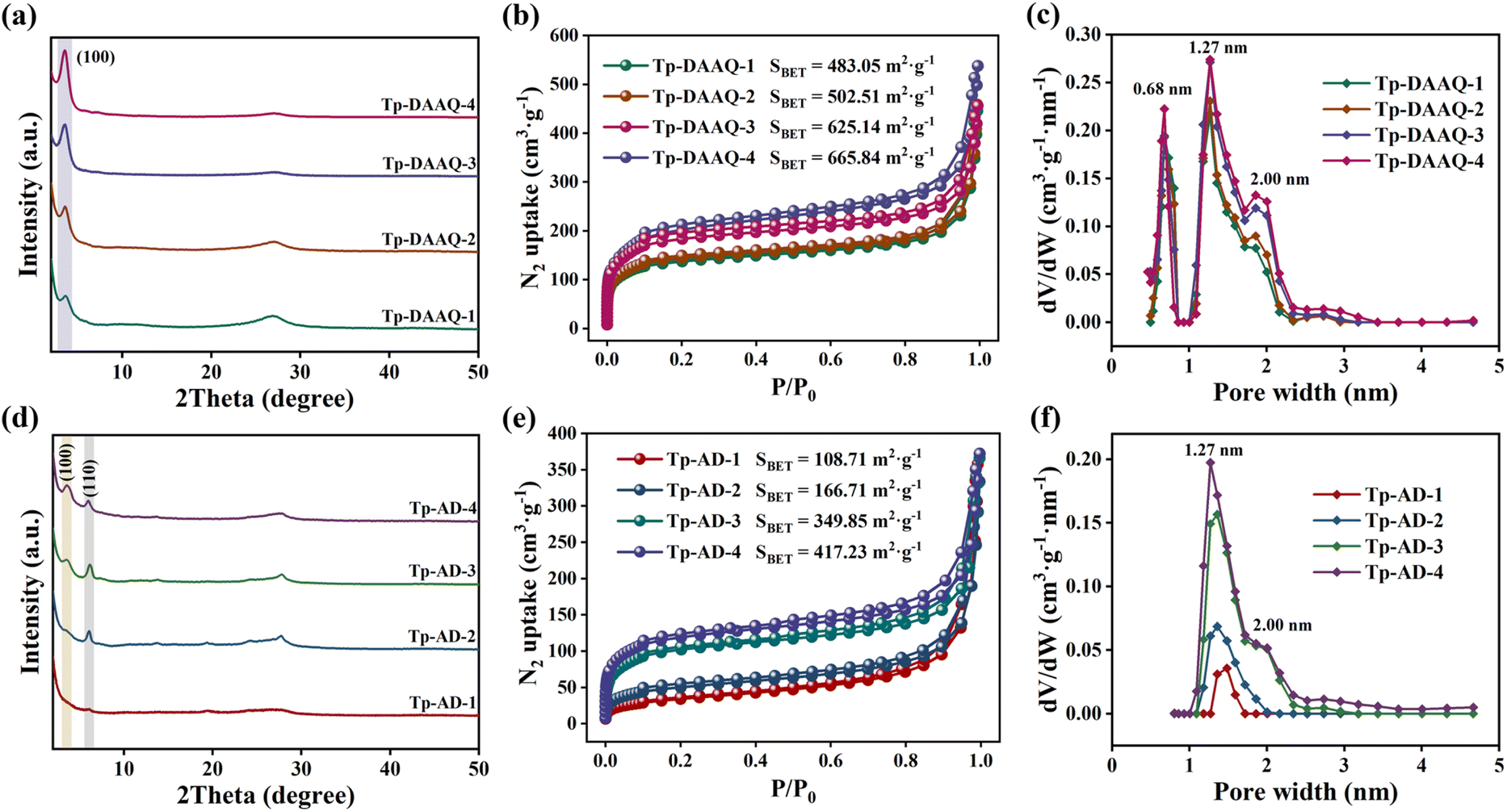 | ||
| Fig. 2 (a) PXRD patterns, (b) N2 adsorption–desorption isotherms and (c) pore size distributions of the defective DAAQ-COFs (Tp-DAAQ-1, Tp-DAAQ-2, Tp-DAAQ-3 and Tp-DAAQ-4). (d) PXRD patterns, (e) N2 adsorption–desorption isotherms and (f) pore size distributions of the defective AD-COFs (Tp-AD-1, Tp-AD-2, Tp-AD-3 and Tp-AD-4). | ||
Solid state 13C nuclear magnetic resonance spectroscopy (13C NMR), Fourier transform infrared spectroscopy (FT-IR) and X-ray photoelectron spectroscopy (XPS) were employed to investigate the chemical structures of the as-synthesized COFs. As displayed in Fig. 1d–f, the distinct characteristic signals of carbon atoms are observed at around 184, 146 and 108 ppm in the three COFs, corresponding to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, C–N and CC,39–41 respectively. These characteristic peaks reveal the existence of β-ketoenamine structure in Tp-DAAQ, Tp-AD and Tp-DAQ. Moreover, the FT-IR spectra show the conspicuous stretching vibrations of C–N (1250 cm−1) and CC (1560 cm−1) in the three COFs (Fig. S5, S15 and S25†),42–44 clearly validating the successful formation of enol-to-keto tautomerism. The XPS spectra suggest that Tp-DAAQ, Tp-AD and Tp-DAQ are composed of C, O and N elements (Table S4†). For the three COFs, the typical characteristic peaks of C–N, CC, CO can be observed at around 286, 284 and 530 eV in the high-resolution XPS spectra of C 1s and O 1s (Fig. S7 and S55–S57†).38,41 The above 13C NMR, FT-IR and XPS results confirm the successful synthesis of Tp-DAAQ, Tp-AD and Tp-DAQ with β-ketoenamine-linked frameworks. Additionally, similar FT-IR spectra were obtained for Tp-DAAQ-1, Tp-DAAQ-2, Tp-DAAQ-3, Tp-DAAQ-4, Tp-AD-1, Tp-AD-2, Tp-AD-3 and Tp-AD-4 (Fig. S6 and S17†), which may indicate the successful construction of defective DAAQ-COFs and AD-COFs.
O, C–N and CC,39–41 respectively. These characteristic peaks reveal the existence of β-ketoenamine structure in Tp-DAAQ, Tp-AD and Tp-DAQ. Moreover, the FT-IR spectra show the conspicuous stretching vibrations of C–N (1250 cm−1) and CC (1560 cm−1) in the three COFs (Fig. S5, S15 and S25†),42–44 clearly validating the successful formation of enol-to-keto tautomerism. The XPS spectra suggest that Tp-DAAQ, Tp-AD and Tp-DAQ are composed of C, O and N elements (Table S4†). For the three COFs, the typical characteristic peaks of C–N, CC, CO can be observed at around 286, 284 and 530 eV in the high-resolution XPS spectra of C 1s and O 1s (Fig. S7 and S55–S57†).38,41 The above 13C NMR, FT-IR and XPS results confirm the successful synthesis of Tp-DAAQ, Tp-AD and Tp-DAQ with β-ketoenamine-linked frameworks. Additionally, similar FT-IR spectra were obtained for Tp-DAAQ-1, Tp-DAAQ-2, Tp-DAAQ-3, Tp-DAAQ-4, Tp-AD-1, Tp-AD-2, Tp-AD-3 and Tp-AD-4 (Fig. S6 and S17†), which may indicate the successful construction of defective DAAQ-COFs and AD-COFs.
The specific surface areas and porosities of all the COFs were evaluated through N2 adsorption/desorption isotherms. As illustrated in Fig. 1g–i, Tp-DAAQ, Tp-AD and Tp-DAQ exhibit typical type I isotherm profiles.45,46 The Brunauer–Emmett–Teller (BET) surface area and pore volume were determined to be 789.09 m2 g−1 and 0.84 cm3 g−1 for Tp-DAAQ, 609.06 m2 g−1 and 0.60 cm3 g−1 for Tp-AD, and 103.13 m2 g−1 and 0.13 cm3 g−1 for Tp-DAQ, respectively. Correspondingly, the pore size distributions were mainly concentrated at 1.27, 1.27 and 1.18 nm for Tp-DAAQ, Tp-AD and Tp-DAQ, respectively, based on non-local density functional theory (NLDFT). Remarkably, the average pore size of Tp-DAQ is smaller than those of Tp-DAAQ and Tp-AD, which is consistent with the simulated result calculated by the AA-stacking model (Fig. S1–S3†). The above results confirm that Tp-DAAQ, Tp-AD and Tp-DAQ possess high surface area, large pore volume and large pore size, offering great potential for the efficient capture of uranium. Furthermore, the specific surface areas, pore volumes and pore sizes of Tp-DAAQ-1, Tp-DAAQ-2, Tp-DAAQ-3, Tp-DAAQ-4, Tp-AD-1, Tp-AD-2, Tp-AD-3 and Tp-AD-4 have different degrees of decrease (Fig. 2b–c, e–f and Table S5†), which implies the successful introduction of defect engineering in DAAQ-COFs and AD-COFs. Apparently, the accessible active sites are favorable for coordinative chelation with UO22+, further leading to an efficient uranium adsorption capability.
The morphologies of the obtained COFs were resolved by scanning electron microscopy (SEM) and high-resolution transmission electron microscopy (HR-TEM). As seen in Fig. S9 and S19,† both Tp-DAAQ and Tp-AD show porous network structures and ordered pore channels, which are favorable for the rapid penetration of UO22+. In addition, the HR-TEM images reveal multi-layer overlapping nano-sheets for Tp-DAAQ and Tp-AD (Fig. S10 and S20†). In the case of Tp-DAQ, spherical morphologies and some rod-like structures are observed (Fig. S27†). Moreover, specific lattice fringes are observed on Tp-DAQ (Fig. S28†), verifying the highly ordered structure. The morphologies of the defective DAAQ-COFs and AD-COFs were also investigated by SEM, as seen in Fig. S11–S14 and S21–S24.† Energy dispersive spectroscopy (EDS) elemental mapping demonstrates that the C, N and O elements are uniformly distributed on these synthesized COFs.
The structural stability of the synthesized COFs was further assessed. Thermogravimetric analysis (TGA) curves manifest that all eleven COFs possess excellent thermal stability up to 300 °C under N2 atmosphere (Fig. S8, S18 and S26†). In addition, the chemical stabilities of Tp-DAAQ, Tp-AD and Tp-DAQ were studied by immersing them in different pH solutions for 24 h. The PXRD patterns show that the three COFs maintain unaltered diffraction peaks after the treatments, providing solid evidence for their remarkable chemical stability (Fig. S29†). Overall, all the prepared COFs have exceptional thermal and chemical stabilities, providing the possibility for long-term and stable U-adsorption performance even under harsh conditions.
3.2 Uranium adsorption performance
Generally speaking, the pH value of an adsorbed solution significantly affects the surface charge of COFs materials and the distribution form of uranium species, leading to difference in the U-uptake capacities of adsorbents. Accordingly, we conducted a series of uranium adsorption tests on DAAQ-COFs, AD-COFs and Tp-DAQ at pH = 3–7. As illustrated in Fig. 3a, 4a and S30,† all COFs exhibit similar pH-dependence and the greatest uranium adsorption performance at pH = 6. In acidic solution, the active sites on the COFs are easily protonated due to the existence of a large number of H+. Besides, H+ can compete with UO22+ to chelate with COFs adsorbents in low pH solutions, which results in poor uranium adsorption performances at pH = 3–5. To elucidate the interaction of COFs and uranium, the zeta potentials of COFs were investigated across different pH levels. As depicted in Fig. S31,† the surfaces of Tp-DAAQ, Tp-AD and Tp-DAQ are negatively charged at pH = 3–8. As reported, the major existing form of U(VI) is (UO2)2CO3(OH)3− at pH = 7 and 8,47 which can bring about strong electrostatic repulsion between the COFs and U(VI). Moreover, a large number of OH− will compete with COFs to coordinate with UO22+ in alkaline conditions, leading to low U-uptake amounts. Taking the above results into account, pH = 6 is considered the optimal pH and all subsequent uranium adsorption tests were carried out at pH = 6. | ||
| Fig. 3 (a) Effect of pH for uranium adsorption on parallel DAAQ-COFs (C0 = 100 mg L−1 and m/V = 1/10 g L−1). (b) Adsorption kinetics and (c) adsorption isotherms of Tp-DAAQ toward uranium (pH = 6 and m/V = 1/10 g L−1). (d) Adsorption capacity and the corresponding distribution coefficient (Kd) (inset) of Tp-DAAQ for various metal ions (the initial concentration of all the metal ions was 100 mg L−1). (e) Regeneration cycle of Tp-DAAQ for uranium adsorption (C0 = 100 mg L−1, pH = 6, m/V = 1/10 g L−1 and t = 8 h). | ||
 | ||
| Fig. 4 (a) Effect of pH for uranium adsorption on parallel AD-COFs (C0 = 100 mg L−1 and m/V = 1/10 g L−1). (b) Adsorption kinetics and (c) adsorption isotherms of Tp-AD toward uranium (pH = 6 and m/V = 1/10 g L−1). (d) Comparison of uranium adsorption of Tp-AD and other previously reported COF adsorbents. (e) Uranium extraction performance of Tp-AD-4 from uranium-spiked tap water, river water, simulated seawater and Bohai seawater (C0 = 10 mg L−1, pH = 6, m/V = 1/2 g L−1 and t = 24 h). | ||
In particular, the highest uranium adsorption capacities are 425.7, 501.5 and 296.6 mg g−1 for Tp-DAAQ, Tp-AD and Tp-DAQ, respectively, at pH = 6 (C0 = 100 mg L−1) (Fig. S30†). Compared to original Tp-DAAQ and Tp-AD, the defective DAAQ-COFs and AD-COFs show higher uranium adsorption capacities (Fig. 3a and 4a), which is attributed the increase of attainable adsorption sites as a consequence of the introduction of defect engineering. It is worth noting that the specific surface area of defective Tp-AD-1 has a conspicuous decrease in comparison with that of Tp-AD, whereas they possess similar uranium adsorption performances. This result indeed confirms the vital contribution of active defect sites, which can effectively coordinate with UO22+ and significantly improve the uranium adsorption capacity.
To evaluate the influence of contact time for uranium adsorption, the uranium adsorption behaviors of Tp-DAAQ, Tp-AD and Tp-DAQ were examined at different time intervals. As depicted in Fig. 3b, 4b and S35,† the U-uptake amounts of the three COFs for UO22+ rapidly increase within 120 min, and the adsorption equilibria are achieved at 360 min for Tp-DAAQ and 240 min for Tp-AD and Tp-DAQ. A higher adsorption capacity and faster adsorption rate for U(VI) are observed on Tp-AD, which may be ascribed to the more accessible active chelating sites. The pseudo-first-order, pseudo-second-order and Weber–Morris (W–M) order models were adopted to examine the adsorption kinetics of Tp-DAAQ, Tp-AD and Tp-DAQ. Compared to pseudo-first-order, the uranium adsorption behavior of all COFs fits better with pseudo-second-order (Table S6†), indicative of the predominant role of chemisorption in the process.48,49 In addition, the fitting results of the W–M model suggest that the adsorption processes of Tp-DAAQ, Tp-AD and Tp-DAQ can be divided into two stages (Fig. S33–S35†). In the first stage, the active sites exposed on the surface of the three COFs easily chelate with UO22+; in the second stage, UO22+ permeates the interior of COFs to facilitate further adsorption. Obviously, the slope of the fitting curve in the second stage is smaller than that in the first stage, which manifests the vital role of active sites on the surface of COFs for uranium adsorption.
To investigate the maximum U-uptake capacity and the effect of temperature on uranium adsorption, uranium adsorption experiments onto the three COFs were conducted at different initial uranium concentrations and temperatures. The U-uptake amounts of the three COFs show a positive correlation with the initial uranium concentration, and the corresponding maximum uranium adsorption capacities of Tp-DAAQ, Tp-AD and Tp-DAQ were 738.4, 834.4 and 382.8 mg g−1 (Fig. 3c, 4c and S38†), respectively. In particular, the saturation adsorption capacities of Tp-DAAQ and Tp-AD were superior to those of most previously reported COFs U-adsorbents (Fig. 4d and Table S9†),8,13,14,19–24,48–56 indicating their exceptional practical application potential. Meanwhile, the Langmuir model, Freundlich model and Dubinin–Radushkevich (D–R) were applied to examine the uranium adsorption processes of the three COFs. We found that the adsorption isotherm curves of the three COFs match well with the Langmuir model (Table S7†), suggesting that the adsorption processes of Tp-DAAQ, Tp-AD and Tp-DAQ are dominated by monolayer adsorption,48,49 which is in good agreement with the adsorption kinetic behavior of the three COFs. Furthermore, the three COFs exhibit higher U-uptake amounts at 323.15 K in comparison with those at 303.15 K (Fig. S39–S41†), which implies that temperature is favorable for uranium adsorption. Correspondingly, the thermodynamic parameters of Tp-DAAQ, Tp-AD and Tp-DAQ were calculated (Table S8†). The positive enthalpy change (ΔH) values (13.36 kJ mol−1 for Tp-DAAQ, 14.29 kJ mol−1 for Tp-AD and 11.51 kJ mol−1 for Tp-DAQ) confirm that the uranium adsorption process is endothermic. In addition, the negative Gibbs free energy (ΔG) at all temperatures indicates the spontaneous uranium adsorption behavior on Tp-DAAQ, Tp-AD and Tp-DAQ.21
In view of the high uranium adsorption capacity, the selectivities of Tp-DAAQ and Tp-AD towards U(VI) were further assessed. As shown in Fig. 3d and S32,† the adsorption capacities of Tp-DAAQ and Tp-AD for U(VI) are higher than 400 mg g−1, whereas they are lower than 40 mg g−1 for other metal ions at the same adsorption conditions. Even in the presence of multiple coexisting ions, the uranium adsorption capacities of Tp-DAAQ and Tp-AD have no obvious decrease (Fig. S61 and S62†). These results confirm the good adsorption selectivity for U(VI), which may be ascribed the synergistic coordination of N and O sites on Tp-DAAQ and Tp-AD. Due to the hard Lewis acid characteristic of UO22+, it is preferential to form a stable complex with hard Lewis bases such as N and O sites.57 Also, the distribution coefficients (Kd) of Tp-DAAQ and Tp-AD for U(VI) were calculated as 7.42 × 103 and 1.03 × 104, respectively. The high Kd values of the two COFs reveal the strong affinity between the frameworks and UO22+, indicating that Tp-DAAQ and Tp-AD can be considered decent materials for uranium adsorption.
Additionally, the cyclic utility of a U-adsorbent is equally paramount for application in practical environments. As displayed in Fig. 3e and S42,† the adsorption capacities of the two COFs for U(VI) show only negligible decreases after six cycles, indicating the excellent recycling and reusability of Tp-DAAQ and Tp-AD. The weak interaction between UO22+ and the frameworks of the two COFs is beneficial for desorption and regeneration. Accordingly, HNO3 solution (0.01 M) was used as the desorption agent. It is worth noting that the slight decrease of adsorption capacity is attributed to the loss of some active adsorption sites in the desorption process. Moreover, the consistent bands in the FT-IR spectra and unaltered PXRD patterns corroborate the undamaged crystal structures after cycles (Fig. S44 and S45†), which suggests that Tp-DAAQ and Tp-AD can serve as reusable adsorbents for uranium extraction.
Encouraged by the ultrahigh uranium adsorption capacities, we evaluated the possibility of Tp-DAAQ-4 and Tp-AD-4 for uranium extraction in four real water systems: tap water, river water (Shanghai, China), simulated seawater and actual Bohai seawater (Dalian, China). To our knowledge, tap water and river water contain a large amount of organic matter and a high concentration of calcium ion. In addition, seawater serves as one of the most complex water environments, containing more than 80 elements, ultrahigh concentration salts and plenty of microorganisms. Accordingly, Tp-DAAQ-4 and Tp-AD-4 were treated with the above-mentioned water solutions spiked with 10 mg L−1 UO22+. Even in the presence of a large number of interfering components, the extraction efficiency reached approximately 80% for Tp-DAAQ-4 and Tp-AD-4 in tap water and river water (Fig. 4e and S46†). More importantly, more than 60% of UO22+ was extracted from the simulated seawater and actual Bohai seawater. Based on these results, we think that Tp-DAAQ-4 and Tp-AD-4 can be used to extract uranium from complex water systems.
3.3 Uranium adsorption mechanism
To gain deep insight into the interaction between the COFs adsorbents and uranium, SEM-EDS, XPS spectra and PXRD were employed to analyze Tp-DAAQ-U, Tp-AD-U and Tp-DAQ-U. As illustrated in Fig. S47–S52,† uranium element is well scattered on the surface of the adsorbents, clearly verifying the successful adsorption of uranium on Tp-DAAQ, Tp-AD and Tp-DAQ. Compared to the original COFs, the full scale XPS spectra of Tp-DAAQ-U, Tp-AD-U and Tp-DAQ-U show two obvious new signals at 392.15 and 381.05 eV (Fig. S53†), which are attributed to U 4f5/2 and U 4f7/2. Moreover, the U 4f peak can be deconvolved into four peaks (Fig. S54†) corresponding to U(VI) 4f5/2 at 392.10 eV, U(IV) 4f5/2 at 391.40 eV, U(VI) 4f7/2 at 381.30 eV and U(IV) 4f7/2 at 380.60 eV.58,59 These results disclose the occurrence of a reduction reaction (UO22+ + 2e− = UO2) during the adsorption process. To confirm our speculation, the PXRD patterns of Tp-DAAQ-U, Tp-AD-U and Tp-DAQ-U were captured. As seen in Fig. S43,† a weak characteristic signal is observed at 27.86° which is ascribed to UO2 (PDF#41-1422).32In the case of Tp-DAAQ, the high-resolution N 1s and O 1s spectra can be divided into four peaks at 399.78, 399.24, 532.15 and 530.66 eV (Fig. S55†), which are assigned to C–N, CN, C–O and CO, respectively. Compared to CN and C–O, the binding energies of C–N and CO show obvious shifts after uranium adsorption. Meanwhile, similar results are observed on Tp-AD and Tp-DAQ (Fig. S56 and S57†), which indicates that C–N and CO may participate in the coordination with UO22+.
Density functional theory (DFT) calculations were used to shed light on the interaction mechanism at the molecular level. As displayed in Fig. 5a, the lowest unoccupied molecular orbitals (LUMO) and highest occupied molecular orbitals (HOMO) of Tp-DAAQ, Tp-AD and Tp-DAQ were calculated, respectively. Compared to Tp-AD, the HOMOs of Tp-DAAQ and Tp-DAQ are mainly distributed on the β-ketoenamine unit, while the LUMOs are exclusively located on the anthraquinone monomer, indicative of better D–A character for Tp-DAAQ and Tp-DAQ. Also, Tp-DAAQ and Tp-DAQ possess smaller energy gaps in comparison with Tp-AD. These results suggest that Tp-DAAQ and Tp-DAQ have better charge separation capabilities,38,60,61 thus promoting the occurrence of U(VI) reduction. Moreover, the electrostatic potential (ESP) of the three COFs was probed to investigate the charge distribution. As illustrated in Fig. 5b, the negative potential of the three COFs is mainly distributed on the oxygen atoms in the β-ketoenamine and anthraquinone units, indicating that oxygen atoms may play a paramount role in the coordination process with UO22+, which is in good agreement with the results of XPS analysis. In addition, Tp-DAAQ possesses the maximum dipole moment of 2.3826 D, demonstrating the strong driving force of Tp-DAAQ to facilitate electron–hole pair separation and the reduction process of U(VI) to U(IV).36,62,63 The adsorption energy was calculated to describe the potential interaction between the N and O sites in COFs and UO22+. As shown in Fig. 5c, Tp-AD exhibits a lower adsorption energy (−12.4 eV) than Tp-DAAQ (−6.9 eV), which explains why Tp-AD possesses a higher uranium adsorption capacity. In comparison with original Tp-DAAQ and Tp-AD, the adsorption energies of defective Tp-DAAQ and Tp-AD are significantly decreased, clearly disclosing the vital role of defect engineering for uranium adsorption.
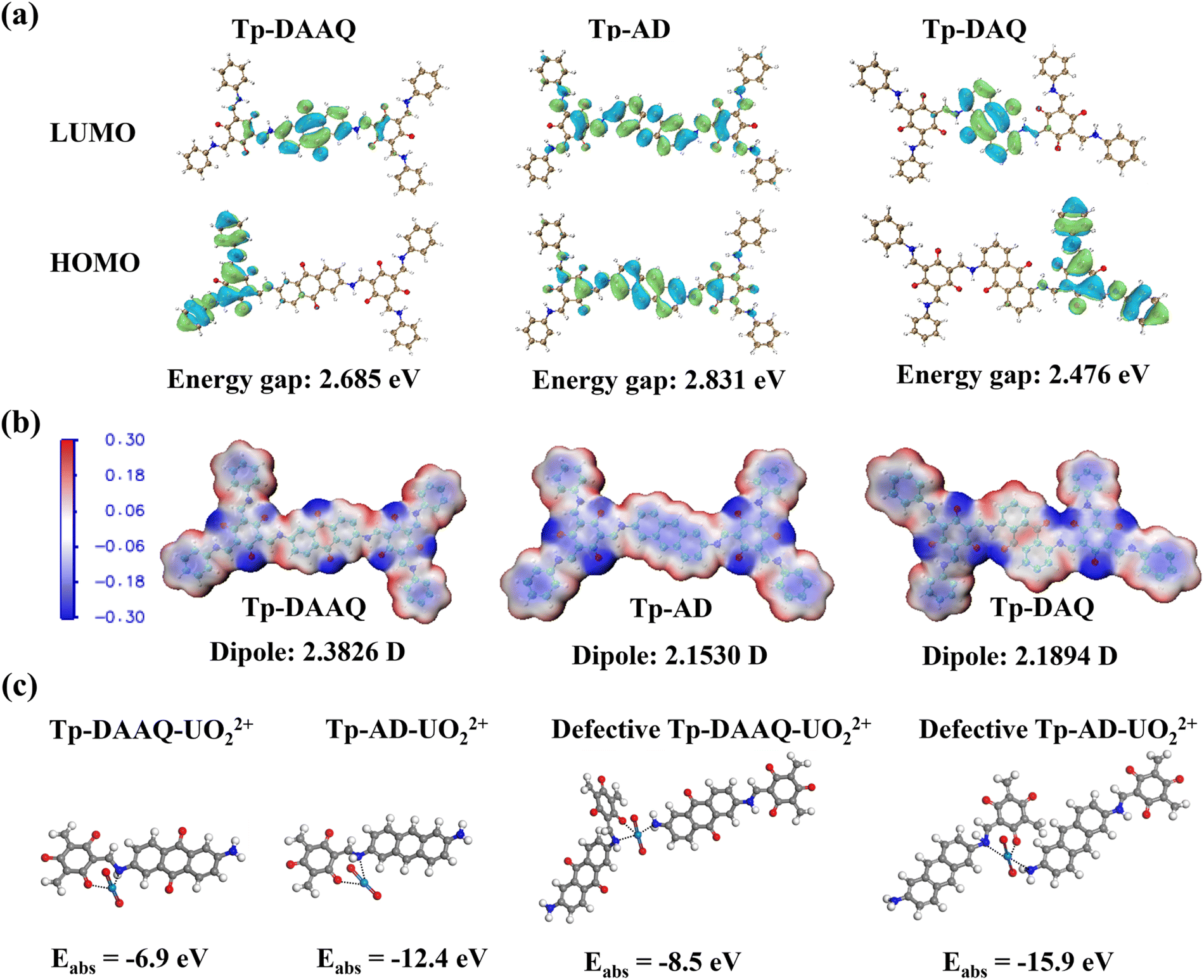 | ||
| Fig. 5 (a) LUMO and HOMO frontier molecular orbitals of the smallest model units of Tp-DAAQ, Tp-AD and Tp-DAQ. (b) Molecular ESP distributions and the corresponding dipole moments of Tp-DAAQ, Tp-AD and Tp-DAQ. (c) Optimal structures and the corresponding adsorption energies of different coordination modes with UO22+. | ||
3.4 Antibacterial performance assay
As is well known, the biofouling caused by microorganisms in various water systems hampers the adsorption capabilities of COFs adsorbents. Accordingly, the antibacterial efficacies of Tp-DAAQ, Tp-AD and Tp-DAQ were evaluated using Gram-negative Escherichia coli (E. coli), Gram-positive Staphylococcus aureus (S. aureus), methicillin-resistant S. aureus (MRSA) and Pseudoalteromonas marina (P. marina) widely found in Bohai seawater (China). After the bacterial suspension with COFs was co-cultured under dark or visible light irradiation conditions, the specific viability of bacteria was decided by the standard plate count method. As displayed in Fig. 6a and S59,† the three COFs can achieve above 90% bacteria killing efficiency for all kinds of bacteria under visible light irradiation for 10 min. Comparatively, no obvious antibacterial phenomena are observed without the addition of COFs under same visible light conditions, which suggests that the dominant antibacterial origination is Tp-DAAQ, Tp-AD and Tp-DAQ. Live/dead bacteria staining assays were conducted to further assess the antibacterial activity under different treatments. Live bacteria were stained by SYTO.9 with green fluorescence, while dead bacteria were stained by propidium iodide (PI) with red fluorescence. As can be seen in Fig. 6b, bright green fluorescence was observed on the blank sample, implying good bacterial viability. After incubation with COFs samples, the presence of obvious red fluorescence indicated that some of the bacteria were killed, which verified the good antibacterial activity. Additionally, similar antibacterial performance was observed with defective DAAQ-COFs and AD-COFs (Fig. S60†).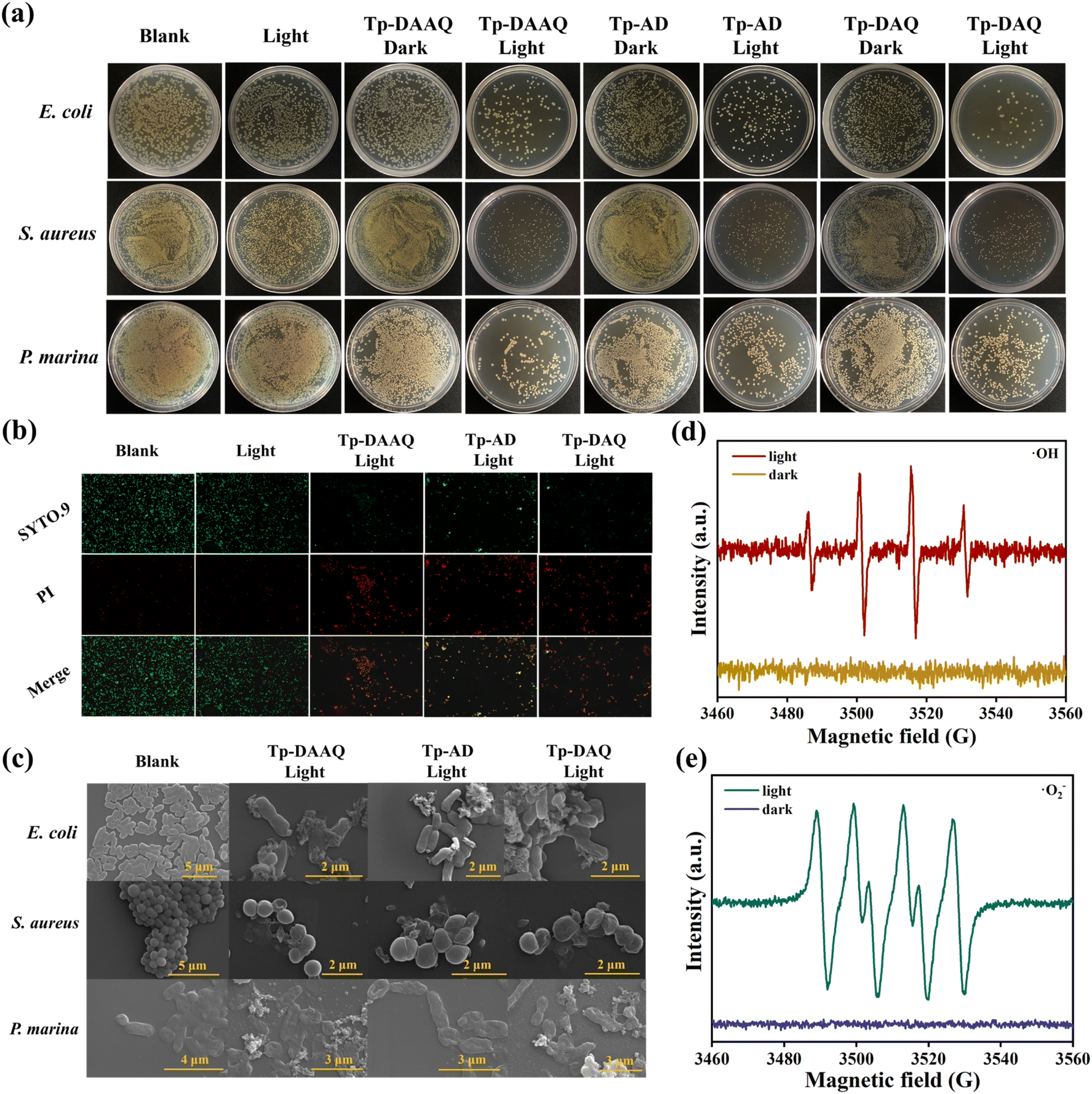 | ||
| Fig. 6 (a) Agar plate images of E. coli, S. aureus and P. marina after being treated with Tp-DAAQ, Tp-AD and Tp-DAQ in different formulations. (b) Live-dead staining images of P. marina co-cultured with Tp-DAAQ, Tp-AD and Tp-DAQ under different treatments. (c) SEM images of E. coli, S. aureus and P. marina incubated under visible light irradiation. ESR spectra of (d) ˙OH−DMPO and (e) ˙O2−DMPO complexes formed by visible light irradiation/no-irradiation of Tp-AD. | ||
To deeply understand the antibacterial behavior, the surface morphologies of bacteria treated with the three COFs samples under visible light irradiation were obtained by SEM tests. In comparison with the blank group, the bacteria treated by COFs samples show obvious wrinkles and membrane damage (Fig. 6c), which are attributed to the typical antibacterial mechanism of ROS release from Tp-DAAQ, Tp-AD and Tp-DAQ.64–67 Furthermore, electron spin resonance (ESR) experiments were conducted under dark and light conditions. As illustrated in Fig. 6d and e, no radical signals were detected under dark conditions, while ˙O2− and ˙OH were trapped by 3,4-dihydro-2,3-dimethyl-2H-pyrrole 1-oxide (DMPO) trapping agent under visible light irradiation for 10 min. These results reveal that the release of ROS is responsible for the outstanding sterilization performance of Tp-DAAQ, Tp-AD and Tp-DAQ under visible light-driven conditions. In addition, the surfaces of the three COFs are negatively charged (Fig. S31†), which can produce repulsion between the COFs and negatively charged bacteria,68,69 resulting in improved antibacterial performance. Collectively, the above-mentioned results demonstrate that all the COFs possess excellent anti-biofouling activity due to the generation of ROS, which is preferential for uranium extraction in complicated water environments.
4. Conclusions
In summary, we prepared a series of β-ketoenamine-linked COFs with good thermal stability and chemical stability for uranium extraction. After the introduction of defect engineering, the uranium adsorption performance was significantly improved, which is ascribed to the exposure of active defect sites. Experimental characterizations and DFT calculations reveal that the coordinative chelation of N and O in the COFs with UO22+ and the reduction of U(VI) to U(IV) are the primary uranium adsorption mechanisms. Meanwhile, the outstanding antibacterial activity of Tp-DAAQ, Tp-AD and Tp-DAQ is attributed to the generation of ROS (˙O2− and ˙OH). Benefiting from high specific affinity toward uranium and good anti-biofouling performance, the extraction efficiencies of Tp-DAAQ-4 and Tp-AD-4 for uranium are approximately 80% in tap water and river water and 60% in simulated seawater and actual Bohai seawater, highlighting their considerable application potential in complex water environments. This work not only provides a paradigm to design coordinative chelating sites in COFs for selective uranium adsorption, but also creatively introduces defect sites to improve the uranium extraction performance.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
B. Y. revised the manuscript. D. M. prepared the materials, measured the adsorption and antibacterial properties, collected pictures and wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by the National Natural Science Foundation of China (22375150) and the Developing Science Funds of Tongji University (1380219059).References
- Y. Wu, Y. Xie, X. Liu, Y. Li, J. Wang, Z. Chen, H. Yang, B. Hu, C. Shen, Z. Tang, Q. Huang and X. Wang, Coord. Chem. Rev., 2023, 483, 215097 CrossRef CAS.
- H. Yang, X. Liu, M. Hao, Y. Xie, X. Wang, H. Tian, G. I. N. Waterhouse, P. E. Kruger, S. G. Telfer and S. Ma, Adv. Mater., 2021, 33, 2106621 CrossRef CAS PubMed.
- H. Wang, W. Yao, Y. Yuan, S. Shi, T. Liu and N. Wang, Adv. Sci., 2024, 11, 2306534 CrossRef CAS PubMed.
- Y. Xie, Z. Liu, Y. Geng, H. Li, N. Wang, Y. Song, X. Wang, J. Chen, J. Wang, S. Ma and G. Ye, Chem. Soc. Rev., 2023, 52, 97–162 RSC.
- X. Ma, K. R. Meihaus, Y. Yang, Y. Zheng, F. Cui, J. Li, Y. Zhao, B. Jiang, Y. Yuan, J. R. Long and G. Zhu, J. Am. Chem. Soc., 2024, 146, 23566–23573 CrossRef CAS PubMed.
- D. Mei, L. Liu and B. Yan, Coord. Chem. Rev., 2023, 475, 214917 CrossRef CAS.
- Y. Sun, R. Leng, X. Ma, J. Zhang, B. Han, G. Zhao, Y. Ai, B. Hu, Z. Ji and X. Wang, Chem. Eng. J., 2023, 459, 141687 CrossRef CAS.
- W. Cui, C. Zhang, W. Jiang, F. Li, R. Liang, J. Liu and J. Qiu, Nat. Commun., 2020, 11, 436 CrossRef CAS PubMed.
- W. Liu, X. Dai, Z. Bai, Y. Wang, Z. Yang, L. Zhang, L. Xu, L. Chen, Y. Li, D. Gui, J. Diwu, J. Wang, R. Zhou, Z. Chai and S. Wang, Environ. Sci. Technol., 2017, 51, 3911–3921 CrossRef CAS PubMed.
- M. Zheng, K. Xuan, S. Yan, Y. Guo, Y. Huang, R. Xu, K. Zhao, Z. Li, X. Li, H. Jiang and Y. Guo, Sep. Purif. Technol., 2024, 339, 126550 CrossRef CAS.
- C. Yu, W. Jiang, M. Lei, M. Yao, X. Sun, Y. Wang, W. Liu and L. Liu, Small, 2024, 20, 2308910 CrossRef CAS PubMed.
- D. Mei, H. Li, L. Liu, L. Jiang, C. Zhang, X. Wu, H. Dong and F. Ma, Chem. Eng. J., 2021, 425, 130468 CrossRef CAS.
- Y. Xu, Z. Yu, Q. Zhang and F. Luo, Adv. Sci., 2023, 10, 2300408 CrossRef CAS PubMed.
- C. Zhang, W. Cui, R. Xu, X. Chen, W. Jiang, Y. Wu, R. Yan, R. Liang and J. Qiu, CCS Chem., 2021, 3, 168–179 CrossRef CAS.
- A. Kaushik, K. Marvaniya, Y. Kulkarni, D. Bhatt, J. Bhatt, M. Mane, E. Suresh, S. Tothadi, K. Patel and S. Kushwaha, Chem, 2022, 10, 2749–2765 Search PubMed.
- S. Mollick, S. Saurabh, Y. D. More, S. Fajal, M. M. Shirolkar, W. Mandal and S. K. Ghosh, Energy Environ. Sci., 2022, 15, 3462–3469 RSC.
- M. Xu, T. Wang, P. Gao, L. Zhao, L. Zhou and D. Hua, J. Mater. Chem. A, 2019, 7, 11214–11222 RSC.
- R. Leng, Y. Sun, C. Wang, Z. Qu, R. Feng, G. Zhao, B. Han, J. Wang, Z. Ji and X. Wang, Environ. Sci. Technol., 2023, 57, 9615–9626 CrossRef CAS PubMed.
- Q. Sun, B. Aguila, L. D. Earl, C. W. Abney, L. Wojtas, P. K. Thallapally and S. Ma, Adv. Mater., 2018, 30, 1705479 CrossRef PubMed.
- Z. Li, R. Zhu, P. Zhang, M. Yang, R. Zhao, Y. Wang, X. Dai and W. Liu, Chem. Eng. J., 2022, 434, 134623 CrossRef CAS.
- D. Mei and B. Yan, Adv. Funct. Mater., 2024, 34, 2313314 CrossRef CAS.
- M. Zhou, S. Wang, F. Liu and B. Hu, Sep. Purif. Technol., 2024, 338, 126501 CrossRef CAS.
- H. Zhang, S. Wang, J. Yu, Z. Li, J. Lan, L. Zheng, S. Liu, L. Yuan, T. Xiu, J. Wang, X. Wang and W. Shi, Chem. Eng. J., 2023, 463, 142408 CrossRef CAS.
- W. Cui, F. Li, R. Xu, C. Zhang, X. Chen, R. Yan, R. Liang and J. Qiu, Angew. Chem., Int. Ed., 2020, 59, 17684–17690 CrossRef CAS PubMed.
- S. Daliran, M. Blanco, A. Dhakshinamoorthy, A. R. Oveisi, J. Alemán and H. García, Adv. Funct. Mater., 2024, 34, 2312912 CrossRef CAS.
- K. Du, L. Xiong, C. Fu, X. Ni, J. Bredas and H. Li, ACS Mater. Lett., 2024, 6, 335–344 CrossRef CAS.
- S. Zhang, X. Yi, G. Hu, M. Chen, H. Shen, B. Li, L. Yang, W. Dai, J. Zou and S. Luo, Coord. Chem. Rev., 2023, 478, 214970 CrossRef CAS.
- X. Sun, X. Zhang and Y. Xie, Matter, 2020, 2, 842 CrossRef.
- J. Xu, X. X. Xue, G. Shao, C. Jing, S. Dai, K. He, P. Jia, S. Wang, Y. Yuan, J. Luo and J. Lu, Nat. Commun., 2023, 14, 7849 CrossRef CAS PubMed.
- S. Hui, X. Zhou, L. Zhang and H. Wu, Adv. Sci., 2024, 11, 2307649 CrossRef CAS PubMed.
- C. Shu, X. Yang, L. Liu, X. Hu, R. Sun, X. Yang, A. I. Cooper, B. Tan and X. Wang, Angew. Chem., Int. Ed., 2024, 63, e202403926 CrossRef CAS PubMed.
- X. Wang, C. Zhang, R. Bi, Z. Peng, A. Song, R. Zhang, H. He, J. Qi, J. Gong, C. Niu, R. Liang and J. Qiu, Adv. Funct. Mater., 2024, 2421623 CrossRef.
- X. Lan, H. Li, Y. Liu, Y. Zhang, T. Zhang and Y. Chen, Angew. Chem., Int. Ed., 2024, 63, e202407092 CrossRef CAS PubMed.
- X. Zhong, Q. Ling, P. Kuang and B. Hu, Chem. Eng. J., 2023, 467, 143415 CrossRef CAS.
- X. Lu, H. Wang, Y. Sun, Y. Xu, W. Sun, Y. Wu, Y. Zhang, C. Yang and Y. Wang, Angew. Chem., Int. Ed., 2024, 63, e202409436 CrossRef CAS PubMed.
- N. Liu, S. Xie, Y. Huang, J. Lu, H. Shi, S. Xu, G. Zhang and X. Chen, Adv. Energy Mater., 2024, 14, 2402395 CrossRef CAS.
- H. Wang, C. Qian, J. Liu, Y. Zeng, D. Wang, W. Zhou, L. Gu, H. Wu, G. Liu and Y. Zhao, J. Am. Chem. Soc., 2020, 142, 4862 CrossRef CAS PubMed.
- Q. Huang, H. Zhang, H. Yang, Z. Yu, Z. Xu, Z. Li, Z. Gao and J. Zou, Appl. Catal., B, 2025, 362, 124721 CrossRef CAS.
- M. A. Khayum, S. Kandambeth, S. Mitra, S. B. Nair, A. Das, S. S. Nagane, R. Mukherjee and R. Banerjee, Angew. Chem., Int. Ed., 2016, 55, 15604 CrossRef CAS PubMed.
- S. Cai, R. Ma, W. Ke, H. Zhang, Y. Liu, M. Jiao, Y. Tian, Y. Fang, M. Wu and Z. Zhou, Chem. Eng. J., 2024, 491, 151979 CrossRef CAS.
- Y. Tao, Y. Hou, H. Yang, Z. Gong, J. Yu, H. Zhong, Q. Fu, J. Wang, F. Zhu and G. Ouyang, Proc. Natl. Acad. Sci. U. S. A., 2024, 121, e2401175121 CrossRef CAS PubMed.
- X. Zhang, J. Zhang, J. Miao, X. Wen, C. Chen, B. Zhou and M. Long, Chem. Eng. J., 2023, 466, 143085 CrossRef CAS.
- C. Yang, L. Hou, H. M. Adeel Sharif, Y. Wang, Y. Cai, C. Li and L. Hou, Sep. Purif. Technol., 2024, 350, 127941 CrossRef CAS.
- M. Liu, Y. Wang, H. Ding, M. Lu, G. Gao, L. Dong, Q. Li, Y. Chen, S. Li and Y. Lan, Sci. Bull., 2021, 66, 1659–1668 CrossRef CAS PubMed.
- X. Huang, Y. Chen, X. Xie and T. Song, Small, 2025, 21, 2408817 CrossRef CAS PubMed.
- G. Liu, Y. Wang, C. Shen, Z. Ju and D. Yuan, J. Mater. Chem. A, 2015, 3, 3051 RSC.
- R. Yao, J. Wang, Y. Li, W. Qu, B. Yang, J. Lu, Z. Wu, Y. Li, Z. Lu, Z. Geng and Z. Wang, Chem. Eng. J., 2025, 503, 158513 CrossRef CAS.
- F. Li, W. Cui, W. Jiang, C. Zhang, R. Liang and J. Qiu, J. Hazard. Mater., 2020, 392, 122333 CrossRef CAS PubMed.
- X. Liu, X. Wang, W. Jiang, C. Zhang, L. Zhang, R. Liang and J. Qiu, Chem. Eng. J., 2022, 450, 138062 CrossRef CAS.
- X. Li, Y. Qi, G. Yue, Q. Wu, Y. Li, M. Zhang, X. Guo, X. Li, L. Ma and S. Li, Green Chem., 2019, 21, 649 RSC.
- X. Xiong, Z. Yu, L. Gong, Y. Tao, Z. Gao, L. Wang, W. Yin, L. Yang and F. Luo, Adv. Sci., 2019, 6, 1900547 CrossRef PubMed.
- W. Cui, C. Zhang, R. Xu, X. Chen, R. Yan, W. Jiang, R. Liang and J. Qiu, ACS EST Water, 2021, 1, 440–448 CrossRef CAS.
- L. Zhong, X. Feng, Q. Zhang, X. Xie and F. Luo, Chem. Sci., 2024, 15, 10882 RSC.
- L. Guo, Z. Huang and F. Luo, Nano Lett., 2024, 24, 14153–14161 CrossRef CAS PubMed.
- J. Zhao, L. Zhao, Q. Gao, G. Fu, F. Li, H. Liu and Y. Fa, Chem. Eng. J., 2024, 500, 157017 CrossRef CAS.
- Z. Li, H. Zhang, X. Xiong and F. Luo, J. Solid State Chem., 2019, 277, 484–492 CrossRef CAS.
- H. Li, J. Sun, S. Qin, Y. Song, Z. Liu, P. Yang, S. Li, C. Liu and C. Shen, Adv. Funct. Mater., 2023, 33, 2301773 CrossRef CAS.
- D. Mei, L. Liu, H. Li, Y. Wang, F. Ma, C. Zhang and H. Dong, J. Hazard. Mater., 2022, 422, 126872 CrossRef CAS PubMed.
- L. Wang, H. Song, L. Yuan, Z. Li, Y. Zhang, J. K. Gibson, L. Zheng, Z. Chai and W. Shi, Environ. Sci. Technol., 2018, 52, 10748–10756 CrossRef CAS PubMed.
- Z. Zhang, Q. Zhang, Y. Hou, J. Li, S. Zhu, H. Xia, H. Yue and X. Liu, Angew. Chem., Int. Ed., 2024, 63, e202411546 CrossRef CAS PubMed.
- Z. Gao, S. Lv, Y. Wang, Z. Xu, Y. Zong, Y. Tao, Y. Zhao, X. Liu, S. Yu, M. Luo, N. Khaorapapong, R. Zhang and Y. Yamauchi, Adv. Sci., 2024, 11, 2406530 CrossRef CAS PubMed.
- C. Han, Z. Wang, S. Sun, J. Guo, X. Huang and X. Liu, ACS Mater. Lett., 2025, 7, 393–400 CrossRef CAS.
- F. Yu, C. Li, W. Li, Z. Yu, Z. Xu, Y. Liu, B. Wang, B. Na and J. Qiu, Adv. Funct. Mater., 2024, 34, 2307230 CrossRef CAS.
- Z. Chen, J. Wang, M. Hao, Y. Xie, X. Liu, H. Yang, G. I. N. Waterhouse, X. Wang and S. Ma, Nat. Commun., 2023, 14, 1106 CrossRef CAS PubMed.
- S. Liu, L. Yao, X. Wang, G. Zhao, F. Li, B. Yao and Y. Dong, J. Mater. Chem. A, 2025, 13, 4530–4537 RSC.
- X. Lin, M. Zhang, W. Lv, J. Li, R. Huang and Y. Wang, Adv. Funct. Mater., 2024, 34, 2310845 CrossRef CAS.
- C. Zhang, J. Guo, X. Zou, S. Guo, Y. Guo, R. Shi and F. Yan, Adv. Healthcare Mater., 2021, 10, 2100775 CrossRef CAS PubMed.
- A. Bigham, V. Rahimkhoei, P. Abasian, M. Delfi, J. Naderi, M. Ghomi, F. Dabbagh Moghaddam, T. Waqar, Y. Nuri Ertas, S. Sharifi, N. Rabiee, S. Ersoy, A. Maleki, E. Nazarzadeh Zare, E. Sharifi, E. Jabbari, P. Makvandi and A. Akbari, Chem. Eng. J., 2022, 432, 134146 CrossRef CAS.
- M. Chen, D. Liu, T. Liu, T. Wei, Q. Qiao, Y. Yuan and N. Wang, Small, 2024, 20, 2401528 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: The refinement details and additional figures. See DOI: https://doi.org/10.1039/d5ta01632k |
| This journal is © The Royal Society of Chemistry 2025 |