
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A novel mixed-conducting network in all-oxide composites: overcoming traditional percolation constraints†
Fanlin
Zeng
 *a,
Ke
Ran
bc,
Christian
Dellen
a,
Hartmut
Schlenz
a,
Joachim
Mayer
bc,
Ruth
Schwaiger
ade,
Wilhelm Albert
Meulenberg
af and
Stefan
Baumann
a
*a,
Ke
Ran
bc,
Christian
Dellen
a,
Hartmut
Schlenz
a,
Joachim
Mayer
bc,
Ruth
Schwaiger
ade,
Wilhelm Albert
Meulenberg
af and
Stefan
Baumann
a
aForschungszentrum Jülich GmbH, Institute of Energy Materials and Devices (IMD), Jülich, 52425, Germany. E-mail: f.zeng@fz-juelich.de
bCentral Facility for Electron Microscopy GFE, RWTH Aachen University, 52074 Aachen, Germany
cErnst Ruska-Centre for Microscopy and Spectroscopy with Electrons (ER-C), Forschungszentrum Jülich GmbH, 52425 Jülich, Germany
dRWTH Aachen University, Chair of Energy Engineering Materials, Aachen, 52056, Germany
eJülich Aachen Research Alliance, JARA-Energy, Jülich, 52425, Germany
fUniversity of Twente, Faculty of Science and Technology, Inorganic Membranes, P. O. Box 217, Enschede, 7500 AE, The Netherlands
First published on 10th April 2025
Abstract
Mixed-conducting composites play pivotal roles in ceramic devices for advancing efficient and environment-friendly energy consumption and conversion processes. Conventionally, these materials are synthesized via the blending of distinct conducting phases, where grain percolation of each phase is considered essential. This approach inevitably leads to intertwined networks interspersed with inactive regions, limiting the overall performance. This study challenges this conventional paradigm by proposing an alternative percolation mechanism that circumvents the need for strict grain connectivity. The mechanism is demonstrated in composites of doped ceria with iron–cobalt oxide additives, where grains of the doped ceria constitute over 80 vol% and are nearly completely percolated for efficient and rapid ionic conduction. Remarkably, even though the additive-induced electronic conducting grains occupy less than 20 vol% and are distributed as islands, the observed electronic conductivity far surpasses conventional predictions. This anomaly is attributed to the accumulation of charge carriers at ceria grain boundaries, which facilitates electronic conduction. Through extensive structural and compositional analyses at micro- and nanoscale levels, the study unveils novel insights into the intricate architecture of this advanced percolation network. Furthermore, the optimization of these composites is achieved by enriching iron and cobalt cations at ceria grain boundaries, while inhibiting grain coarsening. This delicate balance culminates in excellent and sustainable mixed conductivity for oxygen permeation, thus advancing the potential of mixed-conducting composites for applications in clean and efficient energy technologies.
Introduction
Mixed ionic and electronic conducting materials are indispensable in deploying electrochemical ceramic devices to supplant high carbon-emitting industrial processes and facilitate energy conversion from green sources.1,2 For instance, these materials are widely applied as dense membranes in catalytic membrane reactors for partial oxidation of hydrocarbons,3 hydrogen purification,4 and water splitting,5,6 as well as porous electrodes in solid oxide fuel cells (SOFCs) for green power conversion.7Perovskite oxide compounds such as Ba0.5Sr0.5Co0.8Fe0.2O3−δ and La0.6Sr0.4Co0.2Fe0.8O3−δ are widely recognized for their excellent mixed ionic and electronic conductivity.8,9 However, these materials suffer from poor chemical and dimensional stability in application-relevant atmospheres involving CO2, H2O, CH4, H2S, and/or SO2.10,11
By contrast, the fluorite-type compounds, such as ZrO2 stabilized by Y or/and Sc,12–14 as well as CeO2 doped with trivalent rare earth elements (Gd, Sm, and Nd), demonstrate superior chemical compatibility with corrosive gas species.15,16 Unfortunately, Y- or Sc-stabilized ZrO2 shows negligible electronic conductivity within the application temperature range of 800–1000 °C.17 Similarly, trivalent lanthanide (Gd, Sm, and Nd) doped CeO2 functions predominantly as a pure ionic conductor across a broad range of oxygen partial pressure from 0.21 atm to ∼10−15 atm in the same temperature window.18 To introduce mixed conductivity in CeO2, lanthanide dopants with mixed valence states, e.g., Pr and Tb, can be utilized.19–21 In addition, the electronic conductivity can be enhanced by adding a single or binary oxide containing redox-active transition metal element(s).22–27 At sufficiently high concentrations, these additives form an interconnected grain network supporting substantial electronic conductivity. However, excessive additive content leads to a volumetric dilution of the fluorite matrix and disrupts the contiguity of fluorite grains,28 both of which are detrimental to the already sluggish ionic conduction. Thus, it is essential to minimize the additive content while ensuring effective electronic percolation. Experimental findings suggest that even a minor addition (∼2 mol%) of Fe, Co or Cu cations to doped CeO2 can feasibly induce p-type electronic conduction under air condition.29,30
These additives do not form separate electronic conducting grains but predominantly segregate at ceria grain boundaries, existing as point defects or a thin amorphous layer.30,31 In this case, the electron/hole conduction occurs along ceria grain boundaries, as illustrated by schematic I in Fig. 1(a). The corresponding electronic conductivity (line I in Fig. 1(b)) for 2 mol% Co-modified Ce0.8Gd0.2O2−δ exhibits a high activation energy and remains approximately an order of magnitude lower than the ionic conductivity.29 Consequently, the resultant electronic conductivity is insufficient for high-performance electrochemical applications, particularly when compared to CeO2 doped with mixed-valence cations such as Pr. A likely cause is the low concentration of charge carriers at ceria grain boundaries.
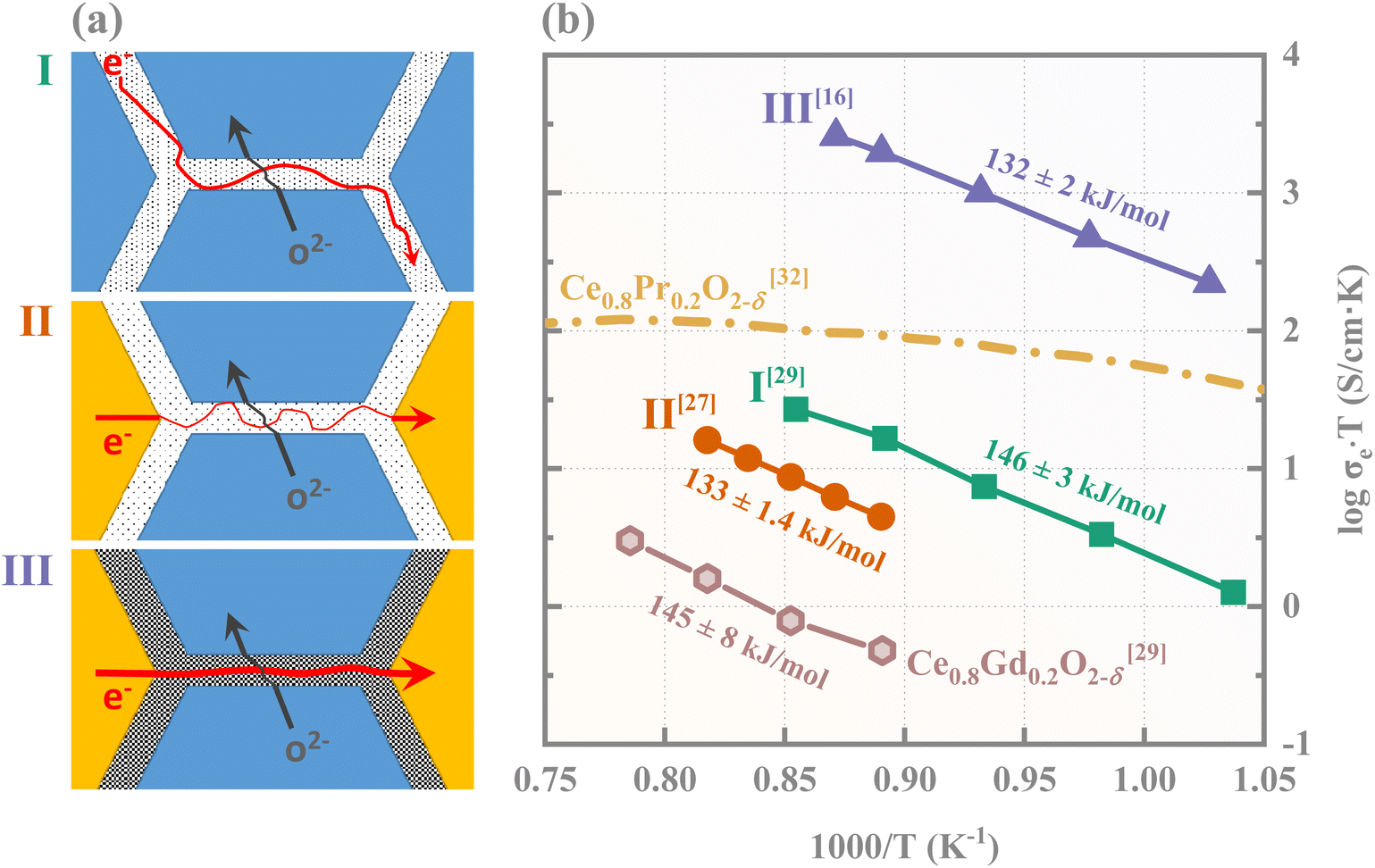 | ||
| Fig. 1 Schematic illustrations (a) and the corresponding electronic conductivities (b) of microstructural configurations where ceria grain boundaries serve as critical pathways for mixed ionic and electronic conduction. In (a), blue and yellow polygons represent grains of the ionic and electronic conductor, respectively, with intergranular gaps denoting grain boundaries. Black dots of varying densities indicate charge carriers at different concentrations. Red and black line arrows represent electronic and ionic pathways, respectively, with thicker ones signifying faster transport and thinner ones indicating slower conduction. In (b), lines I, II, and III correspond to the electronic conductivities of 2 mol% Co-modified Ce0.8Gd0.2O2−δ,29 20 vol% CoFe2O4–80 vol% Ce0.8Gd0.2O2−δ,27 and 18.5 vol% FeCo2O4–81.5 vol% Ce0.8Gd0.2O2−δ,16 respectively. The electronic conductivity values of line II are estimated to be equal to ambipolar conductivities, given that the ionic conductivity values are approximately one order of magnitude higher.27 For comparison, the electronic conductivity values of pure Ce0.8Gd0.2O2−δ29 and Ce0.8Pr0.2O2−δ32 are also presented. The respective activation energies, expressed in kJ mol−1, are annotated next to the plotted data. | ||
An alternative and underexplored composite architecture is proposed herein, as illustrated by schematics II and III in Fig. 1(a). In this structure, the transition metal oxide additive(s) not only forms islands of electronic conducting grains within the ceria matrix but also decorates ceria grain boundaries with transition metal element(s). This configuration enables ceria grain boundaries to act as an “electronic conducting shuttle” between adjacent electronic conducting grains. As compared to the system in schematic I where ceria grain boundaries serve as the sole electronic conducting channels, this arrangement can offer physically shortened paths for electrons/holes along ceria grain boundaries without blocking ionic conduction pathways across ceria grain boundaries. However, experimental results reveal striking conductivity differences between two similar structure representatives: 80 vol% Ce0.8Gd0.2O2−δ–20 vol% CoFe2O4 (ref. 27) and 81.5 vol% Ce0.8Gd0.2O2−δ–18.5 vol% FeCo2O4.16 Their electronic conductivities, plotted as lines II and III in Fig. 1(b), differ by over two orders of magnitude. The 20 vol% CoFe2O4 additives fail to overcome the electronic conductivity bottleneck of Ce0.8Gd0.2O2−δ. A likely cause is the insufficient transition metal segregation at ceria grain boundaries (schematic II in Fig. 1(a)), which results in highly tortuous electronic conducting paths. By contrast, the addition of 18.5 vol% FeCo2O4 leads to enriched transition metal segregation at ceria grain boundaries (schematic III in Fig. 1(a)),33 significantly accelerating electronic transport along ceria grain boundaries.16
Given these findings, further optimization of this composite architecture is imperative to maximize concurrent ionic and electronic conduction. The network geometry and ceria grain boundary composition are expected to have critical effects. To the best of our knowledge, the impact of network geometry has not yet been thoroughly investigated and discussed. While the ceria grain boundary composition is known to govern electronic percolation, its precise modulation and characterization have proven challenging. Additionally, its influence on ionic conductivity remains unclear.
In this work, we systematically address these open questions and challenges using FeCo2O4 modified Ce0.8Gd0.2O2−δ mixed conductors as a model system. The FeCo2O4 content is limited to 15 wt% (∼19.05 vol% or ∼11.44 mol%), i.e., a concentration insufficient for realizing grain percolation. Notably, these two components exhibit strong chemical interactions, leading to the formation of a new electronic conducting GdFeO3-type perovskite, in which Ce substitutes for Gd and Co for Fe.16,28,34,35 This strong interaction provides an excellent opportunity to tailor grain boundary composition and properties. Our results demonstrated that sintering conditions significantly influence the grain boundary structure and segregation. We further introduce a novel approach, wherein Gd2O3 as an additional Gd source is incorporated to enhance phase interactions and optimize the grain boundary environment.
To elucidate the fundamental mechanisms governing mixed conduction, grain boundary characterization is performed at the nanoscale using scanning transmission electron microscopy, combined with energy-dispersive X-ray spectroscopy. Microscale analysis is conducted using scanning electron microscopy and electron backscatter diffraction to quantitatively assess the microstructural evolution and geometric effects on conduction pathways. Finally, the advances in grain arrangement and grain boundary composition in mixed ionic and electronic conduction are evaluated through electrochemical impedance spectroscopy and performance testing in oxygen separation/purification applications.
Experimental
Material preparation
The FeCo2O4 modified Ce0.8Gd0.2O2−δ (FC-CGO) mixed conductors were synthesized via a solid-state reactive sintering process,34,36 wherein 85 wt% of Ce0.8Gd0.2O2−δ (Treibacher Industrie AG, 99%) was ball-milled in ethanol with 15 wt% FeCo2O4. The latter was prepared from a raw material mixture of Fe2O3 (Merck, 99%) and Co3O4 (Merck, 99%) in a 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4 mole ratio. Although FeCo2O4 spinel was not pre-synthesized, high-temperature XRD analysis revealed that a complete chemical reaction between Co3O4 and Fe2O3 occurs at ∼900 °C, yielding FeCo2O4 spinel.36 The dried and sieved powders were pressed into pellets at ∼19 MPa and densified after a single-step heating in air at a rate of 3 K min−1 to either 1050 °C or 1200 °C for 10 h. Details of sample preparation and treatment that are not specific here can be found in a previous study.36 The samples are denoted as FC-CGO_LT and FC-CGO_HT, with “LT” and “HT” referring to the sintering temperature of 1050 °C and 1200 °C, respectively.
:4 mole ratio. Although FeCo2O4 spinel was not pre-synthesized, high-temperature XRD analysis revealed that a complete chemical reaction between Co3O4 and Fe2O3 occurs at ∼900 °C, yielding FeCo2O4 spinel.36 The dried and sieved powders were pressed into pellets at ∼19 MPa and densified after a single-step heating in air at a rate of 3 K min−1 to either 1050 °C or 1200 °C for 10 h. Details of sample preparation and treatment that are not specific here can be found in a previous study.36 The samples are denoted as FC-CGO_LT and FC-CGO_HT, with “LT” and “HT” referring to the sintering temperature of 1050 °C and 1200 °C, respectively.
To promote phase interaction, the total Gd content in the FC-CGO powder was further increased by adding Gd2O3 (Sigma-Aldrich, 99.9%) until the overall Gd/Ce ratio from 2:8 to 3:7. This resulted in a composition of approximately 8.1 wt% Gd2O3, 13.8 wt% FeCo2O4, and 78.1 wt% Ce0.8Gd0.2O2−δ. The same synthesis procedures were then applied to this composition, and the resulting sample is referred to as FCG-CGO_HT.
Additionally, four other conductors were synthesized for comparison analysis. A pure ionic conductor (IC), sintered from Ce0.8Gd0.2O2−δ with minor Fe2O3 addition, represents the fluorite-type ionic conducting phase resulting from phase interaction in the FeCo2O4 modified Ce0.8Gd0.2O2−δ composites.28
A pure electronic conductor (EC), synthesized as a composite containing spinel, rock-salt, and perovskite phases, represents the collective performance of all electronic conducting phases present in the FeCo2O4 modified Ce0.8Gd0.2O2−δ.28
Two mixed conductors (40FC-CGO_HT and CF-CGO_HT) were prepared from 40 wt% FeCo2O4–60 wt% Ce0.8Gd0.2O2−δ and 15 wt% CoFe2O4–85 wt% Ce0.8Gd0.2O2−δ, respectively.
The preparation steps for these additional samples are detailed in ESI Note 1.†
Phase and microstructure investigation
Microstructural characterization and phase analysis were performed on polished sample cross-sections using scanning electron microscopy (SEM; Merlin, Carl Zeiss Microscopy, Oberkochen, Germany) and electron backscatter diffraction (EBSD; NordlysNano, Oxford Instruments, Wiesbaden, Germany). The volume fraction of each phase was estimated by assuming area fraction equivalence,37 determined via image analysis using the HKL Channel 5 software package based on the EBSD results. Grain percolation and tortuosity calculations were performed using the open-source toolkit PoreSpy.38Grain boundary characterization
Cross-sectional samples were prepared via focused ion beam (FIB) milling using a FEI Strata 400 system equipped with Ga ion beam. Further thinning and cleaning were applied to the lamellas using an Ar ion beam in a Fischione Nanomill 1040 at 900 eV and 500 eV beam energy, respectively. Transmission electron microscopy (TEM) and energy-filtered TEM (EFTEM) imaging were conducted using a FEI Tecnai F20 at 200 kV. High resolution high-angle annular dark-field (HAADF) imaging, energy-dispersive X-ray spectroscopy (EDXS) chemical mapping, and electron energy loss spectroscopy (EELS) spectrum imaging were performed using a FEI Titan G2 80-200 ChemiSTEM microscope. This system was equipped with an XFEG, a probe Cs corrector, a Super-X EDXS system, and a Gatan Enfinium ER (model 977) spectrometer with DUAL EELS acquisition capability.39 A convergence semi-angle of 22 mrad was approximated for scanning transmission electron microscopy (STEM) imaging and EDXS mapping with a typical time of around 10 minutes. Background subtraction was applied to improve signal clarity. The collection semi-angles were 80–220 mrad for HAADF imaging. The image quality was enhanced by application of an iterative rigid alignment algorithm, followed by smoothing using a non-linear filtering algorithm.40Performance assessment
The material performance was assessed via total conductivity measurements and oxygen permeation experiments.Total conductivity (σtot), defined as the sum of ionic (σi) and electronic conductivity (σe) (see eqn (1)), was measured under air and argon (Ar) conditions using electrochemical impedance spectroscopy (EIS) at frequencies from 0.1 Hz to 10 MHz.
Measurements were conducted every 50 K from 900 °C to 750 °C. Prior to measurements, silver paste was brushed on the polished sample surfaces, and fired at 900 °C for 1 h to form a thin current-collecting silver layer. Each sample was saturated at the target temperature for 1 h before measurement.
| σtot = σi + σe | (1) |
Oxygen permeation was measured using a 4-end in-house built setup. To eliminate surface exchange limitations, the sintered pellets were polished and screen printed with a ∼4 μm thick porous La0.58Sr0.4Co0.2Fe0.8O3−δ coating on both sides.34,41 The coated pellets were then sealed with gold rings and mounted in glass tubes at ∼1020 °C under pressure. One side of each pellet was fed with air at a flow rate of 250 ml min−1, while the other side was swept by Ar at a flow rate of 50 ml min−1. Oxygen flux was recorded at steady-state conditions every 50 °C from ∼1000 °C down to ∼850 °C, and then normalized by the oxygen partial pressure gradient across the pellet to determine oxygen permeance. If the permeation process is limited by bulk diffusion rather than surface exchange, oxygen permeance can be described by eqn (2).34,41
 | (2) |
 | (3) |
The partial conductivity values (σi and σe) were calculated by combining eqn (1) and (3).
The temperature dependence of conductivity was assessed using activation energy, determined from:
 | (4) |
Results and discussion
Material characterization
 | ||
| Fig. 2 Microstructural characterization via SEM (left column) and EBSD phase mappings (right column) for FC-CGO_LT (a and b), FC-CGO_HT (c and d) and FCG-CGO_HT (e and f). In these images, black dots and lines correspond to pores and grain boundaries, respectively. The annotation given below delineates the specific phases corresponding to the numbered and color-coded grains, with their respective volume fractions provided alongside the individual phase mappings. (a, c and d) were adapted with permission from the previously published studies.34,36 | ||
Further analysis via EBSD on randomly selected cross-sections, as shown in Fig. 2(b, d and f), confirms the crystallographic nature of these grains. The grains of the Ce–Gd oxide and the Gd, Fe-rich oxide adopt a Ce1−xGdxO2−δ fluorite and a GdFeO3-type perovskite structure, respectively, while the Fe–Co oxide grains exhibit either a spinel or a rock-salt structure. Notably, the Ce1−xGdxO2−δ fluorite phase, which serves as the ionic conducting phase, constitutes over 80 vol% in every sample due to the 85 wt% Ce0.8Gd0.2O2−δ precursor input. Whereas, the electronic conducting phases, possessing perovskite, spinel, and rock-salt structure,28 collectively remain below the theoretical percolation threshold value of 30 vol%.
A comparative assessment reveals that FC-CGO_LT and FC-CGO_HT contain similar proportions of individual electronic conducting phases. However, FCG-CGO_HT exhibits a higher concentration of perovskite and rock-salt phases, coupled with a reduced fraction of the spinel phase. The increased perovskite formation in FCG-CGO_HT suggests an enhanced phase interaction due to the incorporation of Gd2O3.
Fig. 3 provides an in-depth illustration of the grain percolation characteristics of ionic and electronic conducting phases, using FC-CGO_HT as an example. As depicted in the right column of Fig. 3(a), nearly all grains of the ionic conducting phase contribute to forming a percolating network. In contrast, as shown in the middle column of Fig. 3(b), the grains of electronic conducting phases fail to form a percolating network due to their insufficient volume. Similar results are observed for FC-CGO_LT and FCG-CGO_HT samples, as shown in Fig. S1(a–d).†
 | ||
| Fig. 3 Overall grain/phase distribution maps (left column), non-percolation maps (middle column) and percolation maps (right column) of ionic conducting phase (a) and electronic conducting phases (b) in FC-CGO_HT. | ||
The effective geometry factor for ionic conduction is derived from the apparent geometry factor (L/S) through eqn (5),43,44 which is schematically depicted in the inset of Fig. 4(a).
 | (5) |
 | ||
| Fig. 4 Critical geometry effects on ionic and electronic conduction reflected by the MacMullin number (ratio of tortuosity to volume fraction) of ionic conducting grains (a) and the intergranular distance distribution profiles for electronic conducting grains (b). | ||
The calculated NM values, presented in Fig. 4(a), follow an increasing trend: FC-CGO_HT < FC-CGO_LT < FCG-CGO_HT. This trend is consistent with the volumetric increase of the ionic blocking but electronic conducting phases, as well as the rise in tortuosity (from 1.503 to 1.566, and to 1.581) of the ionic conducting network. These NM values are more than one order of magnitude lower than the one (∼34.55) calculated for 40CF-CGO_HT, where the high concentration (∼41.2 vol%, Fig. S2(a and b)†) of electronic conducting grains leads to severely limited ionic conduction paths with a tortuosity of ∼18.44 (Fig. S1(e and f)†).
 | ||
| Fig. 5 Elemental mapping via EDXS across ceria grain boundaries in FC-CGO_LT (a), FC-CGO_HT (b), and FCG-CGO_HT (c). The upper grain is randomly oriented, while the lower one is along its [101] zone axis. | ||
Based on the EDXS elemental mapping, the cation intensity ratios (Gd/Ce, Fe/Ce, and Co/Ce) are detailed in Fig. 6. The Gd/Ce ratio exhibits a greater increase from the grain interior to the boundary in FC-CGO_HT than in FC-CGO_LT, suggesting that Gd segregation is enhanced at higher sintering temperatures. Additionally, in FC-CGO_HT, the Gd/Ce ratio exhibits a relaxation at the grain boundary core, with its maximum values occurring at positions adjacent to the core. This observation corroborates previous atomic-scale investigations, which revealed ordered Gd segregation at two atomic planes adjacent to the core.33,35 Analogous phenomena have been reported in Y-stabilized zirconia, where ordered Y segregation at grain boundary vicinities has been observed experimentally and simulated computationally.49
 | ||
| Fig. 6 Elemental intensity ratios across ceria grain boundaries in FC-CGO_LT (a), FC-CGO_HT (b), and FCG-CGO_HT (c). The left grain is randomly oriented, while the right one is along its [101] zone axis. The EDXS scanned areas and directions are indicated in the HAADF images obtained by STEM as shown in the first column of Fig. 5. | ||
In contrast, the Fe/Ce and Co/Ce ratios show a considerably weaker increase from the grain interior to the boundary in FC-CGO_HT than in FC-CGO_LT, suggesting that a higher sintering temperature inhibits Fe and Co segregation. This trend aligns with prior studies on Co-modified Ce0.8Gd0.2O2−δ50,51 and Ce0.8Tb0.2O2−δ,20 which indicates that significant Co enrichment at ceria grain boundaries is only feasible below ∼1100 °C.
Following the incorporation of Gd2O3, FCG-CGO_HT exhibits the smallest increase in all cation intensity ratios from the grain interior to the boundary. In some cases, ceria grain boundaries appear nearly devoid of Fe and Co segregation (see Fig. S4†). This suggests that Gd2O3 acts as a grain boundary scavenger.
 with
with  leading to the formation of dopant–vacancy complexes.
leading to the formation of dopant–vacancy complexes.
 | ||
| Fig. 7 STEM-EELS analysis across the ceria grain boundary in FC-CGO_LT: (a) the simultaneously recorded ADF (annular dark field) image; (b) elemental intensity maps from Ce M4,5, Gd M4,5, Fe L2,3, Co L2,3 and O K edges based on EELS spectrum imaging; (c) representative Ce M4,5-edge EELS spectra collected from different positions, including the grain boundary core (C) and the adjacent grain interior regions (R and L) as indicated in the inset ADF image; (d) The O intensity and the calculated Ce3+ fraction profiles across the region shown in the inset. The left grain is randomly oriented, while the right one is along its [101] zone axis. | ||
The oxidation state of Ce was determined using Ce M4,5-edge EELS spectra. As shown in Fig. 7(c), the spectra feature two characteristic peaks – Ce M4 and M5. The overall peak intensities are weaker at the grain boundary core (C) than in the grain bulk regions (R and L), reflecting either a lower Ce density or a reduced grain boundary thickness. A noticeable increase in the M5/M4 intensity ratio is observed at the grain boundary core compared to the grain interior, indicative of a partial reduction of Ce4+ to Ce3+.33,35,46,52,53 The Ce3+ fraction (x) was quantified using the relationship R = 0.925(1 − x) + 1.25x,33 where R is the M5/M4 intensity ratio in second derivative spectra and 0.925 and 1.25 represent reference R values for Ce4+ and Ce3+, respectively.54 The highest R value of 1.137, determined near the grain boundary core, corresponds to a Ce3+ fraction of ∼65.2%, as shown in Fig. 7(d). This R value falls within the range reported for Ce0.85Gd0.11Pr0.04O2−δ (R = 1.0)55 and Ce0.8Gd0.2O2−δ (R = 1.2),47 confirming a reasonable estimation.
Unlike Ce, Fe cations at ceria grain boundaries are predominately stabilized as Fe3+, as reported in previous studies.33,56 In contrast, the oxidation states of Co can vary among 2+ and 3+, as evidenced by the coexistence of spinel and rock-salt phases. However, precise determination of Co oxidation states remains challenging due to excessive noise in its L2,3 spectra (not shown here).
 exhibit an enhanced concentration at grain boundary cores. This localized enrichment generates positively charged cores that repel positive charge carriers (
exhibit an enhanced concentration at grain boundary cores. This localized enrichment generates positively charged cores that repel positive charge carriers ( and electronic holes) while attracting negatively charged ones (electrons and acceptor dopants like
and electronic holes) while attracting negatively charged ones (electrons and acceptor dopants like  ). This redistribution of charge carriers leads to the formation of space charge layers in the vicinity of the core region, which contributes to partially or fully neutralizing the space charge potential.
). This redistribution of charge carriers leads to the formation of space charge layers in the vicinity of the core region, which contributes to partially or fully neutralizing the space charge potential.
However, for FC-CGO_LT, the O intensity monotonously decreases from the ceria grain interior to the ceria grain boundary (see Fig. 7(d)), with no evident O-enriched or  depleted region adjacent to the grain boundary core. This observation aligns with previous studies on ceria and zirconia that reported O depletion peaks at grain boundary cores without evidence of adjacent O-enriched or
depleted region adjacent to the grain boundary core. This observation aligns with previous studies on ceria and zirconia that reported O depletion peaks at grain boundary cores without evidence of adjacent O-enriched or  depleted layers,47,58–60 suggesting that directly picturing oxygen vacancy profiles remains challenging. An additional analysis of cations' valence states and occupation sites is therefore conducted.
depleted layers,47,58–60 suggesting that directly picturing oxygen vacancy profiles remains challenging. An additional analysis of cations' valence states and occupation sites is therefore conducted.
The presence of Ce3+ at the grain boundary indicates the absence of bounded oxygen, which is consistent with the observed decrease in O intensity (Fig. 7(d)). Since only ∼0.8% of CeO2 can be thermally reduced to Ce2O3 even at 1420 °C,61 and considering FC-CGO_LT's low sintering temperature (1050 °C), the observed high Ce3+ fractions cannot be attributed to thermal reduction. Instead, it likely arises from the grain boundary structure, where  formation and accumulation are energetically favorable or structurally necessary.52,53
formation and accumulation are energetically favorable or structurally necessary.52,53
The absence of interstitial cations in Fig. 7(a) suggests Fe and Co primarily substitute the Ce sites, though their precise atomic positions remain unresolved. Thus, the Fe and Co cations (average valence < 4+) appear to act as heterogeneous acceptor dopants localized almost exclusively at ceria grain boundaries. They diffuse externally from the additive sources rather than internally from the ceria grain bulk.
Given the high concentrations of 

 and
and  for charge compensation, it is surprising that there is still an excess of oxygen vacancies, necessitating Ce4+ reduction. We believe that the high Ce3+ faction is associated with the emergence of a grain boundary superstructure,62 which possesses a lower oxygen coordination as compared to the CeO2 fluorite structure. Consequently, the investigated ceria grain boundary is not expected to exhibit substantial charge imbalance.
for charge compensation, it is surprising that there is still an excess of oxygen vacancies, necessitating Ce4+ reduction. We believe that the high Ce3+ faction is associated with the emergence of a grain boundary superstructure,62 which possesses a lower oxygen coordination as compared to the CeO2 fluorite structure. Consequently, the investigated ceria grain boundary is not expected to exhibit substantial charge imbalance.
This analysis does not provide evidence for the formation of space charge layers for FC-CGO_LT. Instead, it reveals a co-enrichment of acceptors and  at the grain boundary, closely resembling the behavior observed in doped ceria with a high acceptor concentration at grain boundaries.63
at the grain boundary, closely resembling the behavior observed in doped ceria with a high acceptor concentration at grain boundaries.63
With increased sintering temperature for FC-CGO_HT and FCG-CGO_HT, the situation may change and will be discussed later.
At relatively lower temperatures (800–900 °C), transition metal cations preferentially accumulate at ceria particle surfaces and grain boundaries,50,51,64 which is driven by energy minimization mechanisms and promotes densification of ceria particles.65 As temperature increases, the closure of free surfaces and grain boundary coalescence lead to the oversaturation of cation segregation, ultimately triggering the precipitation of secondary phases. The observed Fe and Co segregation profiles for FC-CGO_LT and FC-CGO_HT align well with this framework.
For FCG-CGO_HT, minimal Fe and Co segregation supports the hypothesis that Gd2O3 scavenges Fe and Co segregation. This phenomenon is attributed to the formation of a Gd1−xCexFe1−yCoyO3−δ perovskite phase at temperatures above 1050 °C.36 Consequently, Fe and Co cations, which segregate at low temperatures, are effectively removed upon the formation of this perovskite phase. The reduction in Fe and Co segregation also leads to a decrease in grain boundary mobility at the higher temperature of 1200 °C, as evidenced by the smaller grain size of FCG-CGO_HT relative to FC-CGO_HT (see Fig. S3†).
Regarding Gd segregation, its driving force can be partially attributed to grain boundary energy reduction,63 although the prevailing explanation involves the space charge effect.57,66,67 The contribution of lattice strain relaxation is considered minimal due to the relatively small ionic radius mismatch between Gd and Ce.57,66
As analyzed previously for FC-CGO_LT, the space charge effect is not evident. The relatively low sintering temperature (1050 °C) is likely to result in either incomplete formation or inhibition of space charge layers due to the early-stage accumulation of Fe and Co acceptors alongside  Besides, once the grain boundary is decorated with Fe and Co cations, the overall boundary energy is expected to decrease, facilitating the segregation of Gd from the grain interior, where it exists at a higher energy state.
Besides, once the grain boundary is decorated with Fe and Co cations, the overall boundary energy is expected to decrease, facilitating the segregation of Gd from the grain interior, where it exists at a higher energy state.
As the sintering temperature increases in FC-CGO_HT, it remains uncertain whether the enhanced Gd segregation arises from an emerging space charge effect or is a consequence of grain boundary migration and merging.
For FCG-CGO_HT, the observed minimal Gd segregation may be attributed to inhibited grain boundary migration and/or moderate grain boundary energy reduction, achieved by removing Fe and Co while simultaneously introducing Gd through the Gd2O3 additive. Additionally, the Gd2O3 additive may further suppress space charge layer formation by neutralizing excess core charge through Gd doping directly at the grain boundary.68 This hypothesis is well supported by a low fraction of Ce3+ (∼48.5%, see Fig. S5†), implying less excess of 
Nevertheless, variations among grain boundaries remain, necessitating the following macroscopic assessments.
Material assessments
Under air conduction, see Fig. 8(a), the highest and lowest σtot were measured for the EC and IC samples, respectively. The σtot values of the IC sample closely match those of a reference single-phase fluorite compound Ce0.9Gd0.1O2−δ (densified at 1300 °C for 3 h in air using powders produced by Solvay). This similarity suggests that the IC sample consists of a fluorite phase with a Ce:Gd ratio of approximately 9:1 (rather than the initial 8:2 ratio), while the influence of secondary phases (∼4.5 vol% as given in Fig. S2(c and d)†) is negligible.
 | ||
| Fig. 8 Total conductivity measured in air (a), Ar (c), and Ar-to-air gradient (d) with the corresponding activation energies given in (e). In (b), several conductivity values in air reported for mixed conductors having a comparable content (≤20 vol% or 15 wt%) of electronic conducting phase are shown for comparison.69–71 | ||
The σtot values of the mixed conductors fall between those of the EC and IC samples, following the trend: FC-CGO_LT > FCG-CGO_HT > FC-CGO_HT. Notably, FC-CGO_LT exhibits superior σtot above 800 °C, outperforming other mixed conductors with comparable content (≤20 vol% or 15 wt%) of electronic conducting phase,69–71 as compared in Fig. 8(b).
Under Ar conduction, as shown in Fig. 8(c), the EC sample retains the highest σtot, whereas FC-CGO_HT exhibits the lowest. The σtot of FC-CGO_LT and FCG-CGO_HT are comparable to that of the IC sample, indicating a significantly increased contribution from the ionic conductivity, accompanied by a decrease in the electronic conductivity.
A comparison of the percentage change in σtot values from air to Ar (Fig. S6†) reveal that the IC sample experiences a marginal fluctuation in total conductivity, consistent with its dominant ionic conduction mechanism controlled by the fluorite phase. In contrast, the EC sample and all mixed conductors are subject to a reduction in σtot from air to Ar, indicative of a p-type (hole) conduction mechanism. This reduction is substantially greater for the mixed conductors than for the EC sample, suggesting divergent p-type conduction mechanisms between these two systems. These differences may arise from variations in polaronic defect structures and disparate hopping pathways.
The temperature-dependence conductivity was further analyzed through activation energy, as present in Fig. 8(e). All samples exhibit positive activation energies, confirming a semiconductor-like behavior where conductivity increases with rising temperature.
Under air conditions, a clear distinction in activation energies is observed among the samples, with the IC sample displaying the lowest and FC-CGO_LT the highest.
Under Ar conditions, the activation energies of mixed conductors become comparable to that of the IC sample, indicating a rising contribution from ionic conduction.
For the IC sample, the activation energy remains similar to that of the Ce0.9Gd0.1O2−δ regardless of the atmosphere, reaffirming its pure ionic conduction behavior. However, for the other samples, activation energy decreases when transitioning from air to Ar, with the reduction being more pronounced in mixed conductors than in the EC sample. For instance, FC-CGO_LT exhibits a remarkable 46% decrease in activation energy.
This comprehensive comparison confirms that the mixed conductors indeed exhibit mixed ionic and p-type conductivity. The ionic conductivity is attributed to the fluorite phase, while the p-type conductivity is unlikely to be dominated by the electronic conducting phases. Instead, alternative pathways through the grain boundaries of the ionic conducting phase are likely controlling the overall electronic conduction.
Similar to Pr-doped ceria,72 a small polaron hopping mechanism is proposed to explain the observed electronic conduction along ceria grain boundaries. The conductivity suppression under Ar conditions is attributed to the reduction of Co cations. In other words, a more uniform distribution of Co valence states leads to a reduction of polaronic defect concentrations.
We adopt the rock-salt phase as a tracer for the impact of Ar, and investigate FC-CGO_HT using EBSD at the cross-section after the oxygen permeation measurement. As shown by the phase mappings in Fig. 9(a), a notable variation in the rock-salt phase is observed, with negligible content on the air side but a substantial increase on the Ar side. The spatial distribution of the rock-salt phase across the sample thickness follows a natural exponential decay.
 | ||
| Fig. 9 Gradient variation of the rock-salt phase content (a) and total resistance (b) across the thickness of FC-CGO_HT after the oxygen permeation test, where the two sample surfaces were exposed to Ar and air, respectively, below ∼1020 °C. The rock-salt phase content was quantified from EBSD data with a step width of 5 μm, and fitted using a natural exponential function. The total resistance was calculated assuming a natural exponential decay behavior. | ||
It is assumed that resistance growth with sample thickness also follows a natural exponential function, mathematically expressed as eqn (6) and plotted in Fig. 9(b).
| r(l) = A0(ekl − 1) | (6) |
At l = L (total sample thickness), the total effective resistance under an Ar-to-air gradient, denoted as Refftot, is obtained. This value determines the effective total conductivity (σefftot|Ar→air) via:
 | (7) |
At the sample surface, the slope of r(l) corresponds to the total conductivity in Ar (σArtot) or air (σairtot), as expressed by eqn (8) and (9) (also plotted as the dashed lines in Fig. 9(b)).
 | (8) |
 | (9) |
By combining eqn (6)–(9), the σefftot|Ar→air can be derived as:
 | (10) |
 | (11) |
A key advantage of this formulation is that it eliminates all constants associated with the assumed natural exponential function, allowing σefftot|Ar→air to be determined solely based on separately measured total conductivities in air and Ar. This approach can be extended to other materials, provided that r(l)follows an exponential dependence with only two independent constants.
As shown in Fig. 8(d), the calculated σefftot|Ar→air deviates from σairtot but converges towards σArtot. This trend underscores the dominant influence of the lower oxygen partial pressure on total conductivity. The dominating portion (either ionic or electronic conductivity) of σefftot|Ar→air will be discussed later.
 | ||
| Fig. 10 Oxygen permeances of membranes with thicknesses close to 1 mm. | ||
Notably, substantially lower oxygen permeances have been reported for a similar mixed conductor prepared from 80 vol% Ce0.8Gd0.2O2−δ (nano-sized powder) and 20 vol% CoFe2O4 (in a preformed state rather than a raw Fe2O3 and Co3O4 mixture used here).27 This reference sample (denoted as CF-CGO_ref.) is included in Fig. 10 for comparison. The additive's initial state (preformed spinel or raw oxide mixture) is expected to have limited influence on oxygen permeation, since the spinel phase inherently forms at ∼900 °C.36 However, the additive's Fe:Co ratio (either 1:2 or 2:1) may play a role.
To further investigate this, FC-CGO_HT precursors were modified by increasing the Fe2O3:Co3O4 molar ratio from 3:4 to 3:1, yielding CoFe2O4. Other preparation steps remained unchanged, producing the CF-CGO_HT sample. Its oxygen permeances are comparable to those of FC-CGO_HT but higher than the ones of CF-CGO_ref.,27 indicating that the Fe:Co ratio (1:2 vs. 2:1) has a negligible effect on oxygen permeation.
Another potential factor influencing oxygen permeation is the initial particle size of Ce0.8Gd0.2O2−δ powder, which will be discussed later.
The calculated ambipolar conductivities, see Fig. 11(a), show variations among samples that mirror the trends observed in oxygen permeances. The derived activation energy values closely match previously reported values for the ionic conductivity of pure Gd-doped ceria,27,29,30,73 but are significantly lower than those of CF-CGO_ref.27 This indicates that σamb of the mixed conductors synthesized here is predominantly governed by the ionic conductivity. In contrast, for CF-CGO_ref.,27σamb is likely limited by the electronic conductivity, being associated with the depletion of Fe and Co grain boundary segregation.27
 | ||
| Fig. 11 Ambipolar conductivity (a) derived from oxygen permeation and effective partial conductivities (b) calculated from ambipolar conductivity and effective total conductivity. | ||
By combining the σamb with the previously determined σefftot|A![[r with combining right harpoon above (vector)]](https://www.rsc.org/images/entities/char_0072_20d1.gif) air, the effective partial conductivities under an Ar-to-air gradient were resolved and are compared in Fig. 11(b). The results demonstrate that the effective electronic conductivities substantially exceed the effective ionic conductivities, confirming that ionic transport is the limiting factor for σamb.
air, the effective partial conductivities under an Ar-to-air gradient were resolved and are compared in Fig. 11(b). The results demonstrate that the effective electronic conductivities substantially exceed the effective ionic conductivities, confirming that ionic transport is the limiting factor for σamb.
The highest effective electronic conductivity values observed for FC-CGO_LT can be attributed to: (1) the highest concentration of Fe and Co cations at ceria grain boundaries (see Fig. 6), where Co cations can exist in mixed valence states; (2) the shortest transport paths along ceria grain boundaries, represented by the shortest distance between electronic conducting grains, as indicated in Fig. 4(b).
The lowest effective electronic conductivity for FC-CGO_LT is then explained by the reduced Fe and Co segregation and extended transport paths.
However, this interpretation does not fully account for the moderate electronic conductivity observed for FCG-CGO_HT, where Fe and Co segregation at grain boundaries is nearly absent. This discrepancy suggests an alternative conduction mechanism, such as Fe and Co cations dissolving into the ceria lattice rather than forming grain boundary decorations. Further confirmation was obtained by examining a sample subjected to a 1 minute dwell time at 1200 °C, followed by rapid air quenching. Elemental mappings across the grain boundary (Fig. S8†) revealed Co enrichment in the grain interior near the boundary, representing an initial stage of Co diffusion into the ceria lattice. This diffusion is unlikely for FC-CGO_LT and FC-CGO_HT because: (1) for FC-CGO_LT, the lower sintering temperature (1050 °C) restricts Co incorporation into the lattice; (2) for FC-CGO_HT, the nearly ordered Gd segregation at the grain boundary can inhibit Co diffusion into ceria lattice.
Based on these findings, a distinct electronic conduction mechanism is proposed for FCG-CGO_HT: electronic transport is facilitated by Co-doped ceria grains rather than ceria grain boundary decorations. This accounts for the lowest activation energy of total conductivity (dominated by electronic conductivity) in air, as shown in Fig. 8(e).
Despite the most favorable and lowest NM (see Fig. 4(a)), FC-CGO_HT shows the lowest effective ionic conductivity, which correlates with the presence of strongly ordered Gd segregation (Fig. 5(b) and 6(b)) or a high space charge potential at ceria grain boundaries. Whereas, the partially ordered segregation (Fig. 7(b)) in FC-CGO_LT results in higher effective ionic conductivity.
In FCG-CGO_HT, the reduction in grain boundary core charge due to Gd doping may enhance ionic conduction across the grain boundary.68 Additionally, Co lattice doping increases oxygen vacancy concentrations, further contributing to enhancing lattice ionic conductivity.
As presented in Fig. 12, both oxygen permeance and ambipolar conductivity experience minor fluctuations of ±2–3% without a discernible time-dependent trend. This observation confirms that FC-CGO_LT sustains a steady high-performance state, attributed to the robustness of its mixed ionic and electronic conducting structure. Moreover, effects from a potential elemental diffusion under an oxygen partial pressure gradient are not reflected and might be excluded in this case.
 | ||
| Fig. 12 Time dependence of oxygen permeance and ambipolar conductivity of FC-CGO_LT. | ||
:Co ratio in the additives. We propose that even pure Co3O4 additives could yield comparable performance. However, attempts to prepare pure Co3O4-modified Ce0.8Gd0.2O2−δ were unsuccessful, as samples cracked into pieces after sintering due to thermal mismatch. This suggests that a certain Fe2O3 content is necessary in combination with Co3O4 to improve thermal compatibility with ceria.
Interestingly, Y-stabilized ZrO2 appears to be mechanically compatible with Co3O4. A study by B. Aktas et al.,74 successfully prepared 8 mol% Y-stabilized ZrO2 with up to 15 wt% Co3O4 without cracking. Their findings also noted a slight increase in grain boundary conductivity, though significantly lower than in the present work. To achieve comparable conductivity levels, the high sintering temperature (1400 °C) used in their study should be reduced to prevent Co precipitation from zirconia grain boundaries.
Other transition metal elements, such as Cr, Mn, Ni, and Cu, exhibit high solubility and diffusivity at ceria grain boundaries as well.75 These elements show variable valences upon heating or under different atmospheres, and they are expected to have a similar electronic impact on ceria grain boundaries if low-temperature densification is achieved. However, their effects on ionic conductivity differ. Unlike Fe, Co, Ni, and Cu, elements like Cr and Mn possess valence states higher than 4+. They act as donors with no contribution to balancing excess core charge. As a result, they are not beneficial for ionic conduction.31,76–78
It is noteworthy that transition metal elements leaching from perovskites, such as La0.6Sr0.4CoO3−δ70 and Sr0.4Sm0.6FeO3−δ,71 result in lower conductivity compared to Fe–Co spinel oxides, as shown in Fig. 8(b). This is likely due to: (1) stabilization of Fe and Co cations within the perovskite lattice, preventing them from diffusing to ceria grain boundaries; (2) interdiffusion of more cations between perovskites and ceria,79,80 which can reduce oxygen permeation as reported in multiple studies.81–83
Besides, the grain boundary decoration of transition metal elements is also influenced by the initial surface area of ceria particles. Notably, nanosized Ce0.9Gd0.1O2−δ exhibits no sintering enhancement upon cobalt oxide addition,84 suggesting that only limited Co coverage occurs on particle surfaces and grain boundaries. This implies that a high surface area may hinder grain boundary enrichment of Co cations, a hypothesis that requires further validation.
Conclusions
Contrary to conventional percolation theory, which asserts that optimal conductivity necessitates the continuous connectivity of individual phases, this study establishes an alternative paradigm in which superior mixed conductivities are achieved through novel mechanisms free of strict phase percolation.A case study exemplifying this concept involves mixed ionic and electronic conductors synthesized from Gd-doped ceria with iron–cobalt oxide additions. A superior network for simultaneous ionic and electronic conduction is successfully established by percolating additive (FeCo2O4)-induced electronic conducting grains within the ionic conductor (Gd-doped ceria) through its mixed conductive grain boundaries. The study demonstrates that percolation of the electronic conducting grains within the Gd-doped ceria matrix is not a prerequisite for achieving efficient electronic conduction, thereby significantly mitigating the ion-blocking effect.
Comprehensive structural and compositional analyses, spanning from micro-to nanoscale, reveal that the observed enhancements in conductivity are closely associated with: (1) the geometry of the percolation network, which optimizes conduction pathways; (2) the strategic segregation of iron and cobalt at ceria grain boundaries, which enhances electronic conduction. The synergistic optimizations in both ionic and electronic transport, especially under dual-atmosphere conditions, are achieved through the stabilization of the segregation, alongside the suppression of grain growth, at a significantly lowered densification temperature of 1050 °C.
The optimized MIEC delivers an excellent and sustainable oxygen permeation performance, advancing its applications in clean and efficient energy technologies.
Data availability
The data supporting this study have been included as part of the manuscript and its ESI.†Author contributions
Fanlin Zeng: conceptualization, data curation, formal analysis, investigation, methodology, project administration, resources, software, validation, visualization, writing – original draft; Ke Ran: conceptualization, data curation, formal analysis, investigation, methodology, resources, software, validation, visualization, writing – review & editing; Christian Dellen: data curation, formal analysis, methodology, software, validation, visualization, writing – review and editing; Hartmut Schlenz: conceptualization, supervision, validation, writing – review & editing; Joachim Mayer: resources, supervision, validation, writing – review & editing; Ruth Schwaiger: resources, supervision, validation, writing – review & editing; Wilhelm Albert Meulenberg: resources, supervision, validation, writing – review & editing; Stefan Baumann: conceptualization, data curation, formal analysis, investigation, methodology, project administration, resources, software, supervision, validation, visualization, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge Dr E. Wessel, Dr D. Grüner, Dr Y. J. Sohn, and Mr S. Heinz, for structural characterization, property testing, and sample preparation, as well as Prof. Dr Ing O. Guillon for support.References
- K.-D. Kreuer, Chem. Mater., 2014, 26, 361–380 CrossRef CAS.
- N. Devi, B. Singh and S.-J. Song, in Advanced Ceramics for Versatile Interdisciplinary Applications, ed. S. Singh, P. Kumar and D. P. Mondal, Elsevier, 2022, pp. 201–230, DOI:10.1016/B978-0-323-89952-9.00007-5.
- X. Dong, W. Jin, N. Xu and K. Li, Chem. Commun., 2011, 47, 10886–10902 RSC.
- L. Jia, G. He, Y. Zhang, J. Caro and H. Jiang, Angew. Chem., Int. Ed., 2021, 60, 5204–5208 CrossRef CAS PubMed.
- H. Jiang, H. Wang, S. Werth, T. Schiestel and J. Caro, Angew. Chem., Int. Ed., 2008, 47, 9341–9344 CrossRef CAS PubMed.
- W. Fang, F. Steinbach, Z. Cao, X. Zhu and A. Feldhoff, Angew. Chem., Int. Ed., 2016, 55, 8648–8651 CrossRef CAS PubMed.
- S. C. Singhal, MRS Bull., 2000, 25, 16–21 CrossRef CAS.
- S. Baumann, J. M. Serra, M. P. Lobera, S. Escolástico, F. Schulze-Küppers and W. A. Meulenberg, J. Membr. Sci., 2011, 377, 198–205 CrossRef CAS.
- J. M. Serra, J. Garcia-Fayos, S. Baumann, F. Schulze-Küppers and W. A. Meulenberg, J. Membr. Sci., 2013, 447, 297–305 CrossRef CAS.
- M. Schulz, R. Kriegel and A. Kämpfer, J. Membr. Sci., 2011, 378, 10–17 CrossRef CAS.
- J. Gao, L. Li, Z. Yin, J. Zhang, S. Lu and X. Tan, J. Membr. Sci., 2014, 455, 341–348 CrossRef CAS.
- S. Pirou, J. M. Bermudez, B. T. Na, S. Ovtar, J. H. Yu, P. V. Hendriksen, A. Kaiser, T. R. Reina, M. Millan and R. Kiebach, J. Membr. Sci., 2018, 552, 115–123 CrossRef CAS.
- S. Pirou, J. García-Fayos, M. Balaguer, R. Kiebach and J. M. Serra, J. Membr. Sci., 2019, 580, 307–315 CrossRef CAS.
- V. V. Kharton, F. M. B. Marques and A. Atkinson, Solid State Ionics, 2004, 174, 135–149 CrossRef CAS.
- C. Zhang, J. Sunarso and S. Liu, Chem. Soc. Rev., 2017, 46, 2941–3005 RSC.
- M. Ramasamy, E. S. Persoon, S. Baumann, M. Schroeder, F. Schulze-Küppers, D. Görtz, R. Bhave, M. Bram and W. A. Meulenberg, J. Membr. Sci., 2017, 544, 278–286 CrossRef CAS.
- J. H. Park and R. N. Blumenthal, J. Electrochem. Soc., 1989, 136, 2867 CrossRef CAS.
- S. Wang, T. Kobayashi, M. Dokiya and T. Hashimoto, J. Electrochem. Soc., 2000, 147, 3606 CrossRef CAS.
- C. Chatzichristodoulou and P. V. Hendriksen, J. Electrochem. Soc., 2012, 159, E162 CrossRef CAS.
- D. Ramasamy, A. L. Shaula, A. Gómez-Herrero, V. V. Kharton and D. P. Fagg, J. Membr. Sci., 2015, 475, 414–424 CrossRef CAS.
- P. Shuk and M. Greenblatt, Solid State Ionics, 1999, 116, 217–223 CrossRef CAS.
- V. Esposito, D. Z. de Florio, F. C. Fonseca, E. N. S. Muccillo, R. Muccillo and E. Traversa, J. Eur. Ceram. Soc., 2005, 25, 2637–2641 CrossRef CAS.
- H. Takamura, H. Sugai, M. Watanabe, T. Kasahara, A. Kamegawa and M. Okada, J. Electroceram., 2006, 17, 741–748 CrossRef CAS.
- S. Cheng, M. Søgaard, L. Han, W. Zhang, M. Chen, A. Kaiser and P. V. Hendriksen, Chem. Commun., 2015, 51, 7140–7143 RSC.
- M. Balaguer, J. García-Fayos, C. Solís and J. M. Serra, Chem. Mater., 2013, 25, 4986–4993 CrossRef CAS.
- H. Luo, H. Jiang, K. Efimov, F. Liang, H. Wang and J. r. Caro, Ind. Eng. Chem. Res., 2011, 50, 13508–13517 CrossRef CAS.
- Y. Lin, S. Fang, D. Su, K. S. Brinkman and F. Chen, Nat. Commun., 2015, 6, 6824 CrossRef CAS PubMed.
- F. Zeng, J. Malzbender, S. Baumann, M. Krüger, L. Winnubst, O. Guillon and W. A. Meulenberg, J. Eur. Ceram. Soc., 2020, 40, 5646–5652 CrossRef CAS.
- D. P. Fagg, V. V. Kharton and J. R. Frade, J. Electroceram., 2002, 9, 199–207 CrossRef CAS.
- D. P. Fagg, J. C. C. Abrantes, D. Pérez-Coll, P. Núñez, V. V. Kharton and J. R. Frade, Electrochim. Acta, 2003, 48, 1023–1029 CrossRef CAS.
- S. Taub, R. E. A. Williams, X. Wang, D. W. McComb, J. A. Kilner and A. Atkinson, Acta Mater., 2014, 81, 128–140 CrossRef CAS.
- C. Lenser, F. Gunkel, Y. J. Sohn and N. H. Menzler, Solid State Ionics, 2018, 314, 204–211 CrossRef CAS.
- K. Ran, L. Fischer, S. Baumann, W. A. Meulenberg, K. Neuhaus and J. Mayer, Acta Mater., 2022, 226, 117603 CrossRef CAS.
- F. Zeng, S. Baumann, J. Malzbender, A. Nijmeijer, L. Winnubst, O. Guillon, R. Schwaiger and W. A. Meulenberg, J. Membr. Sci., 2021, 628, 119248 CrossRef CAS.
- K. Ran, F. Zeng, L. Fischer, S. Baumann, W. A. Meulenberg, K. Neuhaus and J. Mayer, Acta Mater., 2022, 234, 118034 CrossRef CAS.
- F. Zeng, J. Malzbender, S. Baumann, A. Nijmeijer, L. Winnubst, M. Ziegner, O. Guillon, R. Schwaiger and W. A. Meulenberg, J. Eur. Ceram. Soc., 2021, 41, 509–516 CrossRef CAS.
- A. J. Schwartz, M. Kumar, B. L. Adams and D. P. Field, Electron Backscatter Diffraction in Materials Science, Springer, New York, USA, 2009 Search PubMed.
- J. T. Gostick, Z. A. Khan, T. G. Tranter, M. D. Kok, M. Agnaou, M. Sadeghi and R. Jervis, J. Open Source Softw., 2019, 4, 1296 CrossRef.
- A. Kovács, R. Schierholz and K. Tillmann, J. Large-Scale Res. Facil., 2016, 2, A43 Search PubMed.
- K. Ran, W. Deibert, M. E. Ivanova, W. A. Meulenberg and J. Mayer, Nanoscale, 2020, 12, 17841–17848 RSC.
- M. Ramasamy, S. Baumann, J. Palisaitis, F. Schulze-Küppers, M. Balaguer, D. Kim, W. A. Meulenberg, J. Mayer, R. Bhave, O. Guillon and M. Bram, J. Am. Ceram. Soc., 2016, 99, 349–355 Search PubMed.
- F. Zeng, J. Malzbender, S. Baumann, W. Zhou, M. Ziegner, A. Nijmeijer, O. Guillon, R. Schwaiger and W. A. Meulenberg, J. Am. Ceram. Soc., 2021, 104, 1814–1830 Search PubMed.
- T.-T. Nguyen, A. Demortière, B. Fleutot, B. Delobel, C. Delacourt and S. J. Cooper, npj Comput. Mater., 2020, 6, 123 Search PubMed.
- J. Landesfeind, J. Hattendorff, A. Ehrl, W. A. Wall and H. A. Gasteiger, J. Electrochem. Soc., 2016, 163, A1373 Search PubMed.
- C. A. Schneider, W. S. Rasband and K. W. Eliceiri, Nat. Methods, 2012, 9, 671–675 CrossRef CAS PubMed.
- J. Bae, Y. Lim, J.-S. Park, D. Lee, S. Hong, J. An and Y.-B. Kim, J. Electrochem. Soc., 2016, 163, F919 Search PubMed.
- Y. Lei, Y. Ito, N. D. Browning and T. J. Mazanec, J. Am. Ceram. Soc., 2002, 85, 2359–2363 CrossRef CAS.
- Z.-P. Li, T. Mori, G. J. Auchterlonie, J. Zou and J. Drennan, Appl. Phys. Lett., 2011, 98, 093104 Search PubMed.
- B. Feng, T. Yokoi, A. Kumamoto, M. Yoshiya, Y. Ikuhara and N. Shibata, Nat. Commun., 2016, 7, 11079 Search PubMed.
- D. Pérez-Coll, P. Núñez, J. C. C. Abrantes, D. P. Fagg, V. V. Kharton and J. R. Frade, Solid State Ionics, 2005, 176, 2799–2805 CrossRef.
- Z. Zhang, W. Sigle, M. Rühle, E. Jud and L. J. Gauckler, Acta Mater., 2007, 55, 2907–2917 CrossRef CAS.
- H. Hojo, T. Mizoguchi, H. Ohta, S. D. Findlay, N. Shibata, T. Yamamoto and Y. Ikuhara, Nano Lett., 2010, 10, 4668–4672 CrossRef CAS PubMed.
- K. Song, H. Schmid, V. Srot, E. Gilardi, G. Gregori, K. Du, J. Maier and P. A. van Aken, APL Mater., 2014, 2, 032104 Search PubMed.
- P. Yan, T. Mori, Y. Wu, Z. Li, G. J. Auchterlonie, J. Zou and J. Drennan, Microsc. Microanal., 2013, 19, 102–110 Search PubMed.
- W. J. Bowman, J. Zhu, R. Sharma and P. A. Crozier, Solid State Ionics, 2015, 272, 9–17 CrossRef CAS.
- E. V. Tsipis, J. C. Waerenborgh and V. V. Kharton, J. Solid State Electrochem., 2017, 21, 2965–2974 CrossRef CAS.
- X. Guo and R. Waser, Solid State Ionics, 2004, 173, 63–67 CrossRef CAS.
- D. R. Diercks, J. Tong, H. Zhu, R. Kee, G. Baure, J. C. Nino, R. O'Hayre and B. P. Gorman, J. Mater. Chem. A, 2016, 4, 5167–5175 RSC.
- M. A. Frechero, M. Rocci, G. Sánchez-Santolino, A. Kumar, J. Salafranca, R. Schmidt, M. R. Díaz-Guillén, O. J. Durá, A. Rivera-Calzada, R. Mishra, S. Jesse, S. T. Pantelides, S. V. Kalinin, M. Varela, S. J. Pennycook, J. Santamaria and C. Leon, Sci. Rep., 2015, 5, 17229 CrossRef CAS PubMed.
- C. Tian and S.-W. Chan, Solid State Ionics, 2000, 134, 89–102 CrossRef CAS.
- P.-L. Chen and I. W. Chen, J. Am. Ceram. Soc., 1996, 79, 1793–1800 CrossRef CAS.
- D. R. Ou, T. Mori, F. Ye, J. Zou, G. Auchterlonie and J. Drennan, Phys. Rev. B:Condens. Matter Mater. Phys., 2008, 77, 024108 Search PubMed.
- W. J. Bowman, M. N. Kelly, G. S. Rohrer, C. A. Hernandez and P. A. Crozier, Nanoscale, 2017, 9, 17293–17302 Search PubMed.
- J. Kim, S. Im, S. H. Oh, J. Y. Lee, K. J. Yoon, J.-W. Son, S. Yang, B.-K. Kim, J.-H. Lee, H.-W. Lee, J.-H. Lee and H.-I. Ji, Sci. Adv., 7, eabj8590 Search PubMed.
- M. Machado, A. L. da Silva, L. P. R. Moraes, L. N. Rodrigues, L. B. Caliman, D. Gouvêa and F. C. Fonseca, CrystEngComm, 2023, 25, 6102–6110 Search PubMed.
- X. Guo and R. Waser, Prog. Mater. Sci., 2006, 51, 151–210 CrossRef CAS.
- A. Tschöpe, Solid State Ionics, 2001, 139, 267–280 CrossRef.
- R. Tanaka, W. S. Oliveira, A. Brandão, J. C. C. Abrantes and J. R. Frade, Electrochim. Acta, 2012, 85, 116–121 CrossRef CAS.
- E.-J. Yi, M.-Y. Yoon, J.-W. Moon and H.-J. Hwang, J. Korean Ceram. Soc., 2010, 47, 199–190 Search PubMed.
- P. Seeharaj, A. Berenov, E. Raj, R. Rudkin and A. Atkinson, Solid State Ionics, 2011, 192, 638–641 CrossRef CAS.
- X. Zhu, M. Li, H. Liu, T. Zhang, Y. Cong and W. Yang, J. Membr. Sci., 2012, 394–395, 120–130 CrossRef CAS.
- S. R. Bishop, T. S. Stefanik and H. L. Tuller, Phys. Chem. Chem. Phys., 2011, 13, 10165–10173 RSC.
- E. Y. Pikalova, A. N. Demina, A. K. Demin, A. A. Murashkina, V. E. Sopernikov and N. O. Esina, Inorg. Mater., 2007, 43, 735–742 Search PubMed.
- B. Aktas and S. Tekeli, Int. J. Mater. Res., 2014, 105, 577–583 Search PubMed.
- N. W. Kwak, D.-K. Lim, S. J. Jeong, P. Byeon, S.-Y. Chung and W. Jung, Adv. Mater. Interfaces, 2020, 7, 2000688 Search PubMed.
- T. Zhang, L. Kong, Z. Zeng, H. Huang, P. Hing, Z. Xia and J. Kilner, J. Solid State Electrochem., 2003, 7, 348–354 CrossRef CAS.
- S.-H. Park and H.-I. Yoo, Solid State Ionics, 2005, 176, 1485–1490 CrossRef CAS.
- S. Taub, K. Neuhaus, H.-D. Wiemhöfer, N. Ni, J. A. Kilner and A. Atkinson, Solid State Ionics, 2015, 282, 54–62 Search PubMed.
- M. Izuki, M. E. Brito, K. Yamaji, H. Kishimoto, D.-H. Cho, T. Shimonosono, T. Horita and H. Yokokawa, J. Power Sources, 2011, 196, 7232–7236 Search PubMed.
- Z.-P. Li, M. Toshiyuki, G. J. Auchterlonie, J. Zou and D. John, ACS Appl. Mater. Interfaces, 2011, 3, 2772–2778 Search PubMed.
- A. Shaula, V. Kharton, F. Marques, A. Kovalevsky, A. Viskup and E. Naumovich, J. Solid State Electrochem., 2006, 10, 28–40 Search PubMed.
- V. Kharton, A. Kovalevsky, A. Viskup, A. Shaula, F. Figueiredo, E. Naumovich and F. Marques, Solid State Ionics, 2003, 160, 247–258 CrossRef CAS.
- J. H. Joo, G. S. Park, C.-Y. Yoo and J. H. Yu, Solid State Ionics, 2013, 253, 64–69 CrossRef CAS.
- E. Jud and L. J. Gauckler, J. Electroceram., 2005, 14, 247–253 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ta06889k |
| This journal is © The Royal Society of Chemistry 2025 |