
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Modeling the role of supramolecular clustering in multivalent assembly†
Nicholas
Sbalbi‡
 ab,
Artem
Petrov‡
b,
Jacob
Sass
b,
Matthew
Ye
a,
Alfredo
Alexander-Katz
a and
Robert J.
Macfarlane
*a
ab,
Artem
Petrov‡
b,
Jacob
Sass
b,
Matthew
Ye
a,
Alfredo
Alexander-Katz
a and
Robert J.
Macfarlane
*a
aDepartment of Materials Science and Engineering, Massachusetts Institute of Technology, Cambridge, MA 02139, USA. E-mail: rmacfarl@mit.edu
bDepartment of Chemical Engineering, Massachusetts Institute of Technology, Cambridge, MA 02139, USA
First published on 25th April 2025
Abstract
In self-assembled systems, a combination of multiple weak supramolecular interactions is often utilized to enable strong yet reversible binding. When modeling the behavior of these multivalent interfaces, it is commonly assumed that binding pairs are independent, i.e., the probability of a pair being bound is unaffected by the bound state of neighboring pairs. Inspired by recent experimental work, we report that for a variety of systems this assumption may not hold, leading to the formation of clusters at the binding interface. Through a series of analytical and numerical models of end-functionalized brushes, we reveal the role of cluster size on binding thermodynamics, detail how entropic contributions from polymer chains provide tunable control of cluster size, and provide predictions for cluster size as a function of system architecture. Investigation of these models yields surprising results: within the melting window, the enthalpy of binding of multivalent interfaces is predicted to depend only on cluster size and not on the overall valency of the multivalent system. Moreover, clustering is predicted to be significant even in systems with only weak dipole and dispersion interactions between neighboring groups. Combined, this work brings to light the potential impacts of clustering on multivalent self-assembly, providing theoretical justification for previous experimental observations and paving the way for future work in this area.
1 Introduction
The term “multivalent interface” can be used to describe any system involving two objects binding simultaneously at multiple sites via non-covalent interactions. Since first brought to light by Mammen et al.,1 the majority of studies involving these interfaces have been in a biological context, where the objects may be cells,2,3 pathogens,4–6 or nanoparticle therapeutics7–9 and individual binding sites are ligand–receptor pairs. Within these systems, multivalency is commonly used to increase binding strength and selectivity while remaining dynamic and reversible. Pursuit of these same properties on the macroscale has led to a recent surge in the study of multivalent soft materials outside of biological contexts, where strong yet reversible interfaces enable functions including hierarchical self-assembly10,11 or self-healing capabilities.12–14 While the size and specificity requirements of biological systems often limits them to lower valencies, synthetic polymers and grafted surfaces are not bound by this restraint, leading to obvious but unanswered questions: is there a limit to multivalency's effect on interface strength? And if so, what is this limit and how is it affected by system architecture?Evidence of an upper limit to multivalent scaling can be found in existing biological systems where plateauing of binding constants can occur at valencies as low as 4.15,16 However, the small size, high curvature, and often restricted ligand flexibility within these systems suggest that their multivalency limit may come from simply steric constraints, i.e., an inability for additional ligands to access the binding interface. Synthetic materials do not necessarily face this same constraint, however, as multivalent scaffolds comprised of polymer chains, colloidal nanoparticles, or macroscopic surfaces can be designed to express hundreds or thousands of supramolecular groups spread over areas that are significantly larger than an individual supramolecular binder. For example, Santos et al. examined the multivalent binding thermodynamics of polymer brush-coated nanoparticles, where the surface of the nanoparticles permitted the grafting of larger numbers of supramolecular groups.17 Despite each nanoparticle presenting a valency of approximately 1000 (i.e., each particle is functionalized with up to 1000 supramolecular binding groups), the reported enthalpies of binding were only 5–20 times that of an individual binding pair. This limited enhancement was hypothesized to equal the size of a so-called “bundle”: a local clustering of interacting chain ends where groups within a cluster readily exchanged with one another. Thus, binding enthalpy was proposed to be controlled not by the number of clusters or the overall system valency, but instead by the number of binding pairs within each cluster. In other words, each cluster acted as an independent multivalent system. Importantly, it was also shown that the size of these clusters was also dependent on the system architecture, specifically the sizes of the nanoparticle core and the height and grafting density of the polymer brushes, as these affected the configurational entropy penalty associated with clustering. Similar clustering behavior has been predicted and observed for mobile receptors within lipid membranes,18,19 but this study marked the first experimental observation for immobile tethers in a polymer brush.
Despite the implications of these observations for multivalent materials design, they remain difficult to probe experimentally, requiring computational investigation to further explore and explain the phenomenon. However, the influence of clustering behavior on multivalent interface strength has not been adequately examined theoretically. In fact, it would be impossible for clustering to emerge in the majority of existing models of multivalent interfaces with immobile tethers due to the common assumption that all ligand–receptor pairs are independent (or in other words, there are no ligand–ligand or receptor–receptor interactions).15,20–22 In this work, we show that it is the very breaking of this assumption that enables the formation of finite-sized clusters, using both a simple analytical theory and numerical simulations. Using these interacting-brush models, we demonstrate that in systems with large overall valencies, the interplay between chain stretching entropy and end group binding enthalpy leads to a wide range of accessible cluster sizes across experimentally-relevant regimes (Fig. 1). We also provide a brief argument to explain the counterintuitive hypothesis that binding enthalpies of massively multivalent systems (e.g., those observed in Santos et al.) can depend only on cluster size, independent of the number of clusters formed.17 Together, these results provide both a justification for the formation of clusters and their effects on interface properties, and establish recommended parameter spaces for future experimental exploration of these predictions. Such designs have potential benefit for a broad materials space where supramolecular chemistry is employed, including adhesives,23,24 biological diagnostics,25,26 therapeutics,7–9 and chemical sensing.27,28
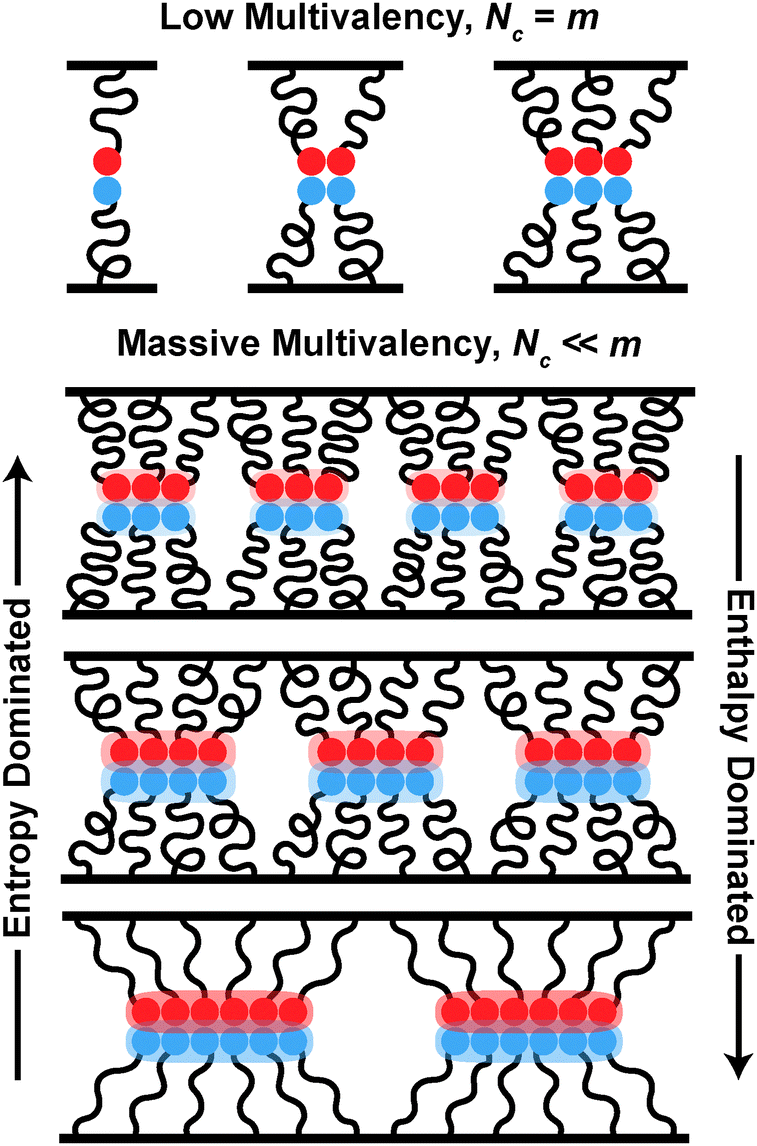 | ||
| Fig. 1 For systems with low multivalency, cluster size Nc is equal to the total number of binding pairs m. For massively multivalent systems, Nc is instead dictated by the balance between the entropic penalty of chain stretching and the enthalpy of binding of end groups, independent of m. | ||
2 Results and discussion
2.1 The role of cluster size in melting
In prior work involving “massively multivalent” systems consisting of polymer-brush-grafted nanoparticles that expressed up to ∼1000 supramolecular groups per particle scaffold, it was hypothesized that multivalent binding enthalpy was restricted by clustering of polymer chain ends. Thus, an upper limit was theorized to exist for the enthalpy of binding of particle interfaces, set by the average number of chain ends within each cluster.17 In follow-up work, similar trends were observed and direct control of cluster size between two nanoparticles was demonstrated using a multivalent polymer.29 In both works, clustering is rationalized by phenomena that would be expected in a physical system consisting of complex chemical components, namely that molecular species tend to self-segregate based on types of intermolecular forces. More simply, “like dissolves like”—the supramolecular groups in these systems possess a large number of polar C–N, C–O, N–H, and O–H bonds, meaning each binding group has an overall net dipole. Conversely, both the polymer chains and solvent are comprised of mostly non-polar C–C and C–H bonds. Thus, lateral interactions between supramolecular groups lead to clustering; similar behavior would be expected of any multivalent system that is comprised of both polar and non-polar components where individual components can self-sort or reorganize (e.g., receptors in lipid membranes,18,19 or elastomeric polymers with multiple tethered supramolecular groups12,14). While these prior works provided significant experimental evidence for this clustering phenomenon, neither proposed a direct reasoning to explain why the multivalent enthalpy of binding depended solely the number of chain ends in a cluster and not the total number of clusters. Here, we provide a brief argument for why this would be true for enthalpies of binding measured around the melting temperature, Tm, as is employed in those studies.For a population of multivalent interfaces, here idealized as two parallel planes each grafted with m end-functionalized polymer chains, melting does not occur at one specific temperature.21,30 As interfaces only become unbound once all binding pairs are broken, melting instead takes place over a temperature range, or window, in between which the fraction of interfaces bound, fbound, transitions from 1 to 0. The specific definition for multivalent melting temperature is a topic of debate,29–31 but here we chose Tm as the temperature at which fbound = 0.5. By this definition, Tm is the temperature at which the concentrations of bound and unbound interfaces are equal, the binding constant is 1, and ΔG of binding is 0. For a given temperature in the melting window, there is a distribution of the number of bound pairs per interface; explaining the unintuitive results of cluster size-dependent multivalency requires solving for the expected value of this distribution for a given fbound and maximum number of pairs m (overall valency). Importantly, the probability of all pairs of a given interface being unbound is equivalent to 1 − fbound. In the simplest case, where binding pairs are assumed to be independent and each ligand has one valid binding partner, each pair's probability of binding must be equal and can therefore be calculated as ppair = 1 − (1 − fbound)1/m (e.g., for m = 4 as shown in Fig. 2a, ppair = 1 − (0.5)1/4 ≈ 16%).
 | ||
| Fig. 2 Probability of binding for independent interface components at Tm. (a) In the case of independent supramolecular binding pairs, the probability of pair binding is calculated from the total system valency and the definition that 50% of interfaces have all pairs unbound. (b) In the case of independent clusters, the probability of cluster binding is calculated from the maximum number of clusters, not the total number of pairs. | ||
This scenario is a Bernoulli process, predicting an average formation of mppair pairs per interface. Combining the equations noted above results in an expected number of pairs formed of m(1 − (1 − fbound)1/m). For fbound = 0.5 and large m, this value converges to exactly ln(2), meaning that at Tm, the average number of pairs formed is independent of the total valency of the system. In fact, convergence occurs for any fbound < 1, resulting in an average of −ln(1 − fbound) pairs. However, the valency at which it converges increases with increasing fbound (e.g., to reach 90% of the asymptote value, m ≥ 4 at fbound = 0.5 vs. m ≥ 22 at fbound = 0.99). This argument relies on the assumption that each ligand can only reach and bind to one receptor, however, existing multivalency models commonly allow ligands to bind to a subset of receptors,20–22 introducing a combinatorial entropy as described by Kitov et al.32 These cases, modeled in ESI,† Section S1, also lead to an average of −ln(1 − fbound) pairs within the melting window.
The above scenarios, which rely on the assumption that pairs are independent, are unable to capture the behavior observed in Santos et al. where the number of pairs formed at Tm is on the order of 5–20.17 To remedy this, the assumption can be broken, allowing pairs to interact with one another (e.g., via self-sorting based on polarity as noted above) and creating clusters. This clustering behavior skews the distribution towards forming a number of pairs that are a multiple of an equilibrium cluster size. States with partially-broken clusters are short-lived because either (1) loose chain ends will rapidly reconnect due to enhanced local concentration within the cluster or (2) complete pair dissociation within the cluster will occur simultaneously to overcome the rebinding effect.33 Repeating the above logic with clusters as the independent component will arrive at a probability of cluster formation of pcluster = 1 − (1 − fbound)Nc/m and an expected number of bound clusters of (m/Nc)(1 − (1 − fbound)Nc/m) where Nc is the cluster size and thus m/Nc is the maximum number of clusters (Fig. 2b). In the limit of large m/Nc, a formation of −ln(1 − fbound) clusters is thus expected, which is equivalent to −Nc![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ln(1 − fbound) expected pairs. A similar conclusion to before can now be made: that at and around Tm, the average number of pairs formed is approximately independent of system valency, though now it is also directly proportional to the system's equilibrium cluster size.
ln(1 − fbound) expected pairs. A similar conclusion to before can now be made: that at and around Tm, the average number of pairs formed is approximately independent of system valency, though now it is also directly proportional to the system's equilibrium cluster size.
For systems with different maximum cluster numbers, a near identical number of clusters are bound on average at and above Tm (Fig. 3a, blue background). When larger fractions of supramolecular groups are in a bound state (i.e., at temperatures below Tm), the system valency begins to have an effect, as its limit on the maximum number of clusters contributes significantly to the average (Fig. 3a, red background). Multiplying the number of clusters by the number of supramolecular groups per cluster gives the average number of pairs bound across an entire multivalent system (Fig. 3b). This value is of significant experimental importance, as it would equal the multiplicative scaling applied to a monovalent binding enthalpy to obtain the multivalent binding enthalpy measured at Tm in the work of Santos et al. and other multivalent nanoparticle studies.17,29 In the valency-independent regime, two systems with vastly different valencies but identical cluster size have the same number of bound pairs at a given fbound, whereas two systems with identical valencies have pairs bound proportional to their cluster size. It is worth noting that the absolute temperatures of these regimes are outside the scope of this work, and are functions of system valency.21
 | ||
| Fig. 3 Average interface behavior in the vicinity of Tm. (a) At and above Tm (blue background, lower fbound), the average number of clusters bound is approximately independent of the maximum number of clusters. Below Tm (red background, higher fbound), the dependence reemerges. (b) For systems with vastly different overall valencies, the same number of bound pairs would be expected at and above Tm. | ||
This result, that cluster size (and not overall valency) controls the number of bound pairs at Tm, provides a theoretical justification for the behavior observed by Santos et al. and their proposed equation for multivalent binding enthalpy in a system assembled with binary A–B supramolecular pairs:17 ΔHbind = NcΔHAB, although with an added prefactor of ln(2). Notably, this relation includes only the enthalpy of the monovalent across-brush interactions (A–B), and not the “like-interactions” between chain ends on the same brush (A–A/B–B). While these “intra-brush” interactions are the driving force for cluster formation and control cluster size (vide infra), their direct contribution towards the interface binding strength is non-obvious, but likely significantly smaller than those “inter-brush”. In relation to the overall interfacial free energy, cluster size also contributes to the entropy of binding, setting topological constraints on the combinatorial entropy often attributed to multivalent binding enhancement.29,32 Derived and further discussed in ESI,† Section S2, this value (found to be proportional to ln(Nc!) under the assumption of unconstrained pairing within a cluster) is similarly independent of m for large m/Nc. Combining these enthalpic and entropic contributions, the overall interfacial free energy would be expected to have a strong dependence on Nc, demonstrating a need to model cluster size as a function of interface design parameters.
Backed by experimental observations, this theory has significant ramifications for supramolecular materials, where systems are often melted and slowly cooled to encourage assembly into thermodynamic products.34 Cluster size would directly control the thermodynamics in this regime, and thus the ability for components to reorganize, the ease of avoiding kinetic traps, and impacting resulting structure.17
2.2 Theoretical cluster size
Given the importance of cluster size in determining the thermodynamics of multivalent systems such as those described above, advancing understanding of these massively multivalent systems requires analysis of how different parameters affect the sizes of these clusters. In the simplest case, one would expect cluster size to be a function of chain length N, grafting density σ (the number of chains grafted per unit area), interaction strength between like and opposing chain ends (εAA = εBB and εAB, respectively), and system temperature T (Fig. 4a). N and σ provide geometric constraints on how much configurational entropy is lost when polymer chain ends are confined to the limited volume of a cluster, and the ε values correspond to the enthalpic benefit gained from forming supramolecular bonds and clusters. These enthalpic and entropic contributions, with their relative weight controlled by T, compete to result in an equilibrium cluster size Nc. Previous work has modeled clustering in a similar telechelic brush system, but did not solve for this value.35 | ||
| Fig. 4 Schematic and cluster size predictions of the analytical theories. (a) Chains are described by a number of Kuhn segments N with a corresponding Kuhn length b and grafting density σ. Chain ends interact within a brush with energy εAA = εBB or across the interface with energy εAB. (b) Plotted against system parameters with f = 0.001, the infinite chain model produces a narrow window of controlled cluster size. (c) The short chain theory (N = 10) predicts a smooth increase in cluster size over all parameter space. | ||
Several important assumptions are required to make the derivation of Nc analytically tractable. Namely, the multivalent interface will be considered infinite and planar, grafted on both sides by end-functionalized, monodisperse brushes. Such an approximation is expected to apply in the case of nanoparticle binding for low curvature systems, where one can approximate the brush locally as planar. For large pairwise interaction strength across the interface, εAB, it can be assumed that each A-functionalized chain end will always form a pair with a B-functionalized chain end, with these A–B pairs resting in a thin contact interface between the two brushes. Under this assumption of “fixed pairs”, which applies to the εAB of ∼12kBT found by Santos et al.,17 the problem becomes analogous to that of a single self-interacting brush experiencing twice the interaction energy per chain end. This assumption can be both justified and relaxed by a concomitant Monte Carlo model (vide infra), but is useful for initial analysis.
Before including the effects of chain stretching on cluster size, the limiting case of N → ∞ was examined, which is analogous to that of clustering in a 2D gas and can be compared to models of clustering of receptors within lipid membranes.18 Mapping the problem onto the Fisher droplet model for 2D gases,36,37 the following set of equations were obtained for average cluster size (derived in ESI,† Section S3),
 | (1) |
 | (2) |
 | (3) |
![[small sigma, Greek, macron]](https://www.rsc.org/images/entities/i_char_e0d2.gif) = a2σ represents the area fraction of chain end groups at the interface, where a2 is the area occupied by a chain end. This result concludes that Nc ≈ 1 for the majority of experimentally relevant system parameters, and that Nc diverges sharply at some critical parameter value (Fig. 4b). Similar behavior was observed in Monte Carlo simulations for the clustering of mobile cell surface receptors.18 However, this behavior contradicts the experimental observations, where Nc was a smoothly varied function of system parameters such as nanoparticle curvature (which affects ) and chain length.17 This gradual behavior can be reproduced through reintroduction of finite chain length and its corresponding entropic contributions.
= a2σ represents the area fraction of chain end groups at the interface, where a2 is the area occupied by a chain end. This result concludes that Nc ≈ 1 for the majority of experimentally relevant system parameters, and that Nc diverges sharply at some critical parameter value (Fig. 4b). Similar behavior was observed in Monte Carlo simulations for the clustering of mobile cell surface receptors.18 However, this behavior contradicts the experimental observations, where Nc was a smoothly varied function of system parameters such as nanoparticle curvature (which affects ) and chain length.17 This gradual behavior can be reproduced through reintroduction of finite chain length and its corresponding entropic contributions.
In the case of short-chain brushes, the deviation of the chain end from its equilibrium position (above the tether point) will lead to an entropic penalty stemming from the reduction of chain conformational entropy. At a fixed value of , increasing the number of supramolecular groups per cluster results in increasingly larger entropic penalties per polymer chain, as added chains would have to stretch further distances to reach the cluster. Thus, to calculate the total entropic penalty of a cluster, the contribution from each chain end would be summed, with these individual contributions being a function of the distance between the chain end's equilibrium position and the cluster center, r (assuming the physical size of the cluster is negligible). If Nc is large, a continuous representation of the grafted surface can be used, where the number of grafting points located in the (r,r + dr) interval equals to 2πrdr × /a2. Assuming only the closest chain ends are recruited to a cluster, the radius of the area that contains all chains forming the cluster (denoted as L) satisfies the normalization condition:  therefore, L = a(Nc/(π))1/2. This continuous representation is valid if L is much greater than the distance between grafting points, which is satisfied when Nc1/2 ≫ 1. Each chain grafted at the distance r∈ [0,L] from the cluster loses conformational entropy TΔS(r) = −1.5kBTr2/(Nb2), where b is the Kuhn segment length, and N is the number of Kuhn segments. The total loss of conformational entropy of all Nc chains forming a cluster equals to
therefore, L = a(Nc/(π))1/2. This continuous representation is valid if L is much greater than the distance between grafting points, which is satisfied when Nc1/2 ≫ 1. Each chain grafted at the distance r∈ [0,L] from the cluster loses conformational entropy TΔS(r) = −1.5kBTr2/(Nb2), where b is the Kuhn segment length, and N is the number of Kuhn segments. The total loss of conformational entropy of all Nc chains forming a cluster equals to  . As clusters are viewed as a dynamic fluid in which chain ends are mobile and can exchange binding partners, we neglect entropic contributions from local chain stretching to accommodate the positioning of pairs within a cluster.
. As clusters are viewed as a dynamic fluid in which chain ends are mobile and can exchange binding partners, we neglect entropic contributions from local chain stretching to accommodate the positioning of pairs within a cluster.
The total interaction potential energy of a cluster, Uc, which counterbalances the entropic penalty of clustering, is calculated in a similar method to the infinite chain case. Namely, each chain end contributes energy ε if it is in the interior of the cluster and energy (1 − f)ε if it is at the cluster edge. Thus, Uc = Ncε − fεπNc1/2 and the total free energy of a cluster Fc can be written as:
 | (4) |
To find the equilibrium cluster size, Fc is minimized with respect to Nc; discussion of why this calculation can be done here but not in the Fisher model is found in ESI,† Section S4. As this expression was derived in the Nc1/2 ≫ 1 approximation, the −fεπNc1/2 term is subdominant with respect to other terms and can be neglected during minimization. With this simplification, the resulting expression for the equilibrium Nc can therefore be written as:
 | (5) |
Eqn (5) predicts Nc behavior that agrees with intuition. Namely, if the interaction strength between end groups |ε| is increased, the chains can overcome a larger entropic penalty of stretching to participate in cluster formation, which in turn increases the cluster size. Increasing and chain length N also increases Nc, with the former increasing the number of chains that can access the cluster without significant stretching and the latter decreasing the entropy of stretching per chain. On the other hand, Nc decreases with increasing temperature T as this leads to a larger weight of the entropic term.
Of significant note is that the short chain theory predicts a gradual change in Nc with all system variables, in contrast to the discontinuous behavior in the infinite chain case (Fig. 4c). Moreover, the Nc values predicted in the range of experimental parameters are exactly on the order of those observed in the nanoparticle self-assembly experiments.17 This confirms that the clustering behavior in end-functionalized brushes is inherently different than that observed for mobile tethers, and that it is exactly the finite chain length that leads to the smooth behavior of Nc.
This short chain theory of cluster size makes a number of assumptions that idealize multivalent brush interfaces in pursuit of a simple model. While some of these assumptions, including disk-like cluster shape and negligible combinatorial entropy effects, can be relaxed with minimal or no change to the resulting power laws (see ESI,† Section S5), limits towards its applicability still remain. Notably, regions with low cluster size, dense chain grafting (1/2 ≲ 1), and moderately long chains remain inaccessible. Thus, to further extend the applicability of this concept to a broader range of systems, a Monte Carlo model was developed to explore a wider range of Nc values, system parameters, and A–B pairing strengths.
2.3 Monte Carlo simulations
The algorithm and parameters for the Monte Carlo model developed here are fully detailed in ESI,† Section S6. Briefly, the interface between the two brushes is discretized into a 2 × Ngrid × Ngrid grid, where A and B chain ends each occupy one Ngrid × Ngrid plane (Fig. 5a). In each iteration, diffusion of the chain ends is modeled by permitting a randomly chosen chain end to move to a neighboring cell, with the probability of moving to state k given by the equation: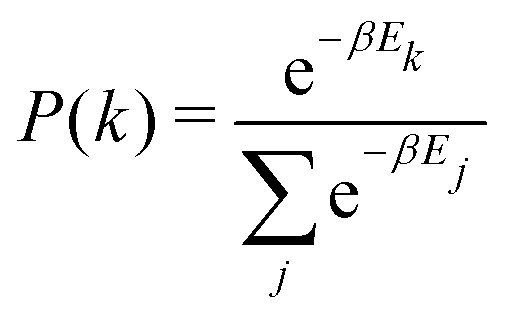 | (6) |
 | (7) |
/a2), and 4εAA = 4εBB = ε (as each chain end contributes half to a maximum of eight interactions with neighbors). As the system is allowed to evolve, the model converges on an equilibrium cluster size, the scaling of which can be easily studied due to the simple nature and low computational cost of the simulation. Both self-interacting brushes (NBB = NAB = 0) and pairs of interacting brushes can be studied (Fig. 5b and Videos S1–S3, Section S7, ESI†). The algorithm varies slightly between the two cases, discussed further in ESI,† Section S6.
 | ||
| Fig. 5 Monte Carlo moves, resulting clustering behavior, and extraction of cluster size. (a) In a given MC move, a chain end, represented by a filled grid cell, moves one cell over in its plane, resulting in an entropic penalty for chain stretching and an enthalpic payoff for interacting with other chain ends. (b) and (c) Representative snapshots of clustering behavior in a single self-interacting brush (b) and two interacting brushes (c). (d) Distributions of cluster sizes for a single brush (N = 10 kDa, σ = 0.0625 chains per nm2) with varying self-interaction strengths. Clustering is indicated by a Gaussian distribution about the average cluster size. | ||
To quantify the behavior within a given simulation, cluster sizes (via 8-connectivity) are collected cumulatively throughout an equilibrated duration (on the order of 1 × 108 MC moves) to produce a cluster size distribution (Fig. 5d for a series of increasing self-interaction strengths within an A-functionalized brush). Two distinct regions are observed in the case of significant clustering, the first being an exponential decay representing the thermal motion of chain ends when diffusing between clusters, and the second being a Gaussian distribution centered around the average cluster size. To eliminate the contribution of thermal motion (an artifact exacerbated by confining chain ends to a two-dimensional interface), the ensemble average maximum cluster size 〈Nc,max〉 was used to evaluate the evolution of these systems over time. This value was chosen over the mean of the Gaussian distribution as it was demonstrated to follow the same scaling laws while being more robust to noise (ESI,† Section S8).
For the majority of simulations, the single self-interaction brush model was utilized, as it could both be directly compared to the analytical theory and was easier to equilibrate than the two-brush model. Results confirmed that the single-brush case is indeed analogous to a two-brush case where it is assumed that all A–B pairs are permanent under the timescale being considered (ESI,† Section 10): cluster sizes for the two-brush model were approximately equivalent to the cluster sizes in a one-brush model with twice the self-interaction strength εAA (Fig. S6, ESI†). Cluster size relations to system variables εAA, N, σ, and T were examined (Fig. 6), with input parameters chosen to span experimentally-relevant values including those reported for cell surface ligands38 and multivalent nanoparticles.17 The value of εAA is on the order of 1kBT to represent non-specific binding between chain ends (Keesom, Debye, and London forces).39 Each data point represents the average maximum cluster size after an equilibration period with error bars representing one standard deviation above and below the mean. The upper limit for each variable examined was determined based on the limits of feasible simulation times.
 | ||
| Fig. 6 Cluster sizes predicted by MC simulation. Simulation parameters are εAA = −1kBT, N = 10, b = 1.8 nm, σ = 0.0625 chains per nm2, and T = 300 K unless varied. Short chain analytical theory results are reported as dashed lines. (a) Clustering does not become significant until after a threshold εAA, after which it approaches linear behavior as the enthalpy of binding overcomes the entropy penalty of stretching. (b) Increasing grafting density increases the number of chain ends that can access a cluster, increasing cluster size almost linearly. (c) Increasing chain length lowers the entropy penalty of stretching, increasing cluster size with slightly slower than the linear behavior. | ||
Based on the prior results from the analytical theory, linear scaling with εAA, N, and σ would be expected (eqn (5)). For variations in εAA this is indeed observed, though this scaling only applies past a minimum strength of self-interaction that varies as a function of chain length (Fig. 6a). Based on the model structure, we hypothesized this minimum should occur at εAA = −3kBT/4Nb2σ, as this is the point where the interaction strength is on the order of the stretching entropy penalty for the meeting of two neighboring chains. Following this hypothesis, clustering would become significant at |εAA| values of roughly 0.74kBT and 0.37kBT for N = 5 and 10 kDa, respectively, which indeed match fairly closely to the onset of linear behavior (Fig. 6a). For interaction strengths below this minimum, the cluster size asymptotically approaches that of encounter frequency in random chain end diffusion (and is exactly equal in the absence of lateral interactions), an artifact of the manner in which the model is designed.
The expected linear scaling is also observed for σ (Fig. 6b). However, variations in N resulted in a lower scaling of ∼2/3 (Fig. 6c). We attribute this discrepancy primarily to discretization effects, as the change in the entropic stretching term on moving between grid cells is large and discontinuous, potentially excluding the true equilibrium position. We would expect that for a large chain or a finer grid, the scaling with N would approach 1, though modifying the model in this manner would be computationally infeasible given the larger number of calculations required. Regardless, these discretization effects would not impact the scaling of other variables with fixed N, as they, along with the entropic spring constant, would be fixed. Temperature, for instance, has the predicted scaling of T−1 for constant N = 10 kDa (Fig. S5, ESI†). Slight inconsistencies aside, the MC model matches the analytical theory surprisingly well, being almost exact in magnitude at low N, though we attribute this primarily to serendipity and would rather emphasize the matching of scaling relations. This agreement suggests that the analytical model accurately represents the underlying entropic and enthalpic contributions towards clustering and can be employed to estimate experimental cluster sizes.
Together, these results predict that a broad range of cluster sizes are accessible within experimentally-relevant regimes, aligning with the magnitudes observed in experiments.17 While in practice some parameters are more straightforward to vary than others, the relatively smooth nature of these relations indicates that cluster size is a well-behaved, tunable quantity. Of particular note is that temperature changes are expected to only have a mild impact on cluster size. In the case of melting and dissociation studies of multivalent complexes, this implies that cluster formation is still significant throughout the melting window, and thus would significantly affect ΔH of binding as argued above. Similarly, clustering would exist in and potentially impact the association/dissociation constants of multivalent systems at room or physiological temperatures.
Moving away from the one brush analogy and outside the high εAB regime enables exploration of systems not immediately applicable to the developed analytical theory (though these studies require slight changes in cluster quantification, see ESI,† Section S11). These explorations include systems not explicitly designed for strong A–B interactions, unlike those studied by Santos et al.17 which are on the order of 10kBT. This low- to intermediate-εAB regime is of particular interest for drug delivery systems, as lowering ligand–receptor bond strength can increase binding selectivity to receptor density, a critical targeting parameter.40,41 Weaker interactions also encourage faster exchange between end groups, which in turn helps self-assembled systems avoid kinetically trapped states.42
Using the two brush model, cluster size can be plotted versus εAB strength for varying εAA = εBB values (Fig. 7). For εAB on the same magnitude as εAA, there is minimal impact on cluster size (Fig. 7a). It is only once εAB increases beyond 1kBT that a significant change in cluster size begins (Fig. 7b); past this point, cluster size increases asymptotically to approximately 1.5 times its original value. It is important to note that this logistic behavior is not observed with variations in εAA. This difference arises from the monovalent nature of A–B interactions in the MC simulation, as once εAB is large enough for these pairs to be “permanent” relative to the lateral A–A and B–B interactions, further increases have little effect. In this regime of high εAB, a chain end must remain paired when leaving the cluster, and thus must break twice the lateral interactions as when leaving independently, leading to larger cluster sizes. These limiting effects are particularly notable for experimental systems such as those used by Santos and coworkers,17 since it indicates that designing systems with increasing monovalent binding strength will asymptotically approach a maximum multivalency, even though these systems' overall binding strengths at Tm would still increase multiplicatively. If A–B interactions were instead modeled as nonspecific (as in the case of general brushes modeled in prior systems35,39), this asymptote would no longer be present. While not required for clustering, A–B interactions promote the formation of larger clusters and directly impact experimental enthalpies of binding, thus providing another lever for tuning interface behavior and binding strength.
 | ||
| Fig. 7 Cluster size in two-brush interface (N = 10 kDa, σ = 0.0625 chains per nm2) as a function of interaction strength between their chain ends, εAB. Cluster size is reported for chain ends of an individual face. (a) At lower εAB values cluster size is almost constant, only increasing by one standard deviation at most. (b) εAB only has a larger effect past 1kBT, where it causes an approximate 50% increase in cluster size before leveling off. | ||
3. Conclusions
Ligand and receptor clustering, as a phenomenon that affects the behavior and strength of multivalent interfaces, has been overlooked in prior examples of supramolecular multivalency under the assumption that ligand–receptor pairs behave independently. Here, we provide an argument that clustering plays a direct role in the binding strength of these systems, with cluster size contributing to (and in the melting window often becoming the sole contributor towards) the multivalent enthalpy of binding. Additionally, through both analytical and numerical models, we reveal the role of chain stretching entropy in controlling cluster size and show that non-negligible cluster sizes are achieved even in systems with minimal dispersion or dipole interactions between end groups. These models provide predictions for cluster sizes over a range of experimentally-relevant system parameters including chain length and grafting density, and predict cluster sizes that match closely in magnitude with experiments. Combined, these results deliver a missing theoretical justification for clustering behavior and its resulting effects on the thermodynamics of binding observed in previous experimental systems.17,29 Armed with these relations, future experimental work will have finer control over interface behavior, enabling precise control of melting behavior in self-assembled systems and their underlying thermodynamics.For future theoretical work, we envision extensions of these models into more complicated systems with fewer simplifications. For instance, the assumption of perfectly Gaussian chain behavior could be relaxed, with a more accurate consideration of solvent effects leading to an improved estimate of the prefactor in eqn (5) and multiple power law regimes. Similarly, implementing curvature effects and capability for imperfect A–B pairing to the theoretical model could lead to improved numerical estimates for the cluster size in the case of brush-coated nanoparticle assemblies. Radical augmentations such as capabilities for out-of-plane motion or kinetic effects could also be explored, with clustering's role on binding dynamics outside the melting window providing applicability for a vast number of systems operating at physiological and room temperature. Extensions could also be made into electrostatic interactions, whose long-range character and additional repulsive interactions would lead to unique and non-obvious ligand distributions at the interface.
Author contributions
All authors were involved in the conception of this research. NS and MY developed the probabilistic theory in the melting window; AP and AAK developed the analytical brush theory; NS wrote the Monte Carlo simulations and JS implemented them on supercomputing resources. NS, AP, and JS wrote the manuscript and all authors edited and reviewed it.Data availability
MATLAB code for Monte Carlo simulations is available at https://github.com/nsbalbi/cluster-mc.Conflicts of interest
There are no conflicts to declare.Acknowledgements
NS acknowledges support from a National Science Foundation Graduate Research Fellowship under Grant No. 2141064. This work was supported with funding from the National Science Foundation (Macromolecular, Supramolecular, and Nanochemistry, Award CHE-2304909). It was also supported by funding from the Department of the Navy, Office of Naval Research, under ONR Award N00014-22-1-2148. AP and AAK acknowledge the support by the National Science Foundation through award number DMREF 2118678. The authors thank the MIT SuperCloud and Lincoln Laboratory Supercomputing Center for providing HPC resources that have contributed to the research results reported within this paper.Notes and references
- M. Mammen, S.-K. Choi and G. M. Whitesides, Angew. Chem., Int. Ed., 1998, 37, 2754–2794 CrossRef PubMed.
- D. Morzy and M. Bastings, Angew. Chem., 2022, 134, e202114167 CrossRef.
- X. Liu, H. Yan, Y. Liu and Y. Chang, Small, 2011, 7, 1673–1682 CrossRef CAS PubMed.
- S. Bhatia, L. C. Camacho and R. Haag, J. Am. Chem. Soc., 2016, 138, 8654–8666 CrossRef CAS PubMed.
- S. Liese and R. R. Netz, ACS Nano, 2018, 12, 4140–4147 CrossRef CAS PubMed.
- N. J. Overeem, P. H. E. Hamming, O. C. Grant, D. Di Iorio, M. Tieke, M. C. Bertolino, Z. Li, G. Vos, R. P. de Vries, R. J. Woods, N. B. Tito, G.-J. P. H. Boons, E. van der Vries and J. Huskens, ACS Cent. Sci., 2020, 6, 2311–2318 CrossRef CAS.
- S. Hong, P. R. Leroueil, I. J. Majoros, B. G. Orr, J. R. Baker and M. M. Banaszak Holl, Chem. Biol., 2007, 14, 107–115 CrossRef CAS PubMed.
- S. Angioletti-Uberti, npj Comput. Mater., 2017, 3, 48 CrossRef.
- M.-H. Li, S. K. Choi, P. R. Leroueil and J. R. Baker, ACS Nano, 2014, 8, 5600–5609 CrossRef CAS PubMed.
- R. L. Li, C. J. Thrasher, T. Hueckel and R. J. Macfarlane, Acc. Mater. Res., 2022, 3, 1248–1259 CrossRef CAS.
- P. J. Santos, P. A. Gabrys, L. Z. Zornberg, M. S. Lee and R. J. Macfarlane, Nature, 2021, 591, 586–591 CrossRef CAS PubMed.
- H. W. Ooi, J. M. M. Kocken, F. L. C. Morgan, A. Malheiro, B. Zoetebier, M. Karperien, P. A. Wieringa, P. J. Dijkstra, L. Moroni and M. B. Baker, BioMacromol., 2020, 21, 2208–2217 CrossRef CAS PubMed.
- G. A. Williams, R. Ishige, O. R. Cromwell, J. Chung, A. Takahara and Z. Guan, Adv. Mater., 2015, 27, 3934–3941 CrossRef CAS PubMed.
- L. Zou, A. S. Braegelman and M. J. Webber, ACS Appl. Mater. Interfaces, 2019, 11, 5695–5700 CrossRef CAS PubMed.
- K. C. Tjandra and P. Thordarson, Bioconjugate Chem., 2019, 30, 503–514 CrossRef CAS PubMed.
- J. Wan, J. X. Huang, I. Vetter, M. Mobli, J. Lawson, H.-S. Tae, N. Abraham, B. Paul, M. A. Cooper, D. J. Adams, R. J. Lewis and P. F. Alewood, J. Am. Chem. Soc., 2015, 137, 3209–3212 CrossRef CAS PubMed.
- P. J. Santos, Z. Cao, J. Zhang, A. Alexander-Katz and R. J. Macfarlane, J. Am. Chem. Soc., 2019, 141, 14624–14632 CrossRef CAS PubMed.
- G. M. Fricke and J. L. Thomas, Biophys. Chem., 2006, 119, 205–211 CrossRef CAS PubMed.
- D. Lingwood and K. Simons, Science, 2010, 327, 46–50 CrossRef CAS PubMed.
- J. Huskens, A. Mulder, T. Auletta, C. A. Nijhuis, M. J. W. Ludden and D. N. Reinhoudt, J. Am. Chem. Soc., 2004, 126, 6784–6797 CrossRef CAS PubMed.
- E. W. Gehrels, W. B. Rogers and V. N. Manoharan, Soft Matter, 2018, 14, 969–984 RSC.
- P. Varilly, S. Angioletti-Uberti, B. M. Mognetti and D. Frenkel, J. Chem. Phys., 2012, 137, 094108 CrossRef PubMed.
- G. Desroches, Y. Wang, J. Kubiak and R. Macfarlane, ACS Appl. Mater. Interfaces, 2022, 14, 9579–9586 CrossRef CAS PubMed.
- G. J. Desroches, P. P. Gatenil, K. Nagao and R. J. Macfarlane, J. Polym. Sci., 2024, 62, 743–752 CrossRef CAS.
- H. Yoon, E. J. Dell, J. L. Freyer, L. M. Campos and W.-D. Jang, Polymer, 2014, 55, 453–464 CrossRef CAS.
- J. Hooper, Y. Liu, D. Budhadev, D. F. Ainaga, N. Hondow, D. Zhou and Y. Guo, ACS Appl. Mater. Interfaces, 2022, 14, 47385–47396 CrossRef CAS PubMed.
- J. J. Reczek, A. A. Kennedy, B. T. Halbert and A. R. Urbach, J. Am. Chem. Soc., 2009, 131, 2408–2415 CrossRef CAS PubMed.
- R. de laRica, R. M. Fratila, A. Szarpak, J. Huskens and A. H. Velders, Angew. Chem., Int. Ed., 2011, 50, 5704–5707 CrossRef CAS PubMed.
- C. J. Thrasher, F. Jia, D. W. Yee, J. M. Kubiak, Y. Wang, M. S. Lee, M. Onoda, A. J. Hart and R. J. Macfarlane, J. Am. Chem. Soc., 2024, 146, 11532–11541 CAS.
- D. Y. Lando, A. S. Fridman, C.-L. Chang, I. E. Grigoryan, E. N. Galyuk, O. N. Murashko, C.-C. Chen and C.-K. Hu, Anal. Biochem., 2015, 479, 28–36 CrossRef CAS.
- R. Jin, G. Wu, Z. Li, C. A. Mirkin and G. C. Schatz, J. Am. Chem. Soc., 2003, 125, 1643–1654 CrossRef CAS PubMed.
- P. I. Kitov and D. R. Bundle, J. Am. Chem. Soc., 2003, 125, 16271–16284 CrossRef CAS.
- M. Weber, A. Bujotzek and R. Haag, J. Chem. Phys., 2012, 137, 054111 CrossRef PubMed.
- S. Y. Park, A. K. R. Lytton-Jean, B. Lee, S. Weigand, G. C. Schatz and C. A. Mirkin, Nature, 2008, 451, 553–556 CrossRef CAS PubMed.
- A. Zilman and S. Safran, Eur. Phys. J. E: Soft Matter Biol. Phys., 2001, 4, 467–473 CrossRef CAS.
- M. E. Fisher, Phys. Phys. Fizika, 1967, 3, 255–283 CrossRef.
- N. Sator, Phys. Rep., 2003, 376, 1–39 CrossRef.
- C.-Z. Zhang and Z.-G. Wang, Langmuir, 2007, 23, 13024–13039 CrossRef CAS PubMed.
- D. Cao and J. Wu, Langmuir, 2006, 22, 2712–2718 Search PubMed.
- F. J. Martinez-Veracoechea and D. Frenkel, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 10963–10968 CrossRef CAS PubMed.
- X. Xia, G. Zhang, M. Pica Ciamarra, Y. Jiao and R. Ni, JACS Au, 2023, 3, 1385–1391 CrossRef CAS PubMed.
- R. J. Macfarlane, R. V. Thaner, K. A. Brown, J. Zhang, B. Lee, S. T. Nguyen and C. A. Mirkin, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 14995–15000 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Detailed analytical derivations, Monte Carlo algorithm, parameter sets, video descriptions, and code (PDF). Representative videos of Monte Carlo simulations (MP4). See DOI: https://doi.org/10.1039/d5sm00163c |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |