
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA priori Design of [Mn(I)-Cinchona] catalyst for Asymmetric Hydrogenation of Ketones and β-Keto carbonyl Derivatives†
Soumen
Paira
a,
Nupur
Jain
b,
Debarsee
Adhikari
a,
Raghavan B.
Sunoj
 *b and
Basker
Sundararaju
*a
*b and
Basker
Sundararaju
*a
aDepartment of Chemistry, Indian Institute of Technology Kanpur, 208016 Kanpur, Uttar Pradesh, India. E-mail: basker@iitk.ac.in
bDepartment of Chemistry, Indian Institute of Technology Bombay, 400076 Mumbai, India. E-mail: rbsunoj@iitb.ac.in
First published on 13th June 2025
Abstract
Herein, we report an efficient [Mncinchona] chiral catalytic system for the asymmetric hydrogenation of ketones, demonstrating a broad substrate scope and high chemoselectivity. The catalyst selectively reduces ketones, while leaving other reducible functional groups, such as olefins, alkynes, nitriles, nitro groups, and esters, unaffected. The solid-state structure of [Mncinchona] complex revealed that the cinchona ligand acts as bi-dentate ligand, providing valuable insights into its coordination environment and reaction mechanism. DFT calculations suggest that hydrogen molecule activation assisted by water molecules occurs via a six-membered transition state, with enantioselectivity driven by preferential hydride transfer to the si face of the prochiral substrate, resulting in the R configuration. This robust catalytic system shows potential for expansion to the reduction of β-ketoesters and γ-ketoamines and is expected to contribute to further advancements in asymmetric hydrogenation.
Introduction
Asymmetric hydrogenation (AH)1 has expanded rapidly since it'’s early developments reported by Knowles2 and Horner.3 groupsThe enantioselective reduction of prochiral molecules by Knowles and Noyori garnered global attention in the form of Nobel Prize in 2001.4 Consequently, the demand for transition metal catalysts for asymmetric synthesis has been steadily increasing both in academia and industry. While efficient catalysts were developed that employ noble metals,5 the associated toxicity of such metals and trace impurity retained in pharmaceuticals obtained through catalytic hydrogenation remains a cause of concern.6 Therefore, the efforts to replace noble metals with earth-abundant, non-toxic base metals for asymmetric hydrogenation have received considerable attention in the last decade.7 In this regard, AH of prochiral compounds to value added chiral target molecules by using chiral iron8 and cobalt9 catalysts is worth exploring. It is equally significant to note that privileged PNP/PNN ligands coupled with noble metals have also been widely employed as hydrogenation and dehydrogenation catalysts.10 The recent success in the use of such achiral pincer ligands in conjunction with manganese as the transition metal for hydrogenation reactions11 has stimulated further developments toward the chiral versions of manganese-pincer catalysts. Inspired by these reports, research groups of Kirchner,12 Clarke,13 Beller,14 and Morris15 have succeeded in realizing enantioselective hydrogenation of ketones based on Mn(I)-PNN or Mn(I)-PNP type catalytic systems. A closer look at these catalytic systems suggests that the chiral amine core based on ferrocene or privileged pincer PNP (chiral phosphine backbone) ligand is necessary to achieve high level enantioinduction.16 More recently, Ding,17 Liu16a,e and Clarke13d have reported much improved catalytic systems based on the PNP or PNN ligands for efficient asymmetric hydrogenation of ketones. The replacement of the phosphine-based Lewis base with bi-dentate or tri-dentate chiral nitrogen-based donor ligands has also been attempted recently through transfer hydrogenation,19 albeit with limited success with enantioselectivities in the moderate-to-good range.12,18,19Given these backgrounds, we envisioned the use of a non-phosphine based chiral bi- or tri-dentate ligands19 belonging to the cinchona family of alkaloids so as to satisfy the coordination environment as well as the metal-ligand cooperativity in chiral Mn(I) catalysis. The long-term goal of this study is to replace the expensive chiral phosphine ligands with more affordable chiral amine based ligands that are likely to reduce the cost of production of chiral drugs and materials.20 Herein, we present an efficient Mn(I)cinchona alkaloid catalyst featuring N,N-donor site21 for highly enantioselective hydrogenation of ketones, β-ketoesters and γ-amino ketones into their chiral secondary alcohols with ees up to 99% using molecular hydrogen as sole reductant.
Results and discussion
We began our work by isolating a series of cinchona-based chiral bi-dentate and tri-dentate ligands from the quinine, quinidine, and cinchonidine-based primary amines in accordance with the literature precedence.21a,b As shown in Table 1, acetophenone (1a) was used as the model substrate, and MnBr(CO)5 was employed as the precatalyst in combination with cinchona-based chiral ligands (L1–L14). The reaction was conducted in a 4:1 water-ethanol mixture (for detailed solvent screening, see the supporting information†), which was identified as the optimal solvent, under 60 bar hydrogen pressure at 40°C. A preliminary screening of cinchona-based bi-dentate ligands (L1–L4), each incorporating two Lewis basic sites (quinuclidine and amine/amide functionalities), under standard conditions, did not result in the formation of the desired hydrogenation product (2a). Subsequent screening of other chiral tri-dentate ligands (L5–L9) led to complete conversion in all the cases, with a maximum ee of 92% with L6 (entry 1). A change from five-membered to six-membered ring upon coordination to metal as in the case of L10/L11 provided significantly low conversion, suggesting that the rigid five-membered coordination might be necessary to achieve the desired results. Surprisingly, the replacement of picolinamide ligand L5 with picolyl amine L12 provided the secondary alcohol 2a in 65% yield with 45% ee. Change of chiral source from quinine-based picolinamide (L6) to cinchonidine-based picolinamide (L13) did not alter much in terms of conversion, however, the former provided slightly higher ee over the latter. Finally, picolinamide with the opposite stereoisomer was examined. As expected, ligand L14 (opposite stereoisomer of L5) derived from quinidine provided the secondary alcohol with opposite stereoselectivity with 90% ee. The absolute stereochemistry of both the isomers is confirmed with the enantiopure compound 2a obtained from the commercial sources indicating that L5 provided (R)-2a and L14 gave (S)-2a. Furthermore, a comprehensive solvent and base screening and control experiments were carried out (entries 2-5, see the supporting information† for more details). The analysis of the results suggests that 10 mol% of sodium carbonate, 6 mol% ligand (L6), 5 mol% of Mn(I) precatalyst under 60 bar hydrogen pressure in water-ethanol (4:1, 0.2M) solvent mixture provide the optimum reaction conditions for asymmetric hydrogenation using molecular hydrogen as sole reductant.|
|
|||
|---|---|---|---|
| Entry | Change in conditions | Yield of 2a (%)b | ee |
a Unless otherwise stated, all reactions were carried out under 60 bar hydrogen pressure using 1a (0.4 mmol), MnBr(CO)5 (5 mol%), L (6 mol%) and Na2CO3 (10 mol%) in a H2O![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :EtOH mixture (4:1, 0.2 M) at 40 °C for 24 h.
b Yields are determined through 1H NMR using anisole as the internal standard.
c Number in parentheses is the isolated yield. :EtOH mixture (4:1, 0.2 M) at 40 °C for 24 h.
b Yields are determined through 1H NMR using anisole as the internal standard.
c Number in parentheses is the isolated yield.
|
|||
| 1 | None | 99 | 92 |
| 2 | Without L | n.r. | n.d. |
| 3 | Without M | n.r. | n.d. |
| 4 | MeOH instead of H2O:EtOH |
25 | 83.5 |
| 5 | H2O instead of H2O:EtOH |
99 | 85 |
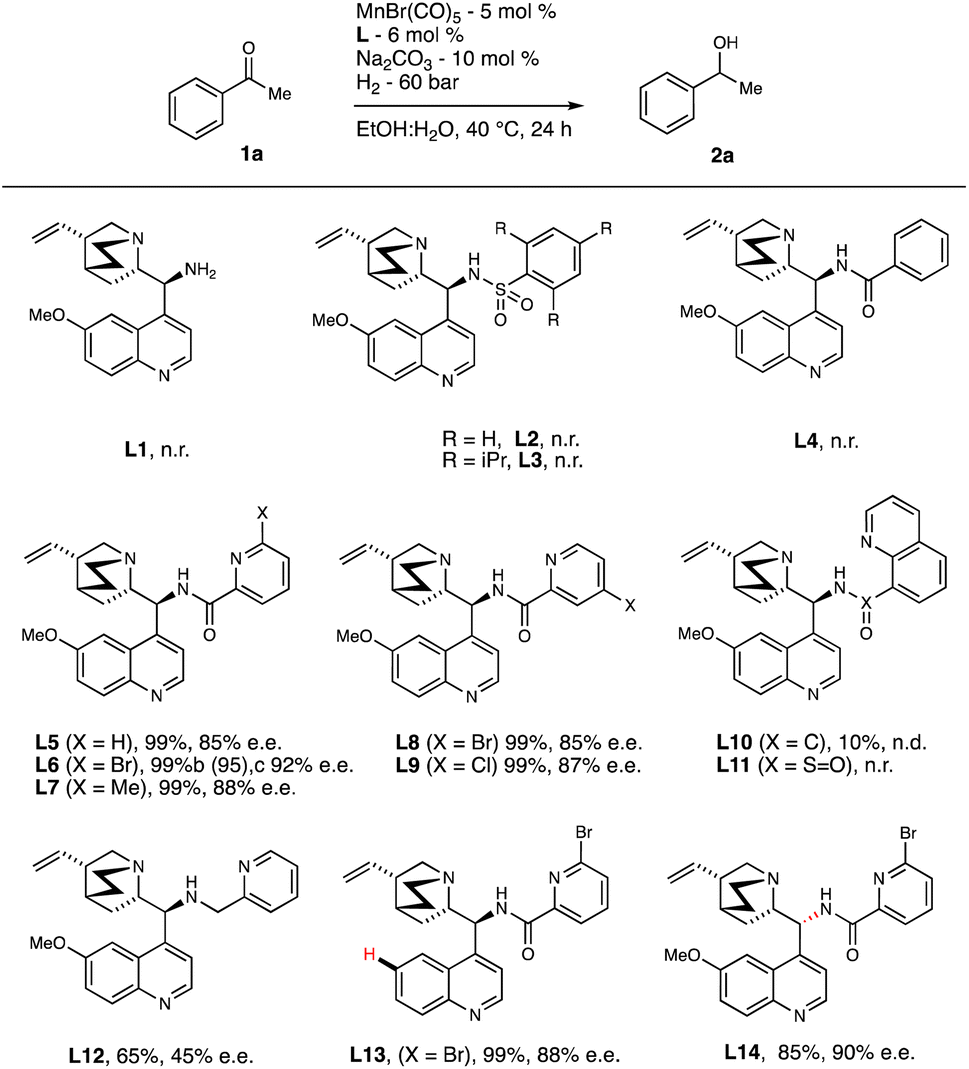
With the optimized reaction conditions in hand, the scope of ketones was explored (Scheme 1). Substrates with both electron-donating groups (OMe-, Me-, tBu-) and electron-withdrawing substituents (Br, Cl, Ph) at the 2-, 3-, and 4-positions on the aryl group were hydrogenated with excellent yields and ees (2b–2k, 86–>99% ee). The absolute configuration of 2f is further confirmed by X-ray.23 Change of the methyl group in 1a with ethyl, isopropyl did not much alter the outcome of the reaction (2l–2m, 92% yield, 95-96% ee) suggesting that steric hinderance at this position may not affect the selectivity. By keeping the ethyl group intact in 1m, further varying electronic parameters of the arene ring did enhance the ee up to 98% with excellent isolated yields of the corresponding secondary alcohols (2n–2q). Even indanone and tetralone underwent smooth reduction with excellent yield of the secondary alcohol although drop in ee was observed in the latter case (2r–2s). 1-acetyl and 2-acetyl naphthalene were examined under the optimized conditions to examine the influence of the peri-hydrogen in the former case for asymmetric hydrogenation. As expected, both 1t and 1u provided the excellent yields of the corresponding secondary alcohols (2t–2u) with 68% and 96% ees respectively.
 | ||
| Scheme 1 Scope of Ketones. Unless otherwise stated, all reactions were carried out under 60 bar hydrogen pressure using 1a (0.4 mmol), MnBr(CO)5 (5 mol%), L6 (6 mol%) and Na2CO3 (10 mol%) in H2O:EtOH mixture (4:1, 0.2 M) at 40°C for 24 h. | ||
Further extension of our catalytic asymmetric hydrogenation to acetyl benzodioxole provided the corresponding alcohol in 94% ee with 89% isolated yield (2v). To probe the potential influence of a trifluoro group next to the carbonyl carbon, we tested 2,2,2-trifluoro-1-phenylethane-1-one (1w) under the optimized conditions, which resulted in 86% ee and an isolated yield of 77%. Heterocycles such as 2-thiophene (1x) did neither hamper the catalytic activity nor ee significantly under the reductive conditions (88% yield, 88% ee). Interestingly, a useful functional group such as a hydroxyl at the alpha-position to the carbonyl group afforded chiral 1,2-diol 2y in 82% yield and 92% ee. This protocol is not only enantioselective but also chemo-selective, leaving other sensitive yet useful functional groups such as amine, cyano, nitro, esters, alkenes, and alkynes unaffected and afforded the secondary alcohols (2z and, 2aa–2af) in excellent yields (87-92%) with enantiomeric excess (ee'’s) between 90% and 99%. However, sterically unbiased substrates such as the aliphatic ketones did not give fruitful catalytic outcome and the provide poor-to-moderate ee'’s (2ag–2aj). Such chemoselectivity had not been observed previously, since [Mn-PNN] complexes are typically known to reduce both ester and nitrile groups, as shown in earlier work by Clarke and colleagues.13a,16b Finally, we examined the substrate/catalyst ratio and found that secondary alcohol 2u was obtained in 70% yield with 97% ee at a substrate-to-catalyst ratio of 2000, yielding a turnover number (TON) of 1400. Further optimization to reduce the catalyst loading and maximize its potential by heterogenizing the catalyst is currently underway.
We next turned our attention to examine the Lewis basic site of the ligand derived from cinchona alkaloid to determine whether it acts as a bi-dentate or tri-dentate ligand. Although, cinchona-based chiral picolinamide ligands were earlier coupled with copper21 for radical coupling reactions, iridium22a–d and ruthenium for asymmetric hydrogenation,22e the corresponding isolated complexes with cinchona ligand or its donor site is not readily known. While the hypothesized tri-dentate binding mode of cinchona ligands with transition metals is commonly proposed in asymmetric catalysis, we believe it is important to investigate these ligands binding mode with manganese in greater detail to have a better understanding of the in situ-generated catalyst.21,22a–c Therefore, it remains unclear at this stage whether L6 functions as a bi-dentate or tri-dentate ligand when it binds to manganese.
To investigate the coordination environment of manganese upon binding to L6, we focused on isolating the in situ-generated [Mncinchona] complex. After several attempts, the optimum conditions were established: combining L6 with MnBr(CO)5 in THF in a sealed tube at 90°C for 20 h resulted in the formation of a yellow precipitate (Scheme 2a). The solvent was evaporated, and the precipitate was then washed with diethyl ether, resulting in the isolation of the expected Mn-cinchona complex (1′) as yellow powder with the yield of 65%. The diffraction of the grown single crystal23 suggested a distorted octahedral geometry of the [Mn(I)cinchona] complex [Mn(I)(L6)Br(CO)3] (1′) (Scheme 2b). A closer look at the structure of the complex indicates that it is in neutral form and adopts an octahedral geometry. In complex 1′, the central metal is coordinated to the picolinamide (Py-CO-NHR) group of L6 in a bi-dentate mode, with three CO ligands arranged in a facial configuration. The bromide ion occupies the axial position. To gain a better understanding into the steric attributes of the isolated active [Mn] complex (1′), a three-dimensional steric effect of the ligand was quantitatively analyzed using the steric maps produced by SambVca 2.1 tool by calculating the % Vbur in total and four different regions around the metal center (Scheme 2b).24 Looking along the Z-axis, the calculation provide more information on how the ligand adapt its shape to the metal environment and the % Vbur in total (43.5%). A careful examination of the quadrants revealed that the isolated precatalyst incredibly buried volumes in three quadrants: NW (55.1%), SW (44.7%), and SE (48.9%), indicating that these are sterically crowded, except for the NE quadrant (25.4%), where the Br-bound to metal site is relatively unhindered. It is likely that the substrate approaches through the less-hindered quadrant (NE) as the preferred direction of approach to the transition metal site. Further confirmation of the new [Mn(Br)(L6)(CO)3] (1′) complex was characterized through spectroscopic techniques including NMR, IR and mass spectrometric characterization. Through IR spectra, the characteristic peak of the three carbonyl stretching frequencies appeared at 1905, 1928 and 2018 cm−1 confirmed the nature of the pi-acceptor CO ligands bound to the manganese. Subsequently, treatment of the isolated [Mn-L6] complex 1′ with 5 equivalents of tert-butoxide in THF at room temperature (25 °C) led to the formation of the active imido complex 1 within a few minutes. This was confirmed by a shift in the characteristic CO stretching frequencies, indicating the successful formation of the imido complex (Scheme 2c).
 | ||
| Scheme 2 (a) Synthesis of [Mn(I)Br(L6)(CO)3] (1′). (b) Solid-state structure of complex (1′) (hydrogen omitted for clarity) and steric map of the isolated Mn(I)cinchona complex. Total % Vbur by the ligand is 43.5%. % Vbur by each quadrants are 44.7 % (SW), 55.1% (NW), 25.4% (NE), 48.9% (SE). (c) IR spectra of active [Mn] complex 1′ (black) and the in situ-generated imido complex 1 (red) recorded in THF at 25°C. | ||
After confirming the structure of the precatalysts and identifying the donor sites from the cinchona ligand L6, we were motivated to investigate the scope of other chelate assisted ketone derivatives, as only two coordination sites of the [Mncinchona] complex are occupied by the ligand. This provided an opportunity to explore a range of functionalized ketone derivatives. Here, the motivation is to exploit the third donor site for substrate binding, which could as well be considered as a mimic of the [Mncinchona] pincer catalysts. In this regard, the efficiency of the [Mncinchona] catalytic system was explored for asymmetric hydrogenation of β-keto ester and γ-amino ketones under our optimized conditions (Scheme 4). It is worth considering that chiral β-hydroxy esters and γ-hydroxy amines are versatile synthons for natural product synthesis and other biologically active compounds.20b
Under the optimized conditions as used for ketones, the model substrate, methyl 3-oxo-3-phenylpropanoate 3a was subjected to asymmetric hydrogenation that resulted in producing the corresponding β-hydroxy ester 4a in 93% yield and 96% ee (Scheme 3). The electronic properties and substitution pattern of the arene ring demonstrate negligible impact on the reactivity and enantioselectivity (4b–4h, 85-93% yield, 92-98% ee). Subsequently, we questioned whether the same protocol can also be extended to the AH of γ-aminoketones into γ-amino alcohols. As evident from literature, the chiral γ-amino alcohols 4i are widely employed in asymmetric catalysis as they can act as chiral ligands, and also can serve as a key intermediate for many drugs such as (S)-duloxetine, (R)-fluoxetine, and (R)-atomoxetine.20b
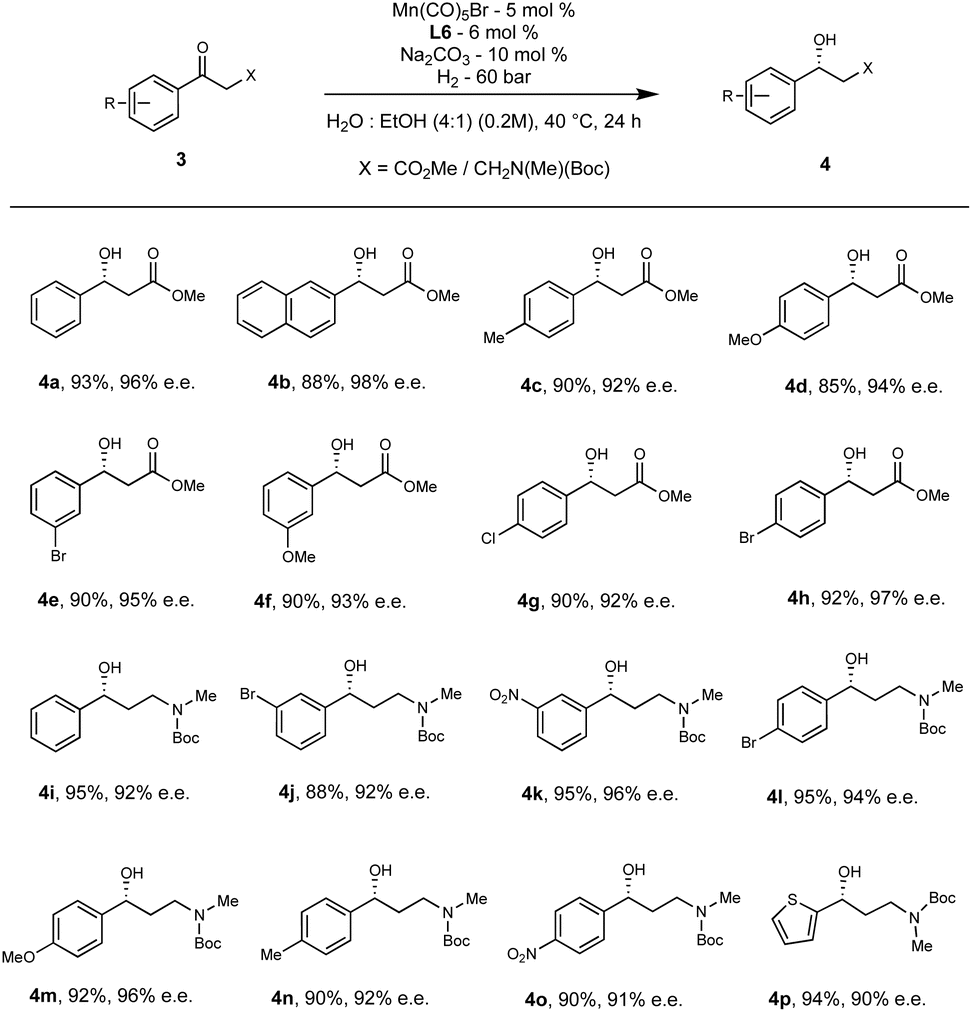 | ||
| Scheme 3 Scope of β-keto esters and β-amino ketones.aaReaction condition for γ-amino ketones remains the same except water used as exclusive solvent. | ||
Brief screening of the ligands and solvent revealed that L6 is still the best ligand and water is a used as exclusive solvent to achieve γ-amino alcohols 4i in 95% yield and 92% ee (Scheme 3). Various electron-withdrawing groups and electron-releasing groups at the meta- and para- positions were tolerated and are also available for further coupling reactions. The reduced γ-amino alcohols 4j–4o were isolated in excellent yields (88-95%) with ees ranging from 90 to 96%. We further demonstrate the utility of this methodology for the analogous intermediate of (R)-duloxetine (4p) in 94% yield with 90% ee. The absolute configuration of 4p is further confirmed by X-rays.23
Computational details
We have considered 4-acetylbiphenyl 2f as a representative ketone (as shown in Scheme 2) and L6 as the chiral ligand in our computational study aimed at investigating the mechanism and origin of enantioselectivity of the catalytic asymmetric hydrogenation reaction.25 The use of a polar protic solvent, such as water, in this reaction prompted us to consider explicit interaction between water and the catalyst as well as the substrates.26 It has been known that continuum solvation models are inadequate in capturing specific interactions between the solvent and solute.27We employ the SMD(water)/UB3LYP-D3/6-31G**,SDD(Mn,Br) level of theory28 to investigate several important aspects of this reaction, such as a) the identification of the most likely catalytic pathway on the basis of computed Gibbs free energies as well as by using the energetic span, b) the identification of the turn- over determining and the selectivity determining steps, c) probing the origin of enantioselectivity in the formation of the chiral alcohol as the product from the achiral ketone, and d) the role of water, ethanol, and sodium carbonate in the catalytic cycle by invoking their explicit participation in the relevant transition states.29
We begin by considering the formation of a Mn-imido complex 1 as shown in Scheme 4. The Mn-imido complex 1 can be obtained from the corresponding pre-catalyst 1′ [MnBr(L6)(CO)3], formed through ligand exchange between [MnBr(CO)5] and L6 by the action of Na2CO3.30 The overall reaction, 1′ + Na2CO3(H2O)6 → 1 + NaBr(H2O)3 + NaHCO3(H2O)3, is found to be exergonic with a Gibbs free energy change of -8.82 kcal/mol−1.31 To evaluate the energetic feasibility of the formation of 1, we have used a combined implicit/explicit solvation approach by systematically considering different binding sites for water molecules.26b,27a We have examined the role of water as a passive participant, engaging it with 1 through intermolecular hydrogen bonding interactions. By strategically positioning water molecules around 1, we could identify notable hydrogen bonding interactions at specific sites as shown in the inset in Scheme 4. The key sites of water binding are denoted as Oa of the carbonyl oxygen of the substrate, Ob of the methoxy group of 6-methoxyquinoline, Nc of 3-vinylquinuclidine, and Nd of 6-methoxyquinoline. The resulting lower energy active catalyst, designated as 1a with four explicitly bound water molecules, is found to be more likely species acting as the catalyst.32 While detailed discussions on each catalytic step are provided in the subsequent sections, we focus on the key events here. The three major mechanistic steps in the catalytic cycle (Scheme 4) are; (i) the dihydrogen activation and the formation of Mn-H intermediate, (ii) the hydride addition across the carbonyl C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O of the substrate leading to the desired alcohol, and (iii) regeneration of the active catalyst and the accompanying release of the chiral alcohol as the final product.33
O of the substrate leading to the desired alcohol, and (iii) regeneration of the active catalyst and the accompanying release of the chiral alcohol as the final product.33
 | ||
| Scheme 4 Catalytic cycle for the Mn-catalyzed asymmetric hydrogenation of 4-acetylbiphenyl by dihydrogen involving the explicit participation of water molecules. | ||
The catalytic cycle can be considered to begin with the coordination of H2 molecule with the active catalyst 1a to form the catalyst–dihydrogen complex, which facilitates hydrogen molecule activation. Among the passive and active modes of water binding considered here, the passive participation refers to water binding to the catalyst or substrate through intermolecular hydrogen bonding. Active participation, on the other hand, involves a more direct engagement of water by being part of the reaction coordinate, particularly in the hydrogen molecule activation step. The catalyst-dihydrogen complex 2 formed as a result of H2 uptake is found to be the most preferred intermediate in the active mode of water participation.34 Subsequently, H2 addition across the Mn-N bond through the transition state [2-3]‡ leads to a Mn-H intermediate 3. In the most preferred mode of hydrogen molecule activation, the explicit water molecule (shown in blue color in Fig. 1) plays two key roles. First, it engages passively through a hydrogen bonding with the imido nitrogen of the catalyst (in 2) and subsequently promotes an active relay transfer.35
 | ||
| Fig. 1 Optimized geometry and corresponding relative Gibbs free energies in parentheses (kcal/mol−1) of the hydrogen molecule activation transition state [2-3]‡. Labels a, b, c, and d represent specific water binding sites. Distances are in Å. Only selected hydrogen atoms are shown for improved clarity. | ||
This mode of participation of water molecule resembles a relay proton transfer often invoked in reaction mechanisms in protic solvents.26,27a,b,36 The relative Gibbs free energy of the H2 activation through [2-3]‡ is found to be 12.3 kcal/mol−1 (Fig. 2).37 Next, the uptake of 4-acetylbiphenyl by the ensuing 3 intermediate can lead to a catalyst-substrate complex 4, wherein the keto group is positioned closer to the Mn-H moiety of the catalyst. Here, the binding of a molecule of water to the carbonyl oxygen (Oe) is found to stabilize intermediate 4. The hydride addition step is found to involve a concerted asynchronous transfer of the Mn-hydride to the carbonyl carbon and the amido proton to the carbonyl oxygen through TS [4-5]‡.13d,14,26b,38 The resulting species 5 is a product bound to the catalyst as shown in Scheme 4. It shall be noted that the stereoinducing hydride addition to the ketone can take place either through the si and the re prochiral face. To locate the energetically most favorable hydride addition transition state, we have considered various conformational possibilities as well as different water binding modes.39 One of the most interesting findings is the critical role played by the explicit water molecule in the stereocontrolling TS [4-5]‡. The inclusion of as many as five explicit water molecules renders the TS energetically more favorable by about 20 to 25 kcal/mol−1 as compared to the unassisted pathway devoid of any water molecules.40 Moreover, the pattern of various noncovalent interactions (NCIs) in the diastereomeric TSs [4-5]si‡ and [4-5]re‡ is found to be different depending on the prochiral face of the ketone involved in the hydride addition (vide infra).
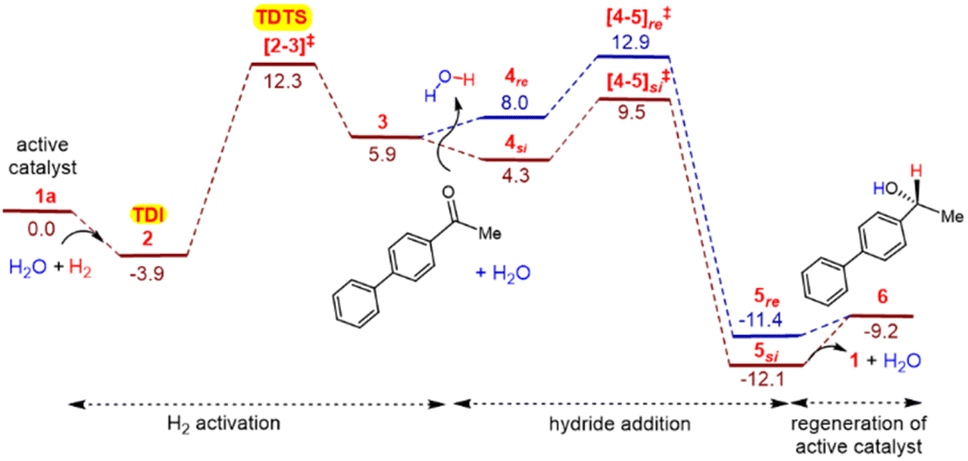 | ||
| Fig. 2 Gibbs free energy profile (in kcal/mol−1) for the catalytic asymmetric hydrogenation of 4-acetylbiphenyl under the explicit participation of five water molecules, as obtained at the SMD(water)/UB3LYP-D3/6-31G**,SDD(Mn,Br) level of theory. The relative Gibbs free energies of intermediates and transition states are calculated with respect to 1a, which is used as the common reference point. The blue and brown lines respectively denote the S-alcohol and R-alcohol product formation. | ||
A perusal of the Gibbs energy profile as provided in Fig. 2 conveys couple of very important aspects of the catalytic cycle. First, the identity of the turn-over determining TS and next is the mechanistic step where the enantioselectivity is determined, which is the hydride addition to the substrate. The application of the energetic span model41 on the Gibbs free energy profile revealed that the catalyst-dihydrogen complex 2 is the turn-over determining intermediate (TDI) and H2 activation transition state [2-3]‡ is the turn-over determining transition state (TDTS) of the catalytic cycle. The energetic span (δE), calculated as the difference in energy between the TDTS and TDI, is 16.2 kcal/mol−1 at 313.15 K.42
The δE value is suggestive of an effective catalytic transformation at 40°C, which is in conformity with the reaction conditions we employed. As far as the enantioselectivity of the reaction is concerned, it is evident that the transition state for the hydride addition to the si prochiral face of 4-acetylbiphenyl [4-5]si‡ is 3.4 kcal/mol−1 lower than [4-5]re‡ for the re face addition. Such an energetic difference between the diastereomeric transition states implies a kinetic advantage for the formation of product 6 with the R configuration at the newly formed stereogenic center.43 This energetic preference is consistent with the high ee of 99% obtained in our experiments.44
After having predicted high enantioselectivity, we sought additional insights into the molecular origin of the energy difference between the stereo controlling hydride addition transition states [4-5]si‡ and [4-5]re‡. In other words, we wish to answer a critical question as to what stereoelectronic factors contribute in rendering [4-5]si‡ lower energy and make it the most preferred mode for the hydride addition. Thus, a careful analysis of the nature and effectiveness of weak noncovalent interactions (NCIs) between the chiral catalysts and reaction partners is undertaken by applying the atoms-in-molecule (AIM) formalism, wherein we focus on the electron densities at the relevant bond critical points (ρbcp).45 The variations in the pattern and efficacy of the NCIs in [4-5]si‡ and [4-5]re‡, as discussed below, are found to be valuable toward understanding the energy difference between them.46 It should be noted the NCIs that are identified between the chiral catalyst and substrate are only shown in Fig. 3,47a whereas a more detailed mapping, including NCIs within the catalyst and those in the substrate, is provided in the Supporting Information.†47b–e Similar NCIs found in both [4-5]si‡ and [4-5]re‡ are assigned same alphabetical notation wherever possible. The important hydrogen bonding as well as other types of NCIs between the catalyst and substrate are identified in these TSs.48 These include CH…π, CH…O, CH…Br, CH…N, and CH…HC interactions as shown in Fig. 3.49 Among these NCIs, we focus on those which are unique to a given TS or those whose strength of interaction is different between the diastereomeric TSs. The CH…HC interactions denoted as a3, a4, and a5 in [4-5]si‡ and a6 and a7 in [4-5]re‡, show similar strengths in both transition states. Other NCIs which are closely similar, such as CH…O (b4 and b5), (l.p)O…π (e1) are not explicitly discussed here, albeit we considered them in estimating the cumulative strength of all the NCIs.
 | ||
| Fig. 3 Optimized transition state geometries for the enantioselective hydride addition: (a). [4-5]re‡ (12.9); (b). [4-5]si‡ (9.5) and showing the important NCIs. Important interatomic distances (in Å) and the corresponding electron densities (ρ×10−2) at the bond critical points are shown in parentheses. The relative Gibbs free energies (in kcal/mol−1) of the TSs computed with respect to 1a are given in parenthesis. Hydrogen atoms not involved in significant interactions are omitted for improved clarity. | ||
Differential NCIs include a CH…O interaction (b8) between the CO ligand of the chiral catalyst and the aryl CH of the substrate in [4-5]si‡, which are absent in the higher energy [4-5]re‡. Additional CH…π interactions (d3 and d4) are found between the methyl of the substrate and the catalyst in the lower energy [4-5]si‡ than those in the higher energy [4-5]re‡ where only one CH…π interaction (d6) is identified. Specifically, two CH…π interactions are noted in [4-5]si‡, one between the methyl group of the ketone and the vinyl moiety of the quinuclidine from the chiral catalyst, and the other between the pyridine group of the Mn-bound chiral catalyst and the biphenyl of the ketone. Distinct NCIs such as a π…π interaction (f1), a CH…Br (g1), and π…Br (h1)50 are noticed in the lower energy [4-5]si‡. In contrast, a CH…N (i1) is unique to the higher energy [4-5]re‡. The most important NCIs in both the TSs include the intermolecular hydrogen bonding interactions, comprising of NH…O (j1, j2) and HO…H (k1, k2, k3, and k4), as well as OH…O (c1, c2, and c3) interactions. Although these NCIs are qualitatively similar and common to both the higher and lower energy transition states, the efficiency of many of the individual interactions is found to be superior in the lower energy [4-5]si‡, as indicated by the corresponding ρbcp values. However, the total number of NCIs is found to be more in the case of the lower energy [4-5]si‡.
Furthermore, the cumulative strength of such NCIs in the most favorable [4-5]si‡ is 4.8 kcal/mol−1 higher as compared to the higher energy [4-5]re‡. The above analysis indicates that the strength of the NCIs also has a notable impact on the overall energy difference between the enantio-controlling TSs. In addition to the differential contribution of the NCI as described above to the energy difference between [4-5]si‡ and [4-5]re‡, we have examined the effect of geometric distortion in the enantio-controlling hydride addition TSs using the activation strain analysis.51,52 We have probed the effect of distortion and interaction between the reaction partners as they move from their respective ground state geometries to that found in the corresponding TS. The origin of chiral induction in the hydride addition step could be better understood based on the relative distortion and interaction. The total distortion energy of the reacting partners is found to be 2.3 kcal/mol−1 lower in the lower energy [4-5]si‡ than in [4-5]re‡. The stabilizing interaction energy between the distorted fragments is however found to be by 1.8 kcal/mol−1 in the lower energy [4-5]si‡. The overall stabilization is still in favor of the lower energy [4-5]si‡. The presence of more effective interactions between the 4-acetylbiphenyl as a substrate and the chiral catalyst can, therefore, be regarded as the major contributing factor rendering the hydride addition to the si prochiral face more preferred over that to the re face of the 4-acetylbiphenyl. In summary, the computed energy difference between the competing diastereomeric TSs can be considered as originating from the differences in the NCIs, distortion and interactions present in them.
Conclusions
We have developed a highly efficient [Mncinchona] chiral catalytic system for the asymmetric hydrogenation of ketones, which exhibits a broad substrate scope and remarkable chemoselectivity. The catalyst selectively reduces ketones while leaving other reducible functional groups, such as olefins, alkynes, nitriles, nitro groups, and esters, unaffected. Notably, nitrile and ester groups that are more prone to reduction under previously reported catalysts. The isolation and characterization of the [Mncinchona] complex 1′ has provided valuable insights into the coordination environment, facilitating a deeper understanding of the reaction mechanism. This, combined with DFT calculations, supports the proposal that hydrogen molecule activation assisted by water molecule forms six-membered transition state which is found to be the turnover-determining transition state, with enantioselectivity arising from preferential hydride transfer to the si face of the prochiral substrate, leading to the formation of the ‘R’ configuration. This newly developed [Mncinchona] catalytic system holds significant potential for broader applications in asymmetric hydrogenation, including the reduction of ketoesters, ketoamines, and amides. Given the versatility and efficiency of this system, we anticipate continued advancements in this dynamic field, further expanding the scope of asymmetric hydrogenation for challenging substrates.Data availability
The data supporting the findings of this study are included in the paper or the Supplementary Information† and are also available upon request from the corresponding author. Crystallographic data for the structures discussed in this article have been submitted to the Cambridge Crystallographic Data Center, Experimental procedures, computational details, and additional DFT results including energy data, and cartesian coordinates for all optimized systems. CCDC 2413245 [MnBr(L6)(CO)3] (1′), CCDC 2413299 (2f) and CCDC 2413301 (4l) contains the supplementary crystallographic data (PDF).Author contributions
B. S. and S. P. conceived the concept. S. P. and D. A. performed all the reaction and analyzed the product. N. J. performed the DFT calculations. R.B.S. supervised the DFT calculations. The manuscript is written through the contributions of all authors.Conflicts of interest
The authors declare a competing financial interest. B.S., S.P and are named as inventors on a patent application filed by the Indian Institute of Technology Kanpur based on the work described in this manuscript; patent granted number 558458. The authors declare no other financial or non-financial competing interests.Acknowledgements
The author gratefully acknowledged SERB (CRG/2020/001282) and PMRF (SP & NJ) for funding this research. The assistance rendered by Dipanti Borah in solving the crystal structure of 1′ is gratefully acknowledged. NJ and RBS thank the generous computing time in the SpaceTime supercomputing facility of IIT Bombay.Notes and references
- (a) The Handbook of Homogeneous Hydrogenation, ed C. J. Elsevier and J. G. De Vries, Wiley-VCH, Weinheim, 2007 Search PubMed; (b) T. Ohkuma, S. Hashaguchi, T. Ikariya and T. Noyori, J. Am. Chem. Soc., 1995, 117, 2675 CrossRef CAS; (c) R. Noyori and T. Ohkuma, Angew. Chem., Int. Ed., 2001, 40, 40 ( Angew. Chem. , 2001 , 113 , 40 ) CrossRef CAS; (d) T. Ohkuma, N. Utsumi, K. Tsutsumi, K. Murata, C. Sandoval and R. Noyori, J. Am. Chem. Soc., 2006, 128, 8724 CrossRef CAS PubMed.
- W. S. Knowles and M. J. Sabacky, Chem. Commun., 1968, 1445–1446 RSC.
- L. Horner, H. Siegel and H. Buthe, Angew Chem. Int. Ed. Engl., 1968, 7, 942 ( Angew. Chem. , 1968 , 80 , 1034–1035 ) CrossRef CAS.
- The Nobel Prize in Chemistry, 2001, https://www.nobelprize.org/prizes/chemistry/2001/summary/.
- J. H. Xie, X.-Y. Liu, J.-B. Xie, L.-X. Wang and Q.-L. Zhou, Angew. Chem., Int. Ed., 2011, 50, 7329–7332 CrossRef CAS PubMed.
- Metal contamination limits in pharmaceutical compounds are 250 ppm (e.g. Mn) compared to 10 ppm for [Ru].
- (a) Z. Zhang, N. A. Butt, M. Zhou, D. Liu and W. Zhang, Chin. J. Chem., 2018, 36, 443–454 CrossRef CAS; (b) J. Wen, F. Wang and X. Zhang, Chem. Soc. Rev., 2021, 50, 3211–3237 RSC; (c) P. Liu and Z. Lu, Synthesis, 2023, 55, 1042–1052 CrossRef.
- (a) W. Zuo, A. Lough, Y. F. Li and R. H. Morris, Science, 2013, 342, 1080–1083 CrossRef CAS PubMed; (b) P. O. Lagaditis, P. E. Sues, J. F. Sonnenberg, K. Y. Wan, A. J. Lough and R. H. Morris, J. Am. Chem. Soc., 2014, 136, 1367–1380 CrossRef CAS PubMed; (c) Y. Y. Li, S. L. Yu, X. F. Wu, J. L. Xiao, W. Y. Shen, R. Z. Dong and J. X. Gao, J. Am. Chem. Soc., 2014, 136, 4031–4039 CrossRef CAS PubMed; (d) P. Gajewski, M. Renom-Carrasco, S. V. Facchini, L. Pignataro, L. Lefort, J. G. De Vries, R. Ferraccioli, A. Forni, U. Piarulli and C. Gennari, Eur. J. Org Chem., 2015, 1887 CrossRef CAS; (e) R. Hodgkinson, A. Del Grosso, G. J. Clarkson and M. Wills, Dalton Trans., 2016, 45, 3992 RSC.
- (a) M. R. Friedfeld, M. Shelvin, J. M. Hoyt, S. W. Krska, M. T. Tudge and P. J. Chirik, Science, 2013, 342, 1076 CrossRef CAS PubMed; (b) M. R. Friedfeld, G. W. Margulieux, B. Schaefer and P. J. Chirik, J. Am. Chem. Soc., 2014, 136, 13178 CrossRef CAS PubMed; (c) P. J. Chirik, Acc. Chem. Res., 2015, 48, 1687 CrossRef CAS PubMed; (d) D. Zhang, E.-Z. Zhu, Z.-W. Lin, Y.-Y. Li and J.-X. Gao, Asian J. Org. Chem., 2016, 5, 1323 CrossRef CAS; (e) M. R. Friedfeld, M. Shelvin, G. W. Margulieux, L. C. Campeau and P. J. Chirik, J. Am. Chem. Soc., 2016, 138, 3314 CrossRef CAS PubMed; (f) J. Chen, C. Chen, C. Ji and Z. Lu, Org. Lett., 2016, 18, 1594 CrossRef CAS PubMed; (g) M. R. Friedfeld, H. Zhong, R. T. Ruck, M. Shelvin and P. J. Chirik, Science, 2018, 360, 888 CrossRef CAS PubMed; (h) T. Chen, Y. Zou, Y. Hu, Z. Zheng, H. Wei, L. Wei and W. Zhang, Angew. Chem., Int. Ed., 2023, 62, e202303488 CrossRef CAS PubMed; (i) S. Chakrabortty, K. Konieczny, F. J. De Zwart, E. O. Bobylev, E. Baráth, S. Tin, B. H. Müller, J. N. H. Reek, B. De Bruin and J. G. De Vries, Angew. Chem., Int. Ed., 2023, 62, e202301329 CrossRef CAS PubMed; (j) S. Chakrabortty, B. De Bruin and J. G. De Vries, Angew. Chem., Int. Ed., 2023, 63, e202315773 CrossRef PubMed.
- (a) J. A. Fuentes, S. M. Smith, M. T. Scharbert, I. Carpenter, D. B. Cordes, A. M. Z. Slawin and M. L. Clarke, Chem.–Eur. J., 2015, 21, 10851 CrossRef CAS PubMed; (b) M. L. Clarke, Synlett, 2014, 25, 1371 CrossRef; (c) H. Nie, G. Zhou, Q. Wang, W. Chen and S. Zhang, Tetrahedron: Asymmetry, 2013, 24, 1567 CrossRef CAS; (d) K. N. Manar, J. Cheng, Y. Yang, X. Yang and P. Ren, ChemCatChem, 2023, 15, e202300004 CrossRef CAS.
- (a) F. Kallmeier and R. Kempe, Angew. Chem., Int. Ed., 2018, 57, 46 ( Angew. Chem. , 2018 , 130 , 48 ) CrossRef CAS PubMed; (b) G. A. Filonenko, G. A. Van Putten, E. J. M. R. Hensen and E. A. Pidko, Chem. Soc. Rev., 2018, 47, 1459 RSC; (c) M. Garbe, K. Junge and M. Beller, Eur. J. Org Chem., 2017, 4344 CrossRef CAS; (d) B. Maji and M. K. Barman, Synthesis, 2017, 49, 3377 CrossRef CAS.
- A. Zirakzadeh, S. R. M. M. de Aguiar, B. Stöger, M. Widhalm and K. Kirchner, ChemCatChem, 2017, 9, 1744–1748 CrossRef CAS.
- (a) M. B. Widegren, G. J. Harkness, A. M. Z. Slawin, D. B. Cordes and M. L. Clarke, Angew. Chem., Int. Ed., 2017, 56, 5825 ( Angew. Chem. , 2017 , 129 , 5919 ) CrossRef CAS PubMed; (b) M. B. Widegren and M. L. Clarke, Org. Lett., 2018, 20, 2654 CrossRef CAS PubMed; (c) M. B. Widegren and M. L. Clarke, Catal. Sci. Technol., 2019, 9, 6047 RSC; (d) C. L. Oates, A. S. Goodfellow, M. Bühl and M. L. Clarke, Angew. Chem., Int. Ed., 2023, 62, e202212479 CrossRef CAS PubMed.
- M. Garbe, K. Junge, S. Walker, Z. Wei, H. Jiao, A. Spannerberg, S. Bachmann, M. Scalone and M. Beller, Angew. Chem., Int. Ed., 2017, 56, 11237 ( Angew. Chem. , 2017 , 129 , 11389 ) CrossRef CAS PubMed.
- (a) K. Z. Demmans, M. E. Olson and R. H. Morris, Organometallics, 2018, 37, 4608–4618 CrossRef CAS; (b) C. S. G. Seo, B. T. H. Tsui, M. V. Gradiski, S. A. M. Smith and R. H. Morris, Catal. Sci. Technol., 2021, 11, 3153–3163 RSC.
- (a) C. Liu, M. Wang, Y. Wang, Y. Peng, Y. Lan and Q. Liu, Angew. Chem., Int. Ed., 2021, 60, 5108 ( Angew. Chem. , 2021 , 133 , 5168 ) CrossRef CAS PubMed; (b) C. L. Oates, M. B. Widegren and M. L. Clarke, Chem. Commun., 2020, 56, 8635 RSC; (c) J. M. Pérez, R. Postolache, M. Castiñeira, R. E. G. Sinnema, D. Vargová, F. de Vries, E. Otten, L. Ge and S. R. Harutyunyan, J. Am. Chem. Soc., 2021, 143, 20071 CrossRef PubMed; (d) F. Ling, H. Hou, J. Chen, S. Nian, X. Yi, Z. Wang, D. Song and W. Zhong, Org. Lett., 2019, 21, 3937 CrossRef CAS PubMed; (e) C. Liu, M. Wang, Y. Xu, Y. Li and Q. Liu, Angew. Chem., Int. Ed., 2022, 61, e202202814 ( Angew. Chem. , 2022 , 134 , e202202814 ) CrossRef CAS PubMed; (f) Z. Wang, X. Zhao, A. Huang, Z. Yang, Y. Cheng, J. Chen, F. Ling and W. Zhong, Tetrahedron Lett., 2021, 82, 153389 CrossRef CAS.
- L. Zhang, Y. Tang, Z. Han and K. Ding, Angew. Chem., Int. Ed., 2019, 58, 4973–4977 CrossRef CAS PubMed.
- (a) K. Z. Demmans, M. E. Olson and R. H. Morris, Organometallics, 2018, 37, 4608–4618 CrossRef CAS; (b) R. van Putten, G. A. Filonenko, A. Gonzalez de Castro, C. Liu, M. Weber, C. Müller, L. Lefort and E. Pidko, Organometallics, 2019, 38, 3187–3196 CrossRef CAS PubMed; (c) J. Schneekönig, K. Junge and M. Beller, Synlett, 2019, 30, 503–507 CrossRef; (d) K. Azouzi, A. Bruneau-Voisine, L. Vendiera, J.-B. Sortais and S. Bastin, Catal. Commun., 2020, 142, 106040 CrossRef CAS; (e) L. Wang, J. Lin, Q. Sun, C. Xia and W. Sun, ACS Catal., 2021, 11, 8033–8041 CrossRef CAS; (f) Y. Su, Z. Ma, J. Wang, L. Li, X. Yan, N. Ma, Q. Liu, G. Solan and G. A. Z. Wang, J. Org. Chem., 2024, 89, 12318–12325 CrossRef CAS PubMed; (g) Q. Sun, B. Wang, X. Meng and W. Sun, J. Catal., 2024, 106, 115680 Search PubMed; (h) J. Yang, L. Yao, Z. Wang, Z. Zuo, S. Liu, P. Gao, M. Han, Q. Liu, G. A. Solan and W.-H. Sun, J. Catal., 2023, 418, 40 CrossRef CAS . For related reviews, see:; (i) D. Fu, Z. Wang, Q. Liu, S. J. Prettyman, G. A. Solan and W.-H. Sun, ChemCatChem, 2024, 16, e202301567 CrossRef CAS; (j) K. Azouzi, D. A. Valyaev, S. Bastin and J.-B. Sortais, Curr. Opin. Green Sustainable Chem., 2021, 31, 100511 CrossRef CAS.
- During the preparation of this manuscript, Mn-transfer hydrogenase based on the biotin–streptavidin technology was appeared, see: (a) W. Wang, R. Tachibana, Z. Zou, D. Chen, X. Zhang, K. Lau, F. Pojer, T. R. Ward and X. Hu, Angew. Chem., Int. Ed., 2023, 62, e202311896 CrossRef CAS PubMed; (b) L. Zhang, P. Dai, X. Meng, Y. Tian, X. Zhou, W. Gou, Y. Wang and C. Li, J. Catal., 2024, 439, 115758 CrossRef CAS; (c) S. Zhang, Z. Ma, Y. Li, Y. Su, N. Ma, X. Guo, L. Li, Q. Liu and Z. Wang, J. Catal., 2024, 437, 115682 CrossRef CAS.
- (a) J. Magano and J. R. Dunetz, Org. Process Res. Dev., 2012, 16, 1156–1184 CrossRef CAS; (b) C. Liu, L. Cao, L. Zhang, Y. Xiong, Y. Ma, R. Cheng and J. Ye, Commun. Chem., 2022, 5, 63 CrossRef CAS PubMed.
- (a) F. Sladojevich, A. Trabocchi, A. Guarna and D. J. Dixon, J. Am. Chem. Soc., 2011, 133, 1710–1713 CrossRef CAS PubMed; (b) M. Hayashi, N. Shiomi, Y. Funahashi and S. Nakamura, J. Am. Chem. Soc., 2012, 134, 19366–19369 CrossRef CAS PubMed; (c) X.-Y. Dong, Y.-F. Zhang, C.-L. Ma, Q.-S. Gu, F.-L. Wang, Z.-L. Li, S.-P. Jiang and X.-Y. Liu, Nat. Chem., 2019, 11, 1158–1166 CrossRef CAS PubMed; (d) S.-P. Jiang, X.-Y. Dong, Q.-S. Gu, L. Ye, Z.-Li. Li and X.-Y. Liu, J. Am. Chem. Soc., 2020, 142, 19652–19659 CrossRef CAS PubMed; (e) X.-L. Su, L. Ye, J.-J. Chen, X.-D. Liu, S.-P. Jiang, F.-L. Wang, L. Liu, C.-J. Yang, X.-Y. Chang, Z.-L. Li, Q.-S. Gu and X.-Y. Liu, Angew. Chem., Int. Ed., 2021, 60, 380–384 CrossRef CAS PubMed; (f) Y.-F. Zhang, X.-Y. Dong, J.-T. Cheng, N.-Y. Yang, L.-L. Wang, F.-L. Wang, C. Luan, J. Liu, Z. L. Li, Q.-S. Gu and X.-Y. Liu, J. Am. Chem. Soc., 2021, 143, 15413–15419 CrossRef CAS PubMed; (g) H. Zhou, Z.-L. Li, Q.-S. Gu and X.-Y. Liu, ACS Catal., 2021, 11, 7978–7986 CrossRef CAS; (h) P.-F. Wang, J. Yu, K.-X. Guo, S.-P. Jiang, J.-J. Chen, Q.-S. Gu, J.-R. Liu, X. Hong, Z.-L. Li and X.-Y. Liu, J. Am. Chem. Soc., 2022, 144, 6442–6452 CrossRef CAS PubMed; (i) X.-Y. Cheng, Y.-F. Zhang, J.-H. Wang, Q.-S. Gu, Z.-L. Li and X.-Y. Liu, J. Am. Chem. Soc., 2022, 144, 18081–18089 CrossRef CAS PubMed.
- (a) L. Zhang, L. Zhang, Q. Chen, L. Li, J. Jiang, H. Sun, C. Zhao, Y. Yang and C. Li, Org. Lett., 2022, 24, 415–419 CrossRef CAS PubMed; (b) L. Li, N. Ma, Q. Chen, H. Sun, J. Tian, Q. Xu, C. Li and L. Zhang, Org. Biomol. Chem., 2022, 20, 7936–7941 RSC; (c) Q. Chen, H. Sun, L. Li, J. Tian, Q. Xu, N. Ma, L. Li, L. Zhang and C. Li, J. Org. Chem., 2022, 87, 15986–15997 CrossRef CAS PubMed; (d) J. Tian, X. Meng, H. Sun, Q. Chen, Q. Xu, P. Dai, L. Li, L. Zhang and C. Li, J. Org. Chem., 2023, 88, 9213–9224 CrossRef CAS PubMed; (e) L. Zhang, Q. Chen, L. Li, J. Jiang, H. Sun, L. Li, T. Liu, L. Zhang and C. Li, RSC Adv., 2022, 12, 14912 RSC.
- CCDC 2413245 [MnBr(L6)(CO3)] (1′), CCDC 2413299 (2f) and CCDC 2413301 (4l) contain the supplementary crystallographic data for this paper.
- (a) L. Falivene, R. Credendino, A. Poater, A. Petta, L. Serra, R. Oliva, V. Scarano and L. Cavallo, Organometallics, 2016, 35, 2286–2293 CrossRef CAS; (b) L. Falivene, Z. Cao, A. Petta, L. Serra, A. Poater, R. Oliva, V. Scarano and L. Cavallo, Nat. Chem., 2019, 11, 872–879 CrossRef CAS PubMed.
- A. Sengupta and R. B. Sunoj, J. Org. Chem., 2012, 77, 10525–10536 CrossRef CAS PubMed.
- (a) Z. Liu, C. Patel, J. N. Harvey and R. B. Sunoj, Phys. Chem. Chem. Phys., 2017, 19, 30647 RSC; (b) M. Anand and R. B. Sunoj, Organometallics, 2012, 31, 6466–6481 CrossRef CAS; (c) D. Roy and R. B. Sunoj, Chem.–Eur. J., 2008, 14, 10530–10534 CrossRef CAS PubMed; (d) D. Roy, C. Patel and R. B. Sunoj, J. Org. Chem., 2009, 74, 6936–6943 CrossRef CAS PubMed; (e) S. Bag, K. Surya, A. Mondal, R. Jayarajan, U. Dutta, S. Porey, R. B. Sunoj and D. Maiti, J. Am. Chem. Soc., 2020, 142, 12453–12466 CrossRef CAS PubMed; (f) Y.-Q. Chen, S. Singh, Y. Wu, Z. Wang, W. Hao, P. Verma, J. X. Qiao, R. B. Sunoj and J.-Q. Yu, J. Am. Chem. Soc., 2020, 142, 9966–9974 CrossRef CAS PubMed.
- (a) M. Anand and R. B. Sunoj, Org. Lett., 2011, 13, 4802–4805 CrossRef CAS PubMed; (b) M. P. Patil and R. B. Sunoj, J. Org. Chem., 2007, 72, 8202–8215 CrossRef CAS PubMed; (c) M. P. Patil and R. B. Sunoj, Chem.–Eur. J., 2008, 14, 10472–10485 CrossRef CAS PubMed; (d) M. Cortes-Clerget, J. Yu, J. R. A. Kincaid, P. Walde, F. Gallou and B. H. Lipshutz, Chem. Sci., 2021, 12, 4237–4266 RSC.
- (a) Full computational details are provided in Section 8.1 in the ESI†.; (b) The Gibbs free energies of important stationary points are provided in Table S16 in the ESI†.; (c) There have been several reports on the use of the UB3LYP-D3 function for studying transition metal catalyzed reactions, see:; (d) E. F. Gérard, V. Yadav, D. P. Goldberg and S. P. de Visser, J. Am. Chem. Soc., 2022, 144, 10752–10767 CrossRef PubMed; (e) C.-C. G. Yeh, S. Ghafoor, J. K. Satpathy, T. Mokkawes, C. V. Sastri and S. P. de Visser, ACS Catal., 2022, 12, 3923–3937 CrossRef CAS; (f) S. K. Das, S. Das, S. Ghosh, S. Roy, M. Pareek, B. Roy, R. B. Sunoj and B. Chattopadhyay, Chem. Sci., 2022, 13, 11817–11828 RSC; (g) S. T. Schneebeli, M. L. Hall, R. Breslow and R. Friesner, J. Am. Chem. Soc., 2009, 131, 3965–3973 CrossRef CAS PubMed; (h) S. Roy, D. A. Vargas, P. Ma, A. Sengupta, L. Zhu, K. N. Houk and R. Fasan, Nat. Catal., 2024, 7, 65–76 CrossRef CAS PubMed.
- (a) M. Das, A. R. Gogoi and R. B. Sunoj, J. Org. Chem., 2022, 87, 1630–1640 CrossRef CAS PubMed; (b) R. B. Sunoj and M. Anand, Phys. Chem. Chem. Phys., 2012, 14, 12715 RSC.
- (a) See Scheme S1 in the ESI† for details of precatalyst formation.; (b) The details of the conformational analysis of the precatalyst complex are provided in Table S1 in the ESI.† The computed endoergic of 18.89 kcal mol−1 for the formation of the precatalyst via the coordination of MnBr(CO)5 with L6 was concomitant with the simultaneous elimination of two CO molecules.; (c) An overlaid image of the computed and crystallographic structures is shown in Fig. S1 in the ESI.† The overall root mean square deviation (rmsd) for all the atomic positions between these two structures is found to be 0.52 Å, suggesting an excellent agreement between the computed and the experimental geometries..
- The details for the formation of a potential Mn-imido complex from the pre-catalyst are provided in Scheme S2 in the ESI.† The Gibbs free energies of such salt-solvent clusters in the solvent continuum are evaluated using the SMD continuum solvation model to arrive at an optimum number of water molecules likely to be present in the first solvation shell (Fig. S2†). The optimized geometries of the lower energy water coordinated ionic salts such as Na2CO3(H2O)6, NaBr(H2O)3, and NaHCO3(H2O)3 are shown in Fig. S3 in the ESI†.
- (a) For additional details of how we could identify the binding of four water molecules as the most likely scenario, see Table S2 in the ESI,† where additional possibilities of active catalysts with varying numbers of explicit water molecules are presented.; (b) The optimized geometries of a lower energy active catalyst are shown in Fig. S4 in the ESI†..
- See Scheme S3 in the ESI† for mechanistic details of Mn-catalyzed asymmetric hydrogenation of acetylbiphenyl under an unassisted pathway.
- (a) The different possibilities considered for water binding modes in the hydrogen molecule activation TS [2-3]w‡ are provided in Tables S3–S5 in the ESI†.; (b) The hydrogen molecule activation transition state [2-3]wabcd‡ with passive water participation is found to be 4.5 kcal mol−1 higher in energy as compared to [2-3]‡ involving active water participation..
- The different possibilities considered for water binding modes in the hydrogen molecule activation TS [2-3]w‡ are provided in Tables S3–S5 in the ESI†.
- L. Chen and D. E. Woon, J. Phys. Chem. A, 2011, 115, 5166–5183 CrossRef CAS.
- The optimized geometries of alternative H2 activation transition states ([2-3]w‡, [2′-3′]w‡, and [2′′-3′′]w‡) with different modes of water binding are provided in Fig. S5–S7 in the ESI†.
- (a) Both stepwise and concerted mechanisms have been considered for the hydride addition step. The stepwise mechanism, wherein the Mn-hydride and amido proton transfer occur sequentially, is proposed in related systems. See; (b) Y. Wang, S. Liu, H. Yang, H. Li, Y. Lan and Q. Liu, Nat. Chem., 2022, 14, 1233–1241 CrossRef CAS; (c) C. Liu, X. Liu and Q. Liu, Chem, 2023, 9, 2585–2600 CrossRef CAS; (d) M. Wang, S. Liu, H. Liu, Y. Wang, Y. Lan and Q. Liu, Nature, 2024, 631, 556–562 CrossRef CAS PubMed; (e) P. A. Dub and T. Ikariya, J. Am. Chem. Soc., 2013, 135, 2604–2619 CrossRef CAS PubMed; (f) P. A. Dub and J. C. Gordon, Nat. Rev. Chem., 2018, 2, 396–408 CrossRef CAS . For examples of concerted mechanisms, see; (g) Y. Wang, L. Zhu, Z. Shao, G. Li, Y. Lan and Q. Liu, J. Am. Chem. Soc., 2019, 141, 17337–17349 CrossRef CAS PubMed; (h) S.-X. Zhang, L. Long, Z. Li, Y.-M. He, S. Li, H. Chen, W. Hao and Q.-H. Fan, J. Am. Chem. Soc., 2025, 147, 5197–5211 CrossRef CAS PubMed; (i) S. Li, G. Li, W. Meng and H. Du, J. Am. Chem. Soc., 2016, 138, 12956–12962 CrossRef CAS PubMed; (j) T. Ohkuma, Proc. Jpn. Acad., Ser. B, 2010, 86, 202–219 CrossRef CAS; (k) X. Guo, Y. Tang, X. Zhang and M. Lei, J. Phys. Chem. A, 2011, 115, 12321–12330 CrossRef CAS PubMed; (l) Y. Zhao, L. Zhang, Y. Tang, M. Pu and M. Lei, Phys. Chem. Chem. Phys., 2022, 24, 13365–13375 RSC; (m) It is found that a concerted asynchronous transfer of Mn-hydride to the carbonyl carbon and the amido proton to the carbonyl oxygen via TS [4-5]si‡ is energetically more favorable. The details are provided in the ESI in Table S11†..
- (a) The hydride addition transition states in various water coordination modes are provided in Section 8.6 in the ESI†.; (b) The Gibbs free energies of the enantiocontrolling hydride addition transition states [4-5]w0-si‡ and [4-5]w0-re‡ are 39.7 and 37.6 kcal mol−1, respectively. The conformational analysis of the hydride addition transition states [4-5]w0-si‡ and [4-5]w0-re‡ in the unassisted mode are provided in Table S6 in the ESI†.; (c) The detailed mapping of the NCIs in [4-5]w0-si‡ and [4-5]w0-re‡ are provided in Fig. S8 in the ESI.† It is noted that the number of NCIs between the catalyst and substrates, as well as their strength, is found to be higher in [4-5]w0-re‡ than in [4-5]w0-si‡..
- (a) The details of the conformational analysis of the hydride addition transition states [4-5]we-si‡ and [4-5]we-re‡ in assisted mode are provided in Table S7 in the ESI.† The detailed mapping of the noncovalent interactions for [4-5]we-si‡ and [4-5]we-re‡ is provided in Fig. S9 in the ESI†.; (b) The Gibbs free energies of the enantiocontrolling hydride addition transition states [4-5]we-si‡ and [4-5]we-re‡ with the explicit water molecules are 30.9 and 32.0 kcal mol−1 lower than those of [4-5]w0-si‡ and [4-5]w0-re‡ without any water molecules bound. The total strength and the number of non-covalent interactions (NCIs) are found to be higher in [4-5]we-si‡ than in [4-5]we-re‡..
- (a) S. Kozuch and S. Shaik, Acc. Chem. Res., 2011, 44, 101–110 CrossRef CAS PubMed; (b) S. Kozuch and J. M. Martin, ACS Catal., 2011, 1, 246–253 CrossRef CAS; (c) S. Kozuch, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 795–815 CAS.
- To obtain maximum δE, different likely combinations of the stationary points involved in the catalytic cycle are considered. The energetic span calculations are shown in Table S12 in the ESI†.
- (a) More details on the conformational analysis of water binding coordination sites for the hydride addition step are given in Table S8 in the ESI†.; (b) For the enantiocontrolling hydride addition [4-5]si‡ and [4-5]re‡ TS, conformational analysis performed using an autosampling program (CREST) is provided, see details in the ESI.† The TS [4-5]si‡ in the 5-water model is lower in energy as compared to other water binding modes, as shown in Table S9†.; (c) Similarly, the hydride addition transition states in different solvent coordination modes are considered, and their details are provided in Table S10 in the ESI.† The enantiocontrolling hydride addition TS ([4-5]wabcde-si‡/[4-5]wabcde-re‡) with water binding is about 4 to 5 kcal mol−1 lower than the corresponding TS ([4-5]wabcdf-si‡/[4-5]wabcdf-re‡) model with a bound ethanol..
- The details of single point energy calculations using the B3LYP and other functionals are provided in the solvent phase in Table S17 in the ESI.† The overall conclusions obtained using different functionals were found to be very similar.
- (a) AIM2000 version 2.0, Buro fur Innovative Software, SBK Software, Bielefeld, Germany, 2002 Search PubMed; (b) R. F. W. Bader, Rev, 1991, 91, 893–928 CAS; (c) C. F. Matta and R. J. Boyd, Quantum Theory of Atoms in Molecules: Recent Progress in Theory and Application, Wiley-VCH, Weinheim, Germany, 2007 CrossRef.
- (a) Y. Dangat, S. Popli and R. B. Sunoj, J. Am. Chem. Soc., 2020, 142, 17079–17092 CrossRef CAS PubMed; (b) M. Pareek and R. B. Sunoj, ACS Catal., 2020, 10, 4349–4360 CrossRef CAS; (c) Y. Reddi, C.-C. Tsai, C. M. Avila, F. D. Toste and R. B. Sunoj, J. Am. Chem. Soc., 2019, 141, 998–1009 Search PubMed; (d) A. Unnikrishnan and R. B. Sunoj, Chem. Sci., 2019, 10, 3826–3835 Search PubMed; (e) B. Bhaskararao and R. B. Sunoj, ACS Catal., 2017, 7, 6675–6685 CrossRef CAS.
- (a) The energetically more favored [4-5]si‡ exhibited a higher cumulative strength of NCIs (−55.7 kcal mol−1) than that in [4-5]re‡ (−50.9 kcal mol−1), calculated on the basis of the NCIs operating between the chiral catalyst and substrate.; (b) Mapping of all the NCIs, including those within the catalyst and in the substrate is provided in Fig. S10 in the ESI†.; (c) The AIM data relevant to the bond critical points along the bond paths found in [4-5]si‡ and [4-5]re‡ are provided in Table S1, in the ESI†.; (d) The strength of the NCIs in the enantiocontrolling TSs, estimated using Espinosa's formulation, is compiled in Table S14 in the ESI.† For details of quantification of NCIs using the topological parameters, see; (e) E. Espinosa, I. Alkorta, J. Elguero and J. E. Molins, Chem. Phys., 2002, 117, 5529–5542 CAS; (f) The energetically more favored [4-5]si‡ exhibited a higher cumulative strength of NCIs than that in [4-5]re‡..
- Some of the NCIs exhibit very similar ρbcp values in both [4-5]si‡ and [4-5]re‡ and are unlikely to contribute to the differential stabilization. The focus is therefore placed on the key differences in the NCI in the enantiocontrolling transition states.
- (a) E. R. Johnson, S. Keinan, P. Mori-Sánchez, J. Contreras-García, A. J. Cohen and W. J. Yang, J. Am. Chem. Soc., 2010, 132, 6498–6506 Search PubMed; (b) R. Parthasarathi, V. Subramanian and N. Sathyamurthy, J. Phys. Chem. A, 2006, 110, 3349–3351 CrossRef CAS PubMed; (c) A. Engdahl and B. Nelander, J. Phys. Chem., 1985, 89, 2860–2864 CrossRef CAS; (d) D. Danovich, S. Shaik, F. Neese, J. Echeverría, G. Aullón and S. J. Alvarez, J. Chem. Theory Comput., 2013, 9, 1977–1991 CrossRef CAS PubMed; (e) J. Contreras-García, E. R. Johnson, S. Keinan, R. Chaudret, J.-P. Piquemal, D. N. Beratan, W. Yang and W. NCIPLOT, J. Chem. Theory Comput., 2011, 7, 625–632 Search PubMed; (f) For an interesting study on the performance of various density functionals in the study of noncovalent interactions, see: A. R. Neves, P. A. Fernandes and M. J. Ramos, J. Chem. Theory Comput., 2011, 7, 2059–2067 Search PubMed; (g) P. L. A. Popelier, J. Phys. Chem. A, 1998, 102, 1873–1878 CrossRef CAS; (h) C. H. Suresh, N. Mohan, P. Vijayalakshmi, K. George and M. J. Mathew, J. Comput. Chem., 2009, 30, 1392–1404 CrossRef CAS PubMed.
- (a) M. B. Shah, J. Liu, Q. Zhang, C. D. Stout and J. R. Halpert, ACS Chem. Biol., 2017, 12, 1204–1210 CrossRef CAS PubMed; (b) H. Matter, M. Nazaré, S. Güssregen, W. D. Will, H. Schreuder, A. Bauer, M. Urmann, K. Ritter, M. Wagner and V. Wehner, Angew Chem. Int. Ed. Engl., 2009, 48, 2911–2916 CrossRef CAS PubMed.
- (a) In our activation strain calculation, the transition state is partitioned into two components, i.e., a catalyst with four bound water molecules and a substrate with one water molecule. See Fig. S11 in the ESI† for additional details.; (b) Summary of distortion and interaction energies obtained from the activation strain analysis is provided in Table S15 in the ESI†..
- (a) F. M. Bickelhaupt, J. Comput. Chem., 1999, 20, 114–128 CrossRef CAS; (b) F. M. Bickelhaupt and K. N. Houk, Angew. Chem., Int. Ed., 2017, 56, 10070–10086 CrossRef CAS PubMed; (c) A. Duan, P. Yu, F. Liu, H. Qiu, F. L. Gu, M. P. Doyle and K. N. Houk, J. Am. Chem. Soc., 2017, 139, 2766–2770 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2413245, 2413299 and 2413301. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5sc02606g |
| This journal is © The Royal Society of Chemistry 2025 |