
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis and reactivity of a parent phosphathioethynolato-borane and a boraarsaketene†
Malte
Jürgensen
ab,
Tanja
Kunz
ab,
Merle
Arrowsmith
 ab,
Maximilian
Dietz
ab,
Stephan
Hagspiel
ab and
Holger
Braunschweig
*ab
ab,
Maximilian
Dietz
ab,
Stephan
Hagspiel
ab and
Holger
Braunschweig
*ab
aInstitute for Inorganic Chemistry, Julius-Maximilians-Universität Würzburg, Am Hubland, 97074 Würzburg, Germany. E-mail: h.braunschweig@uni-wuerzburg.de
bInstitute for Sustainable Chemistry & Catalysis with Boron, Julius-Maximilians-Universität Würzburg, Am Hubland, 97074 Würzburg, Germany
First published on 11th April 2025
Abstract
We present the synthesis and isolation of the first main-group phosphathioethynolates, [LBH2(SCP)] (L = SIMes, CAACMe; SIMes = 1,3-bis(2,4,6-trimethylphenyl)-4,5-dihydroimidazol-2-ylidene, CAACMe = 1-(2,6-diisopropylphenyl)-3,3,5,5-tetramethylpyrrolidin-2-ylidene), in which phosphathioethynolate coordination occurs exclusively through the sulfur atom, giving rise to a phosphaalkyne-type structural motif. This is reflected in the reactivity of the SIMes derivative towards organic azides and [(η2-C2H4)2CoCp] (Cp = C5H5), which mirrors the behavior of 1-adamantylphosphaalkyne, yielding triazaphospholes and a mixed cyclopentadienyl-(1,3-diphosphete) sandwich complex, respectively. Deviations from typical phosphaalkyne reactivity are observed in reactions with boron-containing heterocycles such as pentaphenylborole (PPB) and a carboranyl-substituted 9-borafluorene, which yield an unprecedented bicyclic structure and a zwitterionic spiro compound, respectively. Furthermore, we report the synthesis of the first arsaketenylborane, [(SIMes)BH2(AsCO)], which, although too unstable for isolation, generates an arsinidene in situ, which can be trapped by coordination to a PPB C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond.
C double bond.
Introduction
Since the publication of a reliable multigram synthesis of the phosphaethynolate anion, PCO−, the phosphorus analogue of the cyanate anion, NCO−, by the group of Grützmacher, its reactivity towards main-group compounds has been widely studied.1–6 Its main reactivity patterns include decarbonylation, due to a relatively weak P–CO bond,7,8 and oligomerisation via the P–C multiple bond.4,9 As an ambidentate nucleophile, PCO− can bind via its phosphorus or oxygen atom, depending on its bonding partner.10,11 The most common coordination mode is via the phosphorus atom, leading to phosphaketene compounds, E–PCO (E = main group element or transition metal; e.g.A, Fig. 1).12 In 2021 our group reported the synthesis of the carbene adducts of the parent boraphosphaketene from the salt metathesis of dihydro(triflato)borane precursors with sodium phosphaethynolate.13 The parent boraphosphaketenes exhibit a strong tendency towards dimerisation via the PC double bond, forming 1,3-diphosphetane-2,4-dione rings.
 | ||
| Fig. 1 Selected examples of PCO−-substituted main-group compounds (A and B), as well as the only literature known SCP− complexes (C–D) and the new boron SCP−- and AsCO−-substituted complexes presented herein. Dipp = 2,6-diisopropylphenyl. | ||
Only a small number of phosphaethynolate main-group species, in which PCO− coordinates via the oxygen atom, have been structurally characterised, including the diazaborole B.14,15 While a wide compendium of experimental and computational work has shown that the anions PCO−, NCO− and NCS− share similar net electronic properties,16–18 the higher homologues are less well explored. The equally ambidentate11,16 phosphathioethynolate anion, PCS−, was first synthesised as a highly unstable lithium salt by Becker in 1994.19 In 2016, the Goicoechea group reported the first room-temperature synthesis of PCS− in the form of the potassium salt [K(18-crown-6)][PCS], followed by a general synthesis of phosphorus- and arsenic-containing analogues of the thio- and selenocyanate anions in 2018.16,20 Despite almost 10 years of facile access to stable sodium and potassium salts of PCS−, only spectroscopic and electrochemical investigations of the anion, as well as three transition-metal complexes (but no main-group element species) are known thus far (Fig. 1).16,21–23
The reaction of the neutral tungsten complex [W(CO)5(MeCN)] with PCS− resulted in an almost equimolar mixture of the W–SCP and W–PCS isomers, C and C′, respectively.16 Both isomers decompose in 1,2-dichlorobenzene at room temperature over a period of four days. DFT calculations showed that the [(OC)5W–ECX]− isomer is systematically more stable than the [(OC)5W–XCE]− isomer for E = N, P, and X = O, S, albeit with a mere 0.8 kcal mol−1 energy difference for the phosphathioethynolate derivative, thus reflecting the observation of both C and C′. Hoping to access exclusively S- or P-bound phosphathioethynolate complexes, Weigend, Goicoechea, Tambornino and Hohloch turned to the more Lewis-acidic and polarising La(III) metal center.23 Instead, they isolated complex D, in which the pseudohalide SCP− is η3-bound in an unusual side-on fashion. Unlike C and C′, complex D is stable for weeks in aromatic and for several days in ethereal solvents at room temperature.
While the reactivity of the phosphathioethynolate anion has been scarcely explored, the arsaethynolate anion has been the subject of more extensive studies. Its first synthesis was reported by the group of Goicoechea in 2016.24 Similarly to its lighter homologues, the arsaethynolate anion exhibits ambidentate character,20,24 with DFT calculations indicating a preference for bonding through the arsenic atom.11 The resulting arsaketenes are highly unstable due to their strong tendency to eliminate CO under both photolytic and thermal conditions.7 The highly reactive arsinidenes formed via decarbonylation can be stabilised by coordination with Lewis bases.25
Based on our previous successful synthesis of a parent boraphosphaketene,13 we present the first examples of main-group phosphathioethynolates, [(SIMes)BH2(SCP)] and [(CAACMe)BH2(SCP)] (SIMes = 1,3-bis(2,4,6-trimethylphenyl)-4,5-dihydroimidazol-2-ylidene, CAACMe = 1-(2,6-diisopropylphenyl)-3,3,5,5-tetramethylpyrrolidin-2-ylidene), which display a combination of phosphaalkyne, sulfide and dihydroborane reactivity, enabling the facile generation of novel complex B,P,S-polyheterocycles, as well as the first borylarsaketene, the spontaneous decarbonylation of which generates a highly reactive borylarsinidene, trapped as a rare π-alkene complex.
Results and discussion
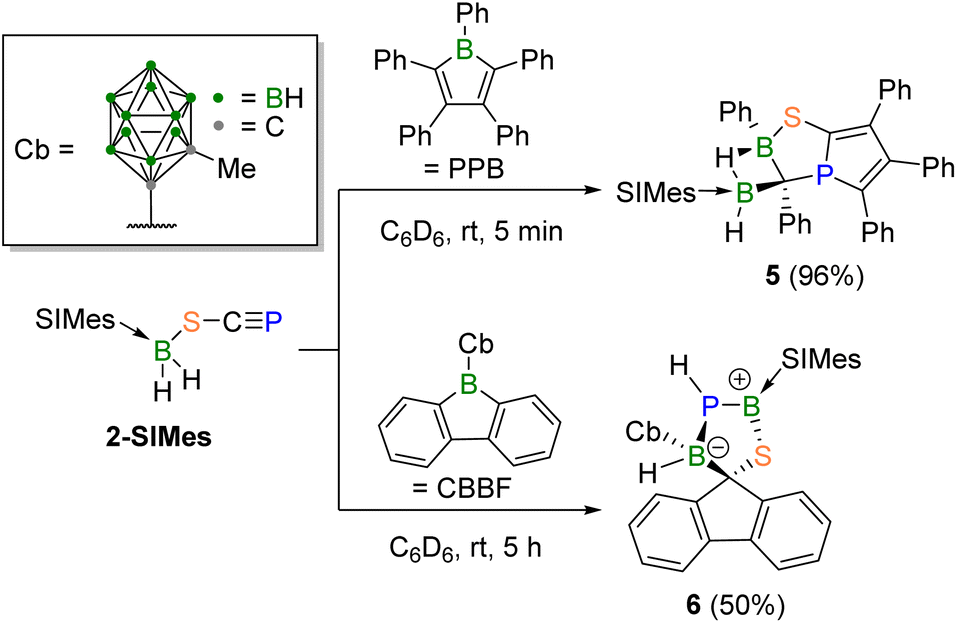
The N-heterocyclic carbene (NHC)- and cyclic alkyl(amino)carbene (CAAC)-stabilised dihydro(triflato)boranes, [(SIMes)BH2(OTf)] (1-SIMes, OTf = trifluoromethylsulfonate = triflate) and [(CAACMe)BH2(OTf)] (1-CAACMe),13 were chosen as starting materials since OTf− is a good leaving group for salt metathesis reactions.26–28 Reactions of 1-SIMes and 1-CAACMe with [Na(diox)3.6][SCP] (diox = 1,4-dioxane) in a benzene/THF mixture (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) afforded the yellow phosphathioethynolato boranes [LBH2(SCP)] (L = SIMes (2-SIMes), CAACMe (2-CAACMe)) in moderate to excellent yields (Scheme 1).
:1) afforded the yellow phosphathioethynolato boranes [LBH2(SCP)] (L = SIMes (2-SIMes), CAACMe (2-CAACMe)) in moderate to excellent yields (Scheme 1).
 | ||
| Scheme 1 Synthesis of the carbene-stabilised phosphathioethynolato boranes 2-SIMes and 2-CAACMe, and crystallography-derived solid-state structure of 2-SIMes. Atomic displacement ellipsoids set at 50% probability. Ellipsoids of ligand periphery and hydrogen atoms omitted for clarity except for boron-bound hydrides. OTf = trifluoromethylsulfonate = triflate; diox = 1,4-dioxane. | ||
While 2-SIMes was stable in the solid state, and for at least 15 minutes under photolytic conditions (or for three hours when heated in benzene at 80 °C) 2-CAACMe underwent unselective decomposition after a few hours in benzene at room temperature. Consequently, further reactivity studies were carried out with 2-SIMes exclusively. NMR-spectroscopic analyses of the salt metathesis reaction mixtures revealed the formation of a single isomer. The greater similarity of the 31P NMR shifts (2-SIMes: δ = −43.7 ppm; 2-CAACMe: δ = −41.2 ppm) to that of C (δ = −92.9 ppm; Fig. 1) rather than C′ (δ = −192.6 ppm) strongly suggests coordination through the sulfur atom.16 The 11B NMR spectra show broad BH2 triplets (2-SIMes: δ = −20.2 (1JHB = 98 Hz) ppm; 2-CAACMe: δ = −17.5 (1JHB = 104 Hz) ppm). The B–SCP bonding mode was confirmed by a C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) P triple bond stretching band at 1508 cm−1 in the solid-state IR spectrum of 2-SIMes, which lies in the range of other phosphaalkyne-containing compounds.29–31
P triple bond stretching band at 1508 cm−1 in the solid-state IR spectrum of 2-SIMes, which lies in the range of other phosphaalkyne-containing compounds.29–31
Single crystals of 2-SIMes suitable for X-ray diffraction (XRD) analysis provided further confirmation of the B–SCP bonding mode (see Scheme 1). The almost linear sulfur-bound SCP− ligand (P1–C1–S1 176.57(13)°) coordinates nearly orthogonally to the boron center (B1–S1–C1 104.13(8)°, C2–B1–S1–C1–96.24°). The P1–C1 distance of 1.5606(19) Å is in the range of related phosphaethynolates PC triple bonds (1.559–1.565 Å) and similar to that of the free SCP− anion (1.579(4) Å).15–17,32,33 The C1–S1 distance (1.6600(19) Å) is similar to that in related thiocyanates, but somewhat longer than in free SCP− (1.613(4) Å),16,34,35 while the B1–S1 distance (1.9670(19) Å) is comparable to that in other boron sulfides and a related NHC-stabilised dihydro(thio)borane.36,37 DFT calculations at the B3LYP/D3(BJ)/def2-SVP level of theory on the S-bound phosphathioethynolate (2-SIMes) and P-bound phosphathioketene (2′-SIMes) isomers provide a Gibbs free energy difference of only 0.7 kcal mol−1 (i.e. within the error margin), with 2-SIMes being the slightly thermodynamically preferred bonding motif. The same is observed for 2-CAACMe and 2′-CAACMe, which show an even smaller energy difference of 0.3 kcal mol−1 (see Table S1 in the ESI†). Since there is no significant thermodynamic preference for the S-bound isomers 2-SIMes and 2-CAACMe, their selective formation must be kinetically driven.‡ Compounds 2-SIMes and 2 CAACMe are the first examples of covalent main-group phosphathioethynolates, and their syntheses the first to induce selective end-on coordination via the SCP sulfur atom.
Given its terminal CP triple bond, compound 2-SIMes is expected to react similarly to other phosphaalkynes, which undergo spontaneous [3 + 2] cycloaddition reactions with azides to yield triazaphospholes.38 To test this hypothesis, compound 2-SIMes was combined with p-(trifluoromethyl)phenyl azide (N3PhCF3). While the 11B NMR shift of the yellow reaction mixture showed almost no change (δ = −21.7 ppm) compared to the precursor 2-SIMes (δ = −20.2 ppm) the 31P NMR spectrum revealed a new resonance at 170.9 ppm, which is drastically downfield-shifted from 2-SIMes (δ = −43.7 ppm) and similar to other known triazaphospholes (δ = 161.8–180.7 ppm).38 Mass spectrometry confirmed the formation of the triazaphosphole 3-PhCF3 (54% yield, Scheme 2, top). In combination with N3Mes (Mes = mesityl = 2,4,6-trimethylphenyl), an analogous sulfur-functionalised triazaphosphole, compound 3-Mes, was formed (δ11B = −22.4 ppm; δ31P = 173.6 ppm) and isolated in 58% yield. Consequently, the (SIMes)BH2S substituent seems to have no significant impact on the reactivity of the phosphaalkyne moiety with organic azides.
 | ||
| Scheme 2 Phosphaalkyne-like reactivity of 2-SIMes. PhCF3 = p-trifluoromethylphenyl. Mes = 2,4,6-trimethylphenyl. | ||
The solid-state structure of 3-PhCF3 is shown in Fig. 2. The (SIMes)BH2S moiety remains intact and the B1–S1–C1 angle of 103.03(14)° is similar to that of the precursor 2-SIMes (104.13(8)°). The five-membered CN3P heterocycle is quasi-planar with endocyclic torsion angles between −0.8(3)° and 0.7(2)°, and bonding parameters (C1–P1 1.712(3), C1–N1 1.372(4), N1–N2 1.290(4), N2–N3 1.354(4), P1–N3 1.719(3) Å) similar to literature-known triazaphospholes.39,40
 | ||
| Fig. 2 Crystallography-derived solid-state structure of 3-PhCF3. Atomic displacement ellipsoids set at 50% probability. Ellipsoids of ligand periphery and hydrogen atoms omitted for clarity except for boron-bound hydrides. | ||
Owing to their highly polarised CP triple bond, phosphaalkynes tend to oligomerise under thermal or photolytic conditions, albeit often in a relatively unselective manner. More selective oligomerisations occur when mediated by transition-metal complexes, low-valent main-group compounds or Lewis acids.38,41–45 The reaction of 2-SIMes with 0.5 equiv. of the so-called “Jonas complex”, [CpCo(η2-C2H4)2] (Cp = cyclopentadienyl), which has been reported to mediate the formation of 1,3-diphosphetes from phosphaalkynes,46–48 yielded complex 4 as a brown solid in 31% yield after workup. The 11B NMR resonance at −23.3 ppm is slightly upfield-shifted from that of 2-SIMes (δ = −20.2 ppm), and the 31P NMR shift of 56.2 ppm is similar to that of the mixed cyclopentadienyl-(1,3-diphosphete) sandwich complex [CpCo{η4-(PCtBu)2}] at 38.1 ppm.48 Mass spectrometry confirmed the formation of the analogous mixed sandwich complex 4 (Scheme 2, bottom). Complex 4 was further characterised by solid-state IR spectroscopy, which evidenced two characteristic weak B–H stretching bands at 2394 and 2380 cm−1.
The known reaction of pentaphenylborole (PPB) with 1-adamantylphosphaalkyne resulted in the formation of a 1-phospha-6-boratricyclo-hept-3-ene, which is the product of the rearranged Diels–Alder adduct.49 Upon combining 2-SIMes with PPB the reaction mixture turned from blue, the colour of PPB, to yellow. While the 11B NMR resonance is merely broadened (ω1/2 ≈ 400 Hz) without discernible shift (δ = −20.2 ppm), the 31P NMR resonance (δ = 101.2 ppm) is considerably downfield-shifted (Δδ = 145 ppm) compared to 2-SIMes (δ = −43.7 ppm), indicating a highly electron-poor phosphorus centre.
A single-crystal XRD analysis (Fig. 3a) revealed the formation of the bent bicyclic compound 5 (96% yield), which is comprised of a phosphole fused to a 1,4,2-thiaphosphaborolane via the phosphaalkyne-derived P1–C1 edge (Scheme 3, top). Owing to its position at the bridgehead of the two fused heterocycles the phosphorus atom adopts an unusual bent T shape (C4–P1–C1 90.57(10), C1–P1–C3 98.43(9), C3–P1–C4 119.04(9)°). This is reminiscent of Uhl's tricyclic phospholo[3,2,1-hi]phosphindoles, which feature a similarly shaped but slightly less bent phosphorus atom (C–P–C ca. 89, 89, 128°) at the bridgehead of two fused phosphole rings, but with a significantly upfield-shifted 31P NMR resonance (δ = 66–69 ppm).50,51 The (SIMes)BH2 moiety of 5 has migrated from the sulfur to the former borole ortho carbon atom C3, now embedded in the C2PBS heterocycle, and bridges to the second borole-derived boron atom B2 via a three-center-two-electron B–H–B σ bond. The single broad 11B NMR resonance of 5 likely results from the resonances of the two boron atoms overlapping.
 | ||
| Fig. 3 Crystallography-derived solid-state structures of (a) 5 and (b) 6. Atomic displacement ellipsoids set at 50% probability. Ellipsoids of ligand periphery and hydrogen atoms omitted for clarity except for B- and P-bound protons. | ||
 | ||
| Scheme 3 Reactivity of 2-SIMes towards PPB and CBBF. | ||
DFT calculations at the B3LYP/D3(BJ)/def2-SVP level of theory were performed to ascertain a possible mechanism for the formation of 5 (Scheme 4). The first two steps were assumed to be the same as for the reaction of PPB with 1-adamantylphosphaalkyne: (i) Diels–Alder adduct formation between the diene backbone of the borole and the PC triple bond of 2-SIMes to yield Int1A, followed by (ii) insertion of the phosphaalkyne carbon atom into the B–C bond of the borole moiety, resulting in the phosphorus-capped BC5 heterocycle Int2A.49 The next step to zwitterionic Int3A involves several bond-breaking and bond-forming steps, initiated by a nucleophilic attack of the endocyclic boron atom by the exocyclic sulfur atom. The final step involves the nucleophilic attack of (SIMes)BH2 unit by the negatively charged carbon atom in Int3A, resulting in B–S bond cleavage to form 5. The overall reaction sequence is thermodynamically favoured with a ΔG value of −38.0 kcal mol−1. While this reaction shows once more that 2-SIMes acts as phosphaalkyne derivative, it also demonstrates the possible involvement of the (SIMes)BH2S moiety as a sulfur nucleophile and boron electrophile.
 | ||
| Scheme 4 Proposed mechanism for the formation of 5 from the reaction of 2-SIMes with PPB. Gibbs free energies in parentheses (kcal mol−1) are referenced to 2-SIMes + PPB (no solvent correction) and calculated at the B3LYP/D3(BJ)/def2-SVP level of theory (solvent not included). | ||
In 2018 Martin and co-workers showed that the reaction of a borafluorene with 1-adamantylphosphaalkyne leads to the insertion of the PC unit into an endocyclic borafluorene B–C bond, forming a central seven-membered 1,3-phosphaborepine.52 While the reaction of 2-SIMes with 9-phenyl-9-borafluorene was unselective at room temperature, the reactivity towards the carboranyl-substituted 9-borafluorene [(C3H13B10)BC12H8] (CBBF)53 yielded a new species displaying a 31P NMR doublet at δ = −105.8 ppm with a coupling constant of 1JPH = 205 Hz, that is drastically upfield-shifted from Martin's boraphosphaalkene (δ = 199 ppm), and indicates the unexpected presence of a proton at phosphorus. The zwitterionic spirofluorene 6 was isolated as a yellow solid in 50% yield (Scheme 3, bottom) and is the first evidence of reactivity entirely divergent from that of simple phosphaalkynes. The borenium ((SIMes)BPS+) centre displays a very broad (ω1/2 ≈ 1000 Hz), low-field 11B NMR resonance at 70.9 ppm, while all other resonances fall within the region typical for four-coordinate boron atoms (δ = −3 to −10 ppm). The crystallography-derived solid-state structure of 6 (Fig. 3b) shows a spiro compound composed of a planar fluorene and an envelope-shaped, zwitterionic 1,3,2,4-thiaphosphadiborole, joined via the central fluorene carbon atom C4. Both hydrides of the former (SIMes)BH2 moiety have migrated, one to the now-adjacent phosphorus atom (hence the 31P NMR PH doublet), the other to the borafluorene-derived sp3-hybridised boron atom B2. The geometry of the borenium center B1 is now trigonal-planar (Σ(∡B1) = 360.00(21)°). The B1–P1 bond length of 1.865(2) is longer than that in both Cowley's (B–P 1.8309(16) Å)54 and Kato's (B–P 1.781(3) Å)55 NHC-stabilised phosphaborenes, which speaks against the presence of a BP double bond in 6. However, DFT calculations provide Wiberg bond indices (WBIs)56 of 1.18 and 1.35 for the B1–P1 and B1–S1 bonds of 6, while a population analysis based on occupation numbers (PABOON)57 yields shared numbers of electrons (SNEs), which provide a more accurate view of bond orders, of 1.53 and 1.65, respectively, suggesting some partial BS and BP double bond character.
The formation of 6 from 2-SIMes and CBBF involves a complex series of bond-breaking and -forming steps, including (in no particular order): (i) cleavage of the phosphaalkyne CP bond, (ii) B–C bond formation between the former borafluorene boron and phosphaalkene carbon atoms, (iii) displacement of the former by the latter from the borafluorene framework to generate an all-carbon fluorene moiety, (iv) B–P bond formation between the former phosphalkene phosphorus and the boron atoms of both 2a and CBBF, and (v) hydride migration from the former (SIMes)BH2 moiety to the phosphorus and borafluorene-derived boron atoms. In analogy to Martin's work,52 the first step of the reaction is likely the insertion of the CP moiety of 2-SIMes into the endocyclic B–C bond of CBBF. To avoid steric clashes between the carboranyl and SIMes ligands, however, the insertion is more likely to yield the 1,2-phosphaborepine, thus establishing both the P1–B2 and C4–Cbiphenyl bonds in Int1B (Scheme 5). This would bring the phosphorus atom and the (SIMes)BH2 moiety into close proximity for B1–P1 bond formation and B1-to-P1 hydride transfer, concomitant with ring size reduction of the central seven-membered C5BP ring to a six-membered borinine ring, yielding the fused 9-boraphenanthrene and PBSC heterocycles of Int2B (presumably via a series of intermediate steps). The latter then undergoes a 1,2-sigmatropic shift to yield the spirofluorene Int3B. Finally, a double tautomerisation, involving a P1-to-B2 proton shift and another B1-to-P1 hydride shift, results in compound 6. The overall reaction is thermodynamically favoured with a highly negative ΔG value of −60.4 kcal mol−1. In this reaction the divergent reactivity from classical phosphaalkyne chemistry is owed to the reactive B–H bonds, as well as the high steric demands and bent geometry of the (SIMes)BH2S moiety.
 | ||
| Scheme 5 Proposed mechanism for the formation of 6 from the reaction of 2-SIMes with CBBF. Gibbs free energies in parentheses (kcal mol−1) are referenced to 2-SIMes + CBBF and calculated at the B3LYP/D3(BJ)/def2SVP level of theory (solvent not included). | ||
Given the versatile reactivity of 2-SIMes, we were also interested in that of its arsaethynolate analogue, AsCO−, another heavier congener of the phosphaethynolate anion. Salt metathesis of 1-SIMes with Na[OCAs] in benzene/THF (5:1) at room temperature yielded a yellow suspension with a new 11B NMR resonance at −32.9 ppm, comparable to that of its boraphosphaketene analogue (δ = −31.1 ppm, Scheme 6, top).13 Compound 7 decomposed rapidly in solution at room temperature (70% in 24 h) and more slowly at −30 °C, thus preventing its clean isolation. Similar observations have been made for other literature-known arsaketenes, owing to their propensity towards decarbonylation and the formation of arsinidene intermediates.25,58,59 While an XRD analysis of 7 provided proof of connectivity via the B–As linkage, as predicted by DFT calculations,11 the data were of insufficient quality for the discussion of bonding parameters due to the rapid decomposition of the crystals (see Fig. S52 in the ESI†).
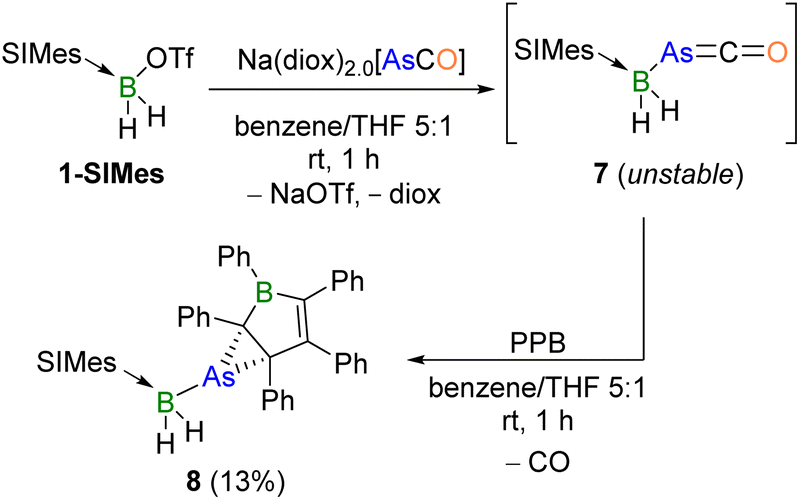 | ||
| Scheme 6 Synthesis of 7 and its reactivity towards PPB. | ||
The arsinidene decomposition products of arsaketenes are known to oligomerise,22 but can also be trapped by adduct formation with Lewis bases.25,58–60 In attempts to trap a potential arsinidene decomposition product of 7, the Lewis bases IMe (=1,3-dimethylimidazol-2-ylidene), PPh3, and the isonitrile Mes*NC (Mes* = 2,4,6-tri-tert-butylphenyl), were added to freshly prepared solutions of 7. Unfortunately, this only resulted in unselective reactions. Inspired by the reaction of the boraphosphaketene (SIMes)BH2(PCO) with boroles, which led to 1,2-phosphaborinines,13 a freshly prepared suspension of 7 in benzene/THF (5:1) was added to a solution of PPB in benzene at room temperature, immediately resulting in a colour change from dark green to red. The new broad 11B NMR resonance at −21.5 ppm was assigned to compound 8,§ which was isolated as a yellow powder in 13% yield (Scheme 6, bottom). The low yield of the otherwise relatively selective reaction (as determined by NMR-spectroscopic analysis of the reaction mixture) is attributed to the numerous crystallisation steps required to obtain the compound in sufficient purity for full characterisation.
The solid-state structure of 8 (Fig. 4a) shows that, instead of the expected arsinidene insertion into the borole ring, the arsinidene [(SIMes)BH2As:] binds to the C2–C3 double bond of the borole. The As–C bonds in 8 (2.110(2), 2.080(2) Å) are slightly longer than typical As–C single bonds (1.97–2.03 Å),61–64 while the C2–C3 bond (1.495(3) Å) is elongated compared to that of PPB (1.43 Å), but significantly shorter than a typical C(sp3)–C(sp3) single bond (1.54 Å).65 Furthermore, despite the coordination of As, the geometry of C2 and C3 remains close to trigonal-planar (Σ∠C2 ca. 358°, Σ∠C3 ca. 351°), suggesting an arsinidene–alkene η2–π interaction rather than a covalent arsirane (or arsacyclopropane) ring. This is also supported by a comparison with Appel's arsirane PhAsC2(TMS)4, which displays shorter endocyclic As–C bonds and a significantly longer C–C bond (As–C 2.021(3), 2.025(4), C–C 1.580(6) Å).66 Conversely, Krossing and Stephan's arsinidenium-cyclopentadiene π-complexes [(η2-Cp*)AsX]+ (Cp* = C5Me5, X = Cl, F) display longer As–C distances but slightly shorter endocyclic C–C bonds (As–C 2.10–2.68 Å; C–C 1.40–1.46 Å), with similar quasi-trigonal-planar geometry at the As-bound carbon atoms as in 8.67,68 Furthermore, the 13C NMR resonance of the As-bound borole carbon nucleus C3 was detected by an HMBC experiment at 115.4 ppm, in the region of alkene resonances, suggesting at least some degree of C2–C3 π bonding, rather than a C2–C3 single bond.¶
 | ||
| Fig. 4 (a) Solid-state structure of 8. Atomic displacement ellipsoids set at 50% probability. Ellipsoids of ligand periphery and hydrogen atoms omitted for clarity except for B-bound protons. (b) Truncated ball-and-stick representation of 8 (Mes and Ph substituents and hydrogen atoms omitted for clarity, except for B-bound protons) with shared numbers of electrons (SNEs) in blue. (c and d) Intrinsic bond orbitals (IBOs) of 8 (stick representations, hydrogen atoms omitted for clarity, except for B-bound protons) involved in As–PPB bonding. Isovalues set at 0.04. | ||
Computational analyses of 8 were carried out to determine the bonding situation within the C2As heterocycle. While the calculated WBIs56 are 0.72 and 0.77 for the As–C bonds and 0.91 for the C–C bond (see Fig. S53 in the ESI†), the SNEs obtained from the PABOON57 analysis are 0.98 and 1.00 for the As–C bonds and 1.52 for the C2–C3 bond (Fig. 4b). This strongly suggests a donor–acceptor bonding situation, in which the PPB π bonds donate into the empty pz orbital at As. An intrinsic bond orbital (IBO) analysis, however, shows that the two IBOs69 contributing to As–PPB bonding are those involving on the one side the 4pz orbital of As1 (41%) and the 2pz orbital of C2 (55%), and on the other side the 2py orbital of As1 (39%) and the 2pz orbital of C3 (51%), with a small but non-negligible contribution of three-centre-two-electron bonding (As1–C2–B2 9%, As1–C3–C4 4%) (Fig. 4c and d, see Table S4 in the ESI†). It thus seems that the bonding situation in the C2As heterocycle is somewhere intermediate between a covalent arsirane and an arsinidene–alkene π complex.
Conclusions
In this work, we have synthesised and isolated the first main-group phosphathioethynolates, [LBH2(SCP)] (L = SIMes (2-SIMes), CAACMe (2-CAACMe)), in which the [SCP]− anion binds to the borane exclusively via the sulfur atom. The phosphaalkyne-type reactivity of 2-SIMes was demonstrated by its [2 + 3] cycloaddition reactions with organic azides and its cobalt-mediated dimerisation. With the borole and borafluorene derivatives PPB and CBBF, respectively, 2-SIMes displayed a combination of phosphaalkyne, sulfide and dihydroborane reactivity, leading to unexpected novel B,P,S-containing polyheterocycles. Furthermore, we have synthesised the first boraarsaketene, [(SIMes)BH2(AsCO)] (7), which was too reactive to isolate, but the decarbonylated arsinidene product of which could be trapped by an unexpected π-complex formation with PPB. This first reactivity study of some of the heavier phosphaethynolato boranes provides a particularly promising glimpse of their usefulness for accessing unprecedented heterocycles with multiple endocyclic main-group elements.Data availability
Synthetic procedures, NMR, IR and UV-vis spectra, X-ray crystallographic and computational details, and coordinates of all DFT-optimised compounds. Crystallographic data have been deposited with the Cambridge Crystallographic Data Center as supplementary publication no. 2428369 (7), 2428370 (6), 2428371 (5), 2428372 (8), 2428373 (3-PhCF3), 2428374 (1-SIMes), and 2428375 (2-SIMes).Author contributions
M. J. conceptualised and carried out the experimental work and wrote the ESI. M. J., M. D. and S. H. carried out the crystallographic experiments and analyses. T. K. and M. A. carried out the computational studies. T. K. wrote the original manuscript draft. M. A. finalised both ESI and manuscript. H. B. provided funding and supervision.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Funding from the Deutsche Forschungsgesellschaft (DFG grants 466754611 and BR1149/33-1) is gratefully acknowledged.Notes and references
- J. M. Goicoechea and H. Grützmacher, Angew. Chem., Int. Ed., 2018, 57, 16968–16994 CrossRef CAS PubMed.
- L. Weber, Eur. J. Inorg. Chem., 2018, 2018, 2175–2227 CrossRef CAS.
- S. Basappa, R. Bhawar, D. H. Nagaraju and S. K. Bose, Dalton Trans., 2022, 51, 3778–3806 RSC.
- D. Heift, Z. Benko and H. Grützmacher, Dalton Trans., 2014, 43, 831–840 RSC.
- F. F. Puschmann, D. Stein, D. Heift, C. Hendriksen, Z. A. Gal, H.-F. Grützmacher and H. Grützmacher, Angew. Chem., Int. Ed., 2011, 50, 8420–8423 CrossRef CAS PubMed.
- A. R. Jupp and J. M. Goicoechea, Angew. Chem., Int. Ed., 2013, 52, 10064–10067 CrossRef CAS PubMed.
- Y. Lu, H. Wang, Y. Xie, H. Liu and H. F. Schaefer, Inorg. Chem., 2014, 53, 6252–6256 CrossRef CAS PubMed.
- A. M. Tondreau, Z. Benkő, J. R. Harmer and H. Grützmacher, Chem. Sci., 2014, 5, 1545–1554 RSC.
- U. Salzner and S. M. Bachrach, J. Am. Chem. Soc., 1994, 116, 6850–6855 CrossRef CAS.
- D. Heift, Z. Benkő and H. Grützmacher, Dalton Trans., 2014, 43, 5920–5928 RSC.
- Á. Horváth, B. D. Lőrincz and Z. Benkő, Chem.–Eur. J., 2023, 29, e202300611 CrossRef PubMed.
- L. Liu, D. A. Ruiz, D. Munz and G. Bertrand, Chem, 2016, 1, 147–153 CAS.
- S. Hagspiel, F. Fantuzzi, R. D. Dewhurst, A. Gärtner, F. Lindl, A. Lamprecht and H. Braunschweig, Angew. Chem., Int. Ed., 2021, 60, 13666–13670 CrossRef CAS PubMed.
- D. W. N. Wilson, A. Hinz and J. M. Goicoechea, Angew. Chem., Int. Ed., 2018, 57, 2188–2193 CrossRef CAS PubMed.
- Y. Mei, J. E. Borger, D.-J. Wu and H. Grützmacher, Dalton Trans., 2019, 48, 4370–4374 RSC.
- A. R. Jupp, M. B. Geeson, J. E. McGrady and J. M. Goicoechea, Eur. J. Inorg. Chem., 2016, 639–648 CrossRef CAS PubMed.
- C. Camp, N. Settineri, J. Lefèvre, A. R. Jupp, J. M. Goicoechea, L. Maron and J. Arnold, Chem. Sci., 2015, 6, 6379–6384 RSC.
- S. Alidori, D. Heift, G. Santiso-Quinones, Z. Benkő, H. Grützmacher, M. Caporali, L. Gonsalvi, A. Rossin and M. Peruzzini, Chem.–Eur. J., 2012, 18, 14805–14811 CrossRef CAS PubMed.
- G. Becker and K. Hübler, Z. Anorg. Allg. Chem., 1994, 620, 405–417 CrossRef CAS.
- F. Tambornino, A. Hinz, R. Köppe and J. M. Goicoechea, Angew. Chem., Int. Ed., 2018, 57, 8230–8234 CrossRef CAS PubMed.
- Q. Yuan, F. Tambornino, A. Hinz, W. T. Borden, J. M. Goicoechea, B. Chen and X.-B. Wang, Angew. Chem., Int. Ed., 2019, 58, 15062–15068 CrossRef CAS PubMed.
- G.-L. Hou, B. Chen, W. J. Transue, Z. Yang, H. Grützmacher, M. Driess, C. C. Cummins, W. T. Borden and X.-B. Wang, J. Am. Chem. Soc., 2017, 139, 8922–8930 CrossRef CAS PubMed.
- F. A. Watt, L. Burkhardt, R. Schoch, S. Mitzinger, M. Bauer, F. Weigend, F. Tambornino, J. M. Goicoechea and S. Hohloch, Angew. Chem., Int. Ed., 2021, 60, 9534–9539 CrossRef CAS PubMed.
- A. Hinz and J. M. Goicoechea, Angew. Chem., Int. Ed., 2016, 55, 8536–8541 CrossRef CAS PubMed.
- S. Yao, Y. Grossheim, A. Kostenko, E. Ballestero-Martínez, S. Schutte, M. Bispinghoff, H. Grützmacher and M. Driess, Angew. Chem., Int. Ed., 2017, 56, 7465–7469 CrossRef CAS PubMed.
- S. Hagspiel, M. Arrowsmith, F. Fantuzzi, A. Hermann, V. Paprocki, R. Drescher, I. Krummenacher and H. Braunschweig, Chem. Sci., 2020, 11, 551–555 RSC.
- S. Hagspiel, M. Arrowsmith, F. Fantuzzi, A. Vargas, A. Rempel, A. Hermann, T. Brückner and H. Braunschweig, Angew. Chem., Int. Ed., 2021, 60, 6446–6450 CrossRef CAS PubMed.
- D. A. Ruiz, G. Ung, M. Melaimi and G. Bertrand, Angew. Chem., Int. Ed., 2013, 52, 7590–7592 CrossRef CAS PubMed.
- J. G. Cordaro, D. Stein and H. Grützmacher, J. Am. Chem. Soc., 2006, 128, 14962–14971 CrossRef CAS PubMed.
- K. Toyota, S. Kawasaki and M. Yoshifuji, J. Org. Chem., 2004, 69, 5065–5070 CrossRef CAS PubMed.
- M. Brym and C. Jones, Dalton Trans., 2003, 3665–3667 RSC.
- C. J. Hoerger, F. W. Heinemann, E. Louyriac, L. Maron, H. Grützmacher and K. Meyer, Organometallics, 2017, 36, 4351–4354 CrossRef CAS.
- S. Bestgen, Q. Chen, N. H. Rees and J. M. Goicoechea, Dalton Trans., 2018, 47, 13016–13024 RSC.
- H. Zhu, M. Ströbele, Z. Yu, Z. Wang, H. J. Meyer and X. You, Inorg. Chem. Commun., 2001, 4, 577–581 CrossRef CAS.
- J. H. Nelson, J. J. MacDougall, N. W. Alcock and F. Mathey, Inorg. Chem., 1982, 21, 1200–1204 CrossRef CAS.
- A. P. M. Robertson, M. F. Haddow and I. Manners, Inorg. Chem., 2012, 51, 8254–8264 CrossRef CAS PubMed.
- S.-H. Ueng, A. Solovyev, X. Yuan, S. J. Geib, L. Fensterbank, E. Lacôte, M. Malacria, M. Newcomb, J. C. Walton and D. P. Curran, J. Am. Chem. Soc., 2009, 131, 11256–11262 CrossRef CAS PubMed.
- M. Regitz, Chem. Rev., 1990, 90, 191–213 CrossRef CAS.
- S. L. Choong, C. Jones and A. Stasch, Dalton Trans., 2010, 39, 5774–5776 RSC.
- J. A. W. Sklorz, S. Hoof, N. Rades, N. De Rycke, L. Könczöl, D. Szieberth, M. Weber, J. Wiecko, L. Nyulászi, M. Hissler and C. Müller, Chem.–Eur. J., 2015, 21, 11096–11109 CrossRef CAS PubMed.
- A. Chirila, R. Wolf, J. Chris Slootweg and K. Lammertsma, Coord. Chem. Rev., 2014, 270–271, 57–74 CrossRef CAS.
- K. M. Szkop, A. R. Jupp, R. Suter, H. Grützmacher and D. W. Stephan, Angew. Chem., Int. Ed., 2017, 56, 14174–14177 CrossRef CAS PubMed.
- G. Hierlmeier, P. Coburger, M. Bodensteiner and R. Wolf, Angew. Chem., Int. Ed., 2019, 58, 16918–16922 CrossRef CAS PubMed.
- L. L. Liu, J. Zhou, Y. Kim, L. L. Caoa and D. W. Stephan, Dalton Trans., 2019, 48, 14242–14245 RSC.
- D. W. N. Wilson, D. D. L. Jones, C. D. Smith, M. Mehta, C. Jones and J. M. Goicoechea, Dalton Trans., 2022, 51, 898–903 RSC.
- K. Jonas, E. Deffense and D. Habermann, Angew. Chem., Int. Ed., 1983, 22, 716–717 CrossRef.
- P. B. Hitchcock, M. J. Maah and J. F. Nixon, J. Chem. Soc., Chem. Commun., 1986, 737–738 RSC.
- P. Binger, R. Milczarek, R. Mynott, M. Regitz and W. Rösch, Angew. Chem., Int. Ed., 1986, 25, 644–645 CrossRef.
- J. H. Barnard, S. Yruegas, S. A. Couchman, D. J. D. Wilson, J. L. Dutton and C. D. Martin, Organometallics, 2016, 35, 929–931 CrossRef CAS.
- A. Hentschel, A. Brand, P. Wegener and W. Uhl, Angew. Chem., Int. Ed., 2018, 57, 832–835 CrossRef CAS PubMed.
- A. Brand, A. Hentschel, A. Hepp and W. Uhl, Eur. J. Inorg. Chem., 2020, 2020, 361–369 CrossRef CAS.
- S. Yruegas, J. H. Barnard, K. Al-Furaiji, J. L. Dutton, D. J. D. Wilson and C. D. Martin, Organometallics, 2018, 37, 1515–1518 CrossRef CAS.
- T. Bischof, X. Guo, I. Krummenacher, L. Beßler, Z. Lin, M. Finze and H. Braunschweig, Chem. Sci., 2022, 13, 7492–7497 RSC.
- A. N. Price, G. S. Nichol and M. J. Cowley, Angew. Chem., Int. Ed., 2017, 56, 9953–9957 CrossRef CAS PubMed.
- A. Rosas-Sánchez, I. Alvarado-Beltran, A. Baceiredo, N. Saffon-Merceron, S. Massou, D. Hashizume, V. Branchadell and T. Kato, Angew. Chem., Int. Ed., 2017, 56, 15916–15920 CrossRef PubMed.
- K. B. Wiberg, Tetrahedron, 1968, 24, 1083–1096 CrossRef CAS.
- C. Ehrhardt and R. Ahlrichs, Theor. Chim. Acta, 1985, 68, 231–245 CrossRef CAS.
- A. Hinz and J. M. Goicoechea, Angew. Chem., Int. Ed., 2016, 55, 15515–15519 CrossRef CAS PubMed.
- E. Ballestero-Martínez, T. J. Hadlington, T. Szilvási, S. Yao and M. Driess, Chem. Commun., 2018, 54, 6124–6127 RSC.
- A. Hinz, M. M. Hansmann, G. Bertrand and J. M. Goicoechea, Chem.–Eur. J., 2018, 24, 9514–9519 CrossRef CAS PubMed.
- M. A. Mardones, A. H. Cowley, L. Contreras, R. A. Jones and C. J. Carrano, J. Organomet. Chem., 1993, 455, C1–C2 CrossRef CAS.
- O. Hegen, A. V. Virovets, A. Y. Timoshkin and M. Scheer, Chem.–Eur. J., 2018, 24, 16521–16525 CrossRef CAS PubMed.
- A. Moezzi, M. M. Olmstead, D. C. Pestana, K. Ruhlandt-Senge and P. P. Power, Main Group Chem., 1996, 1, 197–206 CrossRef CAS.
- M. A. Petrie, M. M. Olmstead, H. Hope, R. A. Bartlett and P. P. Power, J. Am. Chem. Soc., 1993, 115, 3221–3226 CrossRef CAS.
- H. Braunschweig, I. Fernández, G. Frenking and T. Kupfer, Angew. Chem., Int. Ed., 2008, 47, 1951–1954 CrossRef CAS PubMed.
- R. Appel, T. Gaitzsch and F. Knoch, Angew. Chem., Int. Ed., 1985, 24, 419–420 CrossRef.
- A. Kraft, J. Beck and I. Krossing, Chem.–Eur. J., 2011, 17, 12975–12980 CrossRef CAS PubMed.
- J. Zhou, L. L. Liu, L. L. Cao and D. W. Stephan, Angew. Chem., Int. Ed., 2019, 58, 5407–5412 CrossRef CAS PubMed.
- G. Knizia, J. Chem. Theory Comput., 2013, 9, 4834–4843 CrossRef CAS PubMed.
- D. W. N. Wilson, M. P. Franco, W. K. Myers, J. E. McGrady and J. M. Goicoechea, Chem. Sci., 2020, 11, 862–869 RSC.
- D. Auerhammer, M. Arrowsmith, H. Braunschweig, R. D. Dewhurst, J. O. C. Jiménez-Halla and T. Kupfer, Chem. Sci., 2017, 8, 7066–7071 RSC.
- S. K. Mellerup, Y. Cui, F. Fantuzzi, P. Schmid, J. T. Goettel, G. Bélanger-Chabot, M. Arrowsmith, I. Krummenacher, Q. Ye, V. Engel, B. Engels and H. Braunschweig, J. Am. Chem. Soc., 2019, 141, 16954–16960 CrossRef CAS PubMed.
- S. M. I. Al-Rafia, R. McDonald, M. J. Ferguson and E. Rivard, Chem.–Eur. J., 2012, 18, 13810–13820 CrossRef CAS PubMed.
- J. J. Clarke, P. Eisenberger, S. S. Piotrkowski and C. M. Crudden, Dalton Trans., 2018, 47, 1791–1795 RSC.
- D. Prieschl, M. Arrowsmith, M. Dietz, A. Rempel, M. Müller and H. Braunschweig, Chem. Commun., 2020, 56, 5681–5684 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2428369–2428375. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5sc01996f |
| ‡ Goicoechea and co-workers have reported the tert-butylisonitrile-induced isomerisation of a phosphaethynolatoborane (B–OCP) to a boraphosphaketene (B–PCO).70 Attempts to induce the isomerization of 2-L to 2′-L (L = SIMes, CAACMe) using a variety of small Lewis bases (e.g. isonitriles, phosphines, pyridines) led instead to intractable product mixtures. This is likely owed to the propensity of LBH2X systems, where L is a π-acidic carbene ligand, to undergo B-to-Ccarbene hydride migration and/or ring expansion of the NHC/carbene backbone by boron insertion.71–75 |
| § The borole boron nucleus of compound 8 could not be detected in the 11B NMR spectrum owing to excessive line broadening. |
| ¶ The 13C NMR resonance of the As- and B-bound borole carbon nucleus C2 was not detectable in the HMBC experiment. |
| This journal is © The Royal Society of Chemistry 2025 |