
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAromatic cation–π induced multifluorescence tunable two-dimensional co-assemblies for encoded information security†
Zhao
Gao‡
 a,
Jianxiang
Sun‡
a,
Lulu
Shi
a,
Wei
Yuan
b and
Wei
Tian
*a
a,
Jianxiang
Sun‡
a,
Lulu
Shi
a,
Wei
Yuan
b and
Wei
Tian
*a
aShaanxi Key Laboratory of Macromolecular Science and Technology, Xi'an Key Laboratory of Hybrid Luminescent Materials and Photonic Device, MOE Key Laboratory of Material Physics and Chemistry under Extraordinary Conditions, School of Chemistry and Chemical Engineering, Northwestern Polytechnical University, Xi'an 710072, P. R. China. E-mail: happytw_3000@nwpu.edu.cn
bDepartment of Chemistry, National University of Singapore, 3 Science Drive 3, Singapore 117543, Singapore
First published on 11th April 2025
Abstract
The field of light-emitting two-dimensional co-assemblies (2DCAs) is extending rapidly. Nevertheless, multifluorescence tunable 2DCAs are relatively underdeveloped, because the exploration of novel assembly strategies and noncovalent interactions to realize desirable photophysical features is still difficult. Herein, we present the first implementation of an aromatic cation–π interaction induced emissive charge transfer strategy for multifluorescence tunable 2DCAs, which are derived from fluorophore anthracene-based monomers and planar aromatic cations (pyrylium and tropylium). Benefiting from the aromatic cation–π interactions between anthracene and cationic guests, well-regulated 2DCAs are thus successfully obtained. The resultant 2DCAs exhibit a broadened fluorescence tunable range between blue-green and red emission colors, which is simply realized by varying the solvent ratio to turn on/off the aromatic cation–π emission charge transfer in the assembly/disassembly state of 2DCAs. On this basis, the programmable numbers, letters, patterns, and 3D codes with co-assembly encoded information security functions are successfully fabricated on papers, which would have a positive impact on developing supramolecular encryption materials.
Introduction
Luminescent materials, especially those with tunable wavelengths, have drawn great attention recently as promising candidates in sensors, photo-electric devices, and information protection.1–6 Developing new fluorescent compositions at a molecular level with structural diversity, stimuli responsiveness, and photostability is a key point to controlling emission and thus achieving the above applications.7–11 Traditional approaches to regulate luminescence mainly focused on chemical covalent modification of fluorescent chromophores to shift wavelengths.12,13 The method usually suffers from a lack of molecular structures and static emission properties. Moreover, considering the structure–luminescence relationships, desired topological structures are not easy to achieve for this method due to the lack of periodicity. Thus, it is highly desirable to develop advanced light-emitting materials with controlled structures and dynamic emission features.Two-dimensional co-assemblies (2DCAs), defined as lamellar aggregates formed by the co-assembly of two or more different kinds of molecules periodically organized based on noncovalent bonds in two orthogonal directions, have been extensively studied and employed to fabricate functional supramolecular materials.14–18 Although fluorescent supramolecular assemblies are well-known,19–21 multifluorescence tunable 2DCA materials are relatively underdeveloped. The construction of emissive 2DCAs needs the exploration of novel assembly strategies and noncovalent units to realize desirable photophysical properties. Compared to strong covalent bonds, noncovalent bonds such as hydrogen bonds, host–guest interactions, metal–metal interactions, and metal–ligand coordination are regarded as the ideal choices for building smart and well-organized 2DCAs due to their dynamic and self-corrected properties.22–28 Representatively, ubiquitous cation–π interactions between metal cations (K+, Na+, Mg2+, and Ca2+) or cationic amine and aromatic residues in proteins,29–33 have recently attracted researchers' interest in enabling 2D-ordered structures, because of their specific binding directivity and satisfactory association constants.29,30 Our group has recently exploited cation–π interactions to construct a series of 2D supramolecular polymers, showing great potential in the fields of catalysis and separation.34–37 Nonetheless, considering the inherent non-fluorescent nature of metal cations and weak emission and low-wavelength of aromatic residues,38,39 current cation–π units suffer from a non-radiative deactivation process. Hence, cation–π induced fluorescent 2DCAs with the desired wavelength and intensity are still elusive and need to be addressed urgently.
Planar aromatic cations such as pyrylium and tropylium, whose positive charges are delocalized throughout the aromatic rings to form stable π-electron-deficient acceptors, have been reported to be complexed with various electron-rich macrocyclic hosts.40 Benefiting from the narrow energy gap of the charge transfer photophysical process, the host–guest system could be visualized by the naked eye under natural light. Inspired by this, we envision that these planar aromatic cations could be regarded as promising candidates of cationic species for building multifluorescence tunable 2DCAs based on aromatic cation–π interactions. On the other hand, suitable π-conjugated fluorophore monomers are not easy to access. Thus, our group has been committed to the construction of two-component donor–acceptor co-assemblies derived from electron-rich anthracene derivatives and electron-deficient acceptors.34,41–43 Fortunately, the highest occupied molecular orbital (HOMO) of anthracene is near the lowest unoccupied molecular orbital (LUMO) of tropylium and pyrylium, which is the prerequisite for aromatic cation–π electron transitions and thus emissive charge transfer signals.44–47 Therefore, benefiting from these advantages, aromatic cation–π interactions between pyrylium or tropylium and anthracene are greatly promising to enable multifluorescence tunability in 2DCAs.
Based on the above considerations, we herein demonstrate multifluorescence tunable 2DCAs formed via an aromatic cation–π induced emissive charge transfer strategy. 2DCA-C1 and 2DCA-C2 are constructed by using the fluorophore anthracene monomer M1 and two kinds of planar aromatic cations pyrylium C1 and tropylium C2, respectively (Fig. 1). M1 contains an electron-rich 9,10-diethynylanthracene core and two sets of carbamate derivative arms at the termini, which only self-assemble into large aspect ratio nanorods. Upon addition of C1 or C2, it could combine with 9,10-diethynylanthracene of M1via aromatic cation–π interactions, while the terminal carbamate derivatives intermesh with each other. The well-regulated 2DCA-C1 and 2DCA-C2 are thus successfully obtained. More importantly, the tunable emissive charge transfer process is induced by the aromatic cation–π interactions between M1 and C1 or C2, enabling a broader fluorescence tunable range from blue-green to red compared to the self-assemblies of M1. On this basis, the expressive patterns and 3D code encryption system are fabricated on paper, with co-assembly encoded information security functions.
 | ||
| Fig. 1 The construction of aromatic cation–π induced multifluorescence tunable 2DCAs. (a) Chemical structures of monomer M1 and π-conjugated cations C1–C2. The counterion of C1–C2 is BF4−. (b) Schematic representation of the multifluorescence tunability by fuming/removing with good solvent vapor to turn on/off the aromatic cation–π emission charge transfer in the assembly/disassembly state of 2DCAs. | ||
Results and discussion
Morphologies and the proposed stacking mode of 2DCAs
The morphologies of the co-assembly between M1 and C1 were first explored. As shown in the transmission electron microscopy (TEM) and scanning electron microscopy (SEM) images, 2D nanosheets consisting of multiple nanorods connected side by side were observed for 2DCA-C1 (Fig. 2a, b and S1†). The atomic force microscopy (AFM) image also displayed the 2D structures (Fig. 2c). Height profile analysis of the selected area revealed that the average thickness was 5.0 nm (Fig. 2c, inset). The high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) and energy dispersive spectroscopy (EDS) mapping showed that the typical elements of C, N, O, B and F were uniformly distributed in the sample areas (Fig. 2d), proving the uniform mixture of M1 and C1. Similarly, for 2DCA-C2 constructed by using M1 and C2, well-defined 2D structures with a thickness of 5.1 nm were acquired (ESI Fig. S2†). However, in contrast to cationic C1 and C2, when M1 co-assembled with the similar electroneutral molecular mesitylene or cycloheptane, no 2D structures but 1D spindle-like nanorods were observed (ESI Fig. S3†). As for the morphologies of the self-assembly of M1, similar nanorods were obtained, which possess a large aspect ratio and are wide in the middle and narrow at both ends (ESI Fig. S4†). | ||
| Fig. 2 Morphological studies for 2DCAs. (a) and (b) TEM images of 2DCA-C1 and its amplified view. (c) AFM image and the height profile (inset) of 2DCA-C1. The height profile of the AFM image along the white line. (d) HAADF-STEM images and the corresponding EDS for elemental mapping of 2DCA-C1. The sample of 2DCA-C1 was obtained by slowly evaporating their MCH solution ([M1] = [C1] = 40 μM and 2 μM for TEM and AFM experiments, respectively). | ||
Further insights into the stacking mode of 2DCAs were then elucidated. The proposed molecular stacking of 2DCA-C1 was optimized by using the GFN2-xTB method.48 The aligned rodlike aggregates along the y-axis direction connected side by side in the x-axis direction (Fig. 3a). The 2D lamellae along the z-axis direction were formed by the stacking of the sliding aromatic layers (Fig. 3b). The central distances between the adjacent anthracene cores and C1 were calculated to be 4.54 and 3.42 Å (Fig. 3c), which is consistent with that measured results in powder X-ray diffraction (PXRD) experiments (Fig. 3d). Moreover, several strong peaks emerged in the 2θ = 21–32° region compared to that of the self-assembly of M1, indicating the diverse stacking structures in 2DCA-C1. Grazing incidence wide-angle X-ray scattering (GI-WAXS) measurements were further employed to confirm the orientation of 2DCA-C1. The intensities of out-of-plane (qz) Bragg peaks were larger than those of the in-plane ones (qxy) (Fig. 3e and S5†), reflective of a preferred orientation in the qz direction. Two strong diffraction projection areas emerging at qz = 1.27–1.48 Å−1 and 1.70–1.88 Å−1 manifested the horizontal orientation of aromatic stacking and ordered structures of 2DCA-C1. As for 2DCA-C2, a similar stacking mode was confirmed by PXRD, GI-WAXS, morphological analyses and theoretical modeling (Fig. 3d, S2 and S6†). Thus, these results proved that the elaborate monomer M1 could co-assemble with π-conjugated cationic guests to form the highly regular 2D lamellar aggregates.
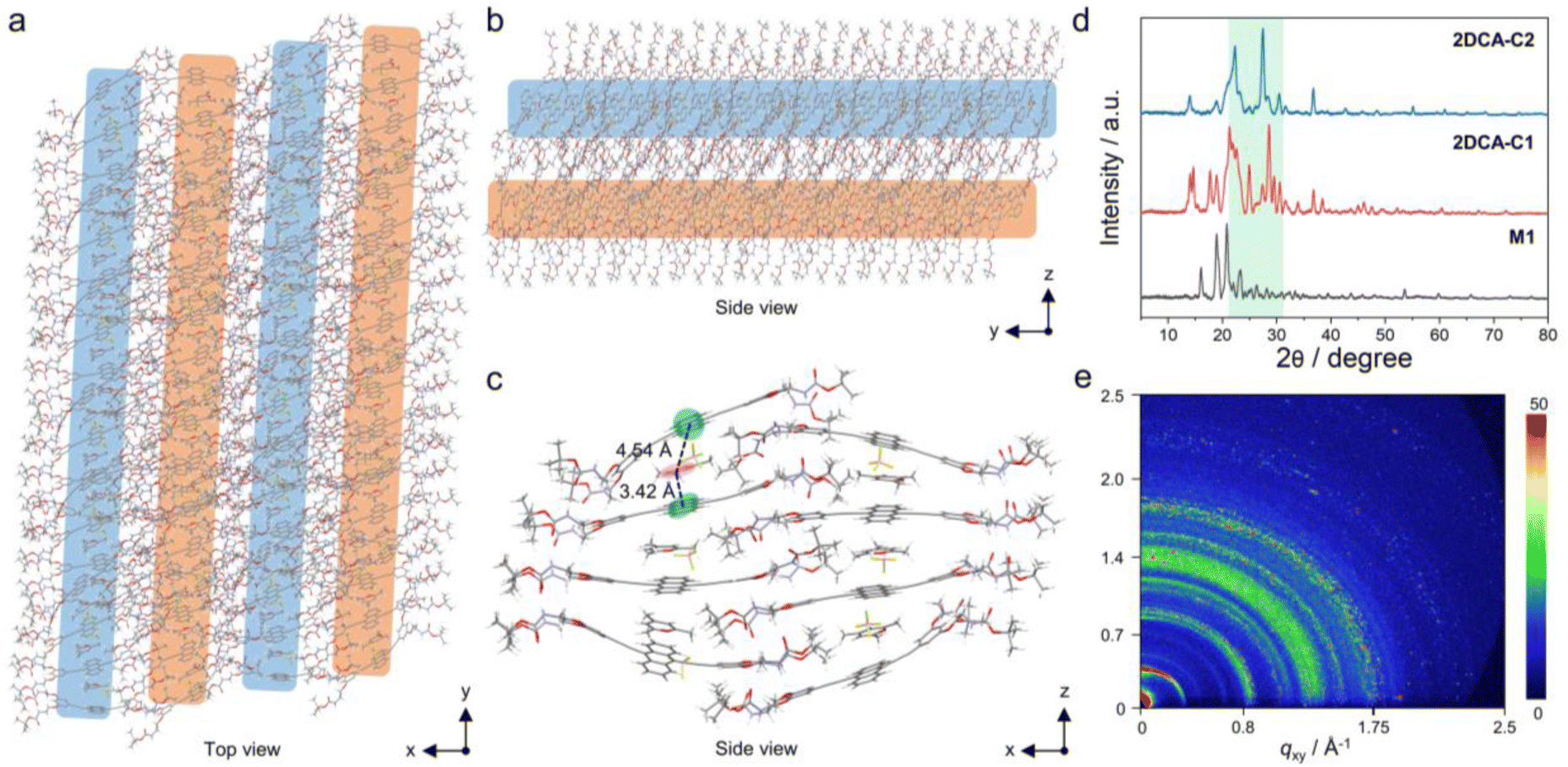 | ||
| Fig. 3 Stacking structures of 2DCA-C1. (a) Top view and (b)–(c) side view of the proposed co-assembly model of 2DCA-C1. The color ribbons in (a) and (b) represent the stacking of 9,10-diethynylanthracene of M1 and C1. Only stacks of eight M1 and six C1 are calculated via the GFN2-xTB method in (c) to give a clear view of the stacking of aromatic layers. (d) PXRD patterns of 2DCA-C1, 2DCA-C2 and M1. (e) GIWAXS pattern of 2DCA-C1 on a Si wafer. | ||
Formation of aromatic cation–π interactions in 2DCAs
Initially, spectral information for aromatic cation–π interactions in 2DCAs was obtained. As we know, chlorinated solvents such as CHCl3 and dichloroethane are common good solvents for π-conjugated neutral monomers,49,50 while polar solvents (methanol or ethanol) are considered as good solvents for cationic C1 and C2. We thus employed the mixed solvent of CHCl3/CH3OH (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v) as the good solvent for M1 and C1–C2. For M1 in dilute CHCl3/CH3OH (1:1, v/v, c = 40 μM), its UV-vis spectra showed two major bands in the regions of 300–325 nm and 375–500 nm with vibronic fine structures (Fig. 4a). With reference to the previous reports,51,52 these two bands separately belonged to the 1Lb and 1La bands of the anthracene moiety in a molecularly dissolved state. Upon adding equal-equivalent C1 in CHCl3/CH3OH (1:1, v/v) of M1, the UV-vis spectra and solution color of M1/C1 were identical to those of M1 (Fig. 4a and inset), indicating no intermolecular interactions but that physical blending occurred between M1 and C1 in the ground state. The same phenomena were observed for M1/C2.
:1, v/v) as the good solvent for M1 and C1–C2. For M1 in dilute CHCl3/CH3OH (1:1, v/v, c = 40 μM), its UV-vis spectra showed two major bands in the regions of 300–325 nm and 375–500 nm with vibronic fine structures (Fig. 4a). With reference to the previous reports,51,52 these two bands separately belonged to the 1Lb and 1La bands of the anthracene moiety in a molecularly dissolved state. Upon adding equal-equivalent C1 in CHCl3/CH3OH (1:1, v/v) of M1, the UV-vis spectra and solution color of M1/C1 were identical to those of M1 (Fig. 4a and inset), indicating no intermolecular interactions but that physical blending occurred between M1 and C1 in the ground state. The same phenomena were observed for M1/C2.
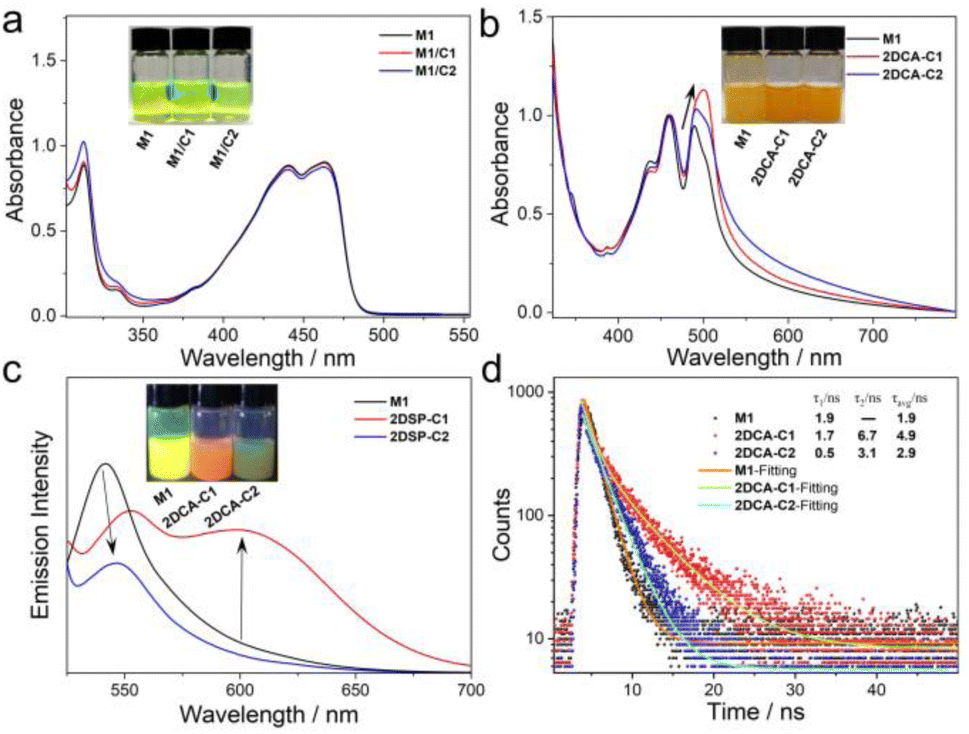 | ||
| Fig. 4 Photophysical investigations for aromatic cation–π interactions in 2DCA-C1 and 2DCA-C2. (a) UV-vis spectra of M1, M1/C1 and M1/C2 in CHCl3/CH3OH (1:1, v/v). c = 40 μM. (Inset) Photographs of M1, M1/C1 and M1/C2 under natural light. (b) UV-vis spectra, (c) fluorescence spectra and (d) fluorescence decay profiles of the self-assembly of M1, 2DCA-C1 and 2DCA-C2 in the poor solvent of MCH. c = 40 μM. Inset of (b) and (c): photographs of the self-assembly of M1, 2DCA-C1 and 2DCA-C2 under natural light and 365 nm UV light, respectively. λex = 500 nm. | ||
Upon switching the solvent from CHCl3/CH3OH (1:1, v/v) to hydrocarbon solvent of methylcyclohexane (MCH, the common poor solvent for rod-coil-type monomers to form aggregation),53,54 the maximum absorption and emission bands of M1 were all redshifted, indicating the supramolecular self-assembly of M1 (Fig. 4b and S7†). This phenomenon is consistent with that in our previously reported studies.41 To achieve effective co-assembly of M1 and C1, equimolar amounts of both compounds were first dissolved in CHCl3/CH3OH (1:1, v/v), followed by solvent evaporation to obtain a homogeneous solid. After the solid was suspended in MCH and subjected to ultrasonication for 3 minutes, 2DCA-C1 was then obtained. As shown in Fig. 4b, the maximum absorption band of M1 at 490 nm redshifted to 502 nm of 2DCA-C1. Simultaneously, 2DCA-C1 showed a structureless emission band located at 605 nm (absolute fluorescence quantum yield, ΦF = 3%), which has a relatively lower energy than the emission of individual M1 (λmax = 542 nm, ΦF = 9%) and C1 (Fig. 4c, S8 and S9†). The emission color of 2DCA-C1 turned from yellow to orange-red (Fig. 4c, inset). These phenomena are attributed to the aromatic cation–π charge transfer transition from the electron-donating π-core of M1 to electron-deficient cation C1 in 2DCA-C1. This conclusion was also confirmed by the time-resolved fluorescence decay profile. The lifetime of M1 was measured to be 1.9 ns, which increased to 4.9 ns for 2DCA-C1 (Fig. 4d), reflective of the charge transfer process. 2DCA-C1 exhibited a double-exponential fluorescence decay, which is attributed to the Sn → CT state transition (τ1) and the CT state → S0 transition (τ2). In contrast, the decay fitting curves revealed a single-exponential decay for M1, belonging to the Sn → S0 transition. The aromatic cation–π charge transfer transition in the analogue of 2DCA-C2 was well confirmed by the spectral characteristics (Fig. 4b–d).
After confirming the aromatic cation–π charge transfer transition of 2DCAs, we then sought to elucidate this non-covalent interaction at the molecular level. For 2DCA-C1 in CDCl3/CD3OH (1:1, v/v), the aromatic resonances of Ha–Hc on M1 and H1 on C1 hardly changed (Fig. 5a). Intriguingly, when switching the solvent to cyclohexane-d12 (the same poor solvent as MCH), the well-defined sharp signals of aromatic protons on 2DCA-C1 became broad and almost disappeared into the baseline (Fig. 5b(i)), reflective of the strong aggregation tendency. Upon increasing the temperature from 308 K to 338 K, the solution color of 2DCA-C1 varied from deep yellow to yellow-green (Fig. 5b, inset), indicating the transition from the aggregated state to the molecularly dissolved state. In the meantime, H1 on C1 shifted downfield, while Ha–Hb and NH protons on M1 shifted upfield (Fig. 5b). Such phenomena indicate the existence of complementary aromatic cation–π interactions between the electron-donating anthracene unit on M1 and the electron-withdrawing C1. The almost unchanged proton Hc also indirectly clarified that the binding site is only the anthracene core of M1.
 | ||
| Fig. 5 Deeper confirmation of the aromatic cation–π interactions. (a) Partial 1H NMR spectra (400 MHz, 298 K, 2 mM) of (i) M1, (ii) 2DCA-C1 and (iii) C1 in CDCl3/CD3OH (1:1, v/v). (b) Temperature-dependent 1H NMR spectra (400 MHz, 2 mM) of 2DCA-C1 in cyclohexane-d12 at (i) 308 K, (ii) 318 K, (iii) 328 K, and (iv) 338 K. (Inset) Photographs of 2DCA-C1 at 308 K (down) and 338 K (up) under natural light. (c) Calculated energy levels and the corresponding frontier molecular orbital diagrams of M1, M1/C1 and M1/C2. (d) Energy distribution mapping of optimized structures with color-mapped IGM isosurface graphs of M1/C1 and M1/C2. The green color isosurfaces in the structures represent the attraction forces. The color scale on the right side denotes the relative strength of noncovalent interactions. | ||
To gain deeper insights into the aromatic cation–π interactions, density functional theory (DFT) calculations were then performed. The simulated electron densities of HOMOs and LUMOs of M1 were evenly distributed throughout the molecules (Fig. 5c), leading to no electron transition. However, the LUMO of M1/C1 was completely localized on cation C1, while the HOMO was distributed on the monomer M1. This differentiation of electron density distribution manifested the occurrence of the aromatic cation–π interactions between M1 and C1. Besides, the calculated HOMO–LUMO gap decreased from M1 (2.66 eV, 466 nm) and C1 (5.22 eV, 237 nm, ESI Fig. S10†) to M1/C1 (2.35 eV, 528 nm), which is consistent with the red-shifted shoulder band in the spectral experiments (Fig. 4b). To directly observe the aromatic cation–π interactions between M1 and C1, the independent gradient model (IGM) method derived from DFT calculations was employed. The green isosurface region in the IGM graph was observed for M1/C1 (Fig. 5d), reflective of the relatively strong binding ability of aromatic cation–π interactions. Quantitatively, the binding energy of M1 and C1 was calculated to be −25.58 kJ mol−1. For analogue C2, it emerged that the aromatic protons downshift with M1 in CDCl3/CD3OH (2:1, v/v) at a high concentration of 20 mM (ESI Fig. S11a†). Moreover, upon increasing the temperature from 308 K to 338 K, Hb and NH protons on M1 shifted upfield, while Hd hardly changed (ESI Fig. S11b†), demonstrating that the aromatic cation–π interaction occurs at the anthracene core position. The calculated electron distribution and IGM results (binding energy: −23.69 kJ mol−1) for M1/C2 all supported the aromatic cation–π interactions between M1 and C2 (Fig. 5c and d).
To further elucidate the supramolecular polymerization mechanism of 2DCAs, temperature-dependent UV-vis spectroscopy experiments were performed. Upon gradually elevating temperature, the 1La bands of anthracene in 2DCA-C1 increased while the assembled bands decreased, accompanying two isosbestic points at 321 and 478 nm (ESI Fig. S12a†). The corresponding color of the solution changed from yellow to green. These phenomena manifest the transition from an assembled state to a molecularly dissolved state. When plotting the fraction of aggregated species (αagg) against temperature via monitoring the spectral variation at 490 nm, a non-sigmoidal curve was acquired (ESI Fig. S12b and S13†), which indicated that the supramolecular polymerization process of 2DCA-C1 follows a nucleation-elongation cooperative mechanism.55–57 The resultant melting curve fitted well with the Meijer–Schenning–van-der-Schoot mathematical model55 (ESI eqn (1) and (2)). A Te (critical elongation temperature) value of 355.1 K and Ka (dimensionless equilibrium constant of the activation step at Te) value of 1.3 × 10−5, together with an he (enthalpy release upon elongation) value of −86.7 kJ mol−1, were obtained. Furthermore, the number-averaged degree of polymerization in the elongation regime, averaged over all active species, 〈Nn〉58 was calculated to be approximately 2900 at room temperature (ESI eqn (3) and Fig. S14a†). Depending on the Van't Hoff plot, Gibbs free energy (ΔG) of 2DCA-C1 was determined to be −38.4 ± 1.8 kJ mol−1 (ESI Fig. S15†). The mechanistic insight into the co-assembly process of 2DCA-C1 was also validated via solvent-dependent UV-vis spectral measurements (ESI eqn (4)–(7) and Fig. S16†). These quantitative thermodynamic parameters reflect a highly cooperative supramolecular polymerization process of 2DCA-C1 (ESI Table S1†). For 2DCA-C2, it complied with the similar nucleation-elongation cooperative supramolecular polymerization mechanism (ESI Fig. S17 and Table S2†).
Aromatic cation–π induced multifluorescence tunability
As the aromatic cation–π interactions in 2DCAs resulted in fascinating emission variations, we then turned to investigate their multifluorescence tunable features. When the volume proportion of CHCl3/CH3OH (1:1, v/v) increased to 19% in the MCH solution of 2DCA-C1, the aromatic cation–π emission band at 605 nm decreased, while the emission band at 552 nm increased and blue-shifted (Fig. 6a). The emission color changed from red to yellow in turn (Fig. 6a, inset). The corresponding Commission Internationale de l’Eclairage (CIE) coordinates varied from (0.53, 0.40) of 0% to (0.42, 0.50) of 19% (Fig. 6c). ΦF values were varied from 3% at 605 nm to 8% at 552 nm. When the volume fraction of CHCl3/CH3OH (1:1, v/v) further increased to 29%, the emission maximum underwent the largest blue-shift to 512 nm with a ΦF value of 75%. The emission colors changed to green with CIE coordinates (0.30, 0.58) (Fig. 6b and c). On continuing to increase the CHCl3/CH3OH (1:1, v/v) content, the emission signals remained the same at the end. Throughout the titration process, the variation range of fluorescence spectra of 2DCA-C1 was up to 93 nm (monitoring the maximum band, Fig. 6d). Thus, 2DCA-C1 could readily realize the variation of emission colors from red to yellow to green by simply changing the ratio of solvents, with a wide fluorescence tunable range.
 | ||
| Fig. 6 Aromatic cation–π induced multifluorescence tunable features. Fluorescence spectral variations of the MCH solution of (a) and (b) 2DCA-C1 (40 μM, λex = 490 nm for the range of 0–19%; 460 nm for the range of 19–29%) and (e) and (f) 2DCA-C2 (40 μM, λex = 440 nm) upon gradually adding the isoconcentration CHCl3/CH3OH (1:1, v/v) solution of 2DCA-C1 and 2DCA-C2, respectively. The corresponding CIE chromaticity diagrams of (c) 2DCA-C1 and (g) 2DCA-C2. Plots of the maximum emission intensity and wavelength of (d) 2DCA-C1 and (h) 2DCA-C2versus the CHCl3/CH3OH (1:1, v/v) fraction in MCH. (Insets) Selected fluorescence photographs of (a) and (b) 2DCA-C1, (d) and (e) 2DCA-C2 under gradually varied solution conditions and a 365 nm UV lamp. | ||
The multifluorescence variation of 2DCA-C2 in MCH with different CHCl3/CH3OH (1:1, v/v) contents was also monitored. When the volume proportion of CHCl3/CH3OH (1:1, v/v) increased to 20%, the emission band at 541 nm strongly increased while the emission band at 481 nm decreased and red-shifted (Fig. 6e). Interestingly, as the volume fraction of CHCl3/CH3OH (1:1, v/v) further increased, the emission band at 541 nm did not increase but decreased gradually, and eventually the emission centered at 512 nm dominated (Fig. 6f). The emission colors of 2DCA-C2 varied from blue to yellow and finally to green (Fig. 6e and f, insets). The corresponding CIE coordinates turned from (0.19, 0.31) through (0.35, 0.45) to (0.21, 0.69) (Fig. 6g). The fluorescence spectra of 2DCA-C2 could vary up to 64 nm (Fig. 6h). Notably, the emission of 2DCA-C2 showed a red shift followed by a blue shift, which probably arises from the superposition of C2 self-aggregation induced emission (ESI Fig. S9d†) and the weak fluorescence of 2DCA-C2. For M1, the fluorescence tunability was also observed, with the emission color changing from yellow to green (ESI Fig. S18†). Additionally, in order to study the effect of anions, we separately obtained other C1-PF6− and C2-Cl− with different counterions by ion exchange from the corresponding compounds C1-BF4− and C2-BF4−. It is found that the multifluorescence tunable features are seldom affected by the counterions of C1 and C2 (ESI Figs. S19 and S20†).
The deeper insight into the multifluorescence tunable mechanism induced by aromatic cation–π interaction in 2DCAs was carefully elucidated. First, the equivalent ratio of CHCl3/CH3OH (1:1, v/v) to 2DCA-C1 in the fluorochromic process was quantitatively determined. A small amount of the CHCl3/CH3OH (1:1, v/v) solution of 2DCA-C1 (8.4 mg, 2 mg mL−1) was dropped onto a filter paper (Fig. 7a, inset). Upon slowly volatilizing the solvent vapor in air, the emission color turned from green to red. The emission signal at 487 nm sharply decreased, and then reached a plateau at around 180 s (Fig. 7a and S21,† the experiments were repeated three times), indicating that the residual solvent at this time is the necessary minimum amount for the vapo-fluorochromic experiments. Simultaneously, the weight change of 2DCA-C1 sample versus the volatilization time was investigated under the same conditions (Fig. 7b). At 180 s, the weight of the residual solvent was measured to be around 3.3 mg, which is 2098 times the molar ratio of 2DCA-C1. Moreover, the fluorescence lifetime of 2DCA-C1 in the solid state decreased from 4.8 ns to 3.3 ns and 2.1 ns after adding 10% and 24% volume of CHCl3/CH3OH (1:1, v/v), respectively (Fig. 7c). These phenomena were attributed to that monomers M1 gradually dominated the emission bands and the aromatic cation–π charge transfer emission disappeared after adding CHCl3/CH3OH (1:1, v/v). Furthermore, the morphological evolution of 2DCA-C1 was analyzed. Upon addition of 5% volume of CHCl3/CH3OH (1:1, v/v) into the MCH solution of 2DCA-C1, the contour of the 2D nanoribbons became blurred (Fig. 7d(i and ii)). Thereafter, 2DCA-C1 was gradually destroyed and its morphology changed to small-sized nanoaggregates (Fig. 7d(iii and iv)). The results indicated that the dissociation of 2DCA-C1 was caused by CHCl3/CH3OH (1:1, v/v). The conclusion was further verified by molecular dynamics (MD) simulations. 2DCA-C1 containing eight monomers 1 and six C1 molecules operated dynamically based on a general AMBER force field at 298 K for 50 ns to equilibrate the initial configuration (Fig. 7e(i)). Upon adding CH2Cl2/CH3OH (1:1, v/v) into 2DCA-C1, the fragments of the initial well-stacked structure were scattered throughout the space (Fig. 7e(ii)). We then exploited the 1H NMR spectral experiments to confirm the solvent-induced disassembly of 2DCA-C1. Upon gradual addition of CDCl3/CD3OH (1:1, v/v) into the cyclohexane-d12 solution of 2DCA-C1, the broad and submerged bands changed to well-defined sharp signals (ESI Fig. S22†), which indicated that the co-assembled 2DCA-C1 turned to molecularly dissolved M1 and C1. We presented a simplified schematic representation for the involved photo-physics pathways to illustrate the effect of solvent vapor on the multifluorescence tunable features (ESI Fig. S23†). Thus, it is unambiguously concluded that the multifluorescence tunability could be easily achieved with the utilization of good solvent by turning on/off the aromatic cation–π emission charge transfer in the assembly/disassembly state of 2DCAs.
 | ||
| Fig. 7 Multifluorescence tunable mechanism studies. (a) Fluorescence spectra and (b) the corresponding weight variation of 2DCA-C1versus the volatilization time of CHCl3/CH3OH (1:1, v/v) at room temperature. Inset of (a): schematic illustration of the sample preparation for quantitatively determining the molar stoichiometry of CHCl3/CH3OH (1:1, v/v) in the vapo-fluorochromic experiments. (c) Fluorescence decay profiles of 2DCA-C1 in MCH upon addition of 0%, 10%, and 24% volumes of CHCl3/CH3OH (1:1, v/v). (d) TEM images of 2DCA-C1 in MCH upon addition of (i) 0%, (ii) 5%, (iii) 10%, and (iv) 24% volumes of CHCl3/CH3OH (1:1, v/v). (e) The stacking modes of 2DCA-C1 (i) in benzene and (ii) in CHCl3/CH3OH (1:1, v/v) obtained from MD simulations at the final state. | ||
Similarly, the multifluorescence tunable mechanism investigation for 2DCA-C2 was also carefully conducted (ESI Fig. S24†). These results indicated that aromatic cation–π emission charge transfer was essential for multifluorescence tunability in 2DCA-C2. In sharp contrast, for the similar electroneutral molecular mesitylene and cycloheptane, no multifluorescence phenomena were observed when mixing with M1, as evidenced by the almost unchanged emission band in the fluorescence spectra (ESI Fig. S25 and S26†). Hence, the aromatic cation–π induced multifluorescence tunability would provide guidance for comprehension of a straightforward approach toward multifluorescence materials.
Co-assembly encoded information security
Benefiting from the aromatic cation–π induced multifluorescence tunable features, we envisioned that the resultant 2DCAs could be used in the field of information security.59,60 In detail, M1 with or without C2 first pre-dissolved in CH2Cl2/CH3OH (1:1, v/v), and the solids were then obtained after evaporating the solvent (Fig. 8a). Upon suspension of the mixture in MCH and ultrasonication for 3 min, the self-assembly of M1 and co-assembly of 2DCA-C2 were obtained. Loading M1 and/or 2DCA-C2 in the given position of the square papers, an encrypted yellow number “8” was assembled under natural light. The encrypted information could be decrypted under UV light. By adjusting the position of M1 and 2DCA-C2, it is easy to achieve the variation from number “0” to “9”. Besides, M1 and 2DCA-C1 with contrasting emission colors could be arranged in a 3-by-5 square unit, presenting the recognized letters of “OPTICAL PROPERTIES” under UV light (Fig. 8b).
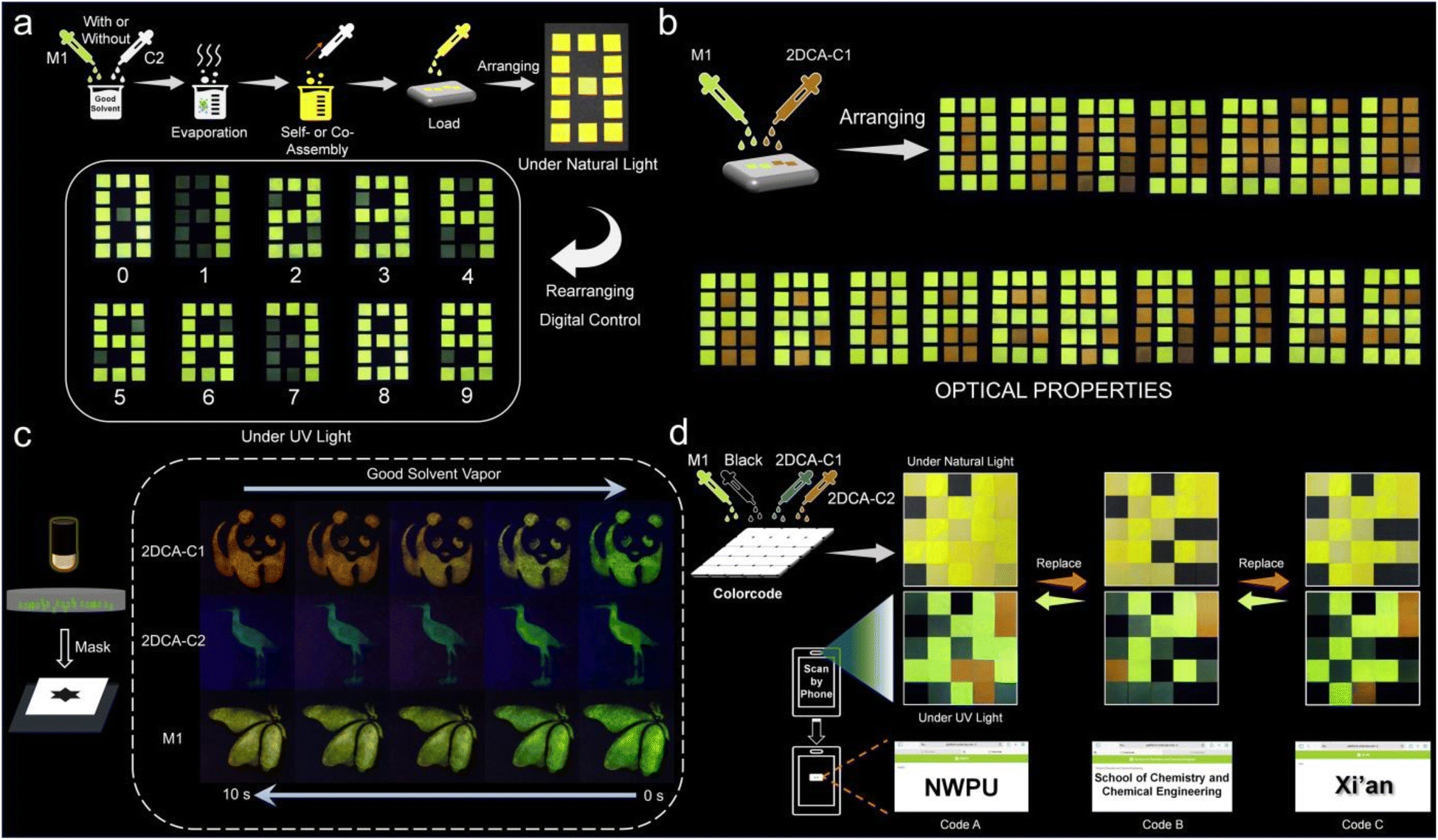 | ||
| Fig. 8 Encoded information security application. (a) Schematic diagram of the arrayed luminescent Arabic numbers derived from M1 and C2. (b) Photographs of the arrayed letters of “OPTICAL PROPERTIES” derived from M1 and 2DCA-C1. (c) Photographs of the assembly-encoded vapo-fluorochromic behaviors of the patterns of “panda”, “bird” and ‘‘butterfly’’ derived from 2DCA-C1, 2DCA-C2 and M1, respectively. (d) Schematic illustration of the prepared multifluorescence 3D codes (size: 5 × 5 cm) derived from M1, 2DCA-C1, 2DCA-C2 and commercial black inks. | ||
Notably, considering that 2DCA-C1, 2DCA-C2 and M1 provided fluorescence color switching between the molecularly dissolved state in good solvent and the aggregated state in the solid (ESI Fig. S27†), various assembly-encoded vapo-fluorochromic patterns have been fabricated via a mask method. For instance, on dropping the MCH solution of 2DCA-C1 on a non-fluorescent paper across a mask plate, a red “panda” pattern was obtained, which is not clear under natural light yet discernible under UV light (Fig. 8c). When this pattern was fumed with CH2Cl2 vapor, the emission color gradually changed to green. Removing the vapor, the red pattern quickly recovered within ten seconds. Likewise, 2DCA-C2 and M1 underwent the assembly-encoded vapo-fluorochromic process. The “bird” pattern derived from 2DCA-C2 could be switched between blue-green and green. The “butterfly” pattern painted from M1 was switched between yellow and green.
Furthermore, a multifluorescence 3D code information encryption system was established. M1, 2DCA-C1, and 2DCA-C2, in combination with commercial black ink, were loaded separately into a 5-by-5 square cell on paper. The prepared 3D code was in the encrypted state under natural light, as the icon failed to be recognized by the application software COLORCODE® on a smartphone (Fig. 8d). When the 3D code was exposed to UV light, the multifluorescence cells emerged at the per-set coordinates. The information of the letters of “NWPU” was then decoded by using a smartphone. Interestingly, according to 3D code theory, the encoded information could only be read when the given multifluorescence cells appear at the right positions. Giving different emission colors to 3D code, different encoding information (Code B: School of Chemistry and Chemical Engineering; Code C: Xi'an) was decoded under UV light. We then performed the fatigue tests on the 3D code patterns, which showed the maintained emission after five cycles (ESI Fig. S28†). The obtained patterns also exhibited good stability under ambient conditions, since the emission colors were scarcely changed for at least two months (ESI Fig. S29†).
Conclusions
In summary, we have successfully obtained the multifluorescence tunable 2DCAs derived from the planar aromatic cations C1–C2 and monomer M1via the aromatic cation–π induced emissive charge transfer approach. The ordered 2D structures of 2DCAs were formed, driven by aromatic cation–π interactions following a nucleation-elongation cooperative mechanism. Furthermore, compared with the self-assemblies of M1, the corresponding co-assemblies of 2DCAs possess a broader fluorescence tunable range by simply varying the solvent ratio to turn on/off the aromatic cation–π emission charge transfer. Benefiting from the multifluorescence switching of 2DCAs between the molecular dissolved state in good solvent and the aggregated state in the solid, a variety of programmable patterns with encoded information encryption functions were prepared on paper. This work provides an important breakthrough in designing luminescent supramolecular assemblies and therefore, will pave the way toward fabricating advanced information protection materials.Data availability
Detailed synthetic procedures, analytical data, and computational methods are provided in the ESI.†Author contributions
Z. G. and W. T. conceived the idea for this project. J. S. and L. S. performed the experiments, analyzed the data, and produced the artwork under the direction of Z. G. and W. T. Z. G. and W. T. wrote the paper. W. Y. was involved in data interpretation. All authors contributed to the manuscript preparation.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (22371230, 22471219 and 22071197), the Postdoctoral Science Foundation of China (2023M732855 and 2022TQ0258), the Shaanxi Fundamental Science Research Project for Chemistry & Biology (22JHQ020 and 23JHZ002), the Fundamental Research Funds for the Central Universities (G2024KY0605 and D5000230114), and the Open Testing Foundation of the Analytical & Testing Center of Northwestern Polytechnical University (2023T013).Notes and references
- H. Wang, X. Ji, Z. Li and F. Huang, Adv. Mater., 2017, 29, 1606117 CrossRef PubMed.
- X. Wang, Z. Gao and W. Tian, Chin. Chem. Lett., 2024, 35, 109757 CrossRef CAS.
- J. Guo, C. Yang and Y. Zhao, Acc. Chem. Res., 2022, 55, 1160–1170 CrossRef CAS PubMed.
- Y. Duo, Y. Yang, T. Xu, R. Zhou, R. Wang, G. Luo and B. Z. Tang, Coord. Chem. Rev., 2023, 482, 215070 CrossRef CAS.
- F. Xu and B. L. Feringa, Adv. Mater., 2023, 35, 2204413 CrossRef CAS PubMed.
- C. Yin, Z.-A. Yan and X. Ma, Chem. Commun., 2023, 59, 13421–13433 RSC.
- X. Yan, T. R. Cook, P. Wang, F. Huang and P. J. Stang, Nat. Chem., 2015, 7, 342–348 CrossRef CAS PubMed.
- Q. Wang, Q. Zhang, Q.-W. Zhang, X. Li, C.-X. Zhao, T.-Y. Xu, D.-H. Qu and H. Tian, Nat. Commun., 2020, 11, 158 CrossRef PubMed.
- V. K. Praveen, B. Vedhanarayanan, A. Mal, R. K. Mishra and A. Ajayaghosh, Acc. Chem. Res., 2020, 53, 496–507 CrossRef CAS PubMed.
- P.-P. Jia, L. Xu, Y.-X. Hu, W.-J. Li, X.-Q. Wang, Q.-H. Ling, X. Shi, G.-Q. Yin, X. Li, H. Sun, Y. Jiang and H.-B. Yang, J. Am. Chem. Soc., 2021, 143, 399–408 CrossRef CAS PubMed.
- J. Song, L. Ma, S. Sun, H. Tian and X. Ma, Angew. Chem., Int. Ed., 2022, 61, e202206157 CrossRef CAS PubMed.
- S. K. Yang, X. Shi, S. Park, T. Ha and S. C. Zimmerman, Nat. Chem., 2013, 5, 692–697 CrossRef CAS PubMed.
- D. Tan, J. Dong, T. Ma, Q. Feng, S. Wang and D.-T. Yang, Angew. Chem., Int. Ed., 2023, 62, e202304711 CrossRef CAS PubMed.
- M. Pfeffermann, R. Dong, R. Graf, W. Zajaczkowski, T. Gorelik, W. Pisula, A. Narita, K. Müllen and X. Feng, J. Am. Chem. Soc., 2015, 137, 14525–14532 CrossRef CAS PubMed.
- R. Dong, M. Pfeffermann, H. Liang, Z. Zheng, X. Zhu, J. Zhang and X. Feng, Angew. Chem., Int. Ed., 2015, 54, 12058–12063 CrossRef CAS PubMed.
- R. Dong, T. Zhang and X. Feng, Chem. Rev., 2018, 118, 6189–6235 CrossRef CAS PubMed.
- L. Hu, X. Zhu, C. Yang and M. Liu, Angew. Chem., Int. Ed., 2022, 61, e202114759 CrossRef CAS PubMed.
- N. Sasaki, J. Kikkawa, Y. Ishii, T. Uchihashi, H. Imamura, M. Takeuchi and K. Sugiyasu, Nat. Chem., 2023, 15, 922–929 CrossRef CAS PubMed.
- G. R. Whittell, M. D. Hager, U. S. Schubert and I. Manners, Nat. Mater., 2011, 10, 176–188 CrossRef CAS PubMed.
- L. Maggini and D. Bonifazi, Chem. Soc. Rev., 2012, 41, 211–241 RSC.
- X. Wang, Z. Gao and W. Tian, Chem. Sci., 2024, 15, 11084 RSC.
- J. Chen, E. Zhu, J. Liu, S. Zhang, Z. Lin, X. Duan, H. Heinz, Y. Huang and J. J. De Yoreo, Science, 2018, 362, 1135–1139 CrossRef CAS PubMed.
- P. Li, M. R. Ryder and J. F. Stoddart, Acc. Mater. Res., 2020, 1, 77–87 CrossRef CAS.
- S. Dhiman, R. Ghosh, S. Sarkar and S. J. George, Chem. Sci., 2020, 11, 12701–12709 RSC.
- Z. Chen and V. W.-W. Yam, Angew. Chem., Int. Ed., 2020, 59, 4840–4845 CrossRef CAS PubMed.
- B. Yang, S.-B. Yu, P.-Q. Zhang, Z.-K. Wang, Q.-Y. Qi, X.-Q. Wang, X.-H. Xu, H.-B. Yang, Z.-Q. Wu, Y. Liu, D. Ma and Z.-T. Li, Angew. Chem., Int. Ed., 2021, 60, 26268–26275 CrossRef CAS PubMed.
- H.-Y. Lin, Y.-T. Wang, X. Shi, H.-B. Yang and L. Xu, Chem. Soc. Rev., 2023, 52, 1129–1154 RSC.
- Z. Gao, J. Sun, L. Shi, W. Yuan, H. Yan and W. Tian, Angew. Chem., Int. Ed., 2025, 64, e202423174 CrossRef CAS PubMed.
- J. C. Ma and D. A. Dougherty, Chem. Rev., 1997, 97, 1303–1324 CrossRef CAS PubMed.
- A. S. Mahadevi and G. N. Sastry, Chem. Rev., 2013, 113, 2100–2138 CrossRef CAS PubMed.
- H. Fan, J. Wang, Z. Tao, J. Huang, P. Rao, T. Kurokawa and J. Ping Gong, Nat. Commun., 2019, 10, 5127 CrossRef PubMed.
- G. Zhao and H. Zhu, Adv. Mater., 2020, 32, 1905756 CrossRef CAS PubMed.
- F. Hu, W. Hao, D. Mücke, Q. Pan, Z. Li, H. Qi and Y. Zhao, J. Am. Chem. Soc., 2021, 143, 5636–5642 CrossRef CAS PubMed.
- X. Xiao, H. Chen, X. Dong, D. Ren, Q. Deng, D. Wang and W. Tian, Angew. Chem., Int. Ed., 2020, 59, 9534–9541 CrossRef CAS PubMed.
- Z. Gao, L. Shi, F. Yan, Y. Han, W. Yuan and W. Tian, Angew. Chem., Int. Ed., 2023, 62, e202302274 CrossRef CAS PubMed.
- W. Chen, Z. Chen, Y. Chi and W. Tian, J. Am. Chem. Soc., 2023, 145, 19746–19758 CrossRef CAS.
- H. Huo, X. Xiao, L. Chang, X. Xiong, M. Shi, J. Wang and W. Tian, Sci. China:Chem., 2023, 66, 2070–2082 CrossRef CAS.
- R. Z. Lange, G. Hofer, T. Weber and A. D. Schlüter, J. Am. Chem. Soc., 2017, 139, 2053–2059 CrossRef CAS.
- G. Chang, L. Yang, J. Yang, M. P. Stoykovich, X. Deng, J. Cui and D. Wang, Adv. Mater., 2018, 30, 1704234 CrossRef PubMed.
- X.-Y. Pang, H. Zhou, H. Yao and W. Jiang, Org. Chem. Front., 2021, 8, 5265–5270 RSC.
- Z. Gao, F. Yan, S. Qiu, Y. Han, F. Wang and W. Tian, Chem. Commun., 2020, 56, 9214–9217 RSC.
- Z. Gao, S. Qiu, F. Yan, S. Zhang, F. Wang and W. Tian, Chem. Sci., 2021, 12, 10041–10047 RSC.
- B. Mu, Z. Gao, C. Liu, X. Xiao and W. Tian, Chem. Commun., 2023, 59, 5514–5530 RSC.
- J. Han, D. Yang, X. Jin, Y. Jiang, M. Liu and P. Duan, Angew. Chem., Int. Ed., 2019, 58, 7013–7019 CrossRef CAS PubMed.
- S. Garain, S. N. Ansari, A. A. Kongasseri, B. C. Garain, S. K. Pati and S. J. George, Chem. Sci., 2022, 13, 10011–10019 RSC.
- J.-R. Wu, G. Wu, D. Li and Y.-W. Yang, Angew. Chem., Int. Ed., 2023, 62, e202218142 CrossRef CAS PubMed.
- F. Wang, R. Liao and F. Wang, Angew. Chem., Int. Ed., 2023, 62, e202305827 CrossRef CAS PubMed.
- S. Grimme, C. Bannwarth and P. Shushkov, J. Chem. Theory Comput., 2017, 13, 1989–2009 CrossRef CAS PubMed.
- J. Matern, I. Maisuls, C. A. Strassert and G. Fernández, Angew. Chem., Int. Ed., 2022, 61, e202208436 CrossRef CAS PubMed.
- Z. Gao, F. Yan, L. Shi, Y. Han, S. Qiu, J.-A. Zhang, F. Wang, S. Wu and W. Tian, Chem. Sci., 2022, 13, 7892–7899 RSC.
- S. Yamane, Y. Sagara and T. Kato, Chem. Commun., 2013, 49, 3839–3841 RSC.
- Y. Li, M. E. Köse and K. S. Schanze, J. Phys. Chem. B, 2013, 117, 9025–9033 CrossRef CAS PubMed.
- R. Liao, F. Wang, Y. Guo, Y. Han and F. Wang, J. Am. Chem. Soc., 2022, 144, 9775–9784 CrossRef CAS PubMed.
- R. M. Veedu, N. Niemeyer, N. Bäumer, K. K. Kalathil, J. Neugebauer and G. Fernández, Angew. Chem., Int. Ed., 2023, 62, e202314211 CrossRef PubMed.
- P. A. Korevaar, C. Schaefer, T. F. A. de Greef and E. W. Meijer, J. Am. Chem. Soc., 2012, 134, 13482–13491 CrossRef CAS PubMed.
- S. Sarkar, A. Sarkar, A. Som, S. S. Agasti and S. J. George, J. Am. Chem. Soc., 2021, 143, 11777–11787 CrossRef CAS PubMed.
- J. Matern, Z. Fernández, N. Bäumer and G. Fernández, Angew. Chem., Int. Ed., 2022, 61, e202203783 CrossRef CAS PubMed.
- M. M. J. Smulders, A. P. H. J. Schenning and E. W. Meijer, J. Am. Chem. Soc., 2008, 130, 606–611 CrossRef CAS PubMed.
- Q. Wang, B. Lin, M. Chen, C. Zhao, H. Tian and D.-H. Qu, Nat. Commun., 2022, 13, 4185 CrossRef CAS PubMed.
- Y.-X. Hu, X. Hao, D. Wang, Z.-C. Zhang, H. Sun, X.-D. Xu, X. Xie, X. Shi, H. Peng, H.-B. Yang and L. Xu, Angew. Chem., Int. Ed., 2023, 62, e202315061 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5sc00007f |
| ‡ Z. Gao and J. Sun contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |