
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLi3.6In7S11.8Cl: an air- and moisture-stable superionic conductor†
Ifeoluwa P.
Oyekunle
 ac,
Erica
Truong
ac,
Tej P.
Poudel
bc,
Yudan
Chen
ac,
Yongkang
Jin
ac,
Islamiyat A.
Ojelade
ac,
Michael J.
Deck
ac,
Bright
Ogbolu
ac,
Md. Mahinur
Islam
ac,
Pawan K.
Ojha
ac,
J. S. Raaj
Vellore Winfred
a,
Dewen
Hou
d,
Hui
Xiong
e,
Chen
Huang
f and
Yan-Yan
Hu
*abc
ac,
Erica
Truong
ac,
Tej P.
Poudel
bc,
Yudan
Chen
ac,
Yongkang
Jin
ac,
Islamiyat A.
Ojelade
ac,
Michael J.
Deck
ac,
Bright
Ogbolu
ac,
Md. Mahinur
Islam
ac,
Pawan K.
Ojha
ac,
J. S. Raaj
Vellore Winfred
a,
Dewen
Hou
d,
Hui
Xiong
e,
Chen
Huang
f and
Yan-Yan
Hu
*abc
aDepartment of Chemistry and Biochemistry, Florida State University, Tallahassee, FL 32306, USA. E-mail: yhu@fsu.edu
bMaterials Science and Engineering Program, Florida State University, Tallahassee, FL 32306, USA
cCenter of Interdisciplinary Magnetic Resonance, National High Magnetic Field Laboratory, Tallahassee, FL 32310, USA
dCenter for Nanoscale Materials Argonne National Laboratory, 9700 S Cass Ave, Lemont, IL 60439, USA
eMicron School of Materials Science and Engineering, Boise State University, Boise, ID 83725, USA
fDepartment of Scientific Computing, Florida State University, Tallahassee, FL 32306, USA
First published on 8th May 2025
Abstract
All-solid-state batteries (ASSBs) have drawn significant attention as future energy storage technologies. Sulfide-based solid electrolytes are promising due to their high ionic conductivity and favorable mechanical properties. However, their reactivity with moisture, leading to decomposition and release of toxic gases such as H2S, poses health and safety risks. In this study, a superionic conductor, Li3.6In7S11.8Cl, which exhibits high structural stability in the presence of water and air, is synthesized. At 25 °C, Li3.6In7S11.8Cl delivers an ionic conductivity of 1.1 mS cm−1, reaching 4.2 mS cm−1 post-exposure to humid air. Multimodal investigations reveal that trapped water inside the Li3.6In7S11.8Cl pellet facilitates ion conduction, which can be reversibly removed without compromising the structural integrity. The structure features a cubic-closed-packed anion sublattice with Li+ ions diffusing via a three-dimensional isotropic network, confirmed by ab initio molecular dynamics simulations. 6Li NMR and relaxometry identify the Wyckoff 16c and 8a as active Li+ sites for ion conduction. The high ionic conductivity, long-term stable cycling performance, and moisture stability of Li3.6In7S11.8Cl make it a preferable electrolyte candidate for high-performance ASSBs.
Introduction
Recent advancements in battery technology have led to the emergence of all-solid-state batteries, which address critical safety concerns associated with conventional Li-ion batteries.1,2 Unlike their liquid-based counterparts, all-solid-state batteries eliminate the risk of electrolyte leakage and ignition attributed to flammable organic solvents.1–5 Consequently, all-solid-state batteries are increasingly recognized as an advantageous alternative to traditional liquid-based systems.3,5–8 Solid electrolytes (SEs) are indispensable for advancing all-solid-state batteries – engendering a need of favorable characteristics for solid electrolytes possessing. Ideally, a solid-state electrolyte should possess compatibility with electrodes, good mechanical properties, low electronic conductivity, good moisture stability, and high ionic conductivity.9Sulfide solid electrolytes are favored due to their high ionic conductivity, rivaling conventional liquid electrolytes.10,11 The lower electronegativity of sulfur, compared to oxygen, weakens the interaction with lithium ions and enhances their mobility within the lattice. Additionally, the larger ionic radius of sulfur creates wider migration pathways for lithium ions, further facilitating their transport.12 Consequently, sulfide solid electrolytes exhibit improved ionic conductivities, rendering them highly promising for all-solid-state batteries.13
However, despite their high ionic conductivity, sulfide electrolytes suffer from poor air and moisture stability,14 causing chemical decomposition and releasing toxic gases such as H2S. Consequently, handling SSEs necessitates stringent safety measures, such as inert atmosphere for preparation and storage.9,12,15–17 This limits their potential for wide-scale applications.9 According to the hard and soft acids and bases (HSAB) theory, phosphorus (a hard acid) in thiophosphates prefers to react with oxygen (a hard base) compared to sulfur (a soft base).17,18 This results in oxygen replacing sulfur during exposure to moisture, leading to rapid hydrolysis of the thiophosphate materials.7,18 Consequently, effective strategies to evaluate and suppress this hydrolysis reaction are critical for the development of stable sulfide-based electrolytes for large-scale applications.18
In this study, we have synthesized a fast ion-conducting thioindate solid electrolyte, Li3.6In7S11.8Cl, with conductivity reaching 1.1 mS cm−1 in the pristine state and 4.2 mS cm−1 when exposed to moisture. We employed a combined approach, utilizing solid-state NMR, synchrotron XRD, and electrochemical impedance spectroscopy (EIS), to characterize the Li+ dynamics and elucidate the short- and long-range structures of Li3.6In7S11.8Cl. Additionally, scanning electron microscopy (SEM) analysis with elemental mapping was performed, in conjunction with multinuclear NMR and XRD, to evaluate the structure integrity of Li3.6In7S11.8Cl upon exposure to humid air. Li3.6In7S11.8Cl demonstrates enhanced moisture stability, likely owing to the strong covalent interaction between In3+ and S2−; this strong interaction effectively prevents oxygen from reacting with In3+ upon exposure to air or moisture, resulting in improved chemical stability of Li3.6In7S11.8Cl. This work provides a robust strategy to improve the stability of sulfide superionic conductors in water and air.
Experimental
Material synthesis
LiCl (Sigma-Aldrich) and LiBr (Sigma-Aldrich) were dried under vacuum at 200 °C for 12 hours before synthesis. Li2S (Alfa Aesar) and In2S3 (Sigma-Aldrich) were received and used without further purification. Stoichiometric amounts of Li2S, In2S3, and LiCl were ground using an agate mortar and pestle in a Li![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :In:S:Cl mole ratio of 4:7:12:1 for 5 minutes. After grinding, the hand-milled powder was transferred into a ZrO2 jar containing two 10-mm balls as a grinding aid. Mechanochemical mixing of the hand-milled powder in a ZrO2 jar sealed under a vacuum was performed using a SPEX® 8000M MIXER/MILL high energy ball mill (SPEX®SamplePrep, USA) for 5 hours. Afterward, the ball-milled powder, typically 100–200 mg, was pressed into a 6-nm pellet under pressure of ∼400 psi inside an argon-filled glovebox. The pellet was transferred into a quartz tube and sintered at 500 °C for 12 hours with a temperature ramping rate of 5 °C min−1, followed by natural cooling under Argon. The resulting pellet had a thickness varying from ∼1 mm to 2 mm, and the pellet appeared light yellow.
:In:S:Cl mole ratio of 4:7:12:1 for 5 minutes. After grinding, the hand-milled powder was transferred into a ZrO2 jar containing two 10-mm balls as a grinding aid. Mechanochemical mixing of the hand-milled powder in a ZrO2 jar sealed under a vacuum was performed using a SPEX® 8000M MIXER/MILL high energy ball mill (SPEX®SamplePrep, USA) for 5 hours. Afterward, the ball-milled powder, typically 100–200 mg, was pressed into a 6-nm pellet under pressure of ∼400 psi inside an argon-filled glovebox. The pellet was transferred into a quartz tube and sintered at 500 °C for 12 hours with a temperature ramping rate of 5 °C min−1, followed by natural cooling under Argon. The resulting pellet had a thickness varying from ∼1 mm to 2 mm, and the pellet appeared light yellow.
Structural characterization
Calculations
Electrochemical measurements
:2 mass ratio and ground. Li6PS5Cl, synthesized according to Patel et al.'s method30 served as the separator. In assembling the half-cells, 12 mg of catholyte was spread onto one side of the Li6PS5Cl pellet to achieve an aerial loading of approximately 1.25 mA h cm2, pressed at 300 MPa for 10 seconds, and then assembled with a Li–In alloy foil on the other side to form Li–In|Li6PS5Cl|2SE:TiS2 (SE:Li3.6In7S11.8Cl) half cells. The cells were sealed with vacuum grease and cycled under controlled conditions at 22 °C with a stack pressure of ∼30 MPa within the voltage window of 1–2.5 V vs. Li–In. For rate performance test, the cell was cycled 5 times at each of the following rates: 0.2C, 0.5C, 1C, and 2C, where C is the theoretical specific capacity of TiS2, 239 mA h g−1. These rates translate to current densities of 0.28 mA cm−2, 0.70 mA cm−2, 1.40 mA cm−2, and 2.80 mA cm−2, respectively. Subsequently, long-term stability testing was performed over 120 cycles at 0.2C.
Results and discussion
Structure
We utilized solid-state reactions to synthesize Li4In7S12Cl (see Experimental section for details). The phase purity was initially determined with X-ray powder diffraction, and the resulting pattern is shown in Fig. 1a. Notably, our pattern displays Bragg reflections that closely match that of Li4In8S16Sn2.31 For precise determination of bulk structural parameters, including atomic coordinates, site occupancies, and thermal parameters of Li4In7S12Cl, a high-resolution X-ray diffraction pattern was collected at the synchrotron beamline at APS (see Experimental section for details). All Bragg peaks were indexed to the Fd![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m space group (Fig. 1b). Rietveld refinement was employed for structural and phase determination, and the refinement results are provided in Table S1.† Rietveld refinement revealed Li3.6In7S11.8Cl as the actual composition, with a trace LiInS2 impurity. This was further validated by SEM-EDX analysis, which showed that the atomic ratio (In:S:Cl) closely matched the refined stoichiometry (Table S2†). This refined composition, Li3.6In7S11.8Cl, will be used hereafter. Attempts have been made to synthesize Li3.6In7S11.8Cl with the stoichiometric ratios of the precursors but yielding a slightly unfavorable ionic conductivity (Fig. S1†) likely due to Li and S loss during the sintering steps.
m space group (Fig. 1b). Rietveld refinement was employed for structural and phase determination, and the refinement results are provided in Table S1.† Rietveld refinement revealed Li3.6In7S11.8Cl as the actual composition, with a trace LiInS2 impurity. This was further validated by SEM-EDX analysis, which showed that the atomic ratio (In:S:Cl) closely matched the refined stoichiometry (Table S2†). This refined composition, Li3.6In7S11.8Cl, will be used hereafter. Attempts have been made to synthesize Li3.6In7S11.8Cl with the stoichiometric ratios of the precursors but yielding a slightly unfavorable ionic conductivity (Fig. S1†) likely due to Li and S loss during the sintering steps.
 | ||
| Fig. 1 (a) Powder X-ray diffraction patterns of the nominal Li4In7S12Cl and the precursors. ICSD patterns of Li4In8S16Sn2 and LiInS2 are also shown as references. (b) High-resolution X-ray diffraction pattern and the corresponding Rietveld refinement of the nominal Li4In7S12Cl, identifying the primary phase composition as Li3.6In7S11.8Cl, which is used instead of Li4In7S12Cl hereafter. (c) and (d) The structure of Li3.6In7S11.8Cl with Fdm space group obtained from the refinement of the high-resolution XRD pattern and viewed from different angles. | ||
Li3.6In7S11.8Cl, like other spinels, features a face-centered cubic (fcc) arrangement of S2−/Cl− anions stabilized by interstitial cations. The anions form a cubic close-packed (ccp) lattice, while the cations are positioned in defined interstitial sites. The cations are located at 8a, 16c, and 16d sites, while anions occupy the 32e positions.32,33 Specifically, the unit cell of Li3.6In7S11.8Cl consists of one tetrahedrally coordinated 8a and two octahedrally coordinated 16c and 16d lithium sites, denoted as Li8a, Li16c, and Li16d, respectively. Sulfur and chlorine atoms co-occupy the 32e anionic site, yielding a disordered anion sublattice (Fig. 1c).
The structure exhibits two distinct planes (Fig. 1d): (0![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 1) and (01). The (01)-plane features a 3D framework consisting of three edge-sharing 16c octahedra that face share with 8a tetrahedra. In addition, Li16c octahedra face-shares with interstitial tetrahedral voids, which provide additional lithium transport pathways. The (01)-plane contains edge-sharing Li16c and Li16d octahedra. With Li occupying 13.2% and In occupying 86.8% of the 16d octahedra, Li-ion transport within this layer becomes less favorable owing to channel blocking and lower anion polarizability that arises from the presence of higher amount of multivalent (M3+) cation within its neighborhood.
1) and (01). The (01)-plane features a 3D framework consisting of three edge-sharing 16c octahedra that face share with 8a tetrahedra. In addition, Li16c octahedra face-shares with interstitial tetrahedral voids, which provide additional lithium transport pathways. The (01)-plane contains edge-sharing Li16c and Li16d octahedra. With Li occupying 13.2% and In occupying 86.8% of the 16d octahedra, Li-ion transport within this layer becomes less favorable owing to channel blocking and lower anion polarizability that arises from the presence of higher amount of multivalent (M3+) cation within its neighborhood.
Local structure and ion dynamics characterized using NMR
High-resolution 6Li NMR spectroscopy was utilized to probe the local Li+ environments in Li3.6In7S11.8Cl, and four major resonances were observed (Fig. 2a). The resonance at 0.17 ppm is assigned to Li8a. The resonances at 1.49 ppm and 2.03 ppm are assigned to Li16c and Li16d, respectively. These assignments correlate with the Li-occupancy determined from the diffraction results. The weak resonance at −0.84 ppm is attributed to a minor impurity phase, LiInS2. This assignment was validated with DFT NMR calculations on LiInS2, confirming a resonance at −0.8 ppm (Table S3†). The quantification from 6Li NMR area integrals is shown in Table S4.† | ||
| Fig. 2 (a) 6Li NMR spectra with resonance assignment. 6Li spectra of LiCl and Li2S are shown as references. (b) 7Li T1 NMR relaxation times of Li3.6In7S11.8Cl as a function of temperature, revealing faster ion dynamics of Li8a and Li16c, compared with Li16d. (c) Raman spectra of as-prepared Li3.6In7S11.8Cl, In2S3, and InCl3. | ||
7Li NMR relaxometry was utilized to study Li+-ion mobility in Li3.6In7S11.8Cl. As shown in Table 1 and Fig. 2b, the 7Li T1 relaxation time is significantly shorter for Li16c (3.1 s) and Li8a (3.0 s), compared to Li16d (11.8 s). The Bloembergen–Purcell–Pound (BPP) model34 provides a framework for understanding spin–lattice relaxation related to ion dynamics. The BPP model is described by  where γ is the gyromagnetic ratio, ħ is the reduced Planck's constant, r0 is the interatomic distance, ω0 = γB0 is the Larmor frequency, and B0 is the external magnetic field strength. In the fast-motion regime (ωoτc ≪ 1), T1 increases with increasing motional rate, while in the slow-motion regime (ωoτc ≫ 1), T1 decreases with increasing motional rate. A motional rate can also lie in the intermediate region where ωoτc ≈ 1.
where γ is the gyromagnetic ratio, ħ is the reduced Planck's constant, r0 is the interatomic distance, ω0 = γB0 is the Larmor frequency, and B0 is the external magnetic field strength. In the fast-motion regime (ωoτc ≪ 1), T1 increases with increasing motional rate, while in the slow-motion regime (ωoτc ≫ 1), T1 decreases with increasing motional rate. A motional rate can also lie in the intermediate region where ωoτc ≈ 1.
| Sample | 7Li, T1 [s] | ||
|---|---|---|---|
| Li8a | Li16c | Li16d | |
| Li3.6In7S11.8Cl (AP) | 3.0 | 3.1 | 11.8 |
| Li3.6In7S11.8Cl (E) | 2.8 | 3.0 | 11.2 |
Variable-temperature 7Li T1 NMR relaxation time measurement reveals a decrease in T1 relaxation time with increasing temperature and thus increasing motional rates for all the resonances, indicating that Li motion lies in the slow-motion regime (ωoτc ≫ 1).34 Therefore, a shorter T1 value will correlate with faster ion mobility. Consequently, Li8a and Li16c exhibit faster Li+ ion motion as the relaxation time is significantly shorter than Li16d. From 7Li NMR line width analysis, the line width of Li8a and Li16c is narrower compared to Li16d (Fig. S2†). A narrower line width may arise from Li+ species with high mobility that averages out homogeneous and inhomogeneous line broadening.
Ion conduction properties determined with electrochemical impedance spectroscopy (EIS)
AC impedance spectroscopy was utilized to evaluate the ion transport properties of Li3.6In7S11.8Cl. The Nyquist plot obtained at 25 °C is presented in Fig. 3a, and a representative Nyquist plot fitting of the data obtained at 0 °C using the equivalent circuit model is presented in Fig. 3b. The high-frequency semicircle with a capacitance of 0.6 pF corresponds to ion transport within the bulk of the solid electrolyte,1,35,36 while the medium-frequency semicircle with a capacitance of 1.2 nF represents the grain boundary contribution.15 The ionic conductivity is calculated from the bulk impedance resistance using the formula σDC = L/(R × A). Here, L represents the pellet thickness, A denotes the surface area of the blocking electrode, and R is the resistance value obtained from the equivalent circuit fitting. An ionic conductivity of 1.1 mS cm−1 is obtained based on the resistance extracted from the fitting.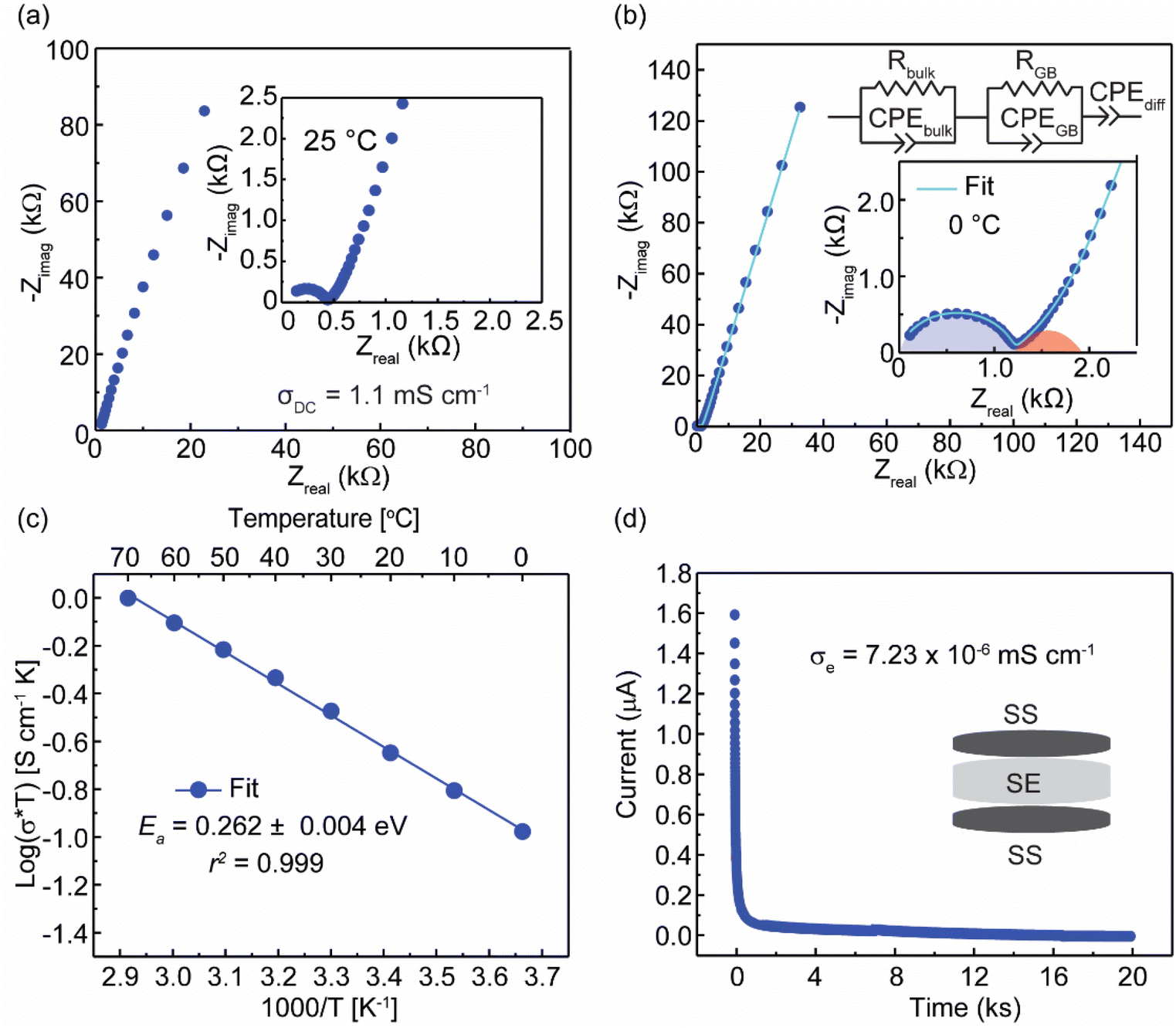 | ||
| Fig. 3 (a) Nyquist plot at 25 °C for Li3.6In7S11.8Cl. (b) Nyquist plot at 0 °C with equivalent circuit fitting (inset) for Li3.6In7S11.8Cl. (c) Arrhenius plot and the extracted activation energy (Ea) for ion transport in Li3.6In7S11.8Cl. (d) DC polarization curve of Li3.6In7S11.8Cl for the cell set up SS|SE|SS for determining the electronic conductivity of the SE, Li3.6In7S11.8Cl. | ||
Variable-temperature EIS was carried out within the frequency range from 1 Hz to 7 MHz and the temperature range from 0 °C to 70 °C. Conductivities at various temperatures were calculated based on the Nyquist plots, and the Arrhenius-type plots were prepared to calculate the activation energy. Fig. 3c depicts the Arrhenius plots of Li3.6In7S11.8Cl. An activation energy barrier (Ea) of 0.26 eV was determined from the linear fit of the Arrhenius plot. This value agrees with a site energy barrier of 0.26 eV obtained from the Bond Valence Site Energy (BVSE) calculation (Fig. S3†). The absence of any discontinuity throughout this temperature range, along with the observed linear Arrhenius behavior, indicates the thermal stability of Li3.6In7S11.8Cl.37 To verify that the conductivity arises from Li+ transport, the electronic conductivity of Li3.6In7S11.8Cl was determined to be 7.23 × 10−6 mS cm−1 using the direct current (DC) polarization method at a constant voltage of 0.1 V (Fig. 3d). The relatively low electronic conductivity indicates negligible electronic contributions to the overall conductivity.
Lithium-ion transport via AIMD simulation
Ab initio molecular dynamics (AIMD) simulations are performed on the relaxed supercell (1 × 1 × 1) of Li3.6In7S11.8Cl. Mean square displacements (MSD) and distribution probability of Li+ at 900 K are extracted from the AIMD simulations (Fig. 4). The MSD of Li+ illustrates the diffusion trajectories along the a, b, and c lattice directions (Fig. 4a). Notably, the MSD of Li+ is similar along the a, b, and c directions, indicating a 3D diffusion for Li+ in the Li3.6In7S11.8Cl. Fig. 4b provides a 3D visualization of Li+ density distribution within the relaxed Li3.6In7S11.8Cl crystal lattice (1 × 1 × 1), highlighting the primary lithium-ion sites labeled as Li8a, Li16c, and Li16d. The yellow isosurfaces represent regions of high Li+ probability density, indicating potential conduction pathways through the crystal lattice. This Li+ density map reveals a complex, interconnected lithium diffusion network, further suggesting a 3D diffusion pathway for Li+. These results underscore the high ionic conductivity of Li3.6In7S11.8Cl, driven by the extensive 3D diffusion network. | ||
| Fig. 4 (a) Mean square displacements of Li+ in Li3.6In7S11.8Cl generated from AIMD simulations at 900 K. (b) Li+ (yellow) probability density distribution in Li3.6In7S11.8Cl in a 1 × 1 × 1 supercell based on the AIMD simulations at 900 K. | ||
Moisture stability of Li3.6In7S11.8Cl
A significant challenge for large-scale production of sulfide solid electrolytes is their poor stability against atmospheric moisture. Even low levels of moisture in the environment have been reported to trigger spontaneous hydrolysis reactions, causing material degradation, compromised performance, and the release of toxic H2S gas.38,39 Broadly, on exposure of solid electrolytes to moisture, decomposition products such as LiCl,40–42 Li2S,40,42,43 LiOH,41–43 LiOH·H2O,39,42 and In2O3 (ref. 41) may be formed. These decomposition compounds exhibit poor ionic conductivity and significantly increase interfacial impedance when formed on the surface of sulfide SSE particles.41,43 Li3.6In7S11.8Cl was exposed to air for 2 hours (relative humidity of 52%) to assess the impact of moisture on its average structure, short-range structure, morphology, and ionic conductivity. Fig. 5c displays the XRD patterns of Li3.6In7S11.8Cl before and after 2 hours of exposure to humid air at room temperature. A comparison of the diffraction pattern of as-prepared and moisture-exposed samples reveals insignificant modification of the average structure. Particularly, no new peaks were formed post-exposure. This is consistent with 6Li NMR results of the moisture-exposed sample (Fig. 5d) – confirming the absence of LiCl, LiOH, or Li2S, post-exposure. | ||
| Fig. 5 (a) Nyquist plot for the as-prepared, humid-air-exposed, and dried Li3.6In7S11.8Cl. (b) Arrhenius plot and the extracted activation energy (Ea) for ion transport in as-prepared and humid-air-exposed Li3.6In7S11.8Cl. (c) SXRD patterns for as-prepared and air/moisture-exposed Li3.6In7S11.8Cl. (d) 6Li MAS NMR spectra of humid-air-exposed and dried Li3.6In7S11.8Cl. | ||
The Hard–Soft Acid–Base (HSAB) theory predicts favorable interactions between soft acids (e.g., Ge4+, Sn4+, In3+) and soft bases (e.g., S2−).38,44–47 These interactions lead to the formation of strong covalent bonds, potentially creating a stable framework with open channels.44 To be specific, In3+, classified as a soft acid, will preferentially interact with the soft base, S2−.47 This preferential bonding with sulfur prevents the hard base oxygen from reacting with In3+ upon exposure to moisture. As a result, Li3.6In7S11.8Cl exhibits improved moisture stability. A decrease in the activation energy (Fig. 5b) is observed for the moisture-exposed pellet (0.24 eV) relative to the as-prepared pellet (0.26 eV). Fig. 5a reveals a significant increase in ionic conductivity to 4.2 mS cm−1 on exposure to moisture – an observation in contrast to what has been reported for most sulfide solid electrolytes,48–50 except layered structures where H2O intercalates between the sheets.51 However, the slightly increased electronic conductivity post-exposure (4.55 × 10−5 mS cm−1, Fig. S4†) may suggest modifications of the local electronic environment and an increase in electron transport.
Raman spectroscopy, a valuable tool for elucidating the local structures of materials,52,53 was employed to investigate the bonding between In3+ and S2−. Fig. 2c presents the Raman spectra of the as-prepared Li3.6In7S11.8Cl and reference samples including In2S3 and InCl3. According to the XRD pattern (Fig. 1a), In2S3 and Li3.6In7S11.8Cl primarily exhibits a disordered cubic spinel-type, α-In2S3 structure, confirmed by the absence of low-intensity peaks prominent in the tetragonal β-In2S3 structure.54,55 Notably, the presence of Raman active modes at 125 cm−1 (Eg), 270, 291, 299 cm−1 (T2g) and 357 cm−1 (Aig) in In2S3 Raman spectrum (Fig. 2c) confirms that it belongs to the α-In2S3 structure type.56,57 The Raman spectrum of Li3.6In7S11.8Cl exhibits a significant shift (blue shift) of the Raman peaks associated with In–S bonds to higher wavenumbers compared with In2S3, specifically to 127, 278, 300, 308 and 360 cm−1.53,58 This shift to higher wavenumbers signifies an increase in the vibrational energy of these bonds, indicating stronger In–S interactions.59 Moreso, compared to the Raman spectrum of In2S3, the peak intensity is significantly higher for Li3.6In7S11.8Cl. More intense peaks suggest increased Raman scattering efficiency, often a result of strong covalent bonding with more polarizable bonds. Therefore, both the blue shift and peak intensity increase support enhanced In–S covalent bonding in Li3.6In7S11.8Cl.
Water uptake can influence cation mobility through solvation or altering the crystallographic structure.60 Although hydration studies on solid Li+ and Na+ conductors are limited, existing research suggests enhanced Li+ and Na+ diffusion with water adsorption.60 The adsorbed water may exist in different forms; for instance, free water clusters and loosely and strongly bound water have been identified in Nafion.61,62 To understand the origin of the improved ionic conductivity of Li3.6In7S11.8Cl post-exposure, we performed 1H and 6,7Li NMR on the as-prepared and moisture-exposed-samples. For the moisture-exposed-sample, a peak at 4.7 ppm was observed in the 1H NMR spectrum; in contrast, only the background resonance (empty rotor) was observed for the as-prepared sample (Fig. S5†). The resonance at ca. 4.7 ppm in moisture-exposed Li3.6In7S11.8Cl indicates that the adsorbed water is more distinctly characterized as a bulk liquid rather than surface-bound. Increased Li+ ion mobility in the moisture-exposed Li3.6In7S11.8Cl is indicated by faster 7Li NMR T1 relaxation (Fig. S6†) with decreased 7Li NMR T1 relaxation time (Table 1), and narrower peak width (Table S5†). Increased ionic conductivity in the moisture-exposed Li3.6In7S11.8Cl is therefore attributed to ion transport facilitated by the adsorbed water.
The SEM images and the EDS elemental mapping of In, S, Cl, and O for the as-prepared Li3.6In7S11.8Cl and moisture-exposed pellet are shown in Fig. 6a and b. Prior to ambient exposure, the as-prepared pellet demonstrates a highly compact structure. The initial SEM image reveals an uneven surface with loose particles; however, it is devoid of major cracks. Notably, the microstructure of the moisture-exposed sample does not change significantly. However, the pellet surface is smoother. Elemental mapping of the as-prepared Li3.6In7S11.8Cl reveals a uniform distribution of In, S, Cl, and a negligible amount of O. The negligible amount of oxygen likely results from the few seconds exposure to humid air during sample transfer into the SEM instrument. Similarly, In, S, Cl are uniformly distributed in the exposed pellet. In addition, a significant oxygen concentration is observed around the particles, likely from the adsorbed water. It is evident that the absorbed water does not disturb the Li3.6In7S11.8Cl structure but rather distributes on the surfaces of the particles. The enhanced microstructural stability of Li3.6In7S11.8Cl may be attributed to the structural stabilization from the strong covalent interaction of In3+ with S2−, effectively preventing hydrolysis reactions.18
 | ||
| Fig. 6 SEM images and EDS elemental mapping of In, S, Cl, and O for (a) as-prepared and (b) moisture-exposed Li3.6In7S11.8Cl. | ||
The hydrolysis resistance of Li3.6In7S11.8Cl was evaluated by full immersion of the solid electrolyte in deionized water (Fig. S7†). Throughout the exposure test, no significant changes were visually observed, indicating that its structure was not severely altered.45 Further quantitative evaluation of hydrolytic resistance was performed by comparing the mass of the electrolytes before and after immersion. The mass remains almost constant at 140 mg – only reaching 139.4 mg after 1 hour, suggesting excellent hydrolysis resistance of Li3.6In7S11.8Cl. TGA and DSC curves of the moisture-exposed sample are shown in Fig. S8.† To simulate non-oxidative environments, thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC) were performed under an inert atmosphere at a constant heating rate from 0 °C to 700 °C. The initial thermal event, observed between 49 °C and 83 °C, reflects dominant endothermic peaks. This corresponds to removing physiosorbed water molecules, resulting in a mass loss of 3.37%. A minor endothermic event observed at ca. 350 °C is attributed to removing bulk water buried inside Li3.6In7S11.8Cl.63 Overall, the mass loss of 3.61% is due to water removal, indicating good thermal stability of Li3.6In7S11.8Cl.
The exposed pellets were dried to investigate the impact of water removal on ion transport and structure. The impedance plot reveals a decrease in ionic conductivity (4.2 mS cm−1vs. 2.06 mS cm−1) of the pellet dried at 150 °C for 12 hours relative to the moisture-exposed pellet. The SEM images and the EDS elemental mapping of the surface and cross-section of the dried pellet are shown in Fig. 7a and b. Elemental mapping reveals a minimal amount of oxygen on the surface of the dried pellet – indicating loss of surface water upon drying at 150 °C. The negligible amount of oxygen may stem from water re-adsorption during sample transfer into the instrument. However, 1H NMR reveals only a slight decrease in the intensity of the bulk water resonance – suggesting that bulk water is retained (Fig. S5†) after extended heating at 150 °C for 12 hours. To confirm this observation, SEM and EDS data were collected on the cross-section of the pellet (Fig. 7b). A high amount of oxygen, evident within the cross-section, validates the presence of bulk water inside the pellet. The presence of bulk water correlates with higher ionic conductivity (Fig. 5a) of the pellet dried at 150 °C compared to the as-prepared pellet (2.06 mS cm−1vs. 1.1 mS cm−1)
 | ||
| Fig. 7 SEM image and EDS elemental mapping of In, S, Cl, and O for the (a) surface (b) cross-section of Li3.6In7S11.8Cl pellet dried at 150 °C. | ||
TGA experiment suggests a thermal event at ca. 350 °C; therefore, to further examine the effect of bulk water loss on the structural stability and ionic conductivity of the moisture-exposed solid electrolyte, the pellet was dried at 350 °C. The corresponding impedance plot is shown in Fig. 5a. A significant decrease in conductivity was observed (2.06 mS cm−1vs. 0.98 mS cm−1) relative to the pellet dried at 150 °C. However, the ionic conductivity does not deviate largely from that of the as-prepared pellet (1.1 mS cm−1vs. 0.98 mS cm−1), suggesting that water can be reversibly removed without compromising the structure or ion transport properties of the solid electrolyte. 1H NMR confirms (Fig. S5†) the complete removal of bulk water after drying at 350 °C, further validated by significantly reduced oxygen distribution from cross-section elemental mapping of the dried pellet (Fig. S9†).
Proposed mechanism of ion transport in moisture-exposed Li3.6In7S11.8Cl
Enhanced ionic conductivity of moisture-exposed polycrystalline solid electrolytes has been linked to the presence of adsorbed water at grain boundaries.64 On exposing Li3.6In7S11.8Cl to moisture, the scanning electron microscopy (SEM) images of Li3.6In7S11.8Cl revealed the distribution of adsorbed water on the surface of- and around the grains (Fig. 7b). 6Li NMR analysis (Fig. 5d) suggests that Li+ in Li3.6In7S11.8Cl is not hydrolyzed or transformed into other chemical phases in the presence of the absorbed water.60 The adsorbed water may effectively soften or plasticize the grain boundaries without compromising its integrity, facilitating Li+ hopping between grains and reducing the energy barrier for Li-ion migration. To assess the impact of the adsorbed-water around grain boundaries on Li+ transport, we performed EIS analysis to separate the resistive contributions from the bulk and grain boundaries. By fitting the impedance spectra to equivalent circuit models, the grain boundary resistance and bulk resistance can be independently extracted. Our EIS analysis (Fig. 8 and Table S6†) revealed a significant reduction in grain boundary resistance (RGB) from 171.7 Ω in the as-prepared sample to 46.6 Ω in the moisture-exposed sample. This confirms that the adsorbed water effectively facilitates Li+ transport along/across grain boundaries. Since grain boundary resistance is often the limiting factor in polycrystalline solid electrolytes, the decrease in grain boundary resistance results in significantly enhanced Li+ transport across/along the grain boundaries and the overall ionic conductivity. | ||
| Fig. 8 Nyquist plots and equivalent circuit fitting of (a) as-prepared Li3.6In7S11.8Cl and (b) moisture-exposed Li3.6In7S11.8Cl. | ||
Electrochemical stability and galvanostatic cycling of Li3.6In7S11.8Cl
Linear sweep voltammetry (LSV) was utilized to assess the electrochemical stability window of Li3.6In7S11.8Cl at 25 °C. The electrochemical stability window signifies the voltage range within which the electrolytes and their components remain electrochemically inert.35 Carbon, utilized as an electronic conductive medium offers a high surface area, facilitating sensitive detection of electric current generated from redox reactions.35,36Fig. 9a shows the linear sweep voltammogram of Li3.6In7S11.8Cl. The anodic peak at ≈0.6 V versus Li–In is assigned to the lithiation of the carbon. Chloride ions (Cl−) may undergo oxidation (2Cl− → Cl2(g) + 2e−) at voltages exceeding 3.74 V versus Li–In, based on their standard electrode potential.65 The anodic peak around ≈3.8 V versus Li–In is indicative of oxidation of Cl species to Cl2.65–67 Therefore, the experimentally determined lower and upper electrochemical stability limit for Li3.6In7S11.8Cl is ca. 1.01–3.39 V versus Li–In. | ||
| Fig. 9 (a) Linear sweep voltammetry (current–voltage) plot of Li3.6In7S11.8Cl (b) the rate performance and long-term cycling stability of a Li–In|Li6PS5Cl|2SE:TiS2 cell (SE = Li3.6In7S11.8Cl). C = 239 mA h g−1. | ||
Galvanostatic cycling at 22 °C was performed to evaluate the half-cell performance across various C-rates, with five cycles per rate, followed by 120 cycles at 0.2C, where C = 239 mA h g−1 (Fig. 9b). A Li–In|Li6PS5Cl|2(Li3.6In7S11.8Cl):TiS2 half-cell was fabricated with TiS2 as the cathode active material (CAM). Li–In anode was utilized due to its stability with solid electrolytes and reduced risk of micropore-induced short-circuiting.2,35 The Li–In|Li6PS5Cl|2(Li3.6In7S11.8Cl):TiS2 half-cell delivered an initial discharge capacity of ca. 240 mA h g−1, eventually stabilizing at ca. 235 mA h g−1. In the second cycle, the cell sustained a high discharge capacity of 226 mA h g−1 and maintained a consistent charge capacity of 225 mA h g−1. Notably, at a high rate of 2C, the cell exhibited a robust performance, delivering a discharge capacity of 150 mA h g−1. Upon reverting to 0.2C after 20 cycles, the Li–In|Li6PS5Cl|2(Li3.6In7S11.8Cl):TiS2 half-cell demonstrated excellent long-term cycling stability, retaining ca. 96.8% of its capacity, over an extended period of 120 cycles.
Conclusion
In this study, an air- and moisture-stable solid electrolyte, Li3.6In7S11.8Cl (space group Fdm), is synthesized, delivering a room-temperature ionic conductivity of 1.1 mS cm−1 with an activation energy of 0.26 eV. Structural characterization reveals a face-centered cubic arrangement of S2−/Cl−, stabilized by interstitial cations in a disordered anion sublattice. Notably, a 3D framework formed by tetrahedra Li8a face-sharing with octahedra Li16c promotes fast Li+ ion diffusion, supported by AIMD simulations. 6,7Li NMR and relaxometry reveal fast ion dynamics of octahedral (16c) and tetrahedral (8a) Li+ sites, responsible for ion transport. Li3.6In7S11.8Cl exhibits stability against air and moisture and shows enhanced ionic conductivity to 4.2 mS cm−1 upon exposure, likely due to ion transport facilitated by the adsorbed water at grain boundaries. The water can be reversely removed upon heating to 350 °C without compromising the structural integrity. The stability is attributed to the structural stabilization provided by the strong covalent bonding between In3+ and S2−.
Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Ifeoluwa P. Oyekunle: data curation: lead; formal analysis: lead; investigation: lead; methodology: lead; software: lead; validation: lead; visualization: lead; writing – original draft: lead. Erica Truong: data curation: supporting; formal analysis: supporting; investigation: supporting; methodology: supporting; validation: supporting; visualization: supporting; writing – original draft: supporting. Tej Prasad Poudel: data curation: supporting; formal analysis: supporting; investigation: supporting; methodology: supporting; validation: supporting; visualization: supporting; writing – original draft: supporting. Yudan Chen: data curation: supporting; formal analysis: supporting; investigation: supporting; methodology: supporting; methodology: supporting; software: supporting; writing – original draft: supporting. Yongkang Jin: data curation: supporting investigation: supporting; validation: supporting; visualization: supporting; writing – original draft: supporting. Islamiyat A. Ojelade: data curation: supporting investigation: supporting; validation: supporting; visualization: supporting; writing – original draft: supporting. Michael J. Deck: data curation: supporting investigation: supporting; validation: supporting; writing – original draft: supporting. Bright Ogbolu: investigation: supporting; validation: supporting; visualization: supporting; writing – review & editing: supporting Md Mahinur Islam: data curation: supporting; validation: supporting; writing – review & editing: supporting Pawan K. Ojha: data curation: supporting; validation: supporting; writing – review & editing: supporting J. S. Raaj Vellore Winfred: data curation: supporting; formal analysis: supporting Dewen Hou: data curation: supporting. Hui Xiong: resources: supporting; supervision: supporting; validation: supporting. Chen Huang: data curation: supporting; software: supporting. Yan-Yan Hu, PhD: conceptualization: lead; formal analysis: lead; funding acquisition: lead; methodology: lead; project administration: lead; resources: lead; supervision: lead; validation: lead; visualization: equal; writing – original draft: equal; writing – review & editing: lead.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The authors acknowledge support from the National Science Foundation under grant no. DMR-1847038. All solid-state NMR experiments were performed at the National High Magnetic Field Laboratory, which is supported by National Science Foundation Cooperative Agreement No. DMR-1644779 and DMR-2128556*. Hou and Xiong thank the support from the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences program under award number DE-SC0019121. Use of the Center for Nanoscale Materials and Advanced Photon Source, both DOE Office of Science user facility, was supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under contract no. DE-AC02-06CH11357.References
- T. P. Poudel, E. Truong, I. P. Oyekunle, M. J. Deck, B. Ogbolu, Y. Chen, P. K. Ojha, T. N. D. D. Gamaralalage, S. V. Patel, Y. Jin, D. Hou, C. Huang, T. Li, Y. Liu, H. Xiong and Y.-Y. Hu, Sliceable, Moldable, and Highly Conductive Electrolytes for All-Solid-State Batteries, ACS Energy Lett., 2024, 10(1), 40–47 CrossRef.
- T. P. Poudel, I. P. Oyekunle, M. J. Deck, Y. Chen, D. Hou, P. K. Ojha, B. O. Ogbolu, C. Huang, H. Xiong and Y. Y. Hu, Li(1.6)AlCl(3.4)S(0.6): a low-cost and high-performance solid electrolyte for solid-state batteries, Chem. Sci., 2025, 16, 2391 RSC.
- H. Yamaguchi, A. Yao, S. Hiroi, H. Yamada, J.-c. Tseng, S. Shimono, F. Utsuno and K. Ohara, Formation process of halogen-rich argyrodite: elemental disordering of atomic arrangement at the 4a and 4d sites in a heat treatment, J. Solid State Electrochem., 2024, 28, 4419–4426 CrossRef CAS.
- S. Cho, Y. Kim, Y. Song, J. Ryu, K. Choi, J. Yang, S. H. Lee, S. G. Im and S. Park, Functional Polymer Thin Films for Establishing an Effective Electrode Interface in Sulfide-Based Solid-State Batteries, Adv. Funct. Mater., 2024, 34(32), 2314710 CrossRef CAS.
- K. H. Kim, M. J. Lee, M. Ryu, T. K. Liu, J. H. Lee, C. Jung, J. S. Kim and J. H. Park, Near-strain-free anode architecture enabled by interfacial diffusion creep for initial-anode-free quasi-solid-state batteries, Nat. Commun., 2024, 15(1), 3586 CrossRef CAS.
- K. G. Naik, D. Chatterjee and P. P. Mukherjee, Solid Electrolyte-Cathode Interface Dictates Reaction Heterogeneity and Anode Stability, ACS Appl. Mater. Interfaces, 2022, 14(40), 45308–45319 CrossRef CAS.
- T. Kim, H. Chang, G. Song, S. Lee, K. Kim, S. Lee, J. Moon and K. T. Lee, Critical Factors Contributing to the Thermal Runaway of Thiophosphate Solid Electrolytes for All-Solid-State Batteries, Adv. Funct. Mater., 2024, 34(42), 2404806 CrossRef CAS.
- H. Liu, Y. Chen, P.-H. Chien, G. Amouzandeh, D. Hou, E. Truong, I. P. Oyekunle, J. Bhagu, S. W. Holder, H. Xiong, P. Gor’kov, J. T. Rosenberg, S. C. Grant and Y.-Y. Hu, Dendrite formation in solid-state batteries arising from lithium plating and electrolyte reduction, Nat. Mater., 2025, 24, 581–588 CrossRef CAS.
- C. Wang, K. Adair and X. Sun, All-Solid-State Lithium Metal Batteries with Sulfide Electrolytes: Understanding Interfacial Ion and Electron Transport, Acc. Mater. Res., 2021, 3(1), 21–32 CrossRef.
- H. Su, J. Li, Y. Zhong, Y. Liu, X. Gao, J. Kuang, M. Wang, C. Lin, X. Wang and J. Tu, A scalable Li-Al-Cl stratified structure for stable all-solid-state lithium metal batteries, Nat. Commun., 2024, 15(1), 4202 CrossRef CAS PubMed.
- P. Wang, H. Liu, S. Patel, J. E. Roberts, Y. Chen, B. Ogbolu, B. E. Francisco and Y. Y. Hu, Dual Polyanion Mechanism for Superionic Transport in BH4-Based Argyrodites, Adv. Energy Mater., 2024, 14(45), 2401549 CrossRef CAS.
- S. Chen, D. Xie, G. Liu, J. P. Mwizerwa, Q. Zhang, Y. Zhao, X. Xu and X. Yao, Sulfide solid electrolytes for all-solid-state lithium batteries: Structure, conductivity, stability and application, Energy Storage Mater., 2018, 14, 58–74 CrossRef.
- A. Hayashi, N. Masuzawa, S. Yubuchi, F. Tsuji, C. Hotehama, A. Sakuda and M. Tatsumisago, A sodium-ion sulfide solid electrolyte with unprecedented conductivity at room temperature, Nat. Commun., 2019, 10(1), 5266 CrossRef CAS.
- R. Rajagopal, Y. Subramanian, S. Kang, J. Park and K.-S. Ryu, Improved interfacial stability of all-solid-state batteries using cation-anion co-doped glass electrolytes, Commun. Mater., 2024, 5(78), 1–13 Search PubMed.
- I. Hanghofer, M. Brinek, S. L. Eisbacher, B. Bitschnau, M. Volck, V. Hennige, I. Hanzu, D. Rettenwander and H. M. R. Wilkening, Substitutional disorder: structure and ion dynamics of the argyrodites Li(6)PS(5)Cl, Li(6)PS(5)Br and Li(6)PS(5)I, Phys. Chem. Chem. Phys., 2019, 21(16), 8489–8507 RSC.
- W. Li, J. Liang, M. Li, K. R. Adair, X. Li, Y. Hu, Q. Xiao, R. Feng, R. Li, L. Zhang, S. Lu, H. Huang, S. Zhao, T.-K. Sham and X. Sun, Unraveling the Origin of Moisture Stability of Halide Solid-State Electrolytes by In Situ and Operando Synchrotron X-ray Analytical Techniques, Chem. Mater., 2020, 32(16), 7019–7027 CrossRef CAS.
- W. D. Jung, M. Jeon, S. S. Shin, J. S. Kim, H. G. Jung, B. K. Kim, J. H. Lee, Y. C. Chung and H. Kim, Functionalized Sulfide Solid Electrolyte with Air-Stable and Chemical-Resistant Oxysulfide Nanolayer for All-Solid-State Batteries, ACS Omega, 2020, 5(40), 26015–26022 CrossRef CAS PubMed.
- X. Lu, O. Camara, Z. Liu, A. Windmüller, C. L. Tsai, H. Tempel, S. Yu, H. Kungl and R. A. Eichel, Tuning the moisture stability of multiphase β-Li3PS4 solid electrolyte materials, Electrochem. Sci. Adv., 2022, 3(2), e2100208 CrossRef.
- G. Kresse and J. Furthmuller, Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54(16), 11169 CrossRef CAS.
- P. E. Blochl, Projector augmented-wave method, Phys. Rev. B Condens. Matter, 1994, 50(24), 17953–17979 CrossRef PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized Gradient Approximation Made Simple, Phys. Rev. Lett., 1996, 77(18), 3865–3868 CrossRef CAS.
- S. P. Ong, W. D. Richards, A. Jain, G. Hautier, M. Kocher, S. Cholia, D. Gunter, V. L. Chevrier, K. A. Persson and G. Ceder, Python Materials Genomics (pymatgen): A robust, open-source python library for materials analysis, Comput. Mater. Sci., 2013, 68, 314–319 CrossRef CAS.
- A. Y. Toukmaji and J. A. Board Jr, Ewald summation techniques in perspective: a survey, Comput. Phys. Commun., 1996, 95, 73–92 CrossRef CAS.
- S. J. Clark, I. Matthew, D. Segall, C. J. Pickard, P. J. Hasnip, M. I. J. Probert, K. Refson and M. C. PayneII, First Principles Methods using CASTEP, Z. Kristallogr. Cryst. Mater., 2005, 220, 567–570 CrossRef CAS.
- M. D. Segall, P. J. D. Lindan, M. J. Probert, C. J. Pickard, P. J. Hasnip, S. J. Clark and M. C. Payne, First-principles simulation: ideas, illustrations and the CASTEP code, J. Phys.: Condens. Matter, 2002, 14, 2717–2744 CrossRef CAS.
- D. C. Liu and J. Nocedal, On the Limited Memory BFGS Method for Large Scale Optimization, Math. Program., 1989, 45, 503–528 CrossRef.
- J. Nocedal, Updating Quasi-Newton Matrices With Limited Storage, Math. Comput., 1980, 35(151), 773–782 CrossRef.
- L. L. Wong, K. C. Phuah, R. Dai, H. Chen, W. S. Chew and S. Adams, Bond Valence Pathway Analyzer—An Automatic Rapid Screening Tool for Fast Ion Conductors within softBV, Chem. Mater., 2021, 33(2), 625–641 CrossRef CAS.
- S. Adams, From bond valence maps to energy landscapes for mobile ions in ion-conducting solids, Solid State Ionics, 2006, 177(19–25), 1625–1630 CrossRef CAS.
- S. V. Patel, S. Banerjee, H. Liu, P. Wang, P.-H. Chien, X. Feng, J. Liu, S. P. Ong and Y.-Y. Hu, Tunable Lithium-Ion Transport in Mixed-Halide Argyrodites Li6–xPS5–xClBrx: An Unusual Compositional Space, Chem. Mater., 2021, 33(4), 1435–1443 CrossRef CAS.
- C. Adenis, J. Olivier-Fourcade, J.-C. Jumas and E. Philippot, Etude structurale par spectroscopie Mössbauer et rayons X de spinelles lacunaires de type In2S3, Rev. Chim. Miner., 1987, 24(1), 10–21 CAS.
- L. Zhou, C. Y. Kwok, A. Shyamsunder, Q. Zhang, X. Wu and L. F. Nazar, A new halospinel superionic conductor for high-voltage all solid state lithium batteries, Energy Environ. Sci., 2020, 13(7), 2056–2063 RSC.
- V. Tsurkan, H.-A. Krug von Nidda, J. Deisenhofer, P. Lunkenheimer and A. Loidl, On the complexity of spinels: Magnetic, electronic, and polar ground states, Phys. Rep., 2021, 926, 1–86 CrossRef CAS.
- N. Bloembergen, E. M. Purcell and R. V. Pound, Relaxation Effects in Nuclear Magnetic Resonance Absorption, Phys. Rev., 1948, 73(7), 679–712 CrossRef CAS.
- T. P. Poudel, M. J. Deck, P. Wang and Y. Y. Hu, Transforming Li3PS4 Via Halide Incorporation: a Path to Improved Ionic Conductivity and Stability in All-Solid-State Batteries, Adv. Funct. Mater., 2023, 34(4), 2309656 CrossRef.
- M. J. Deck, P. H. Chien, T. P. Poudel, Y. Jin, H. Liu and Y. Y. Hu, Oxygen-Induced Structural Disruption for Improved Li+ Transport and Electrochemical Stability of Li3PS4, Adv. Energy Mater., 2023, 14(4), 2302785 CrossRef.
- K. Kaup, K. Bishop, A. Assoud, J. Liu and L. F. Nazar, Fast Ion-Conducting Thioboracite with a Perovskite Topology and Argyrodite-like Lithium Substructure, J. Am. Chem. Soc., 2021, 143(18), 6952–6961 CrossRef CAS PubMed.
- Y. Zhu and Y. Mo, Materials Design Principles for Air-Stable Lithium/Sodium Solid Electrolytes, Angew Chem. Int. Ed. Engl., 2020, 59(40), 17472–17476 CrossRef CAS PubMed.
- J. Sang, K. Pan, B. Tang, Z. Zhang, Y. Liu and Z. Zhou, One Stone, Three Birds: An Air and Interface Stable Argyrodite Solid Electrolyte with Multifunctional Nanoshells, Adv. Sci., 2023, 10(32), e2304117 CrossRef PubMed.
- Y.-T. Chen, M. A. T. Marple, D. H. S. Tan, S.-Y. Ham, B. Sayahpour, W.-K. Li, H. Yang, J. B. Lee, H. J. Hah, E. A. Wu, J.-M. Doux, J. Jang, P. Ridley, A. Cronk, G. Deysher, Z. Chen and Y. S. Meng, Investigating dry room compatibility of sulfide solid-state electrolytes for scalable manufacturing, J. Mater. Chem. A, 2022, 10(13), 7155–7164 RSC.
- M. Yang, L. Chen, H. Li and F. Wu, Air/Water Stability Problems and Solutions for Lithium Batteries, Adv. Energy Mater., 2022, 2022, 1–41 Search PubMed.
- Y. Morino, H. Sano, T. Takahashi, N. Miyashita, A. Sakuda and A. Hayashi, Hydrogen Components of a Sulfide-based Argyrodite-Type Solid Electrolyte after Moisture Exposure, J. Phys. Chem. C, 2023, 127(28), 13616–13622 CrossRef CAS.
- T. A. Yersak, Y. Zhang, F. Hao and M. Cai, Moisture Stability of Sulfide Solid-State Electrolytes, Front. Energy Res., 2022, 10, 1–9 Search PubMed.
- Y. Wang, X. Lu, C. Zheng, X. Liu, Z. Chen, W. Yang, J. Lin and F. Huang, Chemistry Design Towards a Stable Sulfide-Based Superionic Conductor Li(4) Cu(8) Ge(3) S(12), Angew Chem. Int. Ed. Engl., 2019, 58(23), 7673–7677 CrossRef CAS.
- C. Wang, J. Hao, J. Wu, H. Shi, L. Fan, J. Wang, Z. Wang, Z. Wang, L. Yang, Y. Gao, X. Yan and Y. Gu, Enhanced Air Stability and Li Metal Compatibility of Li-Argyrodite Electrolytes Triggered by In2O3 Co-Doping for All-Solid-State Li Metal Batteries, Adv. Funct. Mater., 2024, 34(18), 2313308 CrossRef CAS.
- Y. Fu, Z. Gong, D. Li, Y. Liu, X. Zhou, Y. Yang and Q. Jiao, Zn doping for enhanced sodium-ion conductivity and air stability in Na3SbS4 solid electrolyte, J. Mater. Sci., 2024, 59(7), 3009–3017 CrossRef CAS.
- R. Guo, K. Zhang, W. Zhao, Z. Hu, S. Li, Y. Zhong, R. Yang, X. Wang, J. Wang, C. Wu and Y. Bai, Interfacial Challenges and Strategies toward Practical Sulfide-Based Solid-State Lithium Batteries, Adv. Energy Mater., 2023, 4(1), 1–31 Search PubMed.
- Z. Yu, S. L. Shang, J. H. Seo, D. Wang, X. Luo, Q. Huang, S. Chen, J. Lu, X. Li, Z. K. Liu and D. Wang, Exceptionally High Ionic Conductivity in Na(3) P(0.62) As(0.38) S(4) with Improved Moisture Stability for Solid-State Sodium-Ion Batteries, Adv. Mater., 2017, 29(16), 1605561 CrossRef.
- J. Liang, X. Li, C. Wang, J. T. Kim, R. Yang, J. Wang and X. Sun, Current Status and Future Directions in Environmental Stability of Sulfide Solid-State Electrolytes for All-Solid-State Batteries, Adv. Energy Mater., 2023, 4, 1–14 Search PubMed.
- Y. E. Choi, K. H. Park, D. H. Kim, D. Y. Oh, H. R. Kwak, Y. G. Lee and Y. S. Jung, Coatable Li(4) SnS(4) Solid Electrolytes Prepared from Aqueous Solutions for All-Solid-State Lithium-Ion Batteries, ChemSusChem, 2017, 10(12), 2605–2611 CrossRef CAS.
- A.-K. Hatz, I. Moudrakovski, S. Bette, M. W. Terban, M. Etter, M. Joos, N. M. Vargas-Barbosa, R. E. Dinnebier and B. V. Lotsch, Fast Water-Assisted Lithium Ion Conduction in Restacked Lithium Tin Sulfide Nanosheets, Chem. Mater., 2021, 33(18), 7337–7349 CrossRef CAS.
- M. Deluca, H. Hu, M. N. Popov, J. Spitaler and T. Dieing, Advantages and developments of Raman spectroscopy for electroceramics, Commun. Mater., 2023, 4(1), 78–92 CrossRef CAS.
- Y. Chen, P. Wang, E. Truong, B. Ogbolu, Y. Jin, I. Oyekunle, H. Liu, M. M. Islam, T. Poudel, C. Huang, I. Hung, Z. Gan and Y. Y. Hu, Superionic Conduction in K(3)SbS(4) Enabled by Cl-Modified Anion Lattice, Angew Chem. Int. Ed. Engl., 2024, 63(35), e202408574 CrossRef CAS PubMed.
- P. Pistor, J. M. Merino Alvarez, M. Leon, M. di Michiel, S. Schorr, R. Klenk and S. Lehmann, Structure reinvestigation of alpha-, beta- and gamma-In2S3, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2016, 72(3), 410–415 CrossRef CAS.
- L. A. Wägele, D. Rata, G. Gurieva and R. Scheer, Structural analysis of co-evaporated In2S3 and In2S3:V for solar cell absorber applications, Phys. Status Solidi C, 2017, 14(6), 1600204 CrossRef.
- H. Izadneshana and V. F. Gremenok, Micro Structural Analysis of In2S3 Thin Films by Raman Spectroscopy, J. Appl. Spectrosc., 2014, 81(5), 765–770 CrossRef CAS.
- S. Gallego-Parra, O. Gomis, R. Vilaplana, V. P. Cuenca-Gotor, D. Martinez-Garcia, P. Rodriguez-Hernandez, A. Munoz, A. Romero, A. Majumdar, R. Ahuja, C. Popescu and F. J. Manjon, Pressure-induced order-disorder transitions in beta-In(2)S(3): an experimental and theoretical study of structural and vibrational properties, Phys. Chem. Chem. Phys., 2021, 23(41), 23625–23642 RSC.
- A. I. Ali, M. Ibrahim and A. Hassen, New fabrication method for di-indium tri-sulfuric (In(2)S(3)) thin films, Sci. Rep., 2022, 12(1), 7033 CrossRef CAS.
- Y. Suffren, F.-G. Rollet and C. Reber, Raman Spectroscopy of Transition Metal Complexes: Molecular Vibrational Frequencies, Phase Transitions, Isomers, and Electronic Structure, Comments Inorg. Chem., 2011, 32(5–6), 246–276 CrossRef CAS.
- M. Joos, C. Schneider, A. Münchinger, I. Moudrakovski, R. Usiskin, J. Maier and B. V. Lotsch, Impact of hydration on ion transport in Li2Sn2S5·xH2O, J. Mater. Chem. A, 2021, 9(30), 16532–16544 RSC.
- F. Xu, S. Leclerc and D. Canet, NMR relaxometry study of the interaction of water with a Nafion membrane under acid, sodium, and potassium forms. Evidence of two types of bound water, J. Phys. Chem. B, 2013, 117(21), 6534–6540 CrossRef CAS.
- G. A. H. Ludlam, S. J. P. Gnaniah, R. Degl'Innocenti, G. Gupta, A. J. Wain and H. Lin, Measurement of Water Uptake and States in Nafion Membranes Using Humidity-Controlled Terahertz Time-Domain Spectroscopy, ACS Sustain. Chem. Eng., 2024, 12(20), 7924–7934 CrossRef CAS.
- H. Furusawa, R. Konishi, D. Mori, H. Horino, T. Horiba, Y. Takeda, J. Takada, O. Yamamoto and N. Imanishi, Biogenous iron oxide (L-BIOX) as a high capacity anode material for lithium ion batteries, Electrochim. Acta, 2018, 281, 227–236 CrossRef CAS.
- Z. W. B. Iton, B. C. Lee, A. Y. Jiang, S. S. Kim, M. J. Brady, S. Shaker and K. A. See, Water Vapor Induced Superionic Conductivity in ZnPS(3), J. Am. Chem. Soc., 2023, 145(24), 13312–13325 CrossRef CAS.
- S. Ohno, C. Rosenbach, G. F. Dewald, J. Janek and W. G. Zeier, Linking Solid Electrolyte Degradation to Charge Carrier Transport in the Thiophosphate-Based Composite Cathode toward Solid-State Lithium-Sulfur Batteries, Adv. Funct. Mater., 2021, 31(18), 2010620 CrossRef CAS.
- H. Kwak, S. Wang, J. Park, Y. Liu, K. T. Kim, Y. Choi, Y. Mo and Y. S. Jung, Emerging Halide Superionic Conductors for All-Solid-State Batteries: Design, Synthesis, and Practical Applications, ACS Energy Lett., 2022, 7(5), 1776–1805 CrossRef CAS.
- Y.-T. Chen, D. H. S. Tan, S.-Y. Ham, B. Sayahpour, J. B. Lee, Y. Kim, M.-S. Song, L. H. B. Nguyen, J. A. S. Oh, P. Ridley, A. Cronk, G. Deysher, J. Jang, Z. Chen and Y. S. Meng, Investigating Dry Room Compatibility of Chloride Solid-State Electrolytes for Scalable Manufacturing, J. Electrochem. Soc., 2023, 170(8), 080521 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5sc01907a |
| This journal is © The Royal Society of Chemistry 2025 |