
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A salen-based dinuclear cobalt(II) polymer with direct and indirect synergy for electrocatalytic hydrogen evolution†
Xiao-Mei
Hu‡
,
Wen-Jie
Shi‡
 *,
Jian-Hua
Mei
,
Yu-Chen
Wang
,
Wei-Xue
Tao
,
Di-Chang
Zhong
* and
Tong-Bu
Lu
*
*,
Jian-Hua
Mei
,
Yu-Chen
Wang
,
Wei-Xue
Tao
,
Di-Chang
Zhong
* and
Tong-Bu
Lu
*
Institute for New Energy Materials and Low Carbon Technologies, School of Materials Science and Engineering, Tianjin University of Technology, Tianjin 300384, China. E-mail: lutongbu@tjut.edu.cn; dczhong@email.tjut.edu.cn; wjshi@email.tjut.edu.cn
First published on 2nd May 2025
Abstract
Optimizing the spatial arrangement and geometric configuration of dinuclear metal sites within catalysts to leverage the dinuclear metal synergistic catalysis (DMSC) effect is a promising strategy for enhancing catalytic performance. In this work, we report a salen-based dinuclear cobalt covalent organic polymer (Co2-COP) that exhibits both direct and indirect DMSC synergistic effects, significantly improving catalytic efficiency for the electrocatalytic alkaline hydrogen evolution reaction (HER). Notably, one of the Co atoms in this structural unit features an OH− anion. The OH− anion facilitates both H2O adsorption through p–p orbital overlapping interaction and the subsequent OH* intermediate removal by pre-attracting cations. As a result, Co2-COP exhibits superior HER activity that surpasses its single-atom counterpart by a factor of 36. Control experiments and theoretical calculations revealed that the enhanced catalytic efficiency of Co2-COP is attributed to both the direct DMSC effect between two CoII ions, and the indirect DMSC involving the OH− anion and alkali cations. This synergistic interaction significantly facilitates water activation and accelerates the removal of the OH* intermediate, all of which are intricately linked to the unique dinuclear structure of the material.
Introduction
The dinuclear metal synergistic catalysis (DMSC) effect has emerged as a promising strategy for enhancing catalytic performance in energy-related applications.1–5 This effect is facilitated by the presence of two metal centers within the catalyst, which work synergistically to boost reactivity and selectivity.6 In dinuclear catalysis, two distinct synergistic effects are typically observed: direct and indirect synergistic effects. The direct synergistic effect arises from the interaction between the two metal centers, typically involving electronic transfer,7 metal–metal interactions8 or cooperative coordination,9 which can accelerate substrate activation and facilitate reaction steps. On the other hand, the indirect synergistic effect is mediated by the ligands10 or solvent molecules11 in the reaction environment, which help the active center to stabilize key reaction intermediates or promote efficient charge transfer. While the majority of studies have focused on these effects in isolation,12,13 their coexistence and interplay within a single catalytic system remain largely underexplored.In electrocatalytic hydrogen evolution reactions (HER),14–18 dinuclear metal complexes enhance catalytic performance through both direct metal–metal interactions that facilitate proton reduction and indirect contributions from the ligands that stabilize water and modulate the activation of reactants. Salen-based dinuclear metal coordination complexes are gaining recognition as promising candidates for advanced catalytic systems due to their ability to tune the electronic properties of metal centers and enable cooperative catalysis (Scheme 1a). Moreover, these salen-based complexes have the potential to adsorb OH− anion at one metal site. This OH− anion can form hydrogen bond with H2O, strengthening the adsorption and stabilization of H2O adsorbed at the other metal site through spatial interaction involving the p orbitals of the O atoms (Scheme 1b).19 Furthermore, the adsorbed OH− anions attract alkali metal cation hydrates via electrostatic attraction, thereby facilitating the removal of the OH* intermediate and promoting the continuous progression of the reaction.
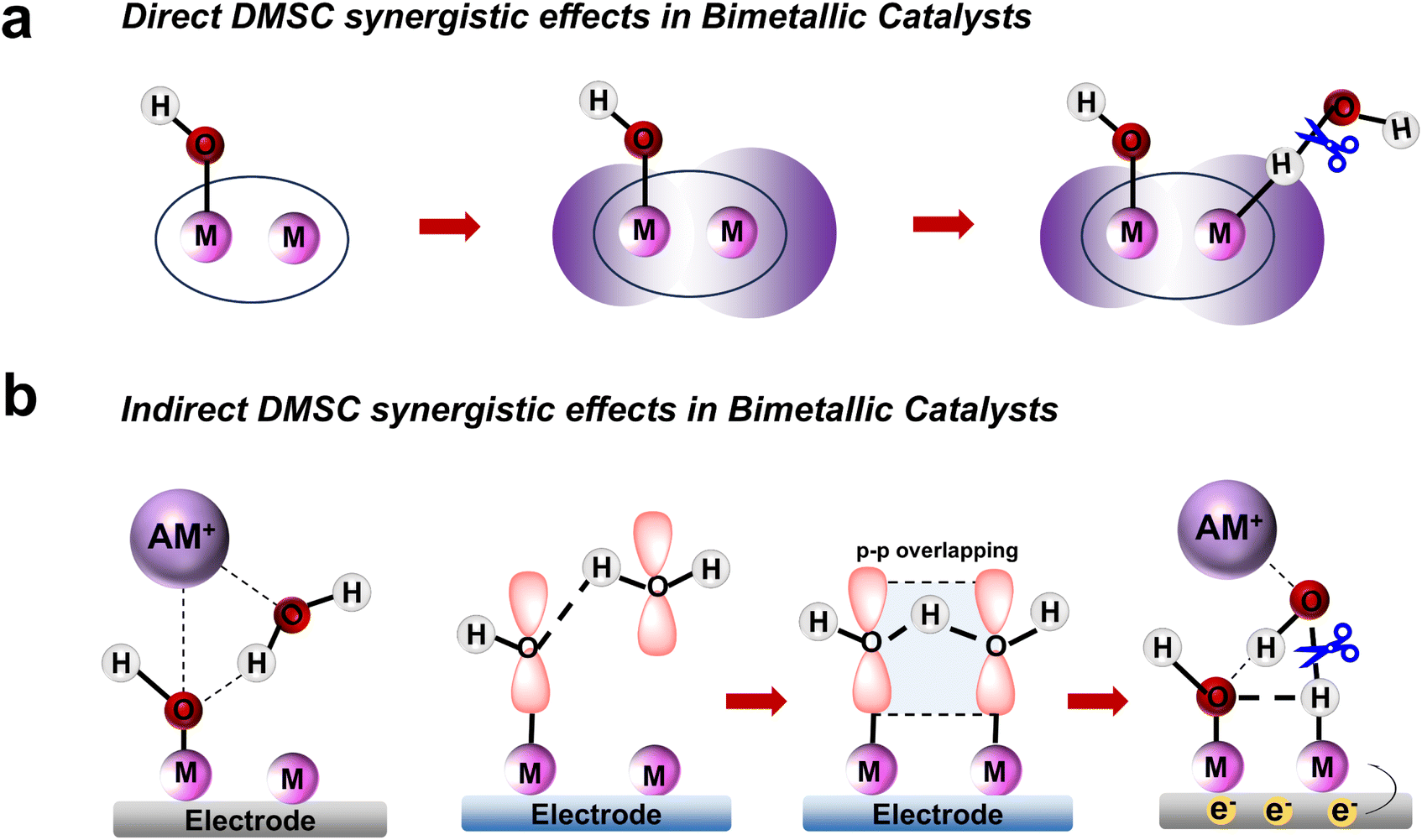 | ||
| Scheme 1 (a) Schematic diagram illustrating the direct DMSC effects in dinuclear catalysts to promote H2O dissociation. (b) Schematic diagram depicting the indirect DMSC effects to enhance H2O dissociation. | ||
Dinuclear cobalt systems represent a key class of DMSC-based catalysts, offering a well-balanced platform that combines electronic flexibility, redox tunability, and cooperative interactions.20–22 Recent studies have focused on engineering ligand environments23 adjusting metal–metal coordination geometry,24 and incorporating secondary interactions11 to enhance both catalytic activity and selectivity. Nevertheless, a systematic understanding of how direct and indirect synergistic effects interplay to enhance catalytic performance in dinuclear cobalt systems remains limited.
Herein, we report a dinuclear cobalt covalent organic polymer (Co2-COP) with well-defined Co-Robson units as an efficient electrocatalyst for alkaline HER (Fig. 1). Co2-COP exhibits significantly higher activity than its single-atom counterpart. Mechanistic studies reveal that adjacent Co atoms modulate the electronic environment to optimize intermediate adsorption (direct synergy), while surface-adsorbed OH− and hydrated K+ promote OH* desorption (indirect synergy). This work offers new insights into dual synergistic mechanisms in dinuclear cobalt catalysis under alkaline conditions.
 | ||
| Fig. 1 Schematic illustration of the synthetic process and structure of Co2-COP and Co-COP. | ||
Results and discussion
Preparation and characterization
The framework of Co2-COP, designated as TFBO-COP, was synthesized by condensing 2,2-dimethyl-1,3-propanediamine (DMPA) with 3,3′,5,5′-tetraformyl-4,4′-biphenyldiol (TFBO) (Fig. S1 and S2†). And Co2-COP was subsequently obtained by metalating TFBO-COP with Co(NO3)2·6H2O (Fig. 1 and S4†).25 Similarly, Co-COP containing mononuclear Co(II)-salen units was synthesized by metalating DHFP-COP, which was prepared by replacing TFBO with 4,4′-dihydroxy-3,3′-diformylbiphenyl (DHFP) in the condensation process (Fig. 1, S3 and S4†). The powder X-ray diffraction (XRD) found that both Co2-COP and Co-COP exhibit amorphous characteristics (Fig. S5†). The Fourier-transform infrared (FT-IR) spectra of TFBO and DHFP exhibit prominent C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibration at 1672 and 1660 cm−1, respectively. These bands disappear in Co2-COP and Co-COP spectra, where a new absorption bands at 1635 cm−1 appears, which can be attributed to the stretching vibration of CN bond (Fig. S6†).26 This shift indicates the successful synthesis of Co2-COP and Co-COP via Schiff-base condensation. In the solid-state 13C cross-polarized magic angle spinning (CP-MAS) NMR spectrum of TFBO-COP, a distinct signal at 166.7 ppm corresponds to the CN group,27 along with signals from aromatic and alkyl carbons (Fig. 2a and S7†), further supporting the formation of these materials through Schiff-base chemistry. Scanning electron microscopy (SEM) (Fig. S8 and S9†) and transmission electron microscopy (TEM) images (Fig. 2b) reveal block-like morphologies for both Co2-COP and Co-COP. Energy-dispersive X-ray spectroscopy (EDX) mapping confirms the homogeneous distribution of Co, N and O elements throughout both materials (Fig. S8 and S9†). The aberration-corrected HAADF-STEM image of Co2-COP shows paired bright dots (Fig. 2c), indicative of diatomic Co. The distance between two Co pair is approximately 2.7 Å, which matches the simulated Co–Co distance (2.7 Å) in Co2-COP (Fig. 2d). In contrast, Co atoms in Co-COP are separated by about 1.1 nm, in agreement with the simulated distance (Fig. S10†).
O stretching vibration at 1672 and 1660 cm−1, respectively. These bands disappear in Co2-COP and Co-COP spectra, where a new absorption bands at 1635 cm−1 appears, which can be attributed to the stretching vibration of CN bond (Fig. S6†).26 This shift indicates the successful synthesis of Co2-COP and Co-COP via Schiff-base condensation. In the solid-state 13C cross-polarized magic angle spinning (CP-MAS) NMR spectrum of TFBO-COP, a distinct signal at 166.7 ppm corresponds to the CN group,27 along with signals from aromatic and alkyl carbons (Fig. 2a and S7†), further supporting the formation of these materials through Schiff-base chemistry. Scanning electron microscopy (SEM) (Fig. S8 and S9†) and transmission electron microscopy (TEM) images (Fig. 2b) reveal block-like morphologies for both Co2-COP and Co-COP. Energy-dispersive X-ray spectroscopy (EDX) mapping confirms the homogeneous distribution of Co, N and O elements throughout both materials (Fig. S8 and S9†). The aberration-corrected HAADF-STEM image of Co2-COP shows paired bright dots (Fig. 2c), indicative of diatomic Co. The distance between two Co pair is approximately 2.7 Å, which matches the simulated Co–Co distance (2.7 Å) in Co2-COP (Fig. 2d). In contrast, Co atoms in Co-COP are separated by about 1.1 nm, in agreement with the simulated distance (Fig. S10†).
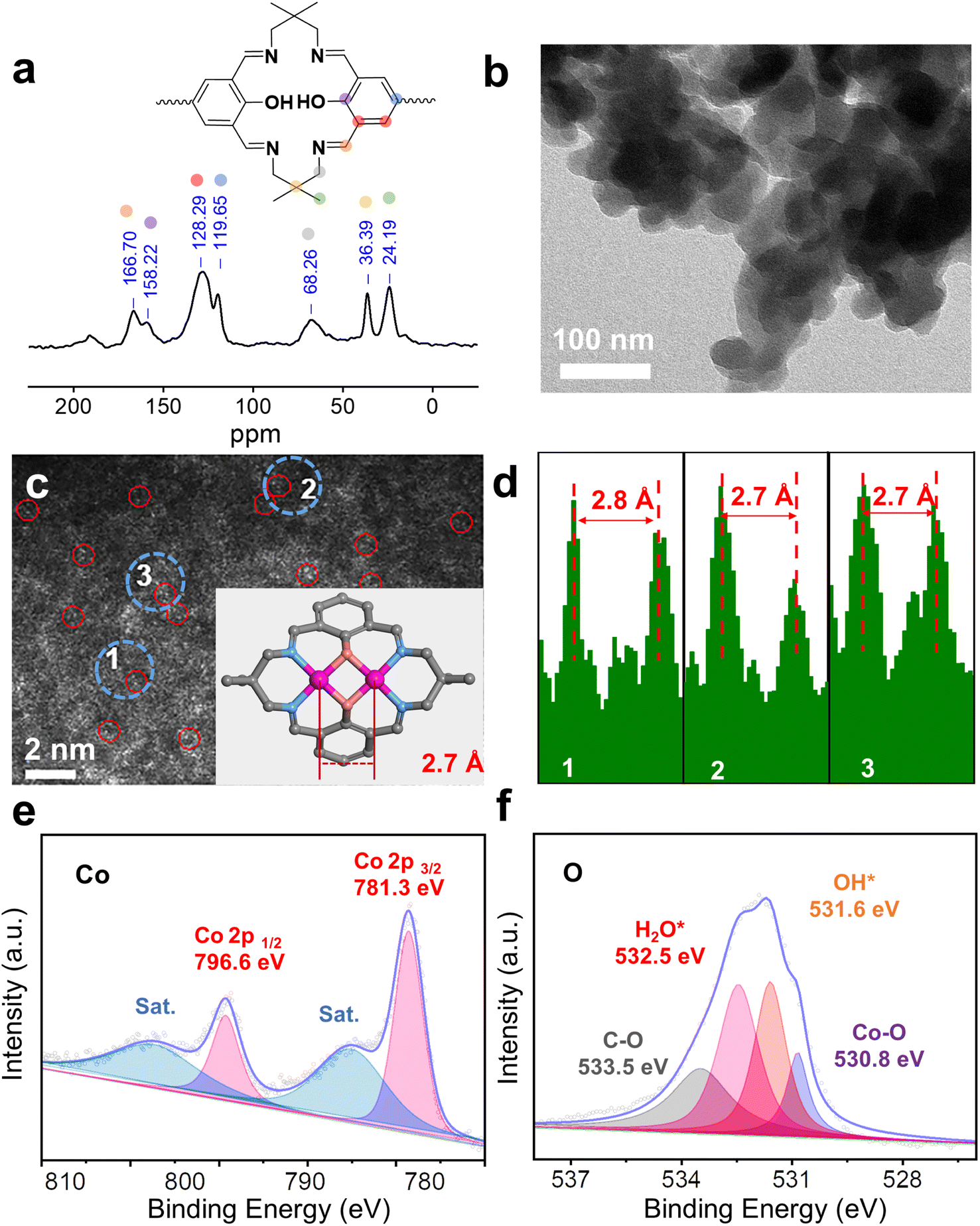 | ||
| Fig. 2 (a) 13C solid-state NMR spectra of TFBO-COP. (b) TEM image of Co2-COP. (c) HAADF-STEM image and the dinuclear cobalt distance in Co2-COP. (d) Intensity profiles corresponding to different areas of dinuclear cobalt in Co2-COP. XPS spectra of Co 2p (e) and O 1s (f) for Co2-COP. | ||
X-ray photoelectron spectroscopy (XPS) analysis confirmed the presence of C, N, O and Co in both polymers (Fig. S11a and S12a†). Co(2p) spectra show peaks at 781.3 and 796.6 eV for Co2-COP, and 781.2 and 796.3 eV for Co-COP, corresponding to Co 2p3/2 and Co 2p1/2, respectively (Fig. 2e and S12b†). These results indicate a +2 oxidation state for Co species in both materials.28 The N 1s spectra of both Co2-COP and Co-COP exhibit peaks at 399.2 and 399.3 eV, associated with Co–N coordination (Fig. S11b and S12d†).29,30 The O 1s spectra of Co2-COP show peaks at 530.8, 531.6, 532.5 and 533.5 eV, assigned to Co–O, OH*, H2O* and C–O,26 with a near 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio of OH* to H2O* (Fig. 2f).31–33 In contrast, the O 1s spectra of Co-COP displays three peaks at 531.0, 532.0, and 533.1 eV, attributed to Co–O, H2O*, and C–O, (Fig. S12c†) indicating the absence of OH* in Co-COP. Inductively coupled plasma optical mass spectrometry (ICP-MS) showed that Co2-COP contains 11.80 ± 0.40 wt% of Co, while Co-COP contains 10.43 ± 0.30 wt%, slightly lower than the theoretical values of 14.80% and 11.99%, respectively (Table S1†).
:1 ratio of OH* to H2O* (Fig. 2f).31–33 In contrast, the O 1s spectra of Co-COP displays three peaks at 531.0, 532.0, and 533.1 eV, attributed to Co–O, H2O*, and C–O, (Fig. S12c†) indicating the absence of OH* in Co-COP. Inductively coupled plasma optical mass spectrometry (ICP-MS) showed that Co2-COP contains 11.80 ± 0.40 wt% of Co, while Co-COP contains 10.43 ± 0.30 wt%, slightly lower than the theoretical values of 14.80% and 11.99%, respectively (Table S1†).
Electrocatalytic HER
The HER activity of Co2-COP and Co-COP were evaluated through linear sweep voltammetry (LSV) in a three-electrode setup using an N2-saturated 1 M KOH solution. Co2-COP demonstrated a catalytic reduction current density of 10 mA cm−2 at an overpotential (η) of 540 mV, which is significantly lower than that of Co-COP (η = 669 mV) (Table S2†). This overpotential corresponds to a specific current of 1233.7 mA mgCo−1 for Co2-COP (Fig. 3a), which is 36 times greater than that of Co-COP (34 mA mgCo−1). The Tafel slope of Co2-COP is 96 mV dec−1, notably lower than Co-COP 135 mV dec−1 (Fig. 3e and S23b†), further indicating its superior electrocatalytic performance. The Tafel slope analysis suggests that the HER mechanism for Co2-COP follows the Volmer–Heyrovsky pathway, whereas Co-COP predominantly follows the Volmer step. | ||
| Fig. 3 (a) HER polarization curves of Co2-COP and Co-COP in 1 M KOH aqueous solution. (b) Visualization of surface electrostatic potentials of Co2-COP. (c) Schematic diagram of FT-IR spectral interpretation. In the absence of AM+, the methyl4N+ adsorbed on the surface of Co2-COP as evidenced by increase in the ∼1488 cm−1 band, in the presence of K+ and Li+, methyl4N* is replaced by K+ or Li+ as indicated by weaken the ∼1488 cm−1 band in the infrared spectrum. HER polarization curves (d) and Tafel plots (e) of Co2-COP in 1.0 M AMOH (AM+ = Li+, Na+, K+ and Cs+). | ||
To further understand the superior HER activity of Co2-COP, electrochemical impedance spectroscopy (EIS) were conducted. The results show that Co2-COP exhibits a charge transfer resistance (Rct) of 89.38, which is significantly lower than that of Co-COP (Rct = 344.5), indicating faster charge transfer kinetics for Co2-COP (Fig. S13 and Table S3†). Additionally, the electrochemically active surface area (ECSA) was estimated by measuring the double-layer capacitance (Cdl) through cyclic voltammetry (CV) in the non-faradaic region (0–0.2 V vs. RHE). Co2-COP shows a Cdl value of 0.47 mF cm−2, similar to that of Co-COP (0.53 mF cm−2; Fig. S14 and Table S2†), suggesting that both materials offer similar accessible surface areas for the HER reaction. This implies that the enhanced HER performance of Co2-COP is due to the synergistic catalytic effect of the diatomic Co centers.
Further electrochemical assessments revealed that Co2-COP exhibits excellent electrocatalytic HER performance over a wide electrochemical window. As shown in Fig. S15a,† Co2-COP demonstrates an averaged faradaic efficiency (FE) of 86.7% in the overpotential range of from 500 to 700 mV, with the turnover frequency (TOF) increasing almost parabolically with η value. At η = 700 mV, the H2 production rate reaches 12.0 s−1, which is double that of Co-COP (Fig. S16†).34 Co2-COP also exhibits good electrochemical stability, as confirmed by long-term cyclic voltammetry and time-dependent potential measurements. After 3000 cycles, the polarization curves of Co2-COP displayed no significant potential decay (Fig. S15b†), and it maintains stable performance during 24 hours of continuous electrolysis at electrolysis at 10 mA cm−2 (inset Fig. S15b†). After the stability measurements, Co2-COP retained the block morphology (Fig. S17†), and no discernible change was observed in the FT-IR (Fig. S18†). The Co species remained in the +2 oxidation state (Fig. S19†). ICP-MS analyses demonstrated negligible leaching of Co (<0.4%) after electrolysis. These observations validate the exceptional stability of Co2-COP.
To identify the active sites in Co2-COP, we performed control experiments using metal-free TFBO-COP, DHFP-COP, and bare GC instead of Co2-COP. The current response of these materials is particularly low, indicating a lack of HER activity (Fig. S20†). This result indicates that Co(II) serves as the catalytic active site in Co2-COP. The differential pulse voltammogram of Co2-COP represented a CoII–CoI redox peak at −0.54 V, which closely resembles that of Co-COP (−0.57 V) (Fig. S21†).35 When 1 mL of H2O was added as the proton source, the CoI/II anodic peak vanished and the current density attributed to catalytic proton reduction increased (Fig. S22†). This observation suggests that the electrocatalytic HER mechanism is similar in Co2-COP and Co-COP, most likely via the Heyrovsky pathway, involving a CoI–H intermediate.36
Given the superior HER activity of Co2-COP, we hypothesis that the presence of AM+ (AM+: alkali metal cations) may play a crucial role in this enhancement. As Fig. 3b shown, the electrostatic potential (ESP) map reveals a blue region surrounding the OH group in Co2-COP, favoring the adsorption of positive cations from the solution. This leads to the hypothesis that AM+ cations can bind to the Co2-COP surface. To verify this hypothesis, we utilized tetramethylammonium (methl4N+) as the vibrational probe to detect AM+ cations adsorption.37,38 As shown in Fig. 3c, the absorbed methyl4N in Co2-COP presented the characteristic peak at ∼1488 cm−1. After treatment with a 100 mM KOH solution, this peak disappeared, whereas treatment with a 100 mM LiOH solution caused only a slight decrease in peak intensity. These results confirm that AM+ cations bind to the Co2-COP surface, with K+ showing higher adsorption capacity than Li+.
We then investigated the HER activity of Co2-COP in alkaline media containing different alkali metal cations (AMOH solution; AM+ = Li+, Na+, K+, and Cs+). As expected, Co2-COP exhibited the highest HER activity in KOH, with an activity order of K+ > Cs+ > Na+ > Li+ (Fig. 3d). This was in contrast to Co-COP, while the HER activity followed the order Cs+ < K+ < Na+ < Li+ (Fig. S23†). The Tafel slope for Co2-COP in different AMOH solutions was consistently below 120 mV dec−1, indicating a Volmer–Heyrovsky mechanism. Notably, the Tafel slope values correlated with the HER activity order, with the lowest value (and highest activity) observed in KOH.
To examine the influence of cation concentration on HER kinetics (Table S4†). The HER activity of Co2-COP and Co-COP increased significantly with higher concentrations of K+, Na+, and Li+ (Fig. S24 and S25†), and the reaction order with respect to cation concentration were positive. Specifically, the HER reaction order for K+ was higher in Co2-COP (around 0.8) compared to Na+ (around 0.6) and Li+ (around 0.5) (Fig. S26†). In contrast, the ranked list in Co-COP is Li+ > Na+ > K+, and their HER reaction orders were approximately 1.3, 0.7, and 0.6, respectively (Fig. S27†). These sequences are also consistent with their activity trends. These findings also suggest that increased cation concentration near the surface of Co2-COP enhances HER kinetics, with K+ being the most effective species for promoting adsorption and modifying the rate-determining step (RDS) of the HER process.
Direct and indirect DMSC effect investigation
Understanding both the direct and indirect DMSC effect of diatomic catalysts on HER is crucial for the design of high-performance electrocatalysts. To explore the direct interaction of diatomic Co in Co2-COP, we performed density functional theory (DFT) calculations. In alkaline media, the HER follows a two-electron transfer process, involving the Volmer, Heyrovsky, and Tafel steps. Water dissociation which is initiated in the Volmer step is crucial for H* generation. Electrochemical HER tests indicate that water dissociation kinetics play a vital role in the activity of both Co2-COP and Co-COP. Hence, we simulated the adsorption of both H2O and H species on the individual structures (Fig. S28†). The energy profile for water adsorption reveals that Co2-COP has a lower energy barrier of −0.29 eV compared to Co-COP's 0.1 eV, indicating that the diatomic Co in Co2-COP significantly enhances water adsorption and dissociation kinetics. Moreover, Co2-COP shows more favorable hydrogen adsorption with a ΔGH value of 0.33 eV, compared to Co-COP's 1.72 eV (Fig. 4a). A comparison of the projected density of states (PDOS, Fig. 4b) further highlights how the adjacent Co atoms in Co2-COP effectively enhances the total d-electron domination of the catalyst near the Fermi level, which will benefit H2O activation and lead to energetically catalytic activity.39 This suggests that the diatomic Co centers in Co2-COP interact synergistically, optimizing reactant and product adsorption to promote efficient HER.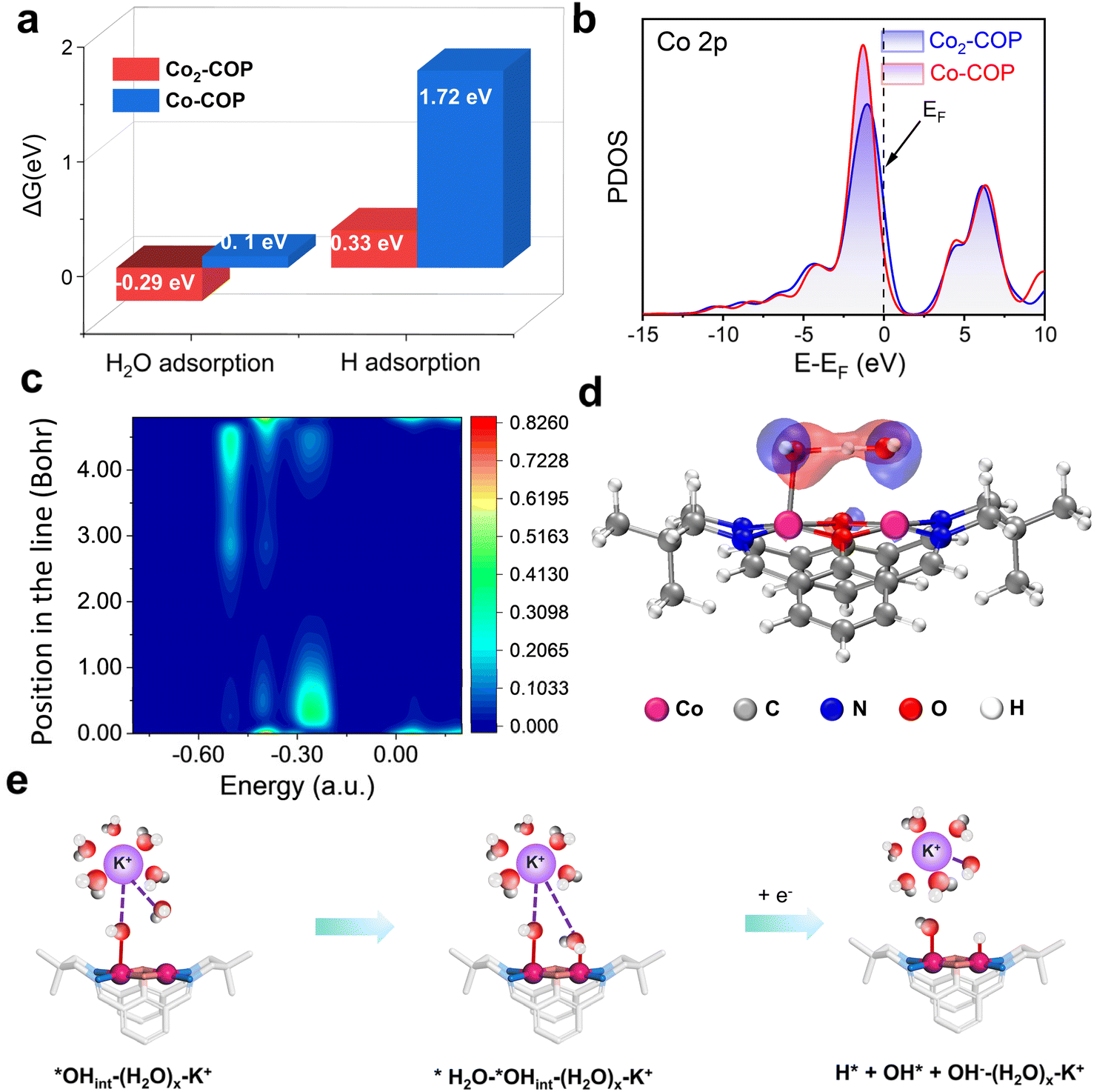 | ||
| Fig. 4 (a) The calculated water adsorption and hydrogen adsorption free-energy diagram (ΔGH2O*) of Co2-COP (red) and Co-COP (blue). (b) PDOS consisting of Co2-COP and Co-COP. The Fermi level is set to 0 eV. LDOS (c) and the orbital interactions (d) between the O atom of the OH− anion and water in Co2-COP, with water adsorbed on the Co site. (e) Mechanism diagram of (H2O)x–K+ synergically promoting alkaline HER for Co2-COP. | ||
Moreover, the adjacent Co atom's OH group in Co2-COP forms a hydrogen bond with H2O, with a bond length of 1.50 Å (Fig. S28†). Notably, the local density of states (LDOS) and orbital spectra of Co2-COP reveal that the adsorbed H2O is stabilized by the OH− anion, which interacts with the H2O at the adjacent Co site through spatial interactions involving the p orbitals of the oxygen atoms (Fig. 4c and d). These results showing the indirect synergistic effect of the diatomic catalyst promotes H2O dissociation. Subsequently, we investigated the indirect influence of cations on HER kinetics in Co2-COP. To understand the pH effect on HER kinetics, we adjusted the pH of the electrolyte and observed the response for both Co2-COP and Co-COP (Table S5†). As shown in Fig. S29a,† no pH dependence for the kinetics of HER in Co2-COP under alkaline conditions on the NHE scale, whereas Co-COP exhibits pH dependence (Fig. S29d†), likely due to differences in the RDS between the two catalysts. The Tafel slope for Co2-COP remained below 120 mV dec−1, consistent with a Volmer–Heyrovsky mechanism (Fig. S29b and e†). Conversely, the RDS of HER by Co-COP remained the Volmer step at all times.40 These observations suggest that the cation preconcentration on the surface of Co2-COP had positively impacts the HER kinetics on the RHE scale. Remarkably, the reaction order of HER in Co2-COP varies with cation concentration, as demonstrated in Fig. S29c and f,† where the reaction order decrease from 0.66 to 0.24 as the voltage shifts from −0.6 to −0.72 V, in contrast to the constant order observed for Co-COP.
Herein we proposed the indirect DMSC effect mechanism for Co2-COP based on the hard–soft acid–base (HSAB) theory established in the literature.41 In this framework, K+ is classified as a Lewis acid, and the OH groups (*OHCo) in the Co2-COP catalyst function as a Lewis base. As a result, the (H2O)x–K+ adduct in the electrical double layer region is attracted to the surface of Co2-COP. The full Volmer mechanism involves the adsorption and dissociation of water molecules. Therefore, the step of Co2-COP involving (H2O)x–K+ can be divided into two separate steps (Fig. 4e):
| H2O* + *OHCo–(H2O)x–K+ → H* + *OHCo + OH−–(H2O)xK+ | (2a) |
| H* + *OHCo + OH−–(H2O)x–K+ → H* + *OHCo + H2O + OH−–(H2O)x–K+ | (2b) |
The affinity of hard Lewis acid K+ towards hard Lewis base OH− is strong, thus promoting OH− desorbing into the bulk and encouraging the Volmer step in a unidirectional manner,42,43 the diatomic Co in Co2-COP worked with cations finished the indirect synergistic catalysis.
The driving force for the OH− desorption weakens in the order of Li+ > Na+ > K+ > Cs+, but the sequential trend for HER activity of Co2-COP is K+ > Cs+ > Na+ > Li+. This apparent anomaly may arise from the interplay between cation hydration and the steric landscape of the dinuclear active sites. The hydrated radii of Li+, Na+, K+, and Cs+ are approximately 3.1, 2.7, 2.4 and 2.1 Å, respectively (Fig. S30†).44–46 By contrast, the salen methylene groups on the Co2-COP surface create a steric pore window, with minimum and maximum separations of ∼2.5 Å and ∼7.3 Å (Fig. S31†). Hydrated K+ and Cs+ cations are comparable to the pore window, so they adsorb loosely on the Co2-COP surface. Since K+ possesses a moderate hydration ability, continuously supplying fresh H2O to the Co sites and giving the highest HER rate. While Cs+ has weaker hydration capacity, delivers water less efficiently and thus shows slightly lower activity than K+. The hydrated Li+ and Na+ clusters are only slightly smaller than the pore window of Co2-COP, form a dense interfacial layer over the Co-OH* sites. Their extensive hydration shells hinder OH− desorption, thereby diminishing the HER performance of Co2-COP.
Conclusion
In summary, we have successfully designed and synthesized a novel dinuclear cobalt catalyst Co2-COP, which incorporates well-defined Co-Robson structural units with a Co–Co distance of 2.7 Å. This design effectively facilitates both water dissociation and OH* intermediate removal, showcasing the power of direct and indirect DMSC effect. Co2-COP exhibits outstanding electrocatalytic performance for the alkaline HER, achieving a mass activity 36 times higher than its single-atom counterpart (Co-COP). Theoretical calculations and experimental data confirm that the enhanced catalytic efficiency of Co2-COP stems from the favorable adsorption energies for both water and hydrogen, as well as the synergistic electronic interactions between the two Co atoms. Furthermore, the alkali-metal-cation-dependent effect, especially with K+, plays a crucial role in optimizing the HER process by stabilizing the OH* intermediate and enhancing charge transfer. These findings provide valuable insights into the combined influence of direct and indirect DMSC effects, paving the way for the design of more efficient dinuclear catalysts for energy conversion applications.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
D.-C. Zhong and W.-J. Shi conceived the idea. X.-M. Hu performed the main experiments. X.-M. Hu analyzed the data. W.-J. Shi and W.-X. Tao performed DFT calculations. J.-H. Mei and Y.-C. Wang performed the FT-IR measurements. W.-J. Shi and X.-M. Hu wrote the manuscript, D.-C. Zhong and T.-B. Lu revised the paper. All authors discussed the results and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Key R&D Program of China (2022YFA1502902), the National Natural Science Foundation of China (22201209, 22271218, 22071182, and 21931007). We appreciate the kind help from Prof. Zhenkun Zhang for the Solid 13C NMR experiments measurements in Nankai University.References
- Z. Chen, Y. Xu, D. Ding, G. Song, X. Gan, H. Li, W. Wei, J. Chen, Z. Li, Z. Gong, X. Dong, C. Zhu, N. Yang, J. Ma, R. Gao, D. Luo, S. Cong, L. Wang, Z. Zhao and Y. Cui, Nat. Commun., 2022, 13, 763 CrossRef CAS PubMed
.
- Q. Hu, K. Gao, X. Wang, H. Zheng, J. Cao, L. Mi, Q. Huo, H. Yang, J. Liu and C. He, Nat. Commun., 2022, 13, 3958 CrossRef CAS
- Y. Zhou, L. Chen, L. Sheng, Q. Luo, W. Zhang and J. Yang, Nano Res., 2022, 15, 7994–8000 CrossRef CAS
- D. Liu, Y. Zhao, C. Wu, W. Xu, S. Xi, M. Chen, L. Yang, Y. Zhou, Q. He, X. Li, B. Ge, L. Song, J. Jiang and Q. Yan, Nano Energy, 2022, 98, 107296 CrossRef CAS
- C. Cai, K. Liu, L. Zhang, F. Li, Y. Tan, P. Li, Y. Wang, M. Wang, Z. Feng, D. Motta Meira, W. Qu, A. Stefancu, W. Li, H. Li, J. Fu, H. Wang, D. Zhang, E. Cortes and M. Liu, Angew. Chem., Int. Ed., 2023, 62, e202300873 CrossRef CAS PubMed
- Z. Yu, G. Xia, V. M. Diaconescu, L. Simonelli, A. P. LaGrow, Z. Tai, X. Xiang, D. Xiong and L. Liu, Chem. Sci., 2024, 15, 9216–9223 RSC
- D. Zu, Y. Ying, Q. Wei, P. Xiong, M. S. Ahmed, Z. Lin, M. M. J. Li, M. Li, Z. Xu, G. Chen, L. Bai, S. She, Y. H. Tsang and H. Huang, Angew. Chem., Int. Ed., 2024, 63, e202405756 CrossRef CAS PubMed
- Y. Li, B. Wei, M. Zhu, J. Chen, Q. Jiang, B. Yang, Y. Hou, L. Lei, Z. Li, R. Zhang and Y. Lu, Adv. Mater., 2021, 33, 2102212 CrossRef CAS PubMed
- T. Ouyang, H. J. Wang, H. H. Huang, J. W. Wang, S. Guo, W. J. Liu, D. C. Zhong and T. B. Lu, Angew. Chem., Int. Ed., 2018, 57, 16480–16485 CrossRef CAS PubMed
- W. Yu, H. Huang, Y. Qin, D. Zhang, Y. Zhang, K. Liu, Y. Zhang, J. Lai and L. Wang, Adv. Energy Mater., 2022, 12, 2200110 CrossRef CAS
- Y.-N. Gong, S.-Q. Zhao, H.-J. Wang, Z.-M. Ge, C. Liao, K.-Y. Tao, D.-C. Zhong, K. Sakai and T.-B. Lu, Angew. Chem., Int. Ed., 2024, 63, e202411639 CrossRef CAS PubMed
- M. A. Deshmukh, A. Bakandritsos and R. Zbořil, Nano-Micro Lett., 2025, 17 DOI:10.1007/s40820-024-01505-2
- D.-C. Zhong, Y.-N. Gong, C. Zhang and T.-B. Lu, Chem. Soc. Rev., 2023, 52, 3170–3214 RSC
- J. A. Turner, Science, 2004, 305, 972–974 CrossRef CAS
- X. Wang, Y. Zheng, W. Sheng, Z. J. Xu, M. Jaroniec and S.-Z. Qiao, Mater. Today, 2020, 36, 125–138 CrossRef CAS
- L. Yu, Q. Zhu, S. Song, B. McElhenny, D. Wang, C. Wu, Z. Qin, J. Bao, Y. Yu, S. Chen and Z. Ren, Nat. Commun., 2019, 10, 5106 CrossRef PubMed
- D. Zhao, Z. Zhuang, X. Cao, C. Zhang, Q. Peng, C. Chen and Y. Li, Chem. Soc. Rev., 2020, 49, 2215–2264 RSC
- F. Sun, J. Qin, Z. Wang, M. Yu, X. Wu, X. Sun and J. Qiu, Nat. Commun., 2021, 12, 4182 CrossRef CAS PubMed
- M. Ding, B.-Q. Shan, B. Peng, J.-F. Zhou and K. Zhang, Phys. Chem. Chem. Phys., 2022, 24, 7923–7936 RSC
- K. Yuan, K. Tao, T. Song, Y. Zhang, T. Zhang, F. Wang, S. Duan, Z. Chen, L. Li, X. Zhang, D. Zhong, Z. Tang, T.-B. Lu and W. Hu, J. Am. Chem. Soc., 2024, 146, 6893–6904 CrossRef CAS PubMed
- H.-F. Wang, W.-J. Shi, Y.-X. Yang, B.-X. Dong, T.-B. Lu and D.-C. Zhong, Sci. China: Chem., 2024, 68, 201–208 CrossRef
- Y.-N. Gong, S.-Y. Lv, H.-Y. Yang, W.-J. Shi, J.-J. Wang, L. Jiang, D.-C. Zhong and T.-B. Lu, CCS Chem., 2024, 6, 3030–3040 CrossRef CAS
- C. J. Lu, W. J. Shi, Y. N. Gong, J. H. Zhang, Y. C. Wang, J. H. Mei, Z. M. Ge, T. B. Lu and D. C. Zhong, Angew. Chem., Int. Ed., 2024, 63, e202405451 CrossRef CAS PubMed
- Y. Wang, W. Shi, W. Tao, J. Zhang, D.-C. Zhong and T. Lu, Sci. China: Chem., 2024, 68, 974–979 CrossRef
- S. M. Elbert and M. Mastalerz, Org. Mater., 2020, 02, 182–203 CrossRef CAS
- B. Zhang, L. Chen, Z. Zhang, Q. Li, P. Khangale, D. Hildebrandt, X. Liu, Q. Feng and S. Qiao, Adv. Sci., 2022, 9, e2105912 CrossRef
- Z. Shan, M. Wu, D. Zhu, X. Wu, K. Zhang, R. Verduzco and G. Zhang, J. Am. Chem. Soc., 2022, 144, 5728–5733 CrossRef CAS PubMed
- J. Zhang, S. Shao, D. Zhou, T. Di and T. Wang, ACS Appl. Energy Mater., 2021, 4, 6340–6347 CrossRef CAS
- F. Kong, X. Fan, X. Zhang, L. Wang, A. Kong and Y. Shan, Carbon, 2019, 149, 471–482 CrossRef CAS
- Z. B. Zhou, P. J. Tian, J. Yao, Y. Lu, Q. Y. Qi and X. Zhao, Nat. Commun., 2022, 13, 2180 CrossRef CAS PubMed
- Y. Wang, W. He, L. Wang, J. Yang, X. Xiang, B. Zhang and F. Li, Chem.–Asian J., 2015, 10, 1561–1570 CrossRef CAS PubMed
- D. Lützenkirchen-Hecht, Surf. Sci. Spectra, 2011, 18, 102–109 CrossRef
- H. Han, Z. Bai, T. Zhang, X. Wang, X. Yang, X. Ma, Y. Zhang, L. Yang and J. Lu, Nano Energy, 2019, 56, 724–732 CrossRef CAS
- D. Cao, H. Xu, H. Li, C. Feng, J. Zeng and D. Cheng, Nat. Commun., 2022, 13, 5843 CrossRef CAS PubMed
- D. Micheroni, G. Lan and W. Lin, J. Am. Chem. Soc., 2018, 140, 15591–15595 CrossRef CAS
- B. Jiang, M. Gil-Sepulcre, P. Garrido-Barros, C. Gimbert-Surinach, J. W. Wang, J. Garcia-Anton, P. Nolis, J. Benet-Buchholz, N. Romero, X. Sala and A. Llobet, Angew. Chem., Int. Ed., 2022, 61, e202209075 CrossRef CAS
- F. He, W. Chen, B.-Q. Zhu, E.-f. Zhen, J. Cai and Y.-X. Chen, J. Phys. Chem. C, 2021, 125, 5715–5722 CrossRef CAS
- V. J. Ovalle, Y.-S. Hsu, N. Agrawal, M. J. Janik and M. M. Waegele, Nat. Catal., 2022, 5, 624–632 CrossRef CAS
- K. L. Zhou, Z. Wang, C. B. Han, X. Ke, C. Wang, Y. Jin, Q. Zhang, J. Liu, H. Wang and H. Yan, Nat. Commun., 2021, 12, 3783 CrossRef CAS
- T. Shinagawa, A. T. Garcia-Esparza and K. Takanabe, Sci. Rep., 2015, 5, 13801 CrossRef
- E. Liu, J. Li, L. Jiao, H. T. T. Doan, Z. Liu, Z. Zhao, Y. Huang, K. M. Abraham, S. Mukerjee and Q. Jia, J. Am. Chem. Soc., 2019, 141, 3232–3239 CrossRef CAS
- N. Danilovic, R. Subbaraman, D. Strmcnik, A. P. Paulikas, D. Myers, V. R. Stamenkovic and N. M. Markovic, Electrocatalysis, 2012, 3, 221–229 CrossRef CAS
- L. Jiao, E. Liu, S. Mukerjee and Q. Jia, ACS Catal., 2020, 10, 11099–11109 CrossRef CAS
- J. Mahler and I. Persson, Inorg. Chem., 2012, 51, 425–438 CrossRef PubMed
- J. Yu, J. Yin, R. Li, Y. Ma and Z. Fan, Chem Catal., 2022, 2, 2229–2252 CrossRef CAS
- K. Zhou, C. Qian and Y. Liu, J. Phys. Chem. B, 2022, 126, 10471–10480 CrossRef CAS PubMed
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5sc02073e |
| ‡ X.-M. Hu and W.-J. Shi contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |