
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUltrahigh photocatalytic hydrogen evolution of linear conjugated terpolymers enabled by an ultra-low ratio of the benzothiadiazole monomer†
Zheng-Hui
Xie‡
ab,
Gang
Ye‡
 c,
Hao
Gong
ab,
Pachaiyappan
Murugan
d,
Can
Lang
ab,
Yi-Fan
Dai
ab,
Kai
Yang
b and
Shi-Yong
Liu
*ab
c,
Hao
Gong
ab,
Pachaiyappan
Murugan
d,
Can
Lang
ab,
Yi-Fan
Dai
ab,
Kai
Yang
b and
Shi-Yong
Liu
*ab
aSchool of Chemical Engineering, Guangdong University of Petrochemical Technology, Maoming, Guangdong 525000, China. E-mail: chelsy@zju.edu.cn
bJiangxi Provincial Key Laboratory of Functional Crystalline Materials Chemistry, Department of Chemistry and Chemical Engineering, Jiangxi University of Science and Technology, Ganzhou 341000, China
cKey Laboratory for the Green Preparation and Application of Functional Materials, Hubei Key Laboratory of Polymer Materials, School of Materials Science and Engineering, Hubei University, Youyi Road 368, Wuhan, 430062, China
dCenter for Global Health Research, Saveetha Medical College and Hospital, Saveetha Institute of Medical and Technical Sciences, Kancheepuram District, Tamil Nadu, India
First published on 29th April 2025
Abstract
Conjugated terpolymers bearing three kinds of π-monomers have been regarded as a promising platform for photocatalytic hydrogen production (PHP). However, the high-performance terpolymers reported so far typically involve large portions (≥20 mol%) of the third monomer. Efficiently modulating the terpolymer by utilizing minimum content of the third component remains a critical challenge. Herein, we report a donor–acceptor linear terpolymer prepared by atom-economical C–H/C–Br coupling with an ultra-low ratio (0.5 mol%) of benzothiadiazole (BT) as the third monomer, which can efficiently modulate properties and afford a hydrogen evolution rate of up to 222.28 mmol h−1 g−1 with an apparent quantum yield of 24.35% at 475 nm wavelength in the absence of a Pt co-catalyst. Systematic spectroscopic studies reveal that even a minimal amount of the BT monomer can effectively tune the light absorption and frontier molecular orbitals of the resulting terpolymers. Compared to the BT-free BSO2–EDOT bi-polymer, the terpolymer BSED–BT0.5% involving 0.5 mol% of BT has a much faster electron transfer (5.76 vs. 1.13 ns) and much lower exciton binding energy (61.35 vs. 32.03 meV), showcasing an important discovery that the BT building block even with an ultra-low ratio enables the effective modulations of terpolymers with ultra-high PHP performance.
Introduction
As the world's fossil energy crisis worsens and global warming intensifies, the search for green and sustainable energies has become one of the key research areas.1,2 Hydrogen gas (H2) has garnered significant attention as a sustainable energy source owing to its high calorific value with carbon-free emission. Photocatalytic hydrogen production (PHP) is recognized as one of the most promising technologies for converting solar energy into green H2.3–6 Since Fujishima and Honda first reported titanium dioxide (TiO2) photocatalysts for PHP application,7 numerous inorganic semiconductor photocatalysts have been explored for hydrogen production8,9 Inorganic photocatalysts, however, struggle with the inefficient utilization of visible light due to their rigid structures, which inherently constrain the tunability of the bandgaps. π-Conjugated polymer (CP)-based photocatalysts, such as graphitic carbon nitride (g-C3N4),10,11 linear conjugated polymers (LCPs),12–14 conjugated microporous polymers (CMPs),15,16 covalent triazine frameworks (CTFs),17,18 and covalent organic frameworks (COFs),19–21 with unique merits such as structural versatility and flexibility, broad tunability of the opto-electrochemical properties, and outstanding capacity for visible light absorption, have attracted significant attention in recent years.22,23a,bThe introduction of donor–acceptor (D–A) architecture into polymeric photocatalysts has emerged as a convenient and pivotal strategy for achieving efficient PHP.24a–c This is due to the interaction between the electron donor and acceptor blocks, which facilitates the formation of a robust internal electric field across the polymeric backbone. This, in turn, triggers intramolecular charge transfer (ICT) from D units to A units.25a–c Upon light irradiation, this ICT can effectively aid the electron (e−)–hole (h+) pair separation—a crucial step toward photocatalysis. Furthermore, the huge library of π-monomers provides enormous versatility in designing CP-based photocatalysts, enabling the fine modulation of the properties for superior performance. Based on this principle, a large number of donor and acceptor building blocks have been explored for building D–A polymer photocatalysts. In 2016, the Wang group26 first reported a variety of D–A type CPs wherein benzothiadizole (BT) served as an acceptor. By optimizing the compositions, an optimal hydrogen evolution rate (HER) of 116 μmol h−1 (50 mg) was achieved. The Jiang group27 developed a D–A-type CP photocatalyst by using 3,7-dibenzothiophene-5,5-dioxide (BTSO2) as an acceptor and achieved an HER of 5.697 mmol g−1 h−1. Recently, our group28 successfully developed an innovative D–A type LCP-based photocatalyst involving 3,4-ethylenedioxythiophene (EDOT) as the hydrophilic donor and BTSO2 as the acceptor through direct C–H arylation polymerization (DArP). This exceptional photocatalyst exhibits a remarkable HER of up to 158.4 mmol h−1 g−1, coupled with an unprecedented apparent quantum yield (AQY) of 13.6% at λ = 550 nm by using 6 mg of photocatalyst without the aid of a Pt cocatalyst.
Compared to the established binary D–A CPs, the ternary strategy by incorporating a third building block into backbones provides a new avenue for the design of CP-based photocatalysts. The pioneering work by Cooper's group15 explored terpolymer-based photocatalysts for hydrogen evolution by polymerizing three kinds of π-monomers. This approach could effectively modulate opto-electrochemical properties and suppress the recombination of charges by leveraging the frontier molecular orbital (FMO) gradients created by the multiple D or multiple A units, thereby enhancing the charge carrier separation and broadening the light absorption.29–32 For example, the Araujo group33 reported a library of D1–D2–A ternary CP photocatalysts with improved PHP performance due to the promoted light absorption by the electron-accepting BT unit. The Jiang group30 reported a library of D–π–A CP photocatalysts by using phenyl as π-linker units, showing broadened light absorption enabled by the elongated π-conjugation, thereby promoting the PHP performance. The Li group34 reported a library of D–π–D–A terpolymer photocatalysts with improved PHP performance due to the accelerated electron transfer by 30 mol% of the pendant BT alongside the main backbone. Very recently, our group35 developed a library of linear terpolymer photocatalysts by using phenyl as a π-linker, among which, the terpolymer involving 25 mol% of the phenyl block exhibits the optimal PHP performance due to improved hydrophilicity and charge separation. It is reasonable to design terpolymers to tune the light absorption and increase the carrier lifetime, thus improving the photocatalytic activity. However, the third building blocks of the terpolymers reported so far typically need to be added more than 20 mol% to sufficiently exert a synergic effect.36–42 It is still a critical challenge to efficiently modulate terpolymers by utilizing a minimum ratio of the third monomers.
In this work, a series of D–A type linear terpolymers with EDOT as a donor and BTSO2 and BT as acceptors were obtained via an atom economical DArP strategy. The feed ratios of the BT unit in the backbone were strategically tuned according to the stoichiometry, leading to formation of ten linear terpolymers (Scheme 1). Note that BT units incorporated even in ultra-low ratios were able to drastically broaden the light absorption, affect the charge transfer pathway, and facilitate the directional charge transport of the terpolymers. The prepared terpolymers exhibited strong absorption in the wavelengths between 300 and 600 nm, which was gradually extended to 800 nm with the increase of BT content. As the BT content increases, the optical bandgaps (Eg) of the terpolymers undergo a precise modulation, narrowing from 2.06 eV to 1.53 eV. These changes were mainly attributed to the precise modulations of ICT, FMOs, and light absorption of the terpolymers by finely tuning the ratios of BT involved in the polymeric backbone, emphasizing the important influence of the BT unit on the opto-electrochemical properties. PHP tests show that the terpolymer BSED–BT0.5% involving 0.5 mol% of BT units exhibits the highest HER up to 222.28 mmol h−1 g−1 under visible light (>420 nm) irradiation without the use of Pt co-catalysts. The highest AQY achieved was 22.73% at 500 nm. Impressively, compared to the BT-free BSO2–EDOT bi-polymer, the terpolymer BSED–BT0.5% shows a much faster non-radiative rate (0.23 vs. 0.69 ns), much lower exciton binding energy (32.03 vs. 61.35 meV), and much faster electron transfer (1.13 vs. 5.76 ns). This work discloses that 0.5 mol% of BT can play a vital role in achieving ultrahigh HERs of the terpolymer, which represents a novel protocol for the design of high-performance terpolymer photocatalysts by using a minimal amount of the third monomer.
 | ||
| Scheme 1 Synthetic routes of bi-polymers BSO2–EDOT and BT–EDOT and terpolymer BSED–BTx (x represents the molar ratio of BT) via DArP polymerization. | ||
Results and discussion
Synthesis and structural characterization
All polymers were prepared via the DArP polymerization of EDOT with dibromo-benzothiophene-5,5-dioxide (BTSO2) and BT by gradually tuning the stoichiometry of BTSO2 and BT building blocks (Scheme 1). The resulting ten binary & ternary LCPs named BSO2–EDOT, BSED–BT0.1%, BSED–BT0.5%, BSED–BT1.0%, BSED–BT2.5%, BSED–BT5.0%, BSED–BT10.0%, BSED–BT25.0%, BSED–BT50.0% and BT–EDOT contain, respectively, zero, 0.1 mol%, 0.5 mol%, 1.0 mol%, 2.5 mol%, 5 mol%, 10 mol%, 25 mol%, 50 mol%, and 1 equiv. of the BT unit.The structures of the resulting LCPs with varied feed ratios of monomers were verified by Fourier transform infrared spectroscopy (FT-IR) and solid-state 13C NMR. Fig. 1a and S1 (ESI†) show the FT-IR spectra of all LCPs. The characteristic peaks at ∼1590 cm−1 and 1080 cm−1 assigned, respectively, to the stretch model of the aryl π-linkage backbone and skeletal vibrations of the C–O–C in the EDOT unit can be found in all LCPs. Except for BTSO2-free BT–EDOT, the other nine LCPs exhibit characteristic peaks at ∼1150 and ∼1300 cm−1 that correspond to the stretching vibrations of the sulfone group (S![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) OS) of the BTSO2 unit. The characteristic peak at 1440 cm−1 corresponds to the stretching vibration of the N–S bond in BT. Notably, the intensity of this peak gradually diminishes as the BT ratio decreases and ultimately vanishes in the BT-free BSO2–EDOT bi-polymer.
OS) of the BTSO2 unit. The characteristic peak at 1440 cm−1 corresponds to the stretching vibration of the N–S bond in BT. Notably, the intensity of this peak gradually diminishes as the BT ratio decreases and ultimately vanishes in the BT-free BSO2–EDOT bi-polymer.
 | ||
| Fig. 1 (a) FT-IR spectra and (b) solid-state 13C NMR spectra of BSO2–EDOT, BSED–BT0.5%, BSED–BT1.0%, BSED–BT50.0%, and BT–EDOT; SEM images of (c) BSO2–EDOT, (d) BSED–BT0.5%, (e) BSED–BT1.00%, (f) BSED–BT50.0%, and (g) BT–EDOT. EDS elemental mapping of (h) N, (i) O and (j) S in BSED–BT0.5%. | ||
Fig. 1b and S2† show the solid-state 13C NMR spectra of the polymers, wherein the carbon atoms were labelled as “a–g”. All polymers have a signal at ∼65 ppm that belongs to the carbons of ethylenedioxy groups in the EDOT unit (carbons a), and the signals at ∼140 and ∼115 ppm are attributed to α and β-quaternary carbons of the EDOT unit (carbons b and c). The signals of the BTSO2 unit can be found at ∼121 ppm (carbons d), ∼129 ppm (carbons f and g) and ∼134 ppm (carbons e). It is worth noting that the intensities of these BTSO2 signals decrease as the contents of BTSO2 decrease and ultimately diminish in the BTSO2-free BT–EDOT bi-polymer. The characteristic signals of the carbons in the BT unit can be found at ∼125 and ∼152 ppm (carbons h and i), and the intensities of these peaks progressively increase as the BT contents increase. The above analyses evidently reveal that the trends of FT-IR and 13C NMR spectra are consistent with the structural evolution of the terpolymers altered from the BT-free BSO2–EDOT bi-polymer to the BTSO2-free BT–EDOT bi-polymer, implying that the target LCPs containing varied amounts of BT have been successfully obtained via the DArP strategy. The elemental analysis (EA) was employed to further confirm the structures of LCPs. The trend of N, C, H and S element contents in the five representative LCPs derived from EA measurement (Table S1†) matched well with the calculated results, demonstrating the successful incorporation of varied monomers into the target LCPs, especially for terpolymer BSED–BT0.5% with a low content of BT.
The morphology and element distributions of the as-prepared LCPs were checked by scanning electron microscopy (SEM) and energy-dispersive spectroscopy (EDS), respectively. SEM images (Fig. 1c–g and S3†) reveal distinct morphological features associated with the varying amounts of BT incorporated. The bi-polymers BSO2–EDOT and BT–EDOT exhibit laminar and sphere stacking morphologies, respectively, and the BT-containing terpolymers display morphologies that are in-between those of the bi-polymers of BSO2–EDOT and BT–EDOT. As shown in Fig. 1c–g and S3,† at an ultra-low ratio of BT content (within 1.0%), the terpolymers exhibited layer stacking morphology. When BT content escalates to 2.5%, the polymers begin to aggregate and adopt the sphere stacking morphologies. Ultimately, in the case of the BT–EDOT bi-polymer, the lamellar stacking completely disappears, and instead, sphere stacking appears. EDS was further employed to check the N, O and S element distributions in the LCPs, whose intensities matched well with the feed ratios of the monomers involved in the polymers (Fig. 1h–j and S4, ESI†), and the N element is accordingly absent in the EDS map of the BT- & N-free bi-polymer BSO2–EDOT (Fig. S4†). Besides SEM, the particle size distribution and average sizes of BSO2–EDOT, BSED–BT0.5%, BSED–BT1.0%, BSED–BT50.0%, and BT–EDOT dispersed in NMP were further examined by the dynamic light scattering (DLS) method, showing size distributions in hundreds of nano-meters (Fig. S11†). This result is consistent with those of our previous study.28,43 The lamellar stacked polymers (Fig. 1c–g) are exfoliated using NMP solvent and are able to be stripped into thin flakes,43,44 which provides more active sites that are accessible to photocatalysis.
Optical & electrochemical properties
Fig. 2a illustrates the photographs of the ten LCPs in the solid state and in the solutions of NMP. The bi-polymers BSO2–EDOT and BT–EDOT appear as red and black powders, respectively, whereas the colors of the terpolymer powders gradually get darkened with the increase of BT contents. All the powdery LCPs are insoluble in common organic solvents including ethanol, dichloromethane, chloroform, and toluene due to the absence of solubilized alkyl side chains. Despite this, these polymers exhibit remarkable dispersibility in aprotic polar solvents, such as N,N-dimethylformamide (DMF) and N-methyl pyrrolidone (NMP), to form stable colloidal solutions (Fig. 2a, below). The colors of the NMP-dispersed bi-polymers BSO2–EDOT and BT–EDOT are red and blue, respectively. As the BT ratios increased, the colors of the terpolymers dispersed in NMP gradually altered from red to brown and lastly to blue. The varied colors of these LCPs in solid & solution imply that light absorption can be facilely tuned by incorporating varied contents of BT into the π-backbones. Meanwhile, the miscibility of NMP with H2O will endow these LCP colloidal dispersions with huge potential to serve as reliable platforms for quasi-homogeneous photocatalysis in aqueous solutions.28,35,43,44 | ||
| Fig. 2 (a) Photographs of all polymers. (b) UV-vis DRS and (c) Tauc-plot of BSO2–EDOT, BSED–BT0.1%, BSED–BT0.5%, BSED–BT1.0%, BSED–BT2.5%, BSED–BT5.0%, and BSED–BT25.0%. (d) Steady state PL spectra, (e) energy band diagrams and (f) transient photocurrents under visible light irradiation of all polymers. | ||
To gain deep insight into the structure–property correlations of polymers involving varied feed ratios of π-monomers, UV-vis diffuse reflectance spectroscopy (DRS), cyclic voltammetry (CV), photoluminescence spectroscopy (PL) and transient photocurrent response (TPR) measurement were systematically carried out to study their opto-electrochemical properties (Fig. 2b–f). As shown in Fig. 2b and S5,† all the LCPs exhibit a strong light absorption between 350 and 700 nm. Among them, the bi-polymer BSO2–EDOT shows the most hypsochromic shift in absorption onset (∼600 nm), while the bi-polymer BT–EDOT exhibits the most bathochromic shift in absorption onset (∼1010 nm, Fig. S5†), indicating a stronger push–pull electron effect in BT–EDOT because BT units have a stronger electron-withdrawing ability than BTSO2 units. As a result of this, compared to the BSO2–EDOT bi-polymer, all terpolymers exhibit a gradual bathochromic shift as the BT ratio increases (Fig. 2b). Impressively, the terpolymer BSED–BT0.1% even with a 0.1 mol% molar ratio of BT blocks has a distinct red-shift compared to the BT-free BSO2–EDOT bi-polymer (inset of Fig. 2b), showing that the push–pull effect of the D–A interaction modulated with the BT unit plays a vital role in narrowing the bandgap and extending the light absorption of the terpolymers, which makes them reliable for light harvesting and photo-driven conversions. The Egs of the LCPs are calculated using the Kubelka–Munk formula and extracted from the Tauc-plot function (Fig. 2c and S5†), showing a decrease in the range of 2.07–1.53 eV with the increase of BT contents (Fig. 2f). Inspired by the perfect dispersibility of the LCPs in NMP, UV-vis was also employed to investigate the light absorption of the polymers in colloidal solutions (see Fig. 2a, below). UV-vis spectra (Fig. S6†) show that the polymers dispersed in NMP exhibit strong absorption in the visible light region between 400 and 700 nm. Like the powdery samples, these NMP-dispersed LCPs also exhibit a gradual bathochromic shift as the BT ratio increases, matching well with the feed ratios of the monomers.
The steady-state PL and TPR were investigated to examine the photogenerated carrier transport and separation in the LCPs. The BT-free bi-polymer BSO2–EDOT exhibits the maximum emission peak (λmaxem) at ∼645 nm, whereas all terpolymers exhibit a noticeable redshift in λmaxem (Fig. 2d). Meanwhile, the terpolymers containing BT exhibit lowered PL intensities with the suppressed e−/h+ recombination, indicating that the incorporation of BT units into the π-backbones provides more probabilities for photon-to-electron conversion, rather than photon-to-photon conversion. As shown in Fig. 2d, the terpolymer BSED–BT0.1% even with 0.1 mol% of BT can drastically weaken the PL intensity compared to the BT-free bi-polymer BSO2–EDOT. As a result, the terpolymer BSED–BT0.5% exhibited the highest photocurrent response (∼1.6 μA cm−2) with four on–off cycles, which is 0.7 μA cm−2 higher than that of BSO2–EDOT (Fig. 2e). When the BT ratio exceeds 0.5 mol%, the TPR of the terpolymers becomes gradually reduced, revealing that 0.5 mol% is an optimal BT content for the photon-to-current conversion, wherein more light induced excitons can be produced.
CV was carried out to investigate the FMOs of the polymers (Fig. S7†). The reduction and oxidation waves of the polymers come respectively from the electron-deficient BTSO2 and BT units and the electron-rich EDOT unit. The calculated lowest unoccupied molecular orbital (LUMO) and highest occupied molecular orbital (HOMO) levels are presented in Fig. 2f, showing that the LUMO levels of all LCPs have sufficient driving forces for proton reduction. Due to the strong electron-deficient character of the BT unit, the LUMOs decrease gradually as the contents of BT increase in low ratios, reaching the deepest LUMO at the BSED–BT5.0% terpolymer that contains 5.0 mol% BT. As the BT contents continue to increase (>5.0 mol%), however, a gradual increase in LUMO levels is observed. The increase of the LUMO at higher BT ratios may be due to the twisted polymeric backbone caused by the BT unit (as can be seen in DFT calculations), which reduces the intramolecular D–A interaction along the conjugated backbone.
Photocatalytic hydrogen production performance
To study the structure–performance correlations of polymers involving varied feed ratios of π-monomers, the PHP of these LCPs was systematically investigated under visible light irradiation (λ > 420 nm) by utilizing the quasi-homogeneous colloidal dispersion of LCPs (6 mg) in NMP/H2O mixed solvent45,46 and ascorbic acid (AA) as a sacrificial electron donor without addition of the Pt co-catalyst. As shown in Fig. 3a, all LCPs demonstrate the capacity to generate H2 gas, albeit with different HERs. The normalized HERs of these ten LCPs are presented in Fig. 3b and Table S2.† The terpolymers BSED–BT0.1% and BSED–BT0.5% exhibited superior PHP to the bi-polymer BSO2–EDOT. Notably, the PHP of BSED–BT0.5% achieved a remarkable 6.67 mmol H2 within 5 h, corresponding to a normalized HER of 222.28 mmol h−1 g−1, which is 1.26 times higher than that of BT-free bi-polymer BSO2–EDOT. However, when the BT content in terpolymers surpasses 0.5 mol%, a gradual decline in HERs is observed and results in the lowest HER in bi-polymer BT–EDOT, demonstrating that a minimal amount (0.5 mol%) of BT plays a vital role in the PHP enhancement. To visualize this high hydrogen productivity, a homemade setup was designed to monitor the PHP reaction by using 3 mg of BSED–BT0.5% photocatalyst. With this setup, the produced H2 was drained from the Schlenk tube under light irradiation, wherein the amount of H2 can be directly displayed by the scales on a cylinder. After 2 hours of irradiation using simulated sunlight, 80 mL of H2 gas was amazingly produced (ESI Video 1†), corresponding to a normalized HER of 595.23 mmol h−1 g−1. To our surprise, the same setup under natural winter sunlight was able to produce 19 mL H2 gas in 2 hours with an HER of 141.37 mmol h−1 g−1 (ESI Video 2 and Fig. S8†).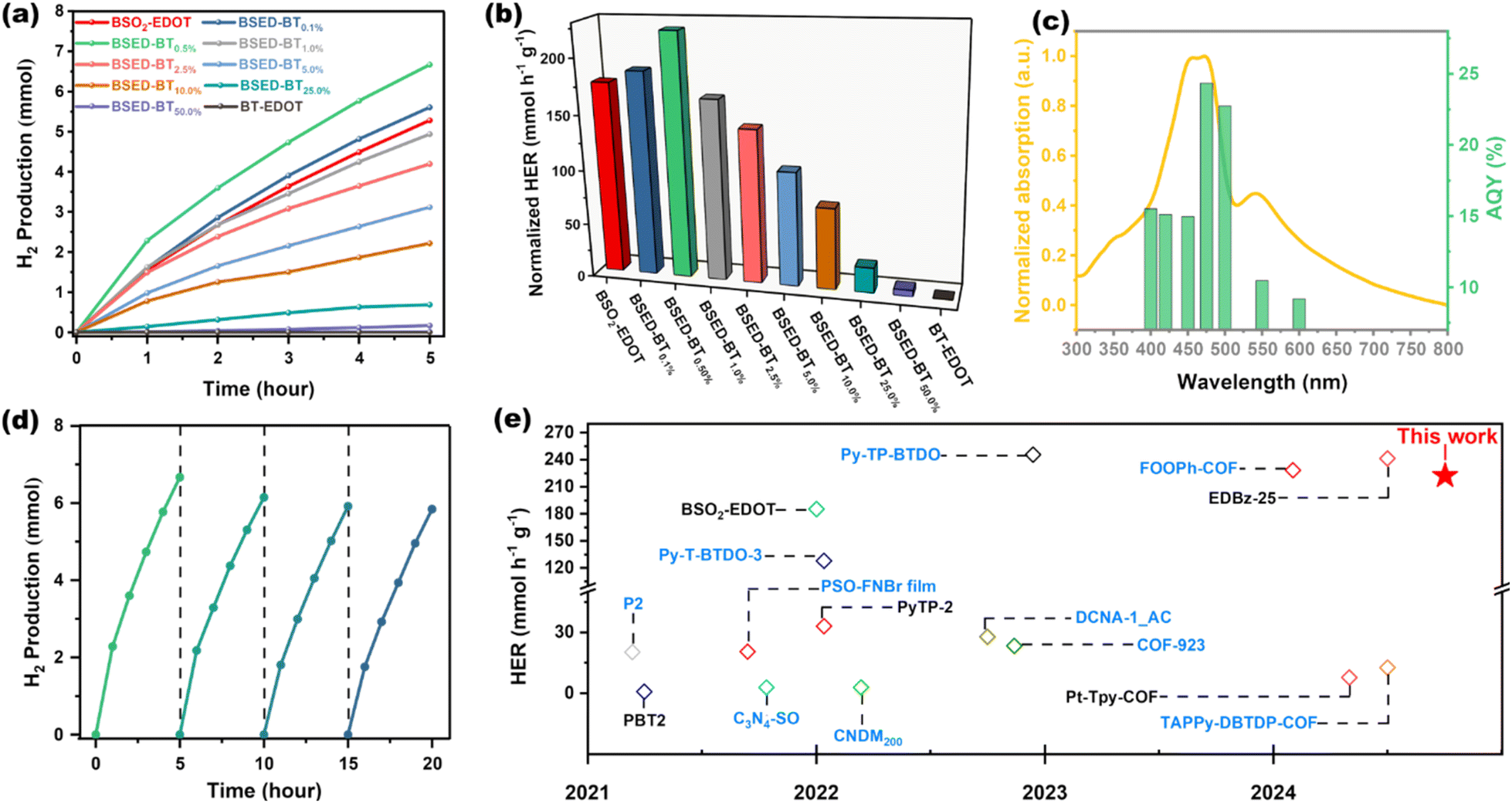 | ||
| Fig. 3 (a) PHP as a function of time for 6 mg polymers under visible light irradiation. (b) Normalized hydrogenolysis reaction rates of all polymers. (c) AQYs of PHP for BSED–BT0.5% at six different incident light wavelengths. (d) Cycling stability test for BSED–BT0.5%. (e) Comparison of the HER of BSED–BT0.5% with those of other recently reported photocatalysts (blue colour indicates that the HER added Pt as a co-catalyst).28,35,38,45–56 | ||
The AQYs of the BSED–BT0.5% photocatalyst were investigated under different monochromatic light sources to check the solar-to-hydrogen conversion efficiency. The AQYs at 400, 420, 450, 475, 500, 550 and 600 nm were 15.51%, 15.12%, 14.98%, 24.35%, 22.73%, 10.46%, and 9.19%, respectively, whose trend is in agreement with the UV-vis spectrum of its colloidal dispersion in NMP (Fig. 3c), manifesting the light-driven process of the proton reduction. It is noteworthy that the highest AQY of 24.35% is achieved at 475 nm, which much exceeds that of the pristine BSO2–EDOT bi-polymer. Next, a cycling test lasting for 20 h in four cycles was carried out to evaluate the stability of BSED–BT0.5% for the PHP reaction. As shown in Fig. 3d, a linear correlation between the H2 evolution and time is well maintained in each cycle. To our delight, albeit with 20 h of consecutive PHP reactions, the polymer retained approximately 88% of the original HER. The decreased HER in the last cycling test was mainly attributed to the consumption of the SED, i.e., AA.28,43
It is known that the photocatalysts for the PHP reaction typically need the aid of a Pt co-catalyst. Herein, the terpolymer BSED–BT0.5% achieves an ultrahigh HER without the addition of a Pt co-catalyst. For a comprehensive comparison, Fig. 3e and Table S3† summarize the HERs of BSED–BT0.5% and the referenced polymeric photocatalysts, showing that BSED–BT0.5% is one of the best performing candidates among the ever-reported polymeric photocatalysts either with or without the aid of Pt cocatalysts (Table S3†).
To check the residual Pd contents of the LCPs and its effect on the HER, three batches of BSED–BT0.5% were synthesized by the DArP strategy, respectively, by using 1 mol%, 3 mol% and 5 mol% Pd2(dba)3, which are accordingly denoted as BSED–BT0.5%–1, BSED–BT0.5%–3 and BSED–BT0.5%–5. The ICP-MS test reveals that all LCPs exhibited almost equal residual Pt loading (∼0.8 wt%, Table S4†), which could be attributed to the saturation of Pd loaded on the LCPs after identical post-treatment procedures. The PHP tests on these three LCPs showed a slight increase in the HER with the increase of Pd pre-catalyst loadings (Table S4†). This may be attributed to the increase in polymerization degrees due to the increased Pd loadings for DArP. The effect of residual Pd and polymerization degree on the photocatalytic performance explored here also coincides with previous studies.28,57 Given that Pt is the most efficient and widely adopted co-catalyst for the PHP reaction (as demonstrated by the majority of cases in Table S3†), the residual Pd remaining in the synthesized polymer photocatalysts is likely to have minimal impact on PHP performance.
Photocatalytic mechanism studies
The above results reveal that even an ultra-low ratio of the BT block incorporated into the backbone of terpolymers can play a pivotal role in the PHP enhancement. To unravel this intriguing phenomenon and understand the mechanism behind this, the time-resolved photoluminescence (TRPL), temperature-dependent PL, and fs-transient absorption spectroscopies (fs-TAS) of the BT-free bi-polymer BSO2–EDOT and terpolymer BSED–BT0.5% (Fig. 4) were systematically conducted to study the effects of BT building blocks on the photo-exciton transition, complexation, separation, and transfer processes. First, TRPL spectroscopy was performed to investigate the charge transfer. By fitting the decay curves (Fig. 4a and Table S5†), the weighted average lifetimes of BSO2–EDOT and BSED–BT0.5% were calculated to be 0.69 and 0.23 ns, respectively. The shortened lifetimes of the excited state indicate a reinforced suppression of the radiative exciton recombination in BSED–BT0.5%, which agrees with the steady-state PL spectra in Fig. 2d. The decreased excited state lifetime and the lowered emission quantum yield suggest that BSED–BT0.5%'s nonradiative rate is more pronounced relative to its radiative rate.58,59 | ||
| Fig. 4 Time-resolved PL (excited with 475 nm pulse) of BSO2–EDOT and BSED–BT0.5% (a); integrated PL (λex = 470 nm) intensities of BSO2–EDOT (b) and BSED–BT0.5% (c) as functions of 1/T (insets show the temperature-dependent PL spectra); contour plots of fs-TAS and TAS with different delay times of BSO2–EDOT (d and e) and BSED–BT0.5% (f and g); attenuation kinetic spectra (h) and fitting data (i) of BSO2–EDOT and BSED–BT0.5% at the indicated wavelengths. | ||
The exciton binding energy (Eb) determines the nonradiative transition of a photocatalyst. Eb is usually used to describe the energy thermodynamically required for exciton dissociation into free e− and h+. The e− present on the HOMO is excited and leaves h+ behind, at which point the e− on its way to the excited leap to h+ through Coulomb forces to form an exciton. A larger energy difference between electrons (e−) and holes (h+) reduces their recombination probability in the bulk phase, thereby facilitating exciton dissociation into free e− and h+. The study of Eb is thus an important means to understand the non-radiative process of BT-based terpolymers. Here, the temperature-dependent PL was checked to reveal the charge separation kinetics and exciton binding energies.60 Owing to the thermally activated non-radiative processes, the integrated PL intensities of BSO2–EDOT and BSED–BT0.5% decrease with the increasing temperatures (Fig. 4b and c). The temperature-dependent PL intensities can be expressed using the Arrhenius equation:
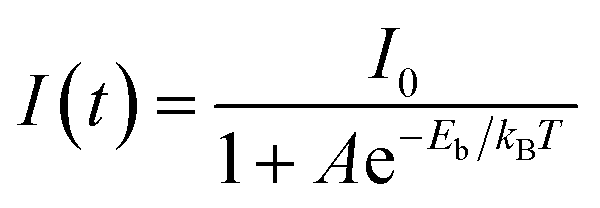
The TRPL and temperature-dependent PL results confirm the thermodynamic contribution of BSED–BT0.5% with ultra-low contents of BT to the charge carrier formation. The reduced Eb of the excitons in BSED–BT0.5% facilitates their dissociation into free charges, thereby increasing non-radiative recombination probability and meanwhile suppressing bulk recombination.
To examine the contribution of BT units to the excited state and carrier dynamics on a picosecond scale, fs-transient absorption spectra (fs-TAS) were measured for BSO2–EDOT and BSED–BT0.5% (Fig. 4d–i) from 0 to 1000 ps by excitation at 470 nm. As shown in Fig. 4d and e, BSO2–EDOT exhibits a distinct negative band in the 420–542 nm range, corresponding to ground-state bleaching (GSB) without the BT-mediated reduction of the Eg. The GSB peaks of both polymers align closely with their absorption edges. Notably, BSED–BT0.5% displays a GSB signal in the 616–693 nm range (Fig. 4f and g), which is absent in BSO2–EDOT, demonstrating that trace BT incorporation effectively broadens the light absorption range of the terpolymer. At 454 nm, the absorption intensity gradually increases for BSO2–EDOT (∼0.5 ps) and BSED–BT0.5% (∼2 ps), corresponding to the electron–electron relaxation process to the LUMO. Here, e− in the HOMO are photoexcited to the LUMO, leaving h+ in the HOMO. Some of these photoexcited e− may recombine with h+via radiative (fluorescence) or nonradiative (heat) pathways, while others survive through rapid electron transfer processes. The suppressed e−/h+ recombination in the BSED–BT0.5% terpolymer leads to a faster decay rate of its GSB peak (∼1000 ps, Fig. 4g) compared to that of the BSO2–EDOT bi-polymer (>1000 ps, Fig. 4e).
To elucidate the effect of the ultra-low ratio of BT on the dynamics of charge carriers, the decay kinetics of BSO2–EDOT and BSED–BT0.5% at specific wavelengths were combined and analyzed (Fig. 4h and i). The decay transient can be described by a double exponential function, and the fitting results yielded time constants τ1 and τ2 with different coefficients, which correspond, respectively, to the electron transfer and capture processes. The τ1 and τ2 are 5.76 and 44.76 ps for BSO2–EDOT and 1.13 and 48.10 ps for BSED–BT0.5%, respectively (Fig. 4i). The TAS kinetics results reveal that, compared to BSO2–EDOT, BSED–BT0.5% has an ultra-fast electron transfer (τ1) and a longer electron capture time (τ2). Meanwhile, unlike BSO2–EDOT, BSED–BT0.5% exhibits additional decay pathways owing to the electron transfer centers provided by the BT segments, enabling rapid electron transfer. This accelerates exciton dissociation and enhances electron–hole pair separation. Overall, the TRPL, temperature-dependent PL, and fs-TAS in Fig. 4 evidently reveal that even with an ultra-low ratio of BT (0.5 mol%), the exciton Eb is significantly reduced, triggering rapid exciton dissociation and increasing the nonradiative transition to generate more free charges. Furthermore, the BT segments create stronger electron transfer centers that rapidly shuttle surrounding electrons, improving carrier dynamics, and ultimately enhancing photocatalytic performance.
The water-wettability of photocatalysts has always played an un-negligible role in the PHP reaction.62–65 Typically, CPs with high water wettability is advantageous to aqueous photo-catalysis. Here, the wettability of the LCPs was investigated by the contact angle (CA) test (Fig. 5a–e and S10†). BTSO2 and EDOT have been demonstrated, respectively, as hydrophilic donor29 and acceptor66 monomers for CP-based photocatalysts.
 | ||
| Fig. 5 Water CAs of BSO2–EDOT (a), BSED–BT0.5% (b), BSED–BT1.0% (c), BSED–BT50.0% (d), and BT–EDOT (e); (f) zeta potentials of BSO2–EDOT and BSED–BT0.5%. | ||
Accordingly, the bi-polymer BSO2–EDOT has a low water CA of 12.0° with a fine wettability. After incorporating a minimal amount of BT unit (0.1 and 0.5 mol%), the terpolymers BSED–BT0.1% and BSED–B0.5% exhibit water CAs of 12.4° and 12.6°, respectively, implying that an ultra-low ratio (<1.0 mol%) of BT has a negligible impact on the wettability. However, as the BT ratio surpasses 1.0 mol%, a gradual increase in the water CA is observed, culminating in a significant increase to 40.5° in the case of bi-polymer BT–EDOT might be due to the low hydrophilicity of the BT unit. The water CA test (Fig. 5a–e and S10†) reveals that the incorporation of BT less than 1.0 mol% has a negligible effect on the water wettability.
The adsorption of H+ by the photocatalyst is a key step for the mass transfer in the PHP process. Zeta potential measurements of BSO2–EDOT and BSED–BT0.5% were carried out to check their surface affinity toward H+. In comparison to BSO2–EDOT, BSED–BT0.5% has a more negative zeta potential (Fig. 5f) and thus a stronger electrostatic attraction toward H+,67,68 which is conducive to the reduction of H+ to H2.
To further disclose the mechanism behind the enhanced performance of the BTSO2–EDOT–BT based terpolymers, the density-functional theory (DFT) calculations at the B3LYP/6-31G (d, p) level using the Beijing density functional (BDF) computational software package were conducted.69a–e Here, the tetramers of the four representative polymers were set as models M1, M2, M3 and M4 for calculations. The calculated molecular geometries, dipole moments (DPs), FMO distributions and electrostatic potentials (ESPs) of the polymers are presented in Fig. 6a–d and S9.† The optimized oligomeric models exhibit twist geometries (Fig. S9†). In bi-polymer BSO2–EDOT, the dihedral angles of BTSO2 with adjacent EDOT are about 8.8° and 11.2°, while in bi-polymer EDOT–BT, the dihedral angles of the BT unit with adjacent EDOT units are 2.4° and 43.8°. Thus, when the BT unit was introduced into the terpolymer to partially replace BTSO2, the backbone became more twisted with a decrease in the planarity.
 | ||
| Fig. 6 DFT-predicated FMO distributions and ESPs of the tetramers M1 (a), M2 (b), M3 (c), and M4 (d); the total energy of protons adsorbed on BT (e) and on BTSO2 (f); the adsorption free energy during H+ reduction (g). | ||
The introduction of built-in electric fields (BEFs) into semiconductors has been regarded as a viable strategy to promote the dissociation of photoinduced excitons into banded e−/h+ pairs, thereby achieving higher carrier concentrations.70 The BEFs mainly rely on the polarization of semiconductors induced by the molecular DPs. The DP values (Fig. S9†) for the models of M1, M2, M3 and M4 are 5.48, 4.87, 10.90, and 2.86 debye (D), respectively. Here, M4 exhibits the lowest DP value, indicating that a higher ratio of BT weakens the BEF intensity of the terpolymer. However, M2 and M3 show distinct dipole moment magnitudes, likely due to the different positions of BT units in their models: the relatively symmetric M2 model has a weaker DP, while the less symmetric M3 model displays a stronger DP. The BT units are statistically distributed in BSED–BT0.5%, which will enhance its DP, thereby strengthening the BEFs. This DFT predication is consistent with the experimental results of the varied optical & electrochemical tests in Fig. 2d, e and 6.
The FMO distributions of the molecular models are shown in Fig. 6. The HOMOs and LUMOs show a partial separation in the bi-polymer model M1. As the third building block BT unit was involved, the LUMOs become localized on the BT units, and the HOMOs become delocalized across the EDOT and BTSO2 units. For the bi-polymer model M4, the LUMOs are mainly located on the electron-withdrawing BT unit, while the HOMOs are distributed across the entire molecule. This suggests that BT units act as relay centers capable of rapidly transferring surrounding electrons. The LUMO level of M4 is more negative than those of M2 and M3, a trend that matches the data obtained from the CV test (Fig. 2f). Consistent with the FMO distribution, positive charges are predominantly localized on the electron-donating EDOT units, while negative charges concentrate on the electron-accepting BTSO2 and BT units (ESP in Fig. 6). This indicates a charge transfer direction from EDOT to BTSO2/BT. The FMOs and ESP distributions suggest that, among the four models, M2 or M3 demonstrate that ultra-low ratios of BT can create stronger electron transfer centers and additional electron transfer pathways, thereby enhancing photocatalytic performance. Furthermore, the proton (H+) adsorption energy was calculated to predicate the affinity of BT and BTSO2 units toward protons (Fig. 6e and f). The results reveal that the N atoms on BT exhibit stronger H+ adsorption ability and lower energy barriers compared to the O atoms on BTSO2 (Fig. 6g), allowing H+ to be more readily reduced on BT units.
The above DFT calculations clarify that the BT units in the polymer create electron transfer centers, generating additional electron transfer pathways. The preferential adsorption and reduction of H+ on BT ensure the efficient utilization of transferred electrons.
Based on the experimental analyses (Fig. 2d and e, 4 and 5f) and theoretical calculation (Fig. 6), the photocatalytic mechanism of BSED–BT0.5% is present as below (Fig. 7). Compared to the BSO2–EDOT bi-polymer, the ultra-low ratio of BT in the BSED–BT0.5% terpolymer has a higher affinity toward H+ and broader light absorption. More photo-induced electrons are quickly transferred to the active sites, produce longer-lived carriers due to its lower Eb, and thus catalytically reduce the adsorbed H+ to H2 in a more efficient way.
 | ||
| Fig. 7 Comparative PHP pathways of the BSO2–EDOT bi-polymer and BSED–BTx terpolymers. | ||
Conclusions
A series of D–A type semiconducting linear conjugated polymers involving varied feed ratios of BTSO2, EDOT and BT monomers were facilely synthesized by an atom-economical DArP strategy. To clarify the structure–property–performance correlations, the LCPs were fully characterized by SEM, EDS, UV-vis, PL, CV, TPR, TRPL, temperature-dependent PL and fs-TAS spectroscopies, CA measurement and DFT calculations and evaluated as photocatalysts for the PHP reaction. The terpolymer BSED–BT0.5% exhibits an HER of up to 222.28 mmol h−1 g−1 under visible light irradiation and a top AQY of 24.35% at 475 nm without the aid of Pt co-catalysts. Systematic mechanism studies reveal that the BSED–BT0.5% terpolymer involving a minimal amount of BT (0.5 mol%) has a negligible impact on the hydrophilicity, but shows outstanding merits over the pristine BSO2–EDOT bi-polymer, including (1) higher affinity toward H+, (2) broader light absorption, (3) drastically lowered Eb (32.03 vs. 61.35 meV), (4) much faster electron transfer (1.13 vs. 5.76 ps), (5) increased electron capture time, and (6) improved carrier dynamics, all of which facilitate the PHP reaction. Our study reveals the significance of the ultra-low ratios of BT in terpolymers and provides unique insight into the design of high-performance CP photocatalysts.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Z. X. performed and analysed the experiments and contributed to the writing of the original draft. G. Y. and P. M. contributed to the writing and partially conceptualized. H. G., Y. D., C. L., and K. Y. performed part of the experiments. Z. X., G. Y. and S.-Y. L. analysed the data and prepared the manuscript. S.-Y. L. supervised and conceptualized the project, performed funding acquisition, provided guidance during all stages, and contributed to the writing of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The National Natural Science Foundation of China (No. 22169009), Jiangxi Provincial Natural Science Foundation (No. 20212ACB204007), and Jiangxi Province Key Laboratory of Functional Crystalline Materials Chemistry (2024SSY05161) are appreciated for financial support. The authors thank Jiangxi Qianvi New Materials Co., Ltd for SEM, EDS and XPS tests. We gratefully acknowledge HZWTECH for providing computation facilities.Notes and references
- S. Chu and A. Majumdar, Nature, 2012, 488, 294–303 CrossRef CAS PubMed.
- M. Grubb, Nature, 2017, 543, 37–38 CrossRef CAS.
- X. Chen, S. Shen, L. Guo and S. S. Mao, Chem. Rev., 2010, 110, 6503–6570 CrossRef CAS PubMed.
- S. Chen, T. Takata and K. Domen, Nat. Rev. Mater., 2017, 2, 17050 CrossRef CAS.
- Q. Wang and K. Domen, Chem. Rev., 2020, 120, 919–985 CrossRef CAS PubMed.
- H. Nishiyama, T. Yamada, M. Nakabayashi, Y. Maehara, M. Yamaguchi, Y. Kuromiya, Y. Nagatsuma, H. Tokudome, S. Akiyama, T. Watanabe, R. Narushima, S. Okunaka, N. Shibata, T. Takata, T. Hisatomi and K. Domen, Nature, 2021, 598, 304–307 CrossRef CAS PubMed.
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CrossRef CAS PubMed.
- W. Zhao, H. Chen, J. Zhang, P. J. Low and H. Sun, Chem. Sci., 2024, 15, 17292–17327 RSC.
- S. Chandrasekaran, L. Yao, L. Deng, C. Bowen, Y. Zhang, S. Chen, Z. Lin, F. Peng and P. Zhang, Chem. Soc. Rev., 2019, 48, 4178–4280 RSC.
- X. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. M. Carlsson, K. Domen and M. Antonietti, Nat. Mater., 2009, 8, 76–80 CrossRef CAS PubMed.
- (a) G. Liao, Y. Gong, L. Zhang, H. Gao, G.-J. Yang and B. Fang, Energy Environ. Sci., 2019, 12, 2080–2147 RSC; (b) Z. Chen, G. Ding, Z. Wang, Y. Xiao, X. Liu, L. Chen, C. Li, H. Huang and G. Liao, Adv. Funct. Mater., 2025, 2423213 CrossRef.
- S. Yanagida, A. Kabumoto, K. Mizumoto, C. Pac and K. Yoshino, Chem. Commun., 1985, 474–475 RSC.
- H. Ye, Z. Wang, Z. Yang, S. Zhang, X. Gong and J. Hua, J. Mater. Chem. A, 2020, 8, 20062–20071 RSC.
- R. J. Lyons, Y. Yang, E. McQueen, L. Luo, A. I. Cooper, M. A. Zwijnenburg and R. S. Sprick, Adv. Energy Mater., 2024, 14, 2303680 CrossRef CAS.
- R. S. Sprick, J. X. Jiang, B. Bonillo, S. Ren, T. Ratvijitvech, P. Guiglion, M. A. Zwijnenburg, D. J. Adams and A. I. Cooper, J. Am. Chem. Soc., 2015, 137, 3265–3270 CrossRef CAS PubMed.
- C. Han, P. Dong, H. Tang, P. Zheng, C. Zhang, F. Wang, F. Huang and J. X. Jiang, Chem. Sci., 2020, 12, 1796–1802 RSC.
- W. Huang, Q. He, Y. Hu and Y. Li, Angew. Chem., Int. Ed., 2019, 131, 8768–8772 CrossRef.
- K. Asokan, T. M. Bhagyasree, G. Devasia, S. Krishnamurty, S. Solim, L. Rueda, D. M. Al-Mohannadi, M. Al-Hashimi, K. Kakosimos and S. S. Babu, Chem. Sci., 2024, 15, 13381–13388 RSC.
- L. Stegbauer, K. Schwinghammer and B. V. Lotsch, Chem. Sci., 2014, 5, 2789–2793 RSC.
- Z. Lin, S. Dai, S. Yao, Q. C. Lin, M. Fu, L. H. Chung, B. Han and J. He, Chem. Sci., 2025, 16, 1948–1956 RSC.
- H. Fan, M. Hu, Y. Duan, L. Zuo, R. Yu, Z. Li, Q. Liu, B. Li and L. Wang, Chem. Sci., 2025, 16, 2316–2324 RSC.
- G. Zhang, Z. A. Lan and X. Wang, Angew. Chem., Int. Ed., 2016, 55, 15712–15727 CrossRef PubMed.
- (a) J.-S. M. Lee and A. I. Cooper, Chem. Rev., 2020, 120, 2171–2214 CrossRef CAS PubMed; (b) J. Kosco, F. Moruzzi, B. Willner and I. McCulloch, Adv. Energy Mater., 2020, 10, 2001935 CrossRef CAS.
- For reviews on D–A polymer for PHP application, see: (a) Z. Q. Sheng, Y. Q. Xing, Y. Chen, G. Zhang, S. Y. Liu and L. Chen, Beilstein J. Nanotechnol., 2021, 12, 607–623 CrossRef CAS PubMed; (b) J. Zhao, J. Ren, G. Zhang, Z. Zhao, S. Liu, W. Zhang and L. Chen, Chem.–Asian J., 2021, 27, 10781–10797 CAS; (c) C. Yang, B. Cheng, J. Xu, J. Yu and S. Cao, EnergyChem, 2024, 6, 10781–10797 CrossRef.
- (a) P. Roy, A. Jha, V. B. Yasarapudi, T. Ram, B. Puttaraju, S. Patil and J. Dasgupta, Nat. Commun., 2017, 8, 1716 CrossRef PubMed; (b) H. Shen, Y. Li and Y. Li, Aggregate, 2020, 1, 57–68 CrossRef; (c) I. S. Demachkie, M. P. Miller, G. I. Warren, J. E. Barker, E. T. Strand, L. N. Zakharov and M. M. Haley, Angew. Chem., Int. Ed., 2025, 64, e202420989 CrossRef CAS PubMed.
- C. Yang, B. C. Ma, L. Zhang, S. Lin, S. Ghasimi, K. Landfester, K. A. Zhang and X. Wang, Angew. Chem., Int. Ed., 2016, 55, 9202–9206 CrossRef CAS PubMed.
- Y. Zhao, W. Ma, Y. Xu, C. Zhang, Q. Wang, T. Yang, X. Gao, F. Wang, C. Yan and J.-X. Jiang, Macromolecules, 2018, 51, 9502–9508 CrossRef CAS.
- Z. R. Tan, Y. Q. Xing, J. Z. Cheng, G. Zhang, Z. Q. Shen, Y. J. Zhang, G. Liao, L. Chen and S. Y. Liu, Chem. Sci., 2022, 13, 1725–1733 RSC.
- A. Jati, S. Dam, S. Kumar, K. Kumar and B. Maji, Chem. Sci., 2023, 14, 8624–8634 RSC.
- C. Shu, C. Han, X. Yang, C. Zhang, Y. Chen, S. Ren, F. Wang, F. Huang and J. X. Jiang, Adv. Mater., 2021, 33, e2008498 CrossRef PubMed.
- F. Yu, Z. Zhu, S. Wang, J. Wang, Z. Xu, F. Song, Z. Dong and Z. Zhang, Appl. Catal., B, 2022, 301, 120819 CrossRef CAS.
- Z. Li, T. Deng, S. Ma, Z. Zhang, G. Wu, J. Wang, Q. Li, H. Xia, S. W. Yang and X. Liu, J. Am. Chem. Soc., 2023, 145, 8364–8374 CrossRef CAS PubMed.
- P. B. Pati, G. Damas, L. Tian, D. L. A. Fernandes, L. Zhang, I. B. Pehlivan, T. Edvinsson, C. M. Araujo and H. Tian, Energy Environ. Sci., 2017, 10, 1372–1376 RSC.
- S. Li, R. Ma, C. Tu, W. Zhang, R. Li, Y. Zhao and K. A. I. Zhang, Angew. Chem., Int. Ed., 2024, e202421040 Search PubMed.
- H. Gong, J. Li, Z.-H. Xie, C. Lang and S.-Y. Liu, Macromolecules, 2024, 57, 7208–7218 CrossRef CAS.
- Y. Xu, N. Mao, C. Zhang, X. Wang, J. Zeng, Y. Chen, F. Wang and J.-X. Jiang, Appl. Catal., B, 2018, 228, 1–9 CrossRef.
- H. He, R. Shen, Y. Yan, D. Chen, Z. Liu, L. Hao, X. Zhang, P. Zhang and X. Li, Chem. Sci., 2024, 15, 20002–20012 RSC.
- C. Han, S. Xiang, S. Jin, C. Zhang and J.-X. Jiang, ACS Catal., 2022, 13, 204–212 CrossRef.
- K. Wu, X.-Y. Liu, M. Xie, P.-W. Cheng, J. Zheng, W. Lu and D. Li, Appl. Catal., B, 2023, 334, 122847 CrossRef CAS.
- X. Xia, J. Feng, Z. Zhong, X. Yang, N. Li, D. Chen, Y. Li, Q. Xu and J. Lu, Adv. Funct. Mater., 2023, 34, 2311987 CrossRef.
- X. Yuan, C. Wang, L. Vallan, A. T. Bui, G. Jonusauskas, N. D. McClenaghan, C. Grazon, S. Lacomme, C. Brochon, H. Remita, G. Hadziioannou and E. Cloutet, Adv. Funct. Mater., 2023, 33, 2211730 CrossRef CAS.
- C. Li, H. Xu, H. Xiong, S. Xia, X. Peng, F. Xu and X. Chen, Adv. Funct. Mater., 2024, 34, 2405539 CrossRef CAS.
- J.-Z. Cheng, L.-L. Liu, G. Liao, Z.-Q. Shen, Z.-R. Tan, Y.-Q. Xing, X.-X. Li, K. Yang, L. Chen and S.-Y. Liu, J. Mater. Chem. A, 2020, 8, 5890–5899 RSC.
- J.-Z. Cheng, Z.-R. Tan, Y.-Q. Xing, Z.-Q. Shen, Y.-J. Zhang, L.-L. Liu, K. Yang, L. Chen and S.-Y. Liu, J. Mater. Chem. A, 2021, 9, 5787–5795 RSC.
- W.-R. Wang, J. Li, Q. Li, Z.-W. Xu, L.-N. Liu, X.-Q. Chen, W.-J. Xiao, J. Yao, F. Zhang and W.-S. Li, J. Mater. Chem. A, 2021, 9, 8782–8791 RSC.
- Q. Wei, X. Yao, Q. Zhang, P. Yan, C. Ru, C. Li, C. Tao, W. Wang, D. Han, D. Han, L. Niu, D. Qin and X. Pan, Small, 2021, 17, e2100132 CrossRef PubMed.
- C. Han, S. Xiang, P. Xie, P. Dong, C. Shu, C. Zhang and J. X. Jiang, Adv. Funct. Mater., 2022, 32, 2109423 CrossRef CAS.
- Y. Hu, Y. Liu, J. Wu, Y. Li, J. Jiang and F. Wang, ACS Appl. Mater. Interfaces, 2021, 13, 42753–42762 CrossRef CAS PubMed.
- S. Xiang, C. Han, C. Shu, C. Zhang and J.-X. Jiang, Sci. China Mater., 2021, 65, 422–430 CrossRef.
- C. Ru, T. Zhou, J. Zhang, X. Wu, P. Sun, P. Chen, L. Zhou, H. Zhao, J. Wu and X. Pan, Macromolecules, 2021, 54, 8839–8848 CrossRef CAS.
- S. Wan, J. Xu, S. Cao and J. Yu, Interd. Mater., 2022, 1, 294–308 CAS.
- J. Yang, S. Ghosh, J. Roeser, A. Acharjya, C. Penschke, Y. Tsutsui, J. Rabeah, T. Wang, S. Y. Djoko Tameu, M. Y. Ye, J. Gruneberg, S. Li, C. Li, R. Schomacker, R. Van De Krol, S. Seki, P. Saalfrank and A. Thomas, Nat. Commun., 2022, 13, 6317 CrossRef CAS PubMed.
- W. Dong, Z. Qin, K. Wang, Y. Xiao, X. Liu, S. Ren and L. Li, Angew. Chem., Int. Ed., 2023, 62, e202216073 CrossRef CAS PubMed.
- C. Liu, D.-L. Ma, P.-J. Tian, C. Jia, Q.-Y. Qi, G.-F. Jiang and X. Zhao, J. Mater. Chem. A, 2024, 12, 16063–16069 RSC.
- N. Liu, S. Xie, Y. Huang, J. Lu, H. Shi, S. Xu, G. Zhang and X. Chen, Adv. Energy Mater., 2024, 14, 2402395 CrossRef CAS.
- L. Hao, R. Shen, G. Liang, M. Kang, C. Huang, P. Zhang and X. Li, Appl. Catal., B, 2024, 348, 123837 CrossRef CAS.
- L. Lianwei, C. Zhengxu, W. Qinghe, L. Wai-Yip, Z. Na and Y. Luping, J. Am. Chem. Soc., 2016, 138, 7681–7686 CrossRef PubMed.
- J. Xu, C. Yang, S. Bi, W. Wang, Y. He, D. Wu, Q. Liang, X. Wang and F. Zhang, Angew. Chem., Int. Ed., 2020, 59, 23845–23853 CrossRef CAS PubMed.
- B. P. Biswal, H. A. Vignolo-Gonzalez, T. Banerjee, L. Grunenberg, G. Savasci, K. Gottschling, J. Nuss, C. Ochsenfeld and B. V. Lotsch, J. Am. Chem. Soc., 2019, 141, 11082–11092 CrossRef CAS PubMed.
- C. Li, J. Liu, H. Li, K. Wu, J. Wang and Q. Yang, Nat. Commun., 2022, 13, 2357 CrossRef CAS PubMed.
- W. Wang, H. Wang, X. Tang, J. Huo, Y. Su, C. Lu, Y. Zhang, H. Xu and C. Gu, Chem. Sci., 2022, 13, 8679–8685 RSC.
- C.-L. Chang, T.-F. Huang, W.-C. Lin, L.-Y. Ting, C.-H. Shih, Y.-H. Chen, J.-J. Liu, Y.-T. Lin, Y.-T. Tseng, Y.-H. Wu, Y.-E. Sun, M. H. Elsayed, C.-W. Chen, C.-H. Yu and H.-H. Chou, Adv. Energy Mater., 2023, 13, 2300986 CrossRef CAS.
- J. Kosco, S. Gonzalez-Carrero, C. T. Howells, W. Zhang, M. Moser, R. Sheelamanthula, L. Zhao, B. Willner, T. C. Hidalgo, H. Faber, B. Purushothaman, M. Sachs, H. Cha, R. Sougrat, T. D. Anthopoulos, S. Inal, J. R. Durrant and I. McCulloch, Adv. Mater., 2021, 34, 2105007 CrossRef PubMed.
- Z. Hu, Z. Wang, X. Zhang, H. Tang, X. Liu, F. Huang and Y. Cao, iScience, 2019, 13, 33–42 CrossRef CAS PubMed.
- Y. Bai, Z. Hu, J. X. Jiang and F. Huang, Chem.–Asian J., 2020, 15, 1780–1790 CrossRef CAS PubMed.
- S. A. J. Hillman, R. S. Sprick, D. Pearce, D. J. Woods, W. Y. Sit, X. Shi, A. I. Cooper, J. R. Durrant and J. Nelson, J. Am. Chem. Soc., 2022, 144, 19382–19395 CrossRef CAS PubMed.
- Y. Xie, F. Mao, Q. Rong, X. Liu, M. Hao, Z. Chen, H. Yang, G. I. N. Waterhouse, S. Ma and X. Wang, Adv. Funct. Mater., 2024, 23, 2411077 CrossRef.
- Y. Mou, X. Wu, C. Qin, J. Chen, Y. Zhao, L. Jiang, C. Zhang, X. Yuan, E. H. Ang and H. Wang, Angew. Chem., Int. Ed., 2023, 62, e202309480 CrossRef CAS PubMed.
- (a) Y. Zhang, B. Suo, Z. Wang, N. Zhang, Z. Li, Y. Lei, W. Zou, J. Gao, D. Peng, Z. Pu, Y. Xiao, Q. Sun, F. Wang, Y. Ma, X. Wang, Y. Guo and W. Liu, J. Chem. Phys., 2020, 152, 064113 CrossRef PubMed; (b) Z. Wang, Z. Li, Y. Zhang and W. Liu, J. Chem. Phys., 2020, 153, 164109 CrossRef CAS PubMed; (c) W. Liu, F. Wang and L. Li, Recent Adv. Relativ. Mol. Theory, 2004, 5, 257–282 CAS; (d) W. Liu, G. Hong, D. Dai, L. Li and M. Dolg, Theor. Chem. Acc., 1997, 96, 75–83 Search PubMed; (e) W. Liu, F. Wang and L. Li, J. Theor. Comput. Chem., 2003, 2, 257–272 CrossRef CAS.
- Z. Deng, H. Zhao, X. Cao, S. Xiong, G. Li, J. Deng, H. Yang, W. Zhang and Q. Liu, ACS Appl. Mater. Interfaces, 2022, 14, 35745–35754 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5sc01438g |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |