
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAzetidinyl Malachite Green: a superior fluorogen-activating protein probe for live-cell and dynamic SIM imaging†
Fei
Deng‡
 abc,
Xiangning
Fang‡
ae,
Qinglong
Qiao
*a,
Guoli
Han
ae,
Lu
Miao
a,
Shuangshuang
Long
ad and
Zhaochao
Xu
*ae
abc,
Xiangning
Fang‡
ae,
Qinglong
Qiao
*a,
Guoli
Han
ae,
Lu
Miao
a,
Shuangshuang
Long
ad and
Zhaochao
Xu
*ae
aDalian Institute of Chemical Physics, Chinese Academy of Sciences, 457 Zhongshan Road, Dalian 116023, China. E-mail: qqlqiao@dicp.ac.cn; zcxu@dicp.ac.cn
bSchool of Chemistry and Chemical Engineering, Jinggangshan University, 28 Xueyuan Road, Ji'an, Jiangxi 343009, China
cKey Laboratory of Jiangxi Province for Special Optoelectronic Artificial Crystal Materials, Jinggangshan University, 28 Xueyuan Road, Ji'an, Jiangxi 343009, China
dSchool of Chemistry and Chemical Engineering, University of South China, 28 Changshengxi Road, Hengyang, Hunan 421001, China
eUniversity of Chinese Academy of Sciences, Beijing 100049, China
First published on 30th April 2025
Abstract
Malachite Green (MG) and its fluorogen-activating protein (FAP) pair are valuable tools for live-cell and super-resolution fluorescence imaging due to their unique near-infrared absorption and signal enhancement. However, the low brightness and photostability of MG have limited its use in dynamic imaging. In this study, we introduce a novel derivative, azetidinly Malachite Green (Aze-MG), which enhances the brightness of the MG-FAP complex by 2.6-fold. This enhancement is achieved by replacing the N,N-dimethylamino group in MG with an azetidine group, which suppresses the twisted intramolecular charge transfer (TICT) effect, leading to improved quantum yield and photostability. Additionally, the reduced binding affinity of Aze-MG for FAP enables a buffering strategy, allowing the reversible exchange of photobleached fluorogens with free fluorogens, thereby ensuring stable fluorescence over time. This combination of improved brightness and buffering capability makes Aze-MG an ideal probe for live-cell and dynamic SIM imaging.
Introduction
Long-term live-cell super-resolution fluorescence imaging is critical for uncovering the dynamic behaviors of cellular nanostructures.1–3 Achieving this level of imaging requires fluorescent probes that offer high specificity, minimal background fluorescence, sufficient brightness, and photostability.4–8 Despite significant advancements in dye chemistry and imaging techniques, designing probes that meet all these criteria remains a challenge.9–13 Fluorogen-activating proteins (FAPs) have emerged as a promising solution for specific and high-contrast labeling in live cells.14–18 These genetically encoded proteins bind to non-fluorescent fluorogens, resulting in significant fluorescence activation, which allows for high signal-to-noise imaging. Malachite Green (MG) and its FAP pair have been widely used due to their unique near-infrared absorption and strong signal enhancement. Waggoner et al. pioneered this system by screening human single-chain antibodies to identify an MG-binding FAP, establishing its foundation in live-cell imaging.19 This FAP encapsulates MG within its binding cavity, constraining the internal rotation of MG's aromatic rings and thereby suppressing non-radiative decay pathways. This interaction results in an 18![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000-fold fluorescence increase in the near-infrared region, ideal for deep tissue imaging due to reduced background and scattering. Building on this work, Waggoner and Bruchez modified MG's pendent ring by introducing extended conjugation and primary amino groups,20,21 creating MG derivatives that bind to the same FAP with emission maxima at 731 and 532 nm, respectively. Additionally, Hu et al. developed a series of 3-indole-MG derivatives exhibiting large “pseudo-Stokes” shifts upon binding to FAPs.22 These studies demonstrate that FAPs exhibit good structural tolerance to modifications in MG, opening up opportunities for further structural modifications of MG. However, MG still faces significant limitations in brightness and photostability, which are critical for sustained dynamic super-resolution imaging.23,24 It is prone to photobleaching and has a limited quantum yield, which complicates long-term super-resolution microscopy and stable tracking of cellular events. Thus, despite the value of MG-FAP pairs, maintaining stable fluorescence for extended imaging remains a major challenge.
000-fold fluorescence increase in the near-infrared region, ideal for deep tissue imaging due to reduced background and scattering. Building on this work, Waggoner and Bruchez modified MG's pendent ring by introducing extended conjugation and primary amino groups,20,21 creating MG derivatives that bind to the same FAP with emission maxima at 731 and 532 nm, respectively. Additionally, Hu et al. developed a series of 3-indole-MG derivatives exhibiting large “pseudo-Stokes” shifts upon binding to FAPs.22 These studies demonstrate that FAPs exhibit good structural tolerance to modifications in MG, opening up opportunities for further structural modifications of MG. However, MG still faces significant limitations in brightness and photostability, which are critical for sustained dynamic super-resolution imaging.23,24 It is prone to photobleaching and has a limited quantum yield, which complicates long-term super-resolution microscopy and stable tracking of cellular events. Thus, despite the value of MG-FAP pairs, maintaining stable fluorescence for extended imaging remains a major challenge.
A promising strategy to maintain stable fluorescence over time involves using a “buffering” approach, where fluorescent dyes can reversibly bind to their targets.25 This allows free, non-fluorescent dyes in the surrounding environment to replace photobleached dyes on the target, sustaining a consistent imaging signal.26 This reversible binding approach has been effectively applied in organelle imaging, where dyes bind in an interchangeable manner to the organelle membrane matrix.27–29 Here, non-specific interactions are sufficient for binding, allowing for probe exchange simply by adjusting the hydrophilic or lipophilic properties of the dye to interact with the organelle membrane. However, designing reversible binding systems for biomolecule–substrate interactions poses greater challenges due to the specificity required. Unlike organelles, biomolecular targets rely on precise molecular recognition; even small structural changes in the dye can disrupt its binding affinity or specificity, making it difficult to achieve both strong binding and rapid exchange.
Rhodamine, which shares significant structural similarity with MG, offers insights into addressing MG's fluorescence limitations.30 In rhodamine dyes, a major nonradiative de-excitation pathway is twisted intramolecular charge transfer (TICT), commonly originating from alkylamino substituents.31,32 Studies show that non-alkylated rhodamine 110 and alkyl-substituted rhodamine 101 have quantum yields of 0.88 and 0.80, respectively, both considerably higher than that of tetramethylrhodamine (TMR) (0.41).33 Notably, Lavis et al. found that replacing TMR's N,N-dimethyl groups with azetidine rings effectively suppressed TICT, doubling its quantum efficiency.34 For MG, crystallographic studies of FAP-bound complexes reveal that the binding cavity is over twice the volume of MG itself.35 While aromatic ring rotations are restricted in this environment, the N,N-dimethyl groups in MG remain capable of free rotation within the cavity, likely contributing to TICT-induced fluorescence quenching and reducing the overall quantum yield.
In this work, we address these limitations by engineering a novel derivative of Aze-MG (MG3 in this paper) to enhance both brightness and dynamic imaging potential. Through strategic modification of MG's structure, specifically by introducing azetidine in place of the dimethylamino groups, we successfully increased the brightness of the MG-FAP complex by threefold. This modification suppresses the twisted intramolecular charge transfer (TICT) effect, resulting in improved quantum yield and photostability. Additionally, we implemented a buffering strategy that enables Aze-MG to reversibly bind with FAP, allowing free fluorogen molecules to replace photobleached ones at the target site. This exchangeable binding capacity supports stable fluorescence over time and enables continuous dynamic imaging under structured illumination microscopy (SIM).
Results and discussion
In alignment with the concept of TICT suppression, we synthesized three derivatives of MG (Fig. 1a)—MG2 with an amino group, MG3 with an azetidine group, and MG4 with a julolidine group—replacing the N,N-dimethyl groups present in the control dye (MG1). The synthesis of MG4 was achieved directly via the condensation of benzaldehyde with julolidine, similar to MG1. In contrast, MG2 and MG3 were synthesized from bisphenol compounds using Pd-catalyzed C–N cross-coupling reactions (Scheme S1†). We first investigated the intrinsic absorption and emission spectra of these MG derivatives. MG3 and MG1 shared almost the same absorption maximum at 606 nm whereas MG2 and MG4 were blue-shifted by 57 nm and red-shifted by 40 nm, respectively, compared to MG1 (Fig. 1b). Notably, the fluorescence of MG2–4 was almost completely quenched, likely due to intense vibrational deactivation (Fig. S2 and S3†), making it difficult to observe. We subsequently performed calculations for MG1–4 using time-dependent density functional theory. It revealed that for all dyes, the lowest energy excitation was attributed solely to the HOMO–LUMO transition, with HOMO–LUMO gaps measured at 6.2092 eV for MG1, 6.4985 eV for MG2, 6.1643 eV for MG3, and 5.9577 eV for MG4, aligning with the observed trends in their absorption maxima (Fig. 1c).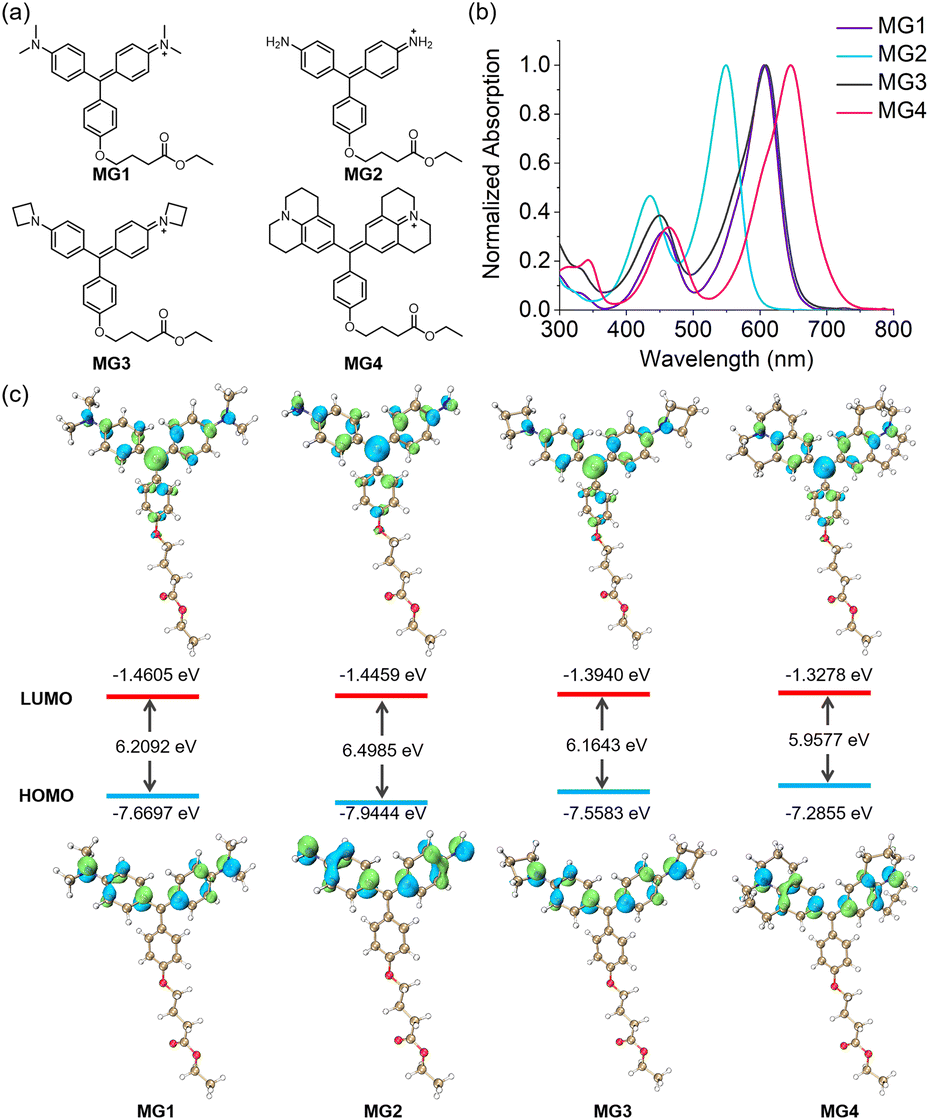 | ||
| Fig. 1 (a) Molecular structures of MG1–4. (b) Absorption spectra of MG1–4 in PBS (pH 7.40). (c) Computed frontier orbitals and energy levels of MG1–4. | ||
Prior research identified dL5 as an ideal FAP to switch on the fluorescence of MG1.36 We then evaluated the activating ability of dL5** to MG2–4. As presented in Table 1, the absorption maxima of all MGs red-shifted upon the addition of 4 fold excess of dL5**.21,35 Specifically, MG2–4 were red-shifted by 20, 32 and 15 nm, respectively, which were much smaller than the 40 nm shift observed for MG1. The absorbance of each MG further increased upon binding with dL5**, with MG3 showing a remarkable 2.6-fold enhancement, significantly surpassing the changes observed in the other derivatives (Fig. 2a). As anticipated, MG1 showed a fluorescence turn-on response to dL5** with a quantum yield of 0.12. Bruchez et al. attributed this phenomenon to the interactions between MG1 and residues located in both the complementarity-determining regions (CDRs) and framework regions of dL5**. Notably, the dimethylamine carbons of MG1 contribute approximately 50% of the total contact area, a feature critical for fluorescence enhancement by restricting intramolecular rotation.35 The negligible fluorescence of MG2/dL5** in our study further supports these findings. The absence of dimethylamine carbons in MG2 results in reduced conformational constraints and a complete loss of fluorescence activation. While the azetidine carbons in MG3 exert a similar effect in the interactions. The MG3/dL5** fluoromodule emits fluorescence in the near-infrared region with a significantly higher quantum yield of 0.28, achieving a 166.7-fold fluorescence enhancement (Fig. 2b). Although MG4 contains julolidine carbons, its substantially larger molecular size likely weakens the binding interaction with dL5**, resulting in only 14.3-fold fluorescence enhancement with a quantum yield of 0.04 upon complex formation. The dissociation constants Kd of MGs to dL5** were measured using a ligand depletion assay at the emission maximum. The Kd values for MG1, MG3 and MG4 were determined to be 3, 16 and 61 nM, respectively (Fig. 2c, d and S4†), showing high affinity to dL5**.
| Dye | λ abs (nm) | ε (M−1 cm−1) | λ em (nm) | Φ | K d (nM) |
|---|---|---|---|---|---|
| MG1 | 606 | 71000 |
n.d. | n.d. | 3 |
| MG1/dL5** | 646 | 102000 |
675 | 0.12 | — |
| MG2 | 549 | 31000 |
n.d. | n.d. | n.d. |
| MG2/dL5** | 569 | 39000 |
n.d. | n.d. | — |
| MG3 | 607 | 43000 |
n.d. | n.d. | 16 |
| MG3/dL5** | 639 | 113000 |
664 | 0.28 | — |
| MG4 | 646 | 54000 |
n.d. | n.d. | 61 |
| MG4/dL5** | 661 | 75000 |
684 | 0.04 | — |
 | ||
| Fig. 2 (a) Absorption and (b) fluorescence spectra of 0.5 μM MGs in the absence and presence of 2 μM dL5** (MG1–4: from left to right). Binding equilibrium analysis of MG1 (c) and MG3 (d) to dL5**. | ||
To investigate the structure–fluorescence relationship of MG-based fluoromodules, we conducted docking simulations. The 3D structure of dL5** (D50E mutation) was constructed using homology modeling based on the MG1-bound L5* (PDB ID: 4K3H) as a template.35 As shown in Fig. 3, the MG derivatives were positioned within the binding cavity formed by a symmetric antiparallel homodimer. MG2 and MG3 oriented approximately orthogonally to the dL5** surface, with their pendent rings extending outward from the cavity, consistent with the orientation observed in MG1. In contrast, MG4 was inserted into the binding cavity with its julolidine moiety protruding outside. The calculated binding affinities for MG1–4 were −10.8, −9.8, −10.4, and −2.5 kcal mol−1, respectively, indicating almost the same affinity for MG1–3. Although the results indicated that dL5** had a high affinity for MG2, fluorescence from the MG2/dL5** complex was not observed. This is likely because the crystal structure of 4K3H revealed that the methyl carbons of MG1 contributed approximately half of the total contact in the MG1-L5* homodimer.35 Therefore, the absence of alkyl groups on the nitrogen atoms of MG2 might allow free rotation of the aromatic rings within the binding cavity, resulting in fluorescence quenching and underscoring the significance of the azetidine group.
 | ||
| Fig. 3 Molecular docking analysis of MGs. (Top) Frontal view of the MGs/dL5** complex. (Bottom) Spatial distribution of amino acid side chains in dL5** that come in contact with MGs. | ||
Briefly, we successfully enhanced the brightness (ε × φ) of the MG1/dL5** fluoromodule by 2.6-fold by replacing the dimethylamino group with a four-membered azetidine ring. While the affinity of MG3 for dL5** was lower than that of MG1, its Kd value of 16 nM was comparable to previous reports and sufficient for fluorescence imaging.21 Notably, MG3 exhibited a substantial absorbance increase of 2.6-fold, compared to a 1.4-fold increase in MG1, thereby enhancing its fluorogenicity. Additionally, the absorption maximum of the MG3/dL5** complex at 639 nm aligns well with the standard 640 nm laser used in fluorescence imaging.
With the enhanced MG3/dL5** fluoromodule, excellent biocompatibility and high stability (Fig. S5 and S6†), we further explored its application in fluorescence imaging. MG3 demonstrates rapid penetration into cells, with the cellular uptake of MG3 primarily mediated by clathrin-dependent endocytosis (Fig. S7†). Using a myc-dL5**-mCer3 fusion protein, we assessed the fluorescence activation capability of this fluoromodule in live cells (Fig. 4a). Similar to the in vitro responses, MG2 showed negligible fluorescence signals in the nucleus, and MG4 exhibited a high background fluorescence. In contrast, MG3 demonstrated excellent fluorogenicity compared to MG1, establishing it as a superior fluorogen-activating protein probe for live-cell imaging. We selected mitochondria and the endoplasmic reticulum as representative organelles for labeling and imaging with MG3. Plasmids containing dL5**, the fluorescent protein mCer3, and a subcellular targeting sequence were transfected into HeLa cells, followed by the addition of 200 nM MG3. As shown in Fig. 4b, MG3 (red) colocalized with mCer3 (green) in the two organelles, indicating that dL5** was appropriately expressed in various subcellular environments, effectively activating the fluorescence of MG3.
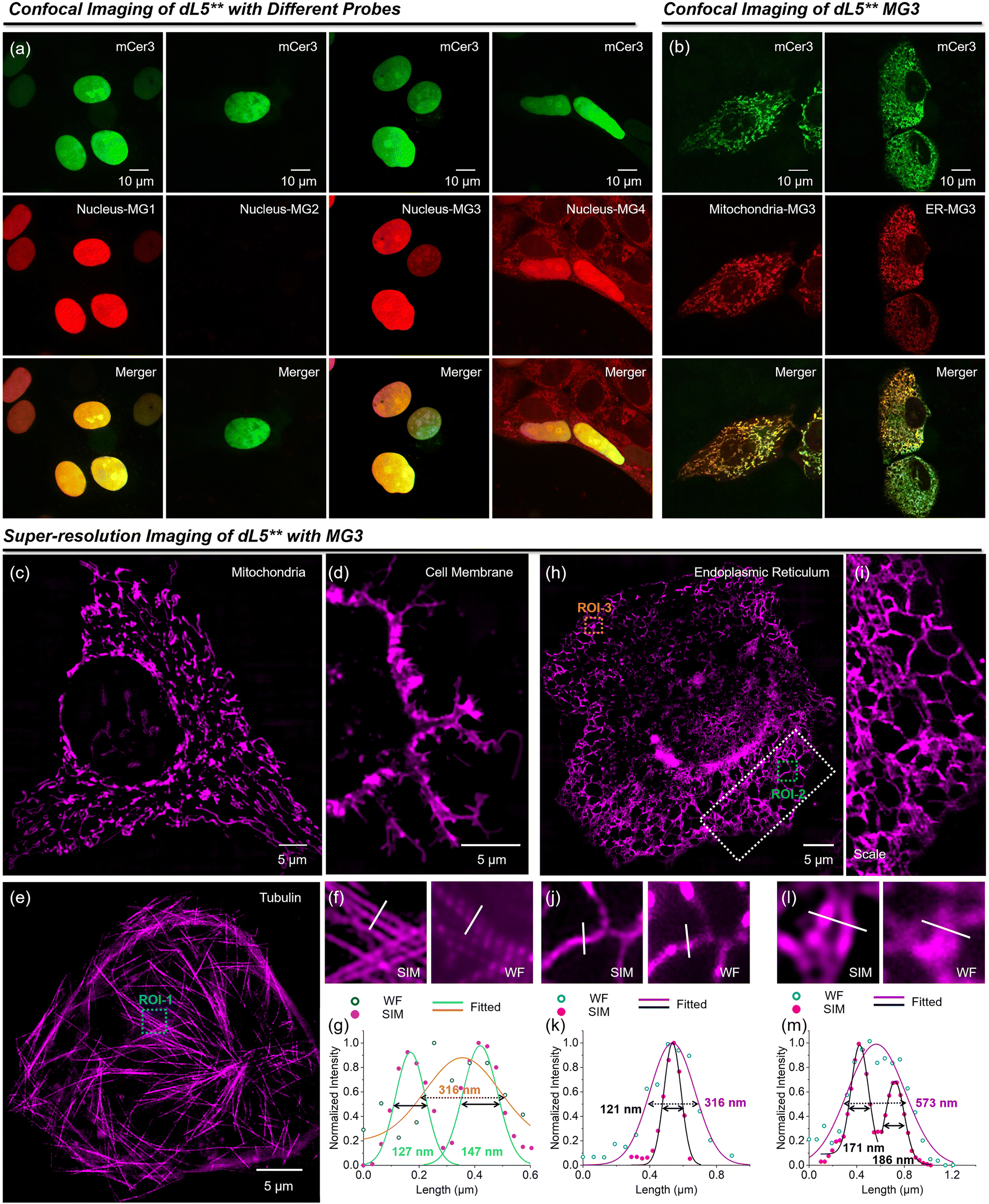 | ||
| Fig. 4 (a) Confocal images of HeLa cells expressing dL5**-mCer3 in the nucleus with MG1–4. (b) Confocal images of HeLa cells expressing dL5**-mCer3 in mitochondria and ER with the addition of 200 nM MG3. Green fluorescence signals from mCer3; red fluorescence signals from MG/dL5**. SIM imaging of mitochondria (c), cell membrane (d), tubulin (e) and endoplasmic reticulum (h). (i) Enlarged view of ROI-2 in (h). (f) SIM and widefield of ROI-1 in (e). (g) Intensity distributions across tubulin (white line) in (f). SIM and widefield of ROI-2 (j) and ROI-3 (l) in (h). (k) and (m) Intensity distributions across ER (white line) in (j) and (l). | ||
To demonstrate the capabilities of MG3 in super-resolution imaging, we first employed SIM to visualize various cellular structures (Fig. 4c–i), including mitochondria, cell membrane, microtubules, and endoplasmic reticulum (ER). MG3 effectively resolved the intricate structures of these organelles at the nanoscale. Adjacent microtubules that appeared indistinguishable under widefield were clearly discernible through super-resolution images, achieving resolutions of 127 nm and 147 nm (Fig. 4f and g). Furthermore, MG3 allowed for the visualization of the fine meshwork of ER, revealing an ER tubule width of 121 nm compared to a width of 316 nm observed with widefield imaging (Fig. 4j and k), further showcasing the high fluorogenicity and brightness of our probes. While widefield identified an ER tubule at 573 nm, super-resolution imaging revealed the finer structural details of the ER mesh, highlighting MG3's substantial potential for super-resolution applications.
Previous reports have indicated that MG1/dL5** is a highly photostable fluoromodule, making it suitable for super-resolution imaging of diverse cellular structures.24 Here, we further evaluated the photostability of both MG1/dL5** and MG3/dL5** in fixed cells under continuous 640 nm laser irradiation during SIM microscopy (Fig. 5). The fluorescence intensity of MG3/dL5** remained at 70% after 600 s, which is slightly higher than the 62% observed for MG1/dL5** (Fig. 5a). These findings were consistent with the outcomes of our confocal microscopy analysis (Fig. S8–S10†). We attribute the enhanced photostability of MG3/dL5** to the buffering effect of MG3 within the fluoromodules. As illustrated in Fig. 5b, continuous irradiation with the 640 nm laser reduced the fluorescence of MG3/dL5** to 58%. Upon cessation of the laser exposure, the fluorescence gradually recovered to 80% after 40 min. This recovery is likely due to the reduced affinity of dL5** for MG3, allowing fresh dyes to replenish those that were photobleached, a mechanism similar to the buffering observed in our previously reported fluorogenic probes.25–29
 | ||
| Fig. 5 (a) Quantification of the relative fluorescent intensity of MG1/dL5** and MG3/dL5** in HeLa cells with continuous irradiation using super-resolution SIM microscopy. (b) Fluorescence recovery of MG3/dL5** in HeLa cells after 20 min of continuous irradiation using a 640 nm laser. | ||
Due to its enhanced photostability, we applied MG3/dL5** for long-term tracking of mitochondrial dynamics through super-resolution imaging. The mitochondrial network was clearly identified under SIM. As shown in Fig. 6a and Movie S1,† we monitored mitochondrial fission over a 30-minute tracking period. At 9 min, the mitochondrion marked by a green arrow gradually extended to establish contact with a neighboring mitochondrion. This contact point underwent continuous changes over the next 10 minutes. Notably, significant deformation of the neighboring mitochondrion was observed at 16 min. By 22 min, the mitochondrion indicated by the green arrow exhibited a boundary and began to produce a small daughter mitochondrion. Subsequently, the mitochondrion marked by the green arrow remained in contact with the daughter mitochondrion, eventually leading to complete separation from the original mitochondrion. Additionally, multiple fission and contact events were recorded in the same region, highlighted by green arrows. Mitochondrial autophagy is also documented in Fig. 6b and Movie S2.† The mitochondrion marked by an orange arrow gradually swelled at the bifurcation point, transitioning from a slender linear shape to a spherical morphology at 8 minutes.
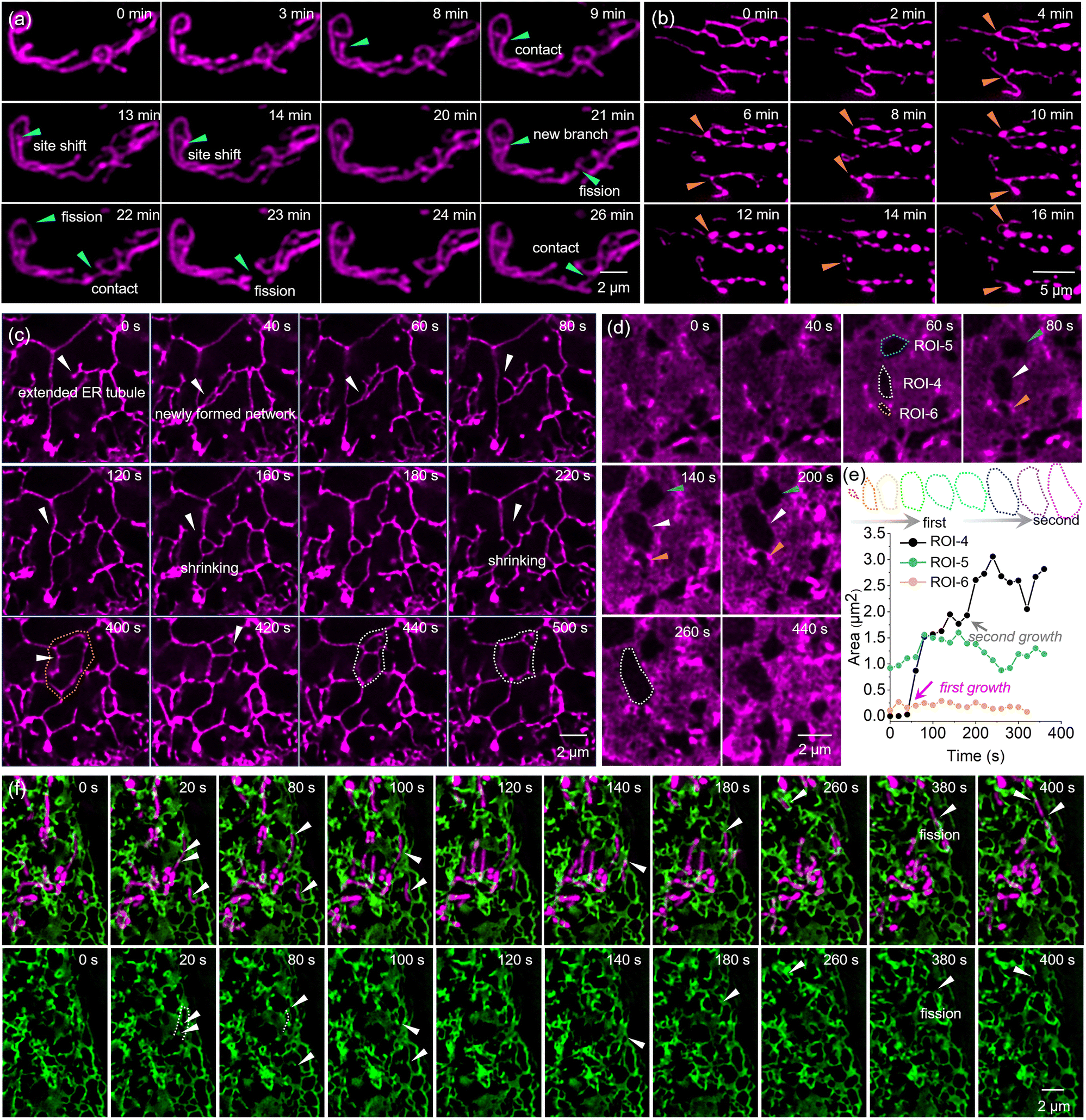 | ||
| Fig. 6 Time-lapse SIM images of (a) mitochondrial fission, (b) mitophagy, ER tubule (c) and RER dynamics (d) in living HeLa cells through MG3/dL5** labeling. (e) Time colored outline of the cavity in ROI-4. And the time dependence of area in ROI-4-6. (f) Dual-color time-lapse SIM images of mitochondria and ER with MG3/dL5** and GFP in living HeLa cells. | ||
Using MG3, we also visualized the dynamic behavior of the ER, including the movement of ER tubules and the formation and disruption of ER networks (Fig. 6c). At 0 s, an ER tubule, marked by a white arrow, gradually extended and formed a new network by 40 seconds. This network subsequently gave rise to a new ER tubule, also indicated by a white arrow, at 80 s, which rapidly moved to form a triangular network by 120 s. The newly formed network exhibited significant dynamics and was not stable, gradually shrinking between 120 and 180 s and ultimately breakingoken apart at 220 s. By 400 s, a new budding point appeared in the same area (marked by a white arrow) and created a new network by 420 s, which divided the original network (outlined by an orange dotted frame) into two smaller networks. In contrast to the previous observation, the upper network gradually shrank while the lower network expanded, ultimately forming a larger network by 500 s.
Additionally, we observed a cell rich in rough endoplasmic reticulum (Fig. S11†) and monitored the formation and morphological changes of hollow regions within the rough endoplasmic reticulum (Fig. 6d). We selected three regions of interest (ROIs) – ROI-4, ROI-5, and ROI-6 – to examine the cavities, which exhibited different dynamic behaviors. The mature cavity in ROI-4 showed rapid growth after 60 s, followed by fluctuations in size, with a gradual reduction occurring between 160 and 260 s (Fig. 6e). Notably, newly formed cavities in ROI-5 displayed two distinct growth phases (Fig. 6e). The outline of cavity was marked with a colored dotted frame and the area was recorded. The first occurred in the initial formation period from 40 to 60 s, during which the cavity area increased from 0.03 μm2 to 1.52 μm2. The second significant growth phase was observed between 160 and 240 s, where the area expanded from 1.77 μm2 to 3.06 μm. This suggested that the expansion of cavities formed within the rough endoplasmic reticulum might involve multiple short-term phases of growth rather than a uniform slow process.
Moreover, we achieved dual-color super-resolution imaging of the ER and mitochondria using MG3, enabling us to monitor the movement of mitochondria being pulled by the ER (Fig. 6f).
At 20 s, a mitochondrion marked by a white arrow was completely enveloped by the green ER, showing stronger fluorescence at the contact sites. This phenomenon was similarly observed at 80 and 100 s. Furthermore, by 80 s, the mitochondrion, which was closely apposed to the ER, exhibited a curved morphology (indicated by a dotted line), reflecting strong interactions. At 140 s, the mitochondrion marked by a white arrow displayed high dynamics, being pulled upward by ER–mitochondria contact sites, leading to division at 380 s, during which the ER participated in this fission event. Notably, ER–mitochondria contact sites were found to define the positions of mitochondrial fission, and ER–mitochondria contacts were maintained throughout the entire division process, with the two compartments bounded by the ER tubule moving back and forth.
Conclusions
In summary, we have enhanced the brightness of the MG/dL5** fluoromodule by replacing the N,N-dimethylamino group in MG with an azetidine structure to suppress the TICT effect. Our probe, MG3, demonstrated a quantum yield of 0.28 in the presence of dL5**, significantly higher than that of the conventional MG1, which showed a quantum yield of 0.12. This modification results in a 2.6-fold brightness enhancement over MG1. Additionally, the optimized MG3/dL5** complex exhibits advantages in fluorogenicity, photostability, and absorption maximum, making it an ideal fluoromodule for live-cell and super-resolution imaging. The enhanced buffering capacity of MG3 allows for reversible binding with the FAP, enabling free fluorogen molecules to replace those that have been photobleached at the target site. This characteristic supports stable fluorescence over time, facilitating continuous dynamic imaging of various cellular structures.Data availability
All data are available in the main text and the ESI† or available from the authors upon reasonable request.Author contributions
Fei Deng: conceptualization, investigation, analysis, writing – original draft. Xiangning Fang: investigation, analysis. Qinglong Qiao: conceptualization, investigation, analysis, writing – original draft. Lu Miao: investigation, analysis. Shuangshuang Long: investigation, analysis. Zhaochao Xu: conceptualization, supervision, writing – reviewing & editing, funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the financial support from the National Natural Science Foundation of China (22225806, 22078314, 22278394, 22468023, 22378385, and 32301060), Jiangxi Provincial Natural Science Foundation (20242BAB25160), Science and Technology Research Project of the Jiangxi Provincial Department of Education (GJJ2201627), and Dalian Institute of Chemical Physics (No. DICPI202227 and DICPI202436).Notes and references
- T. A. Daugird, Y. Shi, K. L. Holland, H. Rostamian, Z. Liu, L. D. Lavis, J. Rodriguez, B. D. Strahl and W. R. Legant, Correlative single molecule lattice light sheet imaging reveals the dynamic relationship between nucleosomes and the local chromatin environment, Nat. Commun., 2024, 15, 4178 CrossRef CAS PubMed.
- Q. Qiao, W. Liu, Y. Zhang, J. Chen, G. Wang, Y. Tao, L. Miao, W. Jiang, K. An and Z. Xu, In Situ Real-Time Nanoscale Resolution of Structural Evolution and Dynamics of Fluorescent Self-Assemblies by Super-Resolution Imaging, Angew. Chem., Int. Ed., 2022, 134, e202208678 CrossRef.
- L. Miao, C. Yan, Y. Chen, W. Zhou, X. Zhou, Q. Qiao and Z. Xu, SIM imaging resolves endocytosis of SARS-CoV-2 spike RBD in living cells, Cell Chem. Biol., 2023, 30, 248–260 CrossRef CAS PubMed.
- S. Samanta, K. Lai, F. Wu, Y. Liu, S. Cai, X. Yang, J. Qu and Z. Yang, Xanthene, cyanine, oxazine and BODIPY: the four pillars of the fluorophore empire for super-resolution bioimaging, Chem. Soc. Rev., 2023, 52, 7197–7261 RSC.
- E. Kozma and P. Kele, Fluorogenic probes for super-resolution microscopy, Org. Biomol. Chem., 2019, 17, 215–233 Search PubMed.
- J. Kwon, M. S. Elgawish and S. H. Shim, Bleaching-Resistant Super-Resolution Fluorescence Microscopy, Adv. Sci., 2022, 9, 2101817 CrossRef PubMed.
- F. M. Jradi and L. D. Lavis, Chemistry of photosensitive fluorophores for single-molecule localization microscopy, ACS Chem. Biol., 2019, 14, 1077–1090 CrossRef CAS PubMed.
- Q. Qiao, W. Liu, W. Chi, J. Chen, W. Zhou, N. Xu, J. Li, X. Fang, Y. Tao and Y. Zhang, Modulation of dynamic aggregation in fluorogenic SNAP-tag probes for long-term super-resolution imaging, Aggregate, 2023, 4, e258 CrossRef CAS.
- A. Salam, K. Kaushik, B. Mukherjee, F. Anjum, G. T. Sapkal, S. Sharma, R. Garg and C. K. Nandi, A zinc metal complex as an NIR emissive probe for real-time dynamics and in vivo embryogenic evolution of lysosomes using super-resolution microscopy, Chem. Sci., 2024, 15, 15659–15669 RSC.
- Z.-H. Wu, X. Zhu, Q. Yang, Y. Zagranyarski, K. Mishra, H. Strickfaden, R. P. Wong, T. Basché, K. Koynov and M. Bonn, Near-infrared perylenecarboximide fluorophores for live-cell super-resolution imaging, J. Am. Chem. Soc., 2024, 146, 7135–7139 CrossRef CAS PubMed.
- X. Ren, C. Wang, X. Wu, M. Rong, R. Huang, Q. Liang, T. Shen, H. Sun, R. Zhang and Z. Zhang, Auxochrome dimethyl-dihydroacridine improves fluorophores for prolonged live-cell super-resolution imaging, J. Am. Chem. Soc., 2024, 146, 6566–6579 CrossRef CAS PubMed.
- Q. Qiao, A. Song, K. An, N. Xu, W. Jia, Y. Ruan, P. Bao, Y. Tao, Y. Zhang and X. Wang, Spontaneously Blinkogenic Probe for Wash-Free Single-Molecule Localization-Based Super-Resolution Imaging in Living Cells, Angew. Chem., Int. Ed., 2025, 64, e202417469 CrossRef CAS PubMed.
- Q. Qiao, W. Liu, J. Chen, X. Wu, F. Deng, X. Fang, N. Xu, W. Zhou, S. Wu and W. Yin, An Acid-Regulated Self-Blinking Fluorescent Probe for Resolving Whole-Cell Lysosomes with Long-Term Nanoscopy, Angew. Chem., Int. Ed., 2022, 134, e202202961 Search PubMed.
- M. Minoshima, S. I. Reja, R. Hashimoto, K. Iijima and K. Kikuchi, Hybrid Small-Molecule/Protein Fluorescent Probes, Chem. Rev., 2024, 124, 6198–6270 CrossRef CAS PubMed.
- E. Gallo, Fluorogen-activating proteins: Next-generation fluorescence probes for biological research, Bioconjugate Chem., 2019, 31, 16–27 CrossRef PubMed.
- L. A. Perkins and M. P. Bruchez, Fluorogen activating protein toolset for protein trafficking measurements, Traffic, 2020, 21, 333–348 Search PubMed.
- S. Xu and H.-Y. Hu, Fluorogen-activating proteins: beyond classical fluorescent proteins, Acta Pharm. Sin. B, 2018, 8, 339–348 CrossRef PubMed.
- Y. Zhang, W. Zhou, N. Xu, G. Wang, J. Li, K. An, W. Jiang, X. Zhou, Q. Qiao and X. Jiang, Aniline as a TICT rotor to derive methine fluorogens for biomolecules: A curcuminoid-BF2 compound for lighting up HSA/BSA, Chin. Chem. Lett., 2023, 34, 107472 CrossRef CAS.
- C. Szent-Gyorgyi, B. A. Schmidt, Y. Creeger, G. W. Fisher, K. L. Zakel, S. Adler, J. A. J. Fitzpatrick, C. A. Woolford, Q. Yan, K. V. Vasilev, P. B. Berget, M. P. Bruchez, J. W. Jarvik and A. Waggoner, Fluorogen-activating single-chain antibodies for imaging cell surface proteins, Nat. Biotechnol., 2008, 26, 235–240 CrossRef CAS PubMed.
- M. Zhang, S. K. Chakraborty, P. Sampath, J. J. Rojas, W. Hou, S. Saurabh, S. H. Thorne, M. P. Bruchez and A. S. Waggoner, Fluoromodule-based reporter/probes designed for in vivo fluorescence imaging, J. Clin. Invest., 2015, 125, 3915–3927 CrossRef PubMed.
- C. P. Pratt, J. J. He, Y. Wang, A. L. Barth and M. P. Bruchez, Fluorogenic Green-Inside Red-Outside (GIRO) Labeling Approach Reveals Adenylyl Cyclase-Dependent Control of BK alpha Surface Expression, Bioconjugate Chem., 2015, 26, 1963–1971 Search PubMed.
- Q. Zhang, Q. Wang, Y. Sun, L. Zuo, V. Fetz and H.-Y. Hu, Superior Fluorogen-Activating Protein Probes Based on 3-Indole–Malachite Green, Org. Lett., 2017, 19, 4496–4499 CrossRef CAS PubMed.
- J. A. Fitzpatrick, Q. Yan, J. J. Sieber, M. Dyba, U. Schwarz, C. Szent-Gyorgyi, C. A. Woolford, P. B. Berget, A. S. Waggoner and M. P. Bruchez, STED nanoscopy in living cells using fluorogen activating proteins, Bioconjugate Chem., 2009, 20, 1843–1847 Search PubMed.
- S. Saurabh, A. M. Perez, C. J. Comerci, L. Shapiro and W. Moerner, Super-resolution imaging of live bacteria cells using a genetically directed, highly photostable fluoromodule, J. Am. Chem. Soc., 2016, 138, 10398–10401 CrossRef CAS PubMed.
- J. Chen, C. Wang, W. Liu, Q. Qiao, H. Qi, W. Zhou, N. Xu, J. Li, H. Piao and D. Tan, Stable Super-Resolution Imaging of Lipid Droplet Dynamics through a Buffer Strategy with a Hydrogen-Bond Sensitive Fluorogenic Probe, Angew. Chem., Int. Ed., 2021, 133, 25308–25317 Search PubMed.
- N. Xu, Q. Qiao, X. Fang, G. Wang, K. An, W. Jiang, J. Li and Z. Xu, Solvatochromic Buffering Fluorescent Probe Resolves the Lipid Transport and Morphological Changes during Lipid Droplet Fusion by Super-Resolution Imaging, Anal. Chem., 2024, 96, 4709–4715 CrossRef CAS PubMed.
- W. Zhou, Y. Tao, Q. Qiao, N. Xu, J. Li, G. Wang, X. Fang, J. Chen, W. Liu and Z. Xu, Cell-Impermeable Buffering Fluorogenic Probes for Live-Cell Super-Resolution Imaging of Plasma Membrane Morphology Dynamics, ACS Sens., 2024, 9, 3170–3177 CrossRef CAS PubMed.
- K. An, Q. Qiao, W. Zhou, W. Jiang, J. Li and Z. Xu, Stable Super-Resolution Imaging of Cell Membrane Nanoscale Subcompartment Dynamics with a Buffering Cyanine Dye, Anal. Chem., 2024, 96, 5985–5991 CrossRef CAS PubMed.
- W. Jiang, Q. Qiao, J. Chen, P. Bao, Y. Tao, Y. Zhang and Z. Xu, Rna Buffering Fluorogenic Probe for Nucleolar Morphology Stable Imaging And Nucleolar Stress-Generating Agents Screening, Adv. Sci., 2024, 11, 2309743 Search PubMed.
- L. Wang, W. Du, Z. Hu, K. Uvdal, L. Li and W. Huang, Hybrid rhodamine fluorophores in the visible/NIR region for biological imaging, Angew. Chem., Int. Ed., 2019, 58, 14026–14043 Search PubMed.
- X. Liu, Q. Qiao, W. Tian, W. Liu, J. Chen, M. J. Lang and Z. Xu, Aziridinyl Fluorophores Demonstrate Bright Fluorescence and Superior Photostability by Effectively Inhibiting Twisted Intramolecular Charge Transfer, J. Am. Chem. Soc., 2016, 138, 6960–6963 CrossRef CAS PubMed.
- A. N. Butkevich, M. L. Bossi, G. v. Lukinavičius and S. W. Hell, Triarylmethane fluorophores resistant to oxidative photobluing, J. Am. Chem. Soc., 2018, 141, 981–989 CrossRef PubMed.
- W. Zhou, X. Fang, Q. Qiao, W. Jiang, Y. Zhang and Z. Xu, Quantitative assessment of rhodamine spectra, Chin. Chem. Lett., 2021, 32, 943–946 CrossRef CAS.
- J. B. Grimm, B. P. English, J. J. Chen, J. P. Slaughter, Z. J. Zhang, A. Revyakin, R. Patel, J. J. Macklin, D. Normanno, R. H. Singer, T. Lionnet and L. D. Lavis, A general method to improve fluorophores for live-cell and single-molecule microscopy, Nat. Methods, 2015, 12, 244–250 CrossRef CAS PubMed.
- C. Szent-Gyorgyi, R. L. Stanfield, S. Andreko, A. Dempsey, M. Ahmed, S. Capek, A. S. Waggoner, I. A. Wilson and M. P. Bruchez, Malachite Green Mediates Homodimerization of Antibody V-L Domains to Form a Fluorescent Ternary Complex with Singular Symmetric Interfaces, J. Mol. Biol., 2013, 425, 4595–4613 CrossRef CAS PubMed.
- J. J. He, Y. Wang, M. A. Missinato, E. Onuoha, L. A. Perkins, S. C. Watkins, C. M. St Croix, M. Tsang and M. P. Bruchez, A genetically targetable near-infrared photosensitizer, Nat. Methods, 2016, 13, 263–268 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5sc01150g |
| ‡ These two authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2025 |