
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMultiple stimulus modulated organic crystal polymorphs with tunable luminescence behavior†
Qian
Zhou
,
Mingxia
Feng
,
Caihong
Shi
,
Mengqiu
Qian
,
Xiurong
Ma
,
Runying
He
,
Xian
Meng
,
Yonggang
Shi
 ,
Qiue
Cao
and
Liyan
Zheng
*
,
Qiue
Cao
and
Liyan
Zheng
*
Key Laboratory of Medicinal Chemistry for Natural Resource of Yunnan University, Ministry of Education, School of Chemical Science and Technology, Yunnan University, Kunming 650091, P. R. China. E-mail: zhengliyan@ynu.edu.cn
First published on 6th May 2025
Abstract
Polymorphism is defined as the ability of a substance to exist in two or more crystalline forms, which provide a unique platform for revealing the relationship between its spatial structure and properties. However, organic crystal polymorphism can be commonly obtained by growing crystals in different solvents or at different temperatures. This study reports a compound named p-An-Br containing carbazole and anthracene chromophores with three multiple stimulus modulated crystal polymorphs with green, yellow and red fluorescence, respectively. Interestingly, switching of p-An-Br between crystal G, crystal Y and crystal R can be achieved through the uptake and release of methanol using different stimuli, which shows dynamically adjustable luminescent colors. Significantly, structure–property investigations via the in-depth analysis of molecular conformations and frameworks of the polymorphic crystals demonstrate that the diverse conformations and abundant noncovalent interactions have a predominant impact on emission behavior. Consequently, the crystals R can be used for the highly sensitive and specific sensing of methanol with a detection limit of 39.35 ppm. This study not only provides a new strategy for crystal polymorphism, but also develops an effective method for the detection of methanol.
1 Introduction
Polymorphism, which is defined as the ability of a substance to exist in two or more crystalline forms, is widely encountered in studies of crystallization, phase transitions, and biomineralization and in the manufacture of specialty chemicals and drugs.1–3 Polymorphs are crucial for providing insights into the crystal packing and structure–property relationships4–7 and especially for providing a perspective to understand luminescence behavior from solid states. Compared to solutions, there is tighter molecular packing within crystals, so tiny changes in molecular configuration may cause a huge difference in luminescent properties.8 Nowadays crystal polymorphism can be obtained under different solvent and temperature conditions (Scheme 1). For example, Yang et al. obtained crystals with different fluorescence emissions through slow evaporation in a mixture of n-hexane/ethyl acetate and methanol/dichloromethane solution.9 Li et al. controlled the temperature to induce temperature responsive color changing phosphorescence in Cs–C5OH crystals. As the temperature increased from 303 K to 403 K, the afterglow color changed from blue to green and then to yellow.10 However, the development of intelligent polymorphic emission materials obtained by other strategies is still highly demanded.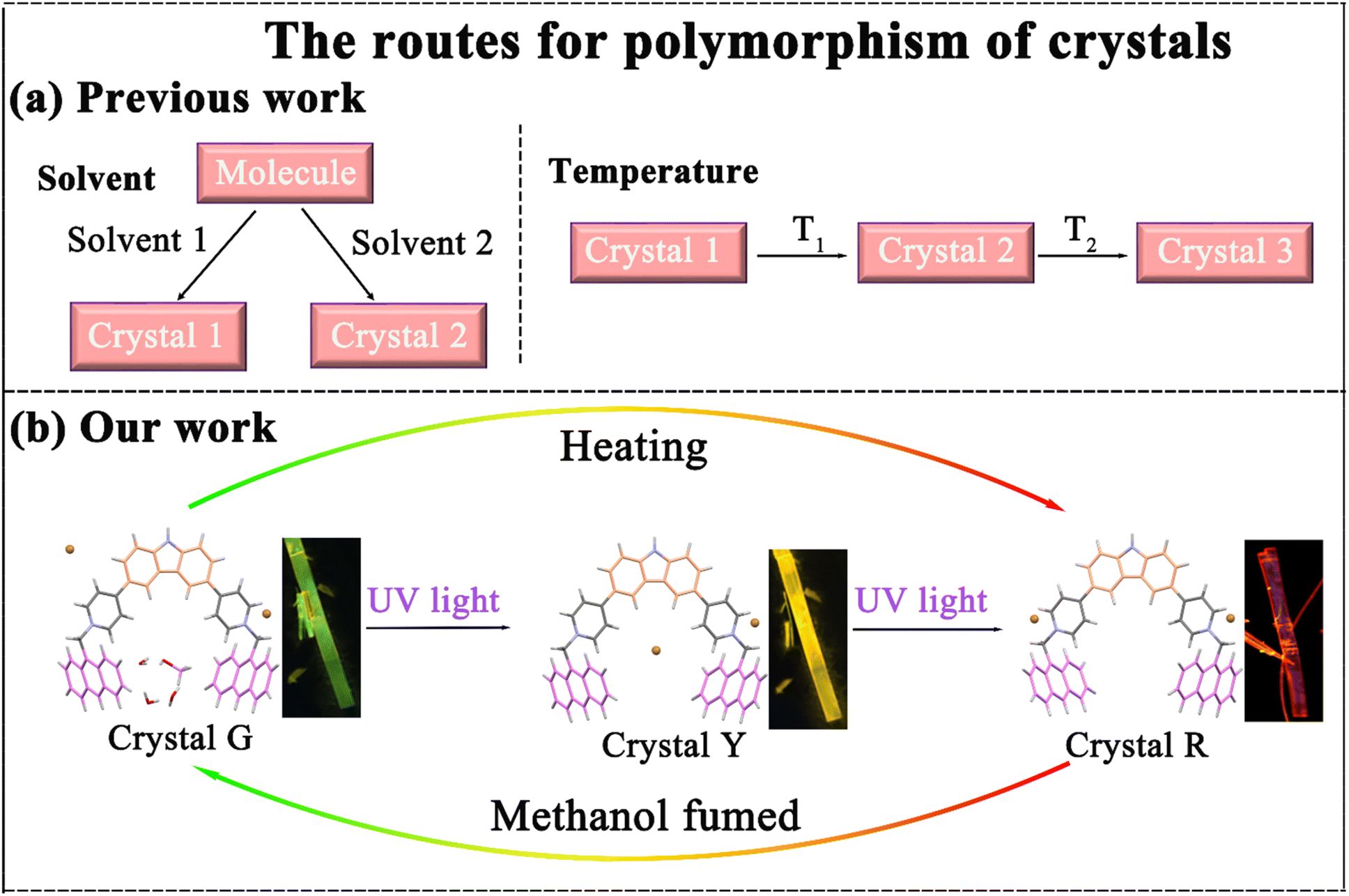 | ||
| Scheme 1 (a) Previous examples of the polymorphism of crystals in the literature. (b) A brief introduction of the polymorphism of p-An-Br crystals with the color switch process in this work. | ||
Luminescent materials that respond to external stimuli (such as light, heat, electricity, and other chemical stimuli) have significant application potential in cutting-edge fields of optoelectronic devices,11 biochemical sensing,12 imaging,13 and anti-counterfeiting encryption,14 because of their dynamic,15 reversible,16 and adaptive conversion characteristics.17 Among various external stimuli, light is considered an ideal candidate due to its ecofriendly and noninvasive nature, spatiotemporal operation, and convenient and precise control.18 These luminescent materials can indicate the stimulus–response behavior through changes in their luminous color, intensity, and lifetimes.19 Among them, the variation of luminescent color has aroused great interest due to the unique advantages of high spatial resolution and in situ/real-time visualization.20 However, there are still large challenges in controlling the emitting color because luminescence wavelength usually depends only on the lowest energy level of the excited state and is independent of the initial excited state.21 Although several strategies, including the introduction of multiple emission centers,22 energy transfer,23 and dynamic chemical reactions,24 have been employed to regulate luminescence color, these approaches require complex molecular designs, matched host–guest energy levels, or even extra reaction reagents. Consequently, developing a fast and dynamic light-color adjustment luminescent material is of great significance.
Herein, a new compound named p-An-Br with three crystal polymorphs displaying different emission colors was reported. Tunable emission colors from green and yellow to red can be achieved through the uptake and release of methanol by illumination, heating and fumigation (Scheme 1). More significantly, the underlying mechanism of crystal polymorphism-dependent fluorescence emission was well revealed by analyzing the different molecular conformations and diverse frameworks of the polymorphic crystals. Methanol vapor can enter crystal R through the host–guest interaction to transform crystal R into crystal G, thus achieving dynamic switching of colorful luminescence between crystal G and crystal R, which can be applied in highly sensitive and selective sensing for methanol vapor.
2 Results and discussion
4,4′-(9H-Carbazole-3,6-diyl)bis(1-(anthracen-9-yl-methyl)pyr-idin-1-ium) (named p-An-Br) was synthesized from 3,6-di(pyridine-4-yl)-9H-carbazole (p-DPC) and 9-(bromomethyl)anthracene via a simple two-step reaction. p-DPC was simply synthesized by the Suzuki cross-coupling reaction. 1H NMR, 13C NMR, and high-resolution mass spectra (HR-MS) measurements verified the structure of p-An-Br (Fig. S1–S6†). The detailed synthetic procedures and characterization are provided in Schemes S1 and S2 (ESI†). The molecular structure of p-An-Br is shown in Fig. 1a.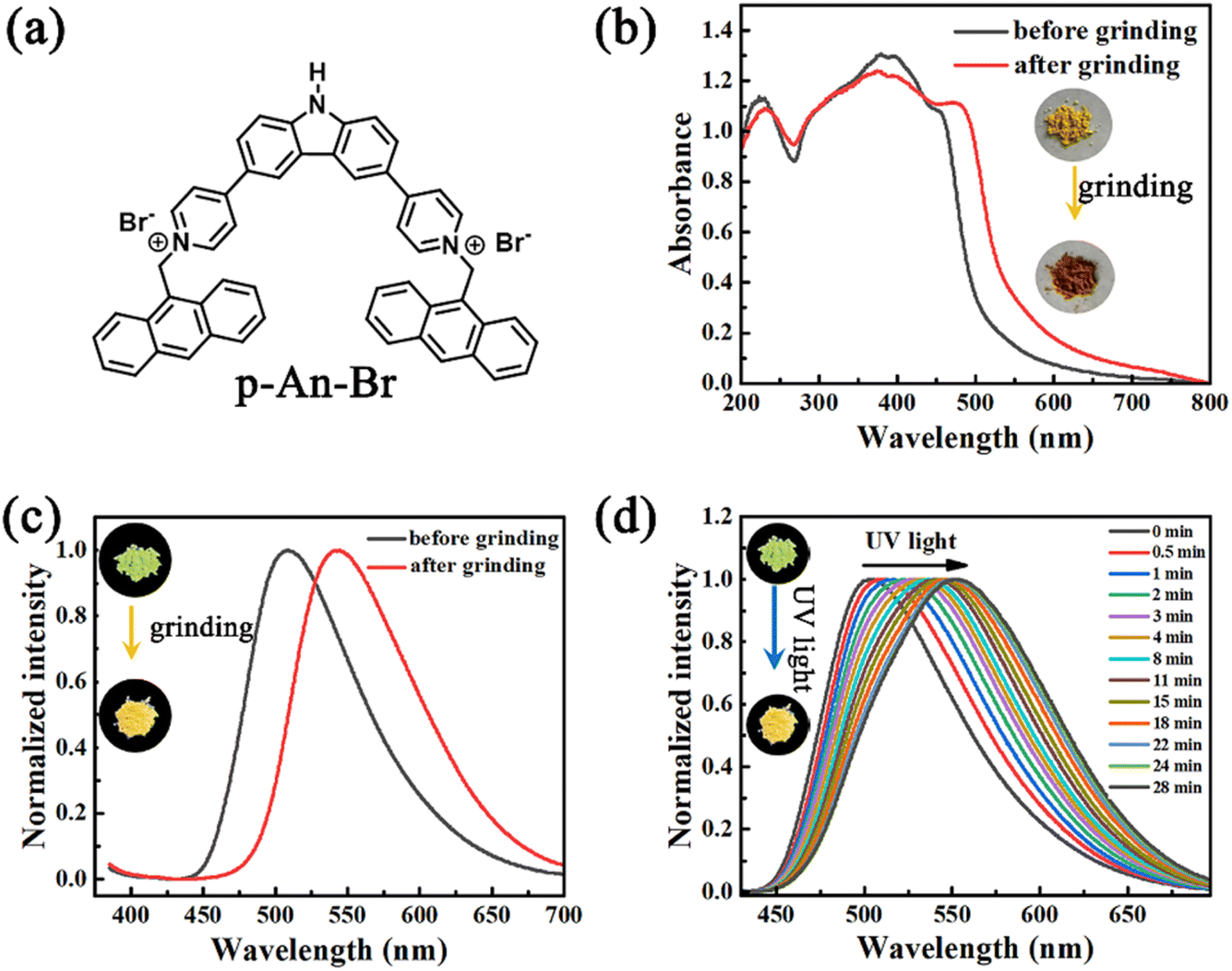 | ||
| Fig. 1 (a) Molecular structure of compound p-An-Br. (b) UV-vis spectra and (c) fluorescence spectra of p-An-Br powder before and after grinding. (d) Fluorescence spectra of solid powders of p-An-Br under continuous UV lamp irradiation time. | ||
2.1 Photophysical properties of the solid state powder
The photophysical properties of p-An-Br were first investigated in solution (Fig. S7†). As a result, p-An-Br demonstrated identifiable absorption bands at 400 nm which belonged to the π–π* transition, and their positions were almost unchanged upon altering the solvent polarity, indicating that the electronic intermolecular interaction in the ground state could be negligible. However, the emission band red-shifted from 495 nm to 530 nm (Dl = 35 nm) by changing the polarity of solvents, such as from CHCl3 to H2O in sequence, which was the typical solvatochromic effect assigned to the intramolecular charge transfer (ICT) transition.Because compounds exhibiting an ICT effect tend to be more responsive to molecular stacking, the solid-state photophysical properties of p-An-Br were further explored. Pristine p-An-Br was a yellow crystalline powder with green fluorescence (508 nm, ΦPL = 14.5%) under UV light irradiation. Upon full grinding with a mortar and pestle, the yellow powder was converted to a brown powder and the absorption band underwent redshift (Fig. 1b). The maximum emission peak redshifted from 508 nm to 544 nm, corresponding to a fluorescence color change to yellow (ΦPL = 12.3%) (Fig. 1c). When ground powder was exposed to methanol for 3 min, it returned to its original state and could be cycled multiple times, indicating that the mechanical-chromatic behavior has good reversibility (Fig. S8†). The fluorescence lifetimes of p-An-Br before and after grinding were also measured (1.13 and 2.30 ns), indicating that the emission belonged to the fluorescence (Fig. S9†). These results indicate that molecular configurations, inter-molecular interactions and packing arrangements can be changed under an external force stimulus. Powder X-ray diffraction (PXRD) measurements were carried out to gain insight into the crystal structure change upon grinding. For the pristine powder, quantities of sharp scattering peaks were observed (Fig. S10†). After grinding, the scattering peaks vanished, indicating a grinding-induced phase transition from the crystal phase to an amorphous state.
In addition, the solid powder of p-An-Br exhibited photochromic fluorescence properties. Upon continuous ultraviolet (UV) light exposure, the fluorescence color red-shifted gradually from green (502 nm) to yellow (553 nm) (Fig. 1d). The yellow fluorescence can revert back to green fluorescence through methanol vapor treatment, and this process can be repeated cyclically, indicating good reversibility of the photochromic process (Fig. S11†). PXRD spectra were measured but no obvious change was found (Fig. S12†), indicating that the molecular structure was unchanged after UV light irradiation. Besides, the fluorescence lifetimes of p-An-Br before and after UV light irradiation were also measured (1.13 and 2.35 ns), revealing that the emission belonged to the fluorescence (Fig. S13†).
2.2 The photophysical properties of the single crystal
As is well-known, it is rather difficult to realize crystal-state photochromism because the tight stacking of molecules significantly hinders molecular isomerization. Out of our expectation, the single crystal of p-An-Br showed excellent reversible photochromic properties. The single crystal exhibited green fluorescence under 365 nm UV light with an intensified fluorescent peak at 520 nm. After irradiation with a UV lamp at 365 nm for 30 minutes, the single crystal color changed to yellow, and as the irradiation time continued to extend up to 24 hours, the single crystal color ultimately turned red (Fig. 2a). And the ultraviolet color of the crystal G did not change at all. It is worth noting that three crystal polymorphs of p-An-Br with different emission colors were achieved through modulating the illumination time. The repeat of irradiation under nitrogen and vacuum atmospheres also resulted in similar color changes, ruling out the involvement of atmospheric oxygen behind the observed photochromism (Fig. S14†). In order to investigate the effect of irradiation wavelength on crystal photochromism, crystal G was irradiated with lamps of different wavelengths for 24 hours. The results are shown in Fig. S15,† where all wavelengths below 520 nm can change crystal G to crystal R, indicating that the photochromism of p-An-Br displayed good application prospects. Furthermore, the crystal R was found to be stable and retained red emission for more than a month under room light or under dark conditions, indicating the stability of the red crystal.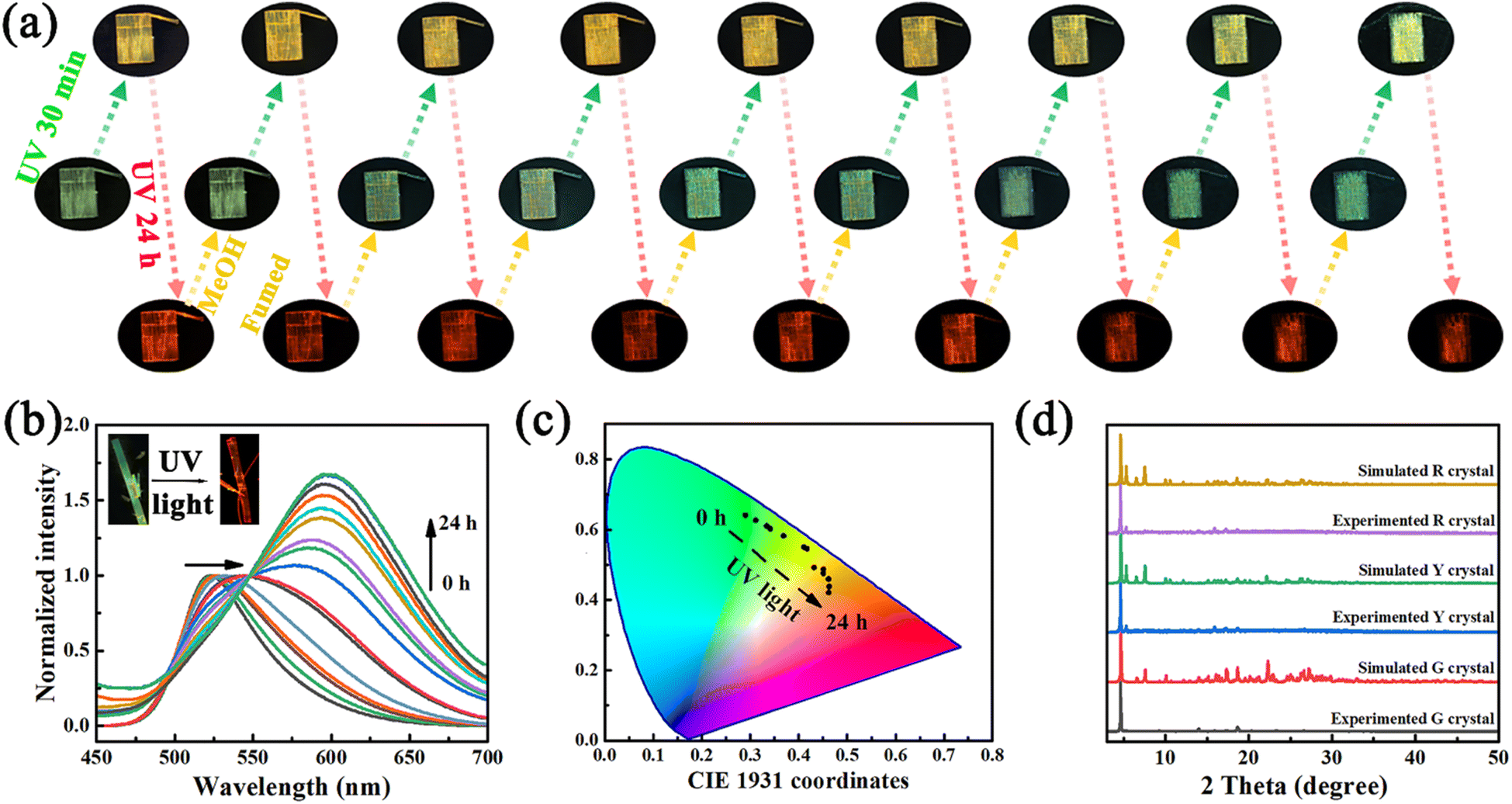 | ||
| Fig. 2 (a) Photoswitchable PL images of the p-An-Br crystal switching 9 times between UV light irradiation and methanol fuming. (b) Fluorescence spectrum changes of crystal G with UV lamp irradiation time. (c) CIE chromaticity coordinates of crystals as a function of UV lamp irradiation time. (d) Simulated PXRD patterns of three single crystals of p-An-Br. | ||
The fluorescence spectra of single crystals at different UV light irradiation times were also recorded. As shown in Fig. 2b, the emission wavelength of the single crystal shifted from 520 nm to 600 nm. Furthermore, the CIE chromaticity coordinate diagram also illustrated this redshift (Fig. 2c). Crystal G exhibited green emission (520 nm) with short lifetimes of several nanoseconds. In contrast, crystal R showed the redshifted, structureless and broadened photoluminescence (PL) spectrum with emission at 600 nm and long lifetime (τ = 34.50 ns), which is in good agreement with the characteristic of the anthracene excimer according to the previous reports (Table 1 and Fig. S16†).25 The photoluminescence quantum yields (PLQYs) of p-An-Br in different aggregation states were measured at room temperature. The rate constants kr and knr were estimated from the experimental PLQYs and the corresponding lifetimes, and the data are summarized in Table S1.† Comparatively, crystal R with long-wavelength emission showed significantly longer τ and lower knr than the other two crystal polymorphs with short-wavelength emission.
| Materials | Obtained conditions | Emission wavelength (nm) | PLQY | Lifetimes (ns) |
|---|---|---|---|---|
| Crystal G | Liquid-phase diffusion (methanol/ethyl acetate) | 520 nm | 12.6% | 3.43 |
| Crystal Y | Illuminating the crystal G with a UV lamp for 30 minutes | 550 nm | 13.8% | 3.26 |
| Crystal R | Illuminating the crystal G with a UV lamp for 24 h | 600 nm | 12.9% | 34.50 |
2.3 The analysis of the single crystal structure
To gain deep insights into the underlying mechanisms for the crystal packing and structure–property relationships of these polymorphs, we cultivated crystals of p-An-Br to study its different packing modes and the corresponding fluorescence. The crystal G was successfully cultivated by diffusion of mixed solutions (methanol/ethyl acetate) at room temperature (additional experimental details in the ESI†). Crystal G was irradiated under ultraviolet light for 30 minutes and 24 hours, resulting in crystal Y and crystal R, respectively. X-ray single crystal analyses of the crystals were carried out and the data are summarized in the ESI, Tables S2.† Three crystal polymorphs of p-An-Br are all in a monoclinic crystal system and crystallize in the space group C2/c.In the crystal G, the torsion angles between the pyridine ring and anthracene were 84.93° and 85.06° (Fig. 3a). Thus, the carbazole unit exhibited a planar quasi-equatorial (QE) conformation, which was favorable for charge delocalization and strengthened the electron-donating ability and ICT effect. Additionally, the carbazole unit presented a crooked QE conformation, and the twisted angles between benzene and pyridine rings of the conformational molecule were larger (5.01° and 4.52°) than those of crystals Y and R (Fig. S17†), resulting in a weak ICT effect of the p-An-Br G crystal. Eight methanol solvent molecules are involved in the unit cell, resulting in a looser packing pattern and weaker intermolecular CT interaction. There are obvious C–H⋯O interactions to be observed between p-An-Br and solvent molecules, corresponding to the monomer emission in crystal G (Fig. S18†). As a comparison, there is no solvent molecule in crystals Y and R. Later, the adjacent molecules were connected through abundant C–H⋯π (2.734–2.884 Å) interactions and C–H⋯Br interactions (2.787–2.840 Å) (Fig. 3b). Therefore, the p-An-Br G crystal showed high excited state energy, thus resulting in a blue-shift emission compared to p-An-Br-Y and p-An-Br-R crystals.
 | ||
| Fig. 3 The single-crystal structure, intramolecular interactions and molecular packing patterns of (a–c) crystal G, (d–f) crystal Y, and (g–i) crystal R. | ||
Comparatively, the carbazole unit of the p-An-Br Y crystal adopted a less crooked QE conformation (Fig. S17†). This could facilitate the conjugation between nitrogen lone pair electrons with the adjacent acceptor, leading to a more localized electronic transition upon excitation. Accordingly, the twisted angles between benzene and pyridine rings were smaller (4.43° and 4.25°) than those of crystal G. Therefore, the ICT effect of the p-An-Br Y crystal was strengthened, which led to a red shift to yellow emission. The more crooked forms (85.66° and 85.19°) of the p-An-Br moiety in crystal Y are considered the other inner reason for its red shift of emission (Fig. 3d). Furthermore, a three-dimensional (3D) supramolecular framework with the cavity units was stacked by the different conformers, which were linked via abundant intermolecular C–H⋯π (2.743 Å) and C–H⋯Br (2.759–2.825 Å) (Fig. 3e and f).
In addition, the torsion angles between the pyridine ring and anthracene in crystal R are 86.71° and 86.48° (Fig. 3g). Moreover, the twisted angles between the benzene and pyridine rings were only 3.80° and 4.02° for crystal R, respectively. Such tiny twisted angles in the acceptor moieties were beneficial for boosting the electron-withdrawing ability. These results led to the bathochromic-shift emission. Compared to crystal G and crystal Y, abundant intermolecular C–H⋯Br (2.821–2.904 Å), C–H⋯π (2.725–2.872 Å) and π⋯π (3.318 Å) interactions existed in the R-crystal (Fig. 3h). There were no π–π interactions between p-An-Br molecules in crystals G and Y due to the highly twisted p-An-Br moiety. Furthermore, more intermolecular π–π interactions in molecular packing of crystal R induce the emission red-shift (Fig. 3i).
Overall, the molecular conformations and packing modes in crystals have a significant influence on the fluorescent properties. The stacking arrangement of crystal G differs slightly from that of crystals Y and R, resulting in different fluorescence colors. As shown in Fig. 3c, f and i, the detailed stacking diagram illustrates the differences among the three polymorphs (G, Y, and R). In G crystals, methanol molecules exist in the framework voids through C–H⋯O interactions, while there are no such interactions in crystals Y and R. The stacking of the three types of crystals depends on the C–H⋯Br and C–H⋯π interactions, especially in crystal R where there is also a π⋯π interaction, resulting in a tighter stacking of crystal R and a red shift in crystal wavelength. When stimulated using pressure, the packing structures are destroyed and the transformation of the molecules from the crystalline state into an amorphous state results in the redshift of the emission. Besides, methanol molecules were found within the pores of single-crystal structures of crystal G, indicating that methanol plays a crucial role in determining the emission of single crystals. When the crystal R is placed in a methanol atmosphere, methanol molecules are absorbed into the pores of the crystal, transforming it into crystal G, thus achieving a reversible change.
To further confirm that the crystal is closely related to the fluorescent emission, PXRD patterns of the three crystals were simulated and are shown in Fig. 2d. The PXRD patterns of the three crystals are consistent with their simulated ones, indicating the high purity of the three crystals (Fig. 2d). The PXRD patterns of crystal Y and crystal R were similar. Compared with crystals Y and R, crystal G has no diffraction peaks at 5.2, 15.8, and 17.2 degrees, indicating a difference in the structure from crystals Y and R and variations in its single crystal structure. These results implied that p-An-Br possessed different aggregation states, and the molecular conformation and stacking arrangement of single crystals could provide a deep insight into the underlying luminescence mechanisms.
2.4 Underlying mechanisms of different polymorphism emissions
Considering the possibility of radical generation upon irradiation, electron paramagnetic resonance (EPR) studies have been conducted to understand the differences between the optical properties of crystal G, crystal Y and crystal R. There was no obvious EPR signal for the crystal G after irradiation; moreover, the appearance of the EPR signal at a g value of 2.0051 in the crystals Y and R after irradiation also demonstrates that EPR silent crystal G turns into the other two polymorphs accompanied by the formation of radicals via charge transfer (Fig. 4a). | ||
| Fig. 4 (a) EPR spectra of the three crystals of p-An-Br. (b) Electron distribution and energy levels of molecular orbitals of the three crystals. (c) Calculated fluorescence emission spectra of the three types of crystals. (d) Dipole moment of the three crystals calculated from DFT calculations in monomer and dimer states. | ||
We further examined the possibility of excimer formation by the concentration-dependent PL spectra in the solutions. The results demonstrated that p-An-Br in high-concentration tetrahydrofuran (THF) solutions (such as 10−2 and 10−1 M) showed new emission bands which were almost the same as that of crystal R (Fig. S19†). Therefore, p-An-Br was capable of forming the excimer with the close AN dimer in solids and solutions. For the individual AN fluorophore without p-DPC, there was no emerging emission band in high-concentration THF solutions (Fig. S20†), also indicating that the molecular design was in favor of excimer formation. We tested the solution sample and the ground powder of p-An-Br, but no photochromism was observed (Fig. S21†), implying that the light-induced molecular motion may play a vital role in the crystal-state photochromism. In situ Fourier transform infrared (FT-IR) spectra were recorded but no change was found (Fig. S22†), indicating that the molecular structure was unchanged after UV lamp irradiation.
Considering the presence of methanol within crystal G, it is possible to remove methanol through heating. Upon heating, the crystal undergoes a process of turning yellow first and then red (Fig. S23†). This should be attributed to the volatilization of methanol in the lattice, leading to the crystalline transition from crystal G into crystal R. In order to verify the specific recognition mechanism of crystal R for methanol, the NMR spectra of crystal R before and after methanol fumigation were measured. As shown in Fig. S24,† additional C and H peaks of methanol appeared in the NMR spectrum after fumigation, indicating that methanol molecules entered the crystal. The thermogravimetric analysis (TGA) data indicate that compared to crystal Y and crystal R, crystal G exhibits an additional weight loss peak in the range of 96–165 °C with a thermal weight loss of 3.37% (Fig. S25†), which corresponds exactly to the relative mass of methanol molecules in the lattice, indicating that the entry of methanol molecules into the lattice of crystal R is the key factor for the recognition of methanol. We attempted to recover crystal G from crystal R by fuming crystal R with methanol vapor (Fig. S26†). As expected, after fuming crystal R for 2 min, the luminous color of the crystal R changes to yellow and then completely turns into green after continuous fuming for 10 min. The fumed crystal can be restored to its initial state by heating for 5 min. The PXRD patterns of the fumed and heated samples are almost consistent with those of crystal G and crystal R (Fig. S27†), implying the single-crystal to single-crystal structural transformations in the process of methanol adsorption. At the same time, it is shown that heating and methanol fumigation can achieve a reversible switch between crystal G and crystal R. While crystal R, which has no solvate, had voids in the crystal structure (Fig. S28†) to promote molecular motions, methanol solvent could occupy the voids through host–guest interaction. These results confirm that the recognition mechanism of methanol is probably induced by the interactions between guest methanol molecules within the host framework. The uptake and release of methanol molecules play a crucial role in the structural switching among these crystal polymorphs.
2.5 Theoretical calculations
To gain insight into the molecular geometries, frontier molecular orbital distributions, and excited-state energy levels of the emitters, theoretical calculations based on time-dependent density functional theory (TD-DFT) were conducted (Tables S4–S6†). The frontier molecular orbitals include the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO). The energy difference of the frontier orbitals (HOMO–LUMO energy gap) is the key factor to determine the emission wavelength. The smaller the energy gap, the longer the emission wavelength. For crystal G, the transition from the HOMO to the LUMO located on the entire molecule mainly contributed to the vertical excitation of crystal G. The transition of crystal G is characterized by the locally excited (LE) state with a short wavelength. The frontier molecular orbitals of this LE state mainly reside at the entire molecule with a minimal degree of charge transfer. Adequate separation and appropriate overlap of the HOMO and the LUMO indicated intramolecular through-bond charge transfer (TBCT) with a band gap of 2.77 eV (Fig. 4b). As for crystal Y and crystal R, it is noteworthy that the electron distribution of molecular orbitals of p-An-Br was much different from that of crystal G. Accordingly, the transition of crystal Y exhibits obvious ICT from the bromide ion to p-DPC with a moderate wavelength. The frontier molecular orbitals of crystal R are characterized by a complete charge separation between the bromide ion and the cation (p-An2+) fragments, with a smaller optical gap (or a longer emission wavelength). Besides, the maximum emission wavelengths of the crystals G, Y and R calculated were located at 489 nm, 530 nm, and 590 nm, which are proximate to the experimental values (Fig. 4c). Hole–electron analysis showed that the crystal R had a stronger charge transfer ability, resulting in a red shift in the emission wavelength (Fig. S29†).Molecular dipole moment is an indicator of charge separation, and large charge separation can cause strong CT effects, resulting in long wavelength emission. Due to the strong CT effect between ICT or TICT states, their dipole moments are greater than those of LE states. When the dipole moment of a molecule changes, its electronic and vibrational energy levels also shift accordingly, which usually leads to a shift in the emission wavelength, depending on the structure of the molecule and the external environment. During the photochromic process, rotation of the pyridine ring and anthracene led to an obvious change in the vector direction of molecular dipole moment. Orientations of the transition dipoles for the monomers of crystals G, Y and R were almost antiparallel and led to a sharp decline for the dipole moment of the dimer. Besides, the photochromic process can also affect the regular arrangement in the crystalline state. Crystallinity is equal to the proportion of the crystalline fraction in the whole, which refers to the degree of structural order of a solid material. Noticeably, each asymmetric unit in the crystal G consisted of a dimer with moderate intermolecular C–H⋯π interactions and its dipole moment was calculated to be 5.23 debye (Fig. 4d). Compared to the dipole moment of the monomer in crystal G (3.01 debye), special packing mode greatly enhanced the total dipole moment of p-An-Br in the crystalline state and promoted intense luminescence. During the process of photochromism, the production of free radicals and the strengthened π–π interaction (crystal R) led to an obvious change in the molecular dipole moment. The formed radicals reduced the molecular charge, while dimers are more tightly packed than monomers, resulting in a greater change in dipole moment. The dipole moment of crystal R drastically decreased from 3.68 D (monomer) to 2.35 D (dimer).
2.6 The detection of methanol
Methanol is one of the most common volatile organic compounds that have a great impact on human health. As reported, working in an environment where the concentration of methanol exceeds health standards can cause metabolic acidosis in humans.26,27 Therefore, rapid and highly sensitive detection of methanol has significant value in life and health, safety production, clinical diagnosis, and other aspects. Herein, considering that crystal R can be recovered to crystal G after methanol vapor fuming, the crystal R was chosen as a sensor to detect volatile solvent vapors. In order to verify the specificity of crystal R in detecting methanol, crystal R was fumigated with different solvent vapors. After fuming with solvent vapors for 10 min, no significant changes in the appearance and luminescence color of the crystal R were observed in these solvents except for methanol (Fig. S30†). As shown in Fig. 5a and S31,† the emission peak of only the methanol-treated sample showed a significant blueshift from 600 nm to 520 nm. The PXRD mode showed that the diffraction peaks did not change significantly after fumigation with other solvents except methanol. And the PXRD mode after fumigation with methanol was consistent with that of crystal G, indicating that the encapsulation of methanol led to structural transformation from crystal R into crystal G (Fig. S32†). On this basis, a control experiment was designed to test the specific recognition of the crystal R for methanol in mixed solvent environments (Fig. 5b and S33†). The experimental results indicate that other solvents do not interfere with the highly specific and selective sensing of methanol. Although alcohol may interact with the crystal R via C–H⋯O interactions, protic solvent molecules larger in size than methanol molecules could not penetrate the crystal matrices. To better illustrate the interaction between the host and solvent molecules, the binding energies of the solvent molecules to the host framework were estimated using DFT calculations on the basis of the single-crystal structures. Notably, the binding energies of methanol, ethanol, propanol, isopropanol, butanol, glycol, benzyl alcohol, phenethyl alcohol, isopentanol, 2-butyl alcohol, diethyl ether, chloroform, dichloromethane and acetonitrile reach −23.34 kJ mol−1, −10.76 kJ mol−1, −9.43 kJ mol−1, −8.35 kJ mol−1, −14.35 kJ mol−1, −9.07 kJ mol−1, −3.48 kJ mol−1, −2.99 kJ mol−1, −6.83 kJ mol−1, −7.16 kJ mol−1, −5.27 kJ mol−1, −4.46 kJ mol−1, −9.06 kJ mol−1 and −13.94 kJ mol−1, respectively, indicating that methanol has the strongest host–guest interaction (Fig. S34†). Being consistent with the strong interactions revealed by the single-crystal structure of crystal G, the strong host–guest binding energy facilitates structural transformation from crystal R into crystal G, resulting in the change of emission color and realizing the specific detection of methanol. The switching between red and green emission can be reliably repeated many times by alternating stimulation of methanol vapor and heating (Fig. S35†).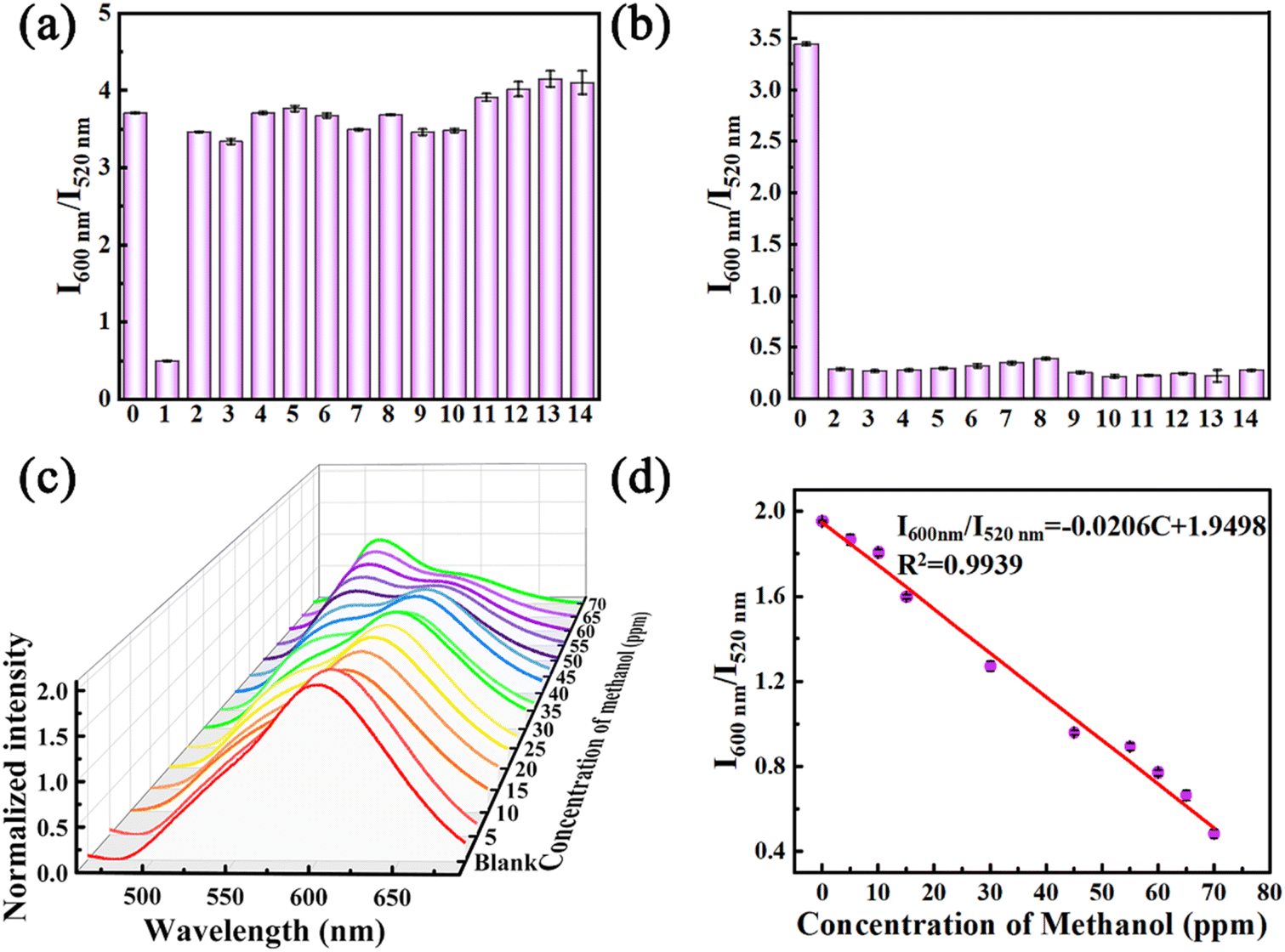 | ||
| Fig. 5 (a) Selective and (b) anti-interference bar charts of crystal R for detecting methanol. (0) Blank, (1) methanol, (2) ethanol, (3) propanol, (4) isopropanol, (5) butanol, (6) glycol, (7) benzyl alcohol, (8) phenethyl alcohol, (9) isopentanol, (10) 2-butyl alcohol, (11) diethyl ether, (12) chloroform, (13) dichloromethane, and (14) acetonitrile. (c) Normalized prompt emission spectra of the crystal R samples treated with different concentrations of methanol solution. (d) Linear relationship between the fluorescence intensity ratio (I600 nm/I520 nm) and the concentration of methanol in the range of 0 to 70 ppm. | ||
To quantitatively detect the concentration of methanol, crystal R was fumigated with different concentrations of methanol vapor for 10 min and then taken out for emission spectroscopy testing. As shown in Fig. 5c, the higher the methanol concentration, the more pronounced the change in the proportion (I600 nm/I520 nm) of the emission peak intensity of crystal R and crystal G. The degree of the proportion of emission peak positively correlates with methanol concentrations in the 5–70 ppm range (Fig. 5d). Based on the methanol concentration and the proportion of the emission peak of the crystal R and crystal G, the standard working curve y = −0.0206x + 1.9498 (R2 = 0.9939) can be obtained to give a limit of detection (LOD) of 39.35 ppm. Compared to other reported chemical sensors, the LOD value is one of the lowest detection limits (Table S7†), which is significantly lower than the threshold limit value (TLV) of methanol (200 ppm), where the TLV is defined as the permitted maximum concentration of a chemical during a working day for continuous exposure without producing adverse health effects.27,28 In addition, the detection sensitivity of crystal R is also far beyond that of most of the previously reported methods for detecting methanol, such as NMR, Raman and fluorescence spectroscopy, with the advantages of strong specificity, fast response, high sensitivity, and visualization.
3 Conclusion
In summary, three crystals of p-An-Br with different emission colors were achieved by multiple stimuli, including light, heating and fumigation. Structure–property investigations in different aggregation states demonstrate that molecular conformations and frameworks of the polymorphic crystals have a significant influence on their emission wavelength. Crystal G crystallized in methanol solvent contains methanol molecules in its framework. Removing methanol from the crystal G lattice through light or heating triggers the transformation from crystal G into crystal R. Conversely, fumigating with methanol vapor can restore crystal G from crystal R. By manipulating the uptake and release of methanol molecules, a repeatable and reversible switch between crystal G and crystal R can be achieved. The unique photophysical properties of crystal G and crystal R are expected to have applications for the highly sensitive and selective detection of methanol. This work provides a good insight into how molecular conformations and supramolecular interactions in the crystal lattice modulate the luminescent properties of the emitters in different aggregation states and would promote the development of novel organic multifunctional emissive materials for related fields.Data availability
The datasets supporting this article have been uploaded as part of the ESI.†Author contributions
Q. Zhou: conceptualization, validation, writing-original draft preparation, writing-reviewing. M. Feng: investigation, validation. C. Shi: software, investigation. M. Qian: validation. X. Ma: software, investigation. R. He: writing-reviewing. X. Meng: writing-reviewing. Y. Shi: project administration, resources. Q. Cao: project administration, resources. L. Zheng: supervision, conceptualization, visualization, project administration, funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This paper was financially supported by the National Natural Science Foundation of China (22464020 and 22164020), the “Ten Thousand Talents Plan” Top Young Talents of Yunnan Province (C6193044), the Yunnan Natural Science Foundation (K264202210049), the Scientific Research Fund Project of the Yunnan Education Department (2025Y0064) and the Scientific Research and Innovation Project of Postgraduate Students in the Academic Degree of Yunnan University (KC-24248765). The authors thank the Advanced Analysis and Measurement Center of Yunnan University for the sample testing service.Notes and references
- L. Yu, Acc. Chem. Res., 2010, 43, 1257 CrossRef CAS PubMed.
- J. Tao, R. Wei, R. Huang and L. Zheng, Chem. Soc. Rev., 2012, 41, 703 RSC.
- J. A. Foster, K. K. Damodaran, A. Maurin, G. M. Day, H. P. G. Thompson, G. J. Cameron, J. C. Bernal and J. W. Steed, Chem. Sci., 2017, 8, 78 RSC.
- (a) Z. Chi, X. Zhang, B. Xu, X. Zhou, C. Ma, Y. Zhang, S. Liu and J. Xu, Chem. Soc. Rev., 2012, 41, 3878 RSC; (b) H. Tsujimoto, D. G. Ha, G. Markopoulos, H. S. Chae, M. A. Baldo and T. M. Swager, J. Am. Chem. Soc., 2017, 139, 4894 CrossRef CAS PubMed; (c) X. Liang, Z. Tu and Y. Zheng, Chem.–Eur. J., 2019, 25, 5623 CrossRef CAS PubMed; (d) P. Data and Y. Takeda, Chem.–Asian J., 2019, 14, 1613 CrossRef CAS PubMed.
- (a) X. He, A. C. Benniston, H. Saarenpaa, H. Lemmetyinen, N. V. Tkachenko and U. Baisch, Chem. Sci., 2015, 6, 3525 RSC; (b) Z. He, L. Zhang, J. Mei, T. Zhang, J. Y. Lam, Z. Shuai, Y. Dong and B. Tang, Chem. Mater., 2015, 27, 6601 CrossRef CAS; (c) X. Cheng, Z. Wang, B. Tang, H. Zhang, A. Qin, J. Sun and B. Tang, Adv. Funct. Mater., 2018, 28, 1706506 CrossRef; (d) H. Liu, Y. Shen, Y. Yan, C. Zhou, S. Zhang, B. Li, L. Ye and B. Yang, Adv. Funct. Mater., 2019, 29, 1901895 CrossRef.
- (a) Y. Zhang, H. Ma, S. Wang, Z. Li, K. Ye, J. Zhang, Y. Liu, Q. Peng and Y. Wang, J. Phys. Chem. C, 2016, 120, 19759 CrossRef CAS; (b) Y. Zhang, Y. Miao, X. Song, Y. Gao, Z. Zhang, K. Ye and Y. Wang, J. Phys. Chem. Lett., 2017, 8, 4808 CrossRef CAS PubMed; (c) J. Liu, T. Hu, Z. Li, X. Wei, X. Hu, H. Gao, G. Liu, Y. Yi, Y. Yamada-Takamura, C. S. Lee, P. Wang and Y. Wang, J. Phys. Chem. Lett., 2019, 10, 1888 CrossRef CAS PubMed.
- B. Huang, W. Chen, Z. Li, J. Zhang, W. Zhao, Y. Feng, B. Tang and C. S. Lee, Angew. Chem., Int. Ed., 2018, 57, 12473 CrossRef CAS PubMed.
- Y. Dong, B. Xu, J. Zhang, X. Tan, L. Wang, J. Chen, H. Lv, S. Wen, B. Li, L. Ye and B. Zou, Angew. Chem., Int. Ed., 2012, 43(51), 10782–10785 CrossRef PubMed.
- F. Hu, W. Yang, L. Li, J. Miao, S. Gong, C. Ye, X. Gao and C. Yang, Chem. Eng. J., 2023, 464, 142678 CrossRef CAS.
- X. Liu, Q. Liao, J. Yang, Z. Li and Q. Li, Angew. Chem., 2023, 135(24), e202302792 CrossRef.
- J. Zhang, S. Xu, L. Zhang, X. Wang, Y. Bian, S. Tang, R. Zhang, Y. Tao, W. Huang and R. Chen, Adv. Mater., 2022, 34, 2206712 CrossRef CAS PubMed.
- X. Wang, S. Wang, Y. Huang, L. Huang, J. Sun and Z. Lin, Chem.–Asian J., 2023, 18, e202201027 CrossRef CAS PubMed.
- K. Y. Zhang, Q. Yu, H. Wei, S. Liu, Q. Zhao and W. Huang, Chem. Rev., 2018, 118, 1770–1839 CrossRef CAS PubMed.
- S. Wang, J. Wang, Q. Huang, X. Zheng, Z. Yao, S. Xiang, Q. Ling and Z. Lin, ACS Appl. Mater. Interfaces, 2022, 14, 14703–14711 CrossRef CAS PubMed.
- Y. Huang, X. Zheng, Z. Yao, W. Lv, S. Xiang, Q. Ling and Z. Lin, Chem. Eng. J., 2022, 444, 136629 CrossRef CAS.
- S. Feng, Q. Huang, S. Yang, Z. Lin and Q. Ling, Chem. Sci., 2021, 12, 14451–14458 RSC.
- S. Feng, Y. Ma, S. Wang, S. Gao, Q. Huang, H. Zhen, D. Yan, Q. Ling and Z. Lin, Angew. Chem., Int. Ed., 2022, 61, e202116511 CrossRef CAS PubMed.
- H. Sun, Z. Yu, C. Li, M. Zhang, S. Shen, M. Li, M. Liu, Z. Li, D. Wu and L. Zhu, Angew. Chem., 2024, e202413827 Search PubMed.
- R. Zhang, Y. Chen and Y. Liu, Angew. Chem., Int. Ed., 2023, 62(52), e202315749 CrossRef CAS PubMed.
- (a) S. Wang, D. Wu, S. Yang, Z. Lin and Q. Ling, Mater. Chem. Front., 2020, 4, 1198–1205 RSC; (b) F. Nie, K. Wang and D. Yan, Nat. Commun., 2023, 14, 1654 CrossRef CAS PubMed; (c) J. Wang, Y. Fang, C. Li, L. Niu, W. Fang, G. Cui and Q. Yang, Angew. Chem., Int. Ed., 2020, 59, 10032–10036 CrossRef CAS PubMed; (d) X. Yang, S. Wang, J. Wu, W. Hu, L. Huang, Q. Ling and Z. Lin, Adv. Opt. Mater., 2023, 12, 2301661 CrossRef.
- (a) G. N. Lewis and M. Kasha, J. Am. Chem. Soc., 1944, 66, 2100–2116 CrossRef CAS; (b) N. Gan, X. Zou, Y. Zhang, L. Gu and Z. An, Appl. Phys. Rev., 2023, 10, 021313 CrossRef CAS.
- (a) M. Wang, F. Li, Y. Lei, F. Xiao, M. Liu, S. Liu, X. Huang, H. Wu and Q. Zhao, Chem. Eng. J., 2022, 429, 132288 CrossRef CAS; (b) W. Li, Q. Huang, Z. Mao, X. He, D. Ma, J. Zhao, J. W. Y. Lam, Y. Zhang, B. Z. Tang and Z. Chi, Nat. Commun., 2022, 13, 7423 CrossRef CAS PubMed.
- (a) S. Kuila and S. J. George, Angew. Chem., Int. Ed., 2020, 59, 9393–9397 CrossRef CAS PubMed; (b) X. Zheng, Q. Han, Q. Lin, C. Li, J. Jiang, Q. Guo, X. Ye, W. Z. Yuan, Y. Liu and X. Tao, Mater. Horiz., 2023, 10, 197–208 RSC.
- (a) X. Lin, C. Xu, Y. Qiu and X. Ma, Ind. Eng. Chem. Res., 2023, 62, 13053–13060 CrossRef CAS; (b) X. Lin, J. Wang, B. Ding, X. Ma and H. Tian, Angew. Chem., Int. Ed., 2021, 60, 3459–3463 CrossRef CAS PubMed; (c) D. Li, Y. Yang, J. Yang, M. Fang, B. Z. Tang and Z. Li, Nat. Commun., 2022, 13, 347 CrossRef CAS PubMed.
- (a) P. Fielding and R. C. Jarnagin, J. Chem. Phys., 1967, 47, 247 CrossRef CAS; (b) L. S. Kaanumalle, C. L. Gibb, B. C. Gibb and V. Ramamurthy, J. Am. Chem. Soc., 2005, 127, 3674 CrossRef CAS PubMed; (c) Z. Zhang, Y. Zhang, D. Yao, H. Bi, I. Javed, Y. Fan, H. Zhang and Y. Wang, Cryst. Growth Des., 2009, 9, 5069 CrossRef CAS; (d) S. Sekiguchi, K. Kondo, Y. Sei, M. Akita and M. Yoshizawa, Angew. Chem., Int. Ed., 2016, 55, 6906 CrossRef CAS PubMed; (e) S. Hisamatsu, H. Masu, M. Takahashi, K. Kishikawa and S. Kohmoto, Cryst. Growth Des., 2015, 15, 2291 CrossRef CAS; (f) H. D. Becker, K. Sandros, B. W. Skelton and A. H. White, J. Phys. Chem., 1981, 85, 2930 CrossRef CAS; (g) H. Qi, C. Zhang, Z. Huang, L. Wang, W. Wang and A. J. Bard, J. Am. Chem. Soc., 2016, 138, 1947 CrossRef CAS PubMed.
- N. Garg, A. Sarkar and B. Sundararaju, Coord. Chem. Rev., 2021, 433, 213728 CrossRef CAS.
- A. J. Paine and A. D. Dayan, Hum. Exp. Toxicol., 2001, 20, 563 CrossRef CAS PubMed.
- (a) J. V. D. Broek, S. Abegg, S. E. Pratsinis and A. T. Güntner, Nat. Commun., 2019, 10, 4220 CrossRef PubMed; (b) A. Mirzaei, S. G. Leonardi and G. Neri, Ceram. Int., 2016, 42, 15119–15141 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2419510, 2419513 and 2419515. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5sc01503k |
| This journal is © The Royal Society of Chemistry 2025 |