
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licenceπ-Expansion as gateway to viologen-based pimers†
Geoffrey
Groslambert‡
 a,
Vivien
Andrieux‡
a,
Malo
Duquesnoy
a,
Raphaël
Rullan
a,
Lhoussain
Khrouz
a,
Sandrine
Denis-Quanquin
b,
Stephan N.
Steinmann
b,
Tangui
Le Bahers
*ac,
Floris
Chevallier
*a,
Denis
Frath
*b and
Christophe
Bucher
*b
a,
Vivien
Andrieux‡
a,
Malo
Duquesnoy
a,
Raphaël
Rullan
a,
Lhoussain
Khrouz
a,
Sandrine
Denis-Quanquin
b,
Stephan N.
Steinmann
b,
Tangui
Le Bahers
*ac,
Floris
Chevallier
*a,
Denis
Frath
*b and
Christophe
Bucher
*b
aENS de Lyon, CNRS, LCH, UMR 5182, 69342, Lyon Cedex 07, France. E-mail: tangui.le_bahers@ens-lyon.fr; floris.chevallier@ens-lyon.fr
bCNRS, ENS de Lyon, LCH, UMR 5182, 69342, Lyon Cedex 07, France. E-mail: denis.frath@ens-lyon.fr; christophe.bucher@ens-lyon.fr
cInstitut Universitaire de France, 5 rue Descartes, 75005 Paris, France
First published on 16th April 2025
Abstract
The present article reports on a new and efficient synthetic strategy towards tetracene-bipyridiniums. On the basis of extensive experimental analyses supported by DFT simulations, we report the first observation of a mixed valence complex formed in solution under standard conditions from an unconstrained bis-viologene derivative.
The π-dimerization or pimerization capabilities of π-conjugated organic compounds have been the subject of increasing interests over the last decade. π-Dimers and pimers are commonly accepted trivial denominations designating sandwich-like, multicenter-bonded, dimeric entities featuring sub-van der Waals intradimer separation distances.1 Even though they are fundamentally different, π-dimers and pimers have often been mixed up and these denominations used inaccurately.2–4 In π-dimers, the non-covalent “chemical bonding” arises from the orbital overlaps occurring between two semi-occupied molecular orbitals (SOMOs) centered on two identical radical ions or neutral species, yielding a diamagnetic complex.4–6 On the other hand pimers, which are in essence the mixed-valence analogues of π-dimers, involve orbital overlaps between an organic π-conjugated system with its own radical, yielding a charged paramagnetic complex.1,2 Kochi and coworkers have greatly contributed to the body of knowledge in this field by demonstrating that pimers are in fact particular cases of charge-transfer complexes wherein both species involved behave as the Donor and Acceptor.7 Both pimer and π-dimer also exhibit distinct spectroscopic signatures, including diagnostic absorption bands observed in the near-infrared (NIR, 1200–2500 nm for pimer, 700–1200 nm for π-dimer) and different electron spin resonance (ESR) signals.7 As a matter of fact, pimers and π-dimers complexes have so far been mainly observed with naphtalenediimides (NDIs),8–10 phenothiazine,11 tetrathiafulvalenes (TTFs)12–15 or oligothiophenes16–18 derivatives. Conversely, viologen-based derivatives form only π dimers under standard conditions.19–23 It is also worth noting that the formation of π dimers has been widely used to date to trigger various molecular and supramolecular metamorphic phenomena24–30 finding applications in magnetic switching4,31–33 or for the development of responsive soft materials26,34 and chiral switches.21,35
As a matter of fact, the rare pimers of viologens mentioned so far in the literature have been observed in the solid state36 or in highly constrained structures.37,38 Failure to observe such species in solution, even with highly preorganized covalently-linked bis-viologen derivatives such as 14+, may arise from the fact that viologens carry two positive charges and adopt a planar conformation only in their cation radical state. Unlike other π-conjugated molecules known to form stable pimers, the association between a twisted bipyridinium dication (V2+) and a planar radical cation (V+˙) is thus most likely disfavored by a combination of steric and electrostatic repulsions (Scheme 1).
 | ||
| Scheme 1 Intramolecular dimerization of standard vs. π-expanded bipyridiums. | ||
We devised a new and efficient synthetic strategy towards tetracene-bipyridiniums (TCBs) and report the first observation of a mixed valence complex formed in solution under standard conditions from an unconstrained bis-viologen derivative. Our first attempts to access the targeted TCB-derivatives consisted of reproducing a procedure described in the literature proceeding by direct photocyclization of the 3,5-diphenyl-4,4′-bipyridine precursor (3).39,40 However, we encountered difficulties reproducing this protocol at a larger scale, which led us to develop an alternative route which is presented in Scheme 2.
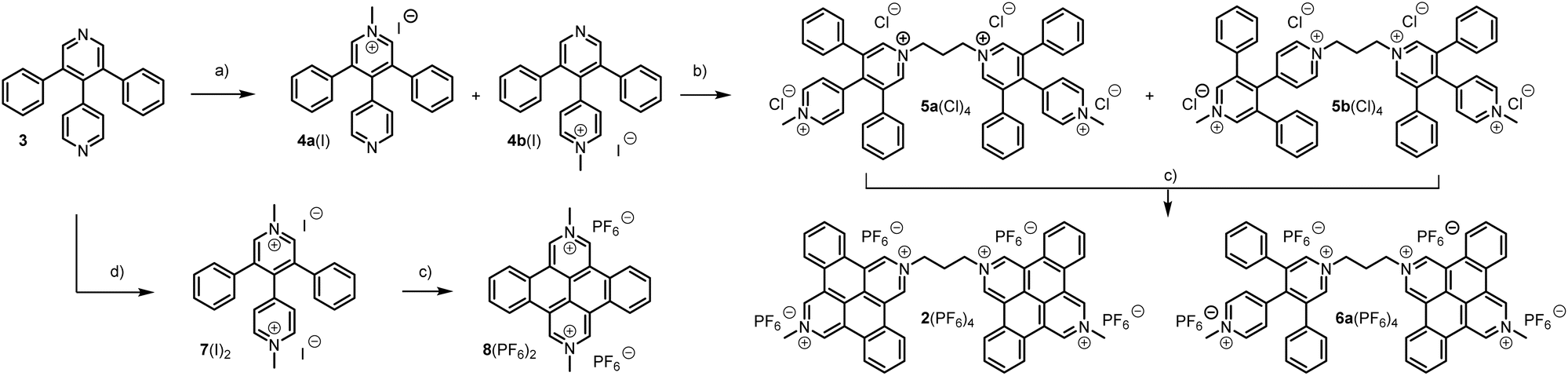 | ||
| Scheme 2 Synthesis of TCBs. (a) MeI (2 eq.), CH2Cl2, RT, 7 days, 75%. (b) (i) 1,3-Diiodopropane, MeCN, refl., 3 days. (ii) H2O, KPF6. (iii) MeCN, TBACl, 74%. (c) (i) hν (350 nm), H2O, 20 h. (ii) H2O, KPF6, 19%. (d) MeI (20 eq.), CH2Cl2, 35 °C, 24 h, 74%. | ||
This strategy relies on the idea that both the solubility and the reactivity of the species involved in the photochemical reaction would be drastically enhanced by a prior quaternization of the pyridine rings. The key bipyridyl precursor 3 was obtained in one step by a modified Hantzsch reaction involving 4-pyridinecarboxaldehyde and two equivalents of phenylacetaldehyde as reactants. Reaction of 3 with 2 equivalents of iodomethane at RT in DCM was then found to afford a mixture of the mono-quaternized isomers 4a+ and 4b+ obtained in a 25/75 ratio, respectively. This regioselectivity can be easily explained by the attractive effect of the phenyl substituents making the neighboring nitrogen atom less reactive to electrophiles. Conversely, carrying out the same reaction with a large excess of methyl iodide at 40 °C led to a quantitative precipitation of the di-quaternized product 7(I)2, which could be isolated by simple filtration. The mixture of isomers 4a+ and 4b+ obtained at RT was subsequently reacted with 1,3-diiodopropane in hot acetonitrile, yielding a precipitate composed of a mixture of the regioisomers 5a4+ and 5b4+ obtained in an 80/20 ratio. The two isomers were finally separated by fractional recrystallization and fully characterized by NMR and electrochemical methods (see ESI section†).
The iodide salts of 5a4+, 5b4+ and 74+ were then subjected to light irradiation. Our initial studies showed that irradiation at 365 nm of an aqueous solution of 7(I)2 results in the precipitation of the targeted tetracene 8(I)2 obtained with 70% yield after washing. Similar experiments carried out from the crude mixture of isomers 5a(I)4 and 5b(I)4 (80/20) conversely led to the precipitation of one single product identified as the mono cyclized isomer 6a(I)4 (the other isomer 6b(I)4 was not observed, see ESI† for details) which could be isolated in 19% yield after 20 hours of irradiation.§ We therefore failed to obtain the targeted cyclization product 24+ under these conditions due to the insolubility of the intermediate product 6(I)4. This problem was eventually circumvented by working with the chloride salts 5a(Cl)4 and 5b(Cl)4, easily obtained by anion metathesis from the corresponding iodide derivates. No precipitation was indeed observed upon irradiation of a 1 mg mL−1 aqueous solution of 5a(Cl)4 and 5b(Cl)4, (80/20 mixture) and a careful monitoring of the reaction allowed to achieve a full conversion of the mixture to the targeted bis tetracene-bipyridinium 24+. Pure samples of 2(PF6)4, showing good solubility in polar solvents like DMSO or DMF, could eventually be obtained by ion metathesis and purification by chromatography.
Variable-temperature NMR studies were carried out with 82+ and 24+ in DMSO to assess the ability of these conjugated molecules to interact in solution (ESI Fig. S2†). These studies involved monitoring the shift of the 1H NMR signals when increasing the temperature of samples at 1 mM in bipyridinium. No signs of aggregation were observed for monomer 82+ under these conditions. Conversely, a significant downfield shift of 0.05 ppm observed for inner core aromatic signals of 24+ when increasing the temperature from 298 to 338 K demonstrate that the two bipyridinium tetracene units interact with each other despite the repulsive electrostatic forces at play.
The electrochemical properties of the π-expanded derivative 2(PF6)4 were then studied by cyclic voltammetry (CV) in DMF and compared with those of known reference compounds, including the 1,1′-dimethyl-4,4′-bipyridinium derivative 9(PF6)2, the propyl linked bis viologen 1(PF6)4 as well as the tetracene-based monomer 8(PF6)2. Selected CV curves and key figures obtained with these compounds are shown in Table 1 and Fig. 1 respectively.
| (E1,c)1 (n, ΔEp) V2+/V+˙ | (E1,c)2 (n, ΔEp) V2+/V+˙ | E 2,c (n, ΔEp) V+˙/V0 | ΔEc | |
|---|---|---|---|---|
| 9 2+ | −0.859 (1, 64) | na | −1.234 (1, 54) | 375 |
| 1 4+ | −0.739 (2, 32) | na | −1.253 (2, 34) | 514 |
| 8 2+ | −0.878 (1, 54) | na | −1.356 (1, 56) | 478 |
| 2 4+ | −0.682 (1, na) | −0.794 (1, na) | −1.439 (2, 44) | 757 |
 | ||
| Fig. 1 CV curves (DMF, nBu4NPF6 0.1 M, VC Ø = 3 mm, E vs. Ag+/Ag, 100 mV s−1) of (A) 92+ (1 mM), (B) 14+ (0.5 mM), (C) 84+ (1 mM) and (D) 24+ (0.5 mM). | ||
Comparing the data collected with the monomers 82+ and 92+ reveals that both compounds are reduced at similar potential but that 8+˙ is significantly more stable than the non-conjugated analog 9+˙, as shown by the 100 mV difference observed between the ΔEc values measured for the two compounds.¶ In this case, this large increase in stability is attributed to an improved delocalization of the radical over the tetracene bipyridinium skeleton. The bis-bipyridiniums 14+ was selected as a second reference in this study for the known ability of the doubly reduced species 12(+˙) to undergo an intramolecular π-dimerization yielding the diamagnetic π-dimer [1Dim]2+ (eqn (5) in Fig. 2 with X = 1). One key feature of the mechanism involved is the instability of the intermediate 13+˙ and its instantaneous disproportionation into 14+ and 12(+˙) (eqn (3) in Fig. 2), which can be easily revealed on the CV curves by the observation of a single two-electron reduction wave at E1/2 = −0.739 V displaying an unusually short peak to peak potential shift of about 32 mV. Another diagnostic feature revealing the stabilization of the doubly reduced species as the π-dimer [1Dim]2+ is the large increase in the ΔEc values reaching more than 500 mV, far above what is measured for the monomer 92+.
 | ||
| Fig. 2 Representation of the stepwise viologen/TCB-centered reductions and the consecutive pimerization, dimerization and disproportionation equilibria. | ||
The CV data displayed in Fig. 1B thus demonstrate that the π-dimer [1Dim]2+ is the only complex formed in solution and that neither the intermediate 13+˙ nor the corresponding pimer (eqn (4) in Fig. 2) are observed under these conditions. As can be seen in Fig. 1D a very different behavior was observed with the tetracene 24+, showing two successive one-electron reduction waves at −682 and −794 mV (Fig. 1D) attributed to the stepwise formation of 23+˙ and 22(+˙) (eqn (1) and (2) in Fig. 2 with X = 2). Such a separation is conversely not observed on the last wave attributed to an undifferentiated two-electron reduction yielding the neutral species 20. Other remarkable features seen on the CV curve include the fact that 24+ is significantly easier to reduce than 14+, and that the doubly reduced species 22(+˙) is also harder to reduce than 12(+˙).
Altogether, these data thus demonstrate that the diamagnetic π-dimer [2Dim]2+ produced in situ after the second electron transfer at −794 mV is thermodynamically more stable than [1Dim]2+, its stability domain being larger, and most importantly that a stable “mixed-valence” pimer intermediate is formed along the way after the first one electron reduction at −682 mV (eqn (4) in Fig. 2 with X = 2). These data suggest that the monoreduced intermediate species 23+˙ is not instantaneously consumed by disproportionation due to its stabilization by folding to form the intramolecular pimer [2Pim]3+˙. Quantum chemical calculations were then undertaken to understand the stabilization of the pimer intermediate in the extended viologen series. The energy associated with folding (intramolecular dimerization) of the propyl-linked derivatives were computed for the fully oxidized species (14+ and 24+), the mixed valence intermediates (13+˙ and 23+˙) and for the fully reduced species (12(+˙) and 22(+˙)) (Table 2). All computational details, including the unusual calculation protocol used to address the strong triplet instability observed for 12(+˙) and 22(+˙) are described in the ESI section.† These data reveal that (i) the folded forms of 14+ and 13+˙ show weak stabilizations of similar amplitude and (ii) only the doubly reduced species 12(+˙) experiences a noticeable stable folded form as a π-dimer [1Dim]2+; (iii) increasing the π-system leads to greater stability of all the folded forms whatever the oxidation state; (iv) comparison of the data calculated for derivatives 1 and 2 reveals that the largest stability gain of more than 50 kJ mol−1, is observed for the intermediate pimer complex [2Pim]3+˙.
| X4+ | X3+˙ | X2(+˙) | |
|---|---|---|---|
| X = 1 | −12 | −19 | −49 |
| X = 2 | −57 | −72 | −92 |
By estimating the dispersion and electrostatic contributions to the dimerization energy for all systems (see ESI†), we demonstrated that the large stabilization observed with the π-expanded viologens is at least partly due to greater delocalization of the positive charge, leading to a reduction in the electrostatic repulsion between the two viologen subunits. It is interesting to note that the π-extension has only a minor effect on the “covalence” between the two radicals in [XDim]2+, as shown by the HOMO orbitals that is very similar for X = 1 and X = 2 (see ESI†). TD-DFT calculations were also performed to predict the spectroscopic signature of the yet unknown pimer intermediate [2Pim]3+˙. In agreement with the experimental data discussed in the following section, we observed that all calculated electronic transitions were more red-shifted with 2 than with 1. This proved to be true for the diagnostic π-dimer band, computed at λmax = 660 nm for [1Dim]2+ and at 756 nm for [2Dim]2+, and even more for the pimers, with a computed lowest transition energy at 1480 nm for [1Pim]3+˙ and at 2130 nm for [2Pim]3+˙ (see ESI† for details). Since TD-DFT has not been well assessed for the simulation of electronic transitions for such radicals, the calculated values of absorption maxima should be used with caution. Still, the trend indicates that the spectrum of [2Pim]3+˙ should exhibit a characteristic absorption band significantly more red-shifted than that of the π-dimer [2Dim]2+. Two ESR spectra recorded during the electrochemical reduction of 24+ are shown in Fig. 3. The spectrum shown in Fig. 3A, recorded after addition of 2 electrons per molecule, is attributed to the paramagnetic open form 22(+˙) in equilibrium with the diamagnetic folded π-dimer [2Dim]2+ (eqn (5) in Fig. 2). In line with this attribution, we found its intensity to decrease with cooling (ESI Fig. S15†) and its multiplicity to result from superhyperfine couplings with 2 14N and 1H nuclear spins, as observed under the same conditions for the reference compound 8+˙ (ESI Fig. S11†). The second spectrum recorded in the early stages of the reduction (Fig. 3B, for Q < 1e− per molecule), shows a broad envelope attributed to exchange processes between radical and non-radical species,7,41 to which is added another signal showing numerous hyperfine couplings much smaller than those observed in Fig. 3A (see ESI Fig. S12† for details), which is in perfect agreement with what is expected for the mixed-valence pimer [2Pim]3+˙, presenting one single electron delocalized over two viologen subunits.
 | ||
| Fig. 3 ESR spectra recorded at RT after electrochemical reduction of 2(PF6)4 in DMF, nBu4NPF6 0.1 M at Eapp = −1 V, (A) spectrum recorded after addition of 2e− per molecule, (B) spectrum recorded after addition of 1e− per molecule. | ||
Spectroelectrochemical studies were carried out to further characterize the intermediate [2Pim]3+˙. Formation of that species was further confirmed by absorption spectroscopy measurement, comparing the signatures of species generated in situ by reduction of 14+, 24+, 92+ and 82+ (ESI section†). As can be seen in Fig. 4, the one-electron reduction of 82+ led to the development of one single set of signals (424, 453, 535, 627 and 691 nm) attributed to the free radical 8+˙. The two-electron reduction of 24+ (one electron per viologen) led conversely to a decrease in the intensity of the bands at 399 and 424 nm attributed to 24+, in favor of new signals at 453, 494, 631 and 960 nm attributed to the π-dimer [2Dim]2+. Formation of the intermediate complex [2Pim]3+˙ is finally revealed on the green solid line curve, recorded after addition of 1e− per molecule, by characteristics that correspond to neither 8+˙, 24+ nor [2Dim]2+, the main difference being the absence of absorption at 533 nm and the observation of an intense signal at 676 nm. The observation of the diagnostic pimer band, could be achieved when the experiment was carried out in acetonitrile (ESI Fig. S8†). In the NIR region above 2000 nm, the absorption increased in the first stages of the reduction, which carried out further completely silences this band and develops a π-dimer band at 973 nm.
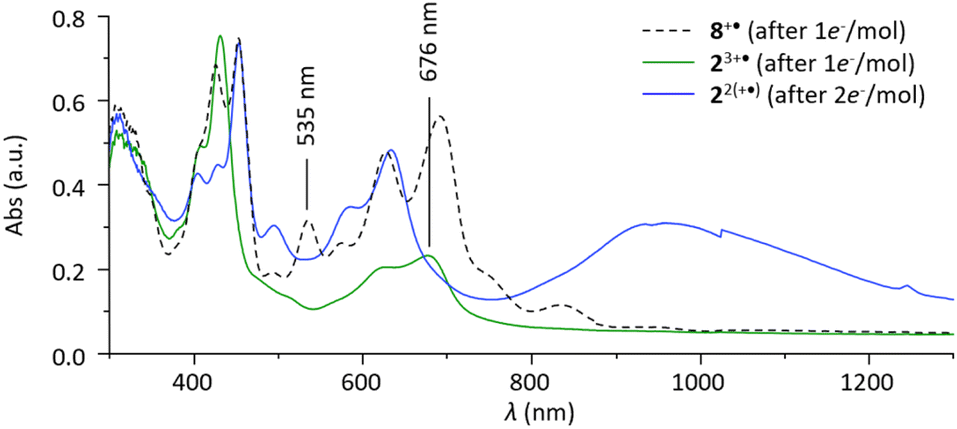 | ||
| Fig. 4 UV/Vis/NIR (l = 1 mm) spectra of the reduced species of 82+ (1 mM) and 24+ (0.5 mM) in DMF (nBu4NPF6 0.1 M). | ||
Conclusions
We have developed a new and efficient synthetic strategy towards π-expanded-bipyridiniums. On the ground of extensive experimental analyses supported by DFT simulations, we report the first observation of a mixed valence complex formed in solution under standard conditions from an unconstrained bis-viologene derivative. As a general statement, mixed-valence species have proved essential for the development of organic electronics.42–46 We therefore believe that extending the π system to 4,4′-bipyridiniums derivatives is an extremely promising strategy, not only for stabilizing species of interest for electronics, but also for conferring emissive properties that open up promising prospects in the field of electrofluorochromism.47Data availability
All the experimental data related to this work can be found in the ESI.†Author contributions
GG and VA contributed equally to this work. GG, VA and MD performed synthetic and analytical experiments. RR and TLB performed DFT calculations. SDQ performed VT-NMR measurements. LK performed ESR measurements. FC, DF and CB conceptualized and supervised the project. GG wrote the original draft. SNS, TLB, FC, DF and CB reviewed the article.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge support from the CBPsmn (PSMN, Pôle Scientifique de Modélisation Numérique) of the ENS de Lyon for the computing resources. The platform operates the SIDUS solution.48 V. A. thanks ENSL for the CDSN PhD Grant. The authors would also like to thank Dr O. Piva for giving us access to its photochemical reactor. The authors thank the ANR for financial support (ANR-21-CE06-0020-01, ANR-22-CE07-0025 and ANR-24-CE06-1543).Notes and references
- C. Kahlfuss, E. Saint-Aman and C. Bucher, in Organic Redox Systems, 2016, pp. 39–88 Search PubMed.
- J. Joseph, M. Berville, J. Wytko, J. Weiss and H.-P. Jacquot de Rouville, Chem.–Eur. J., 2024, e202403115 Search PubMed.
- M. R. Geraskina, A. S. Dutton, M. J. Juetten, S. A. Wood and A. H. Winter, Angew. Chem., Int. Ed., 2017, 56, 9435–9439 CrossRef CAS PubMed.
- A. T. Buck, J. T. Paletta, S. A. Khindurangala, C. L. Beck and A. H. Winter, J. Am. Chem. Soc., 2013, 135, 10594–10597 CrossRef CAS PubMed.
- K. E. Preuss, Polyhedron, 2014, 79, 1–15 CrossRef CAS.
- M. Kertesz, Chem.–Eur. J., 2019, 25, 400–416 CrossRef CAS PubMed.
- S. V. Rosokha and J. K. Kochi, Acc. Chem. Res., 2008, 41, 641–653 CrossRef CAS PubMed.
- A. Takai, T. Yasuda, T. Ishizuka, T. Kojima and M. Takeuchi, Angew. Chem., Int. Ed., 2013, 52, 9167–9171 CrossRef CAS PubMed.
- Y. Wu, M. Frasconi, D. M. Gardner, P. R. McGonigal, S. T. Schneebeli, M. R. Wasielewski and J. F. Stoddart, Angew. Chem., Int. Ed., 2014, 53, 9476–9481 CrossRef CAS PubMed.
- A. H. G. David, M. Roger, O. Alévêque, H. Melnychenko, L. Le Bras, M. Allain, A. Gapin, D. Canevet, O. Ségut, E. Levillain and A. Goujon, Angew. Chem., Int. Ed., 2024, e202413616 Search PubMed.
- D. Sun, S. V. Rosokha and J. K. Kochi, J. Am. Chem. Soc., 2004, 126, 1388–1401 CrossRef CAS PubMed.
- C. Jia, D. Zhang, X. Guo, S. Wan, W. Xu and D. Zhu, Synthesis, 2002, 2002, 2177–2182 Search PubMed.
- K. Nakamura, T. Hashimoto, T. Shirahata, S. Hino, M. Hasegawa, Y. Mazaki and Y. Misaki, Chem. Lett., 2011, 40, 883–885 CrossRef CAS.
- K. Nakamura, T. Takashima, T. Shirahata, S. Hino, M. Hasegawa, Y. Mazaki and Y. Misaki, Org. Lett., 2011, 13, 3122–3125 CrossRef CAS PubMed.
- Š. Lipnická, M. Bělohradský, V. Kolivoška, L. Pospíšil, M. Hromadová, R. Pohl, J. V. Chocholoušová, J. Vacek, J. Fiedler, I. G. Stará and I. Starý, Chem.–Eur. J., 2013, 19, 6108–6121 CrossRef PubMed.
- R. Takita, C. Song and T. M. Swager, Org. Lett., 2008, 10, 5003–5005 CrossRef CAS PubMed.
- J. Casado, K. Takimiya, T. Otsubo, F. J. Ramírez, J. J. Quirante, R. P. Ortiz, S. R. González, M. M. Oliva and J. T. López Navarrete, J. Am. Chem. Soc., 2008, 130, 14028–14029 CrossRef CAS PubMed.
- T. Satou, T. Sakai, T. Kaikawa, K. Takimiya, T. Otsubo and Y. Aso, Org. Lett., 2004, 6, 997–1000 CrossRef CAS PubMed.
- S. J. Atherton, K. Tsukahara and R. G. Wilkins, J. Am. Chem. Soc., 1986, 108, 3380–3385 CrossRef CAS.
- S.-I. Imabayashi, N. Kitamura, S. Tazuke and K. Tokuda, J. Electroanal. Chem., 1988, 243, 143–160 CrossRef CAS.
- C. Gao, S. Silvi, X. Ma, H. Tian, A. Credi and M. Venturi, Chem.–Eur. J., 2012, 18, 16911–16921 CrossRef CAS PubMed.
- P. Dalvand, K. Nchimi Nono, D. Shetty, F. Benyettou, Z. Asfari, C. Platas-Iglesias, M. A. Olson, A. Trabolsi and M. Elhabiri, RSC Adv., 2021, 11, 29543–29554 RSC.
- K. Cai, L. Zhang, R. D. Astumian and J. F. Stoddart, Nat. Rev. Chem., 2021, 5, 447–465 CrossRef CAS PubMed.
- S. Chowdhury, Y. Nassar, L. Guy, D. Frath, F. Chevallier, E. Dumont, A. P. Ramos, G. J.-F. Demets and C. Bucher, Electrochim. Acta, 2019, 316, 79–92 CrossRef CAS.
- C. Kahlfuss, S. Denis-Quanquin, N. Calin, E. Dumont, M. Garavelli, G. Royal, S. Cobo, E. Saint-Aman and C. Bucher, J. Am. Chem. Soc., 2016, 138, 15234–15242 CrossRef CAS PubMed.
- C. Kahlfuss, T. Gibaud, S. Denis-Quanquin, S. Chowdhury, G. Royal, F. Chevallier, E. Saint-Aman and C. Bucher, Chem.–Eur. J., 2018, 24, 13009–13019 CrossRef CAS PubMed.
- C. Kahlfuss, S. Chowdhury, A. F. Carreira, R. Grüber, E. Dumont, D. Frath, F. Chevallier, E. Saint-Aman and C. Bucher, Inorg. Chem., 2021, 60, 3543–3555 CrossRef CAS PubMed.
- C. K. Lee, C. Gangadharappa, A. C. Fahrenbach and D. J. Kim, Adv. Mater., 2024, 36, 2408271 CrossRef CAS PubMed.
- A. Iordache, M. Oltean, A. Milet, F. Thomas, B. Baptiste, E. Saint-Aman and C. Bucher, J. Am. Chem. Soc., 2012, 134, 2653–2671 CrossRef CAS PubMed.
- A. Iordache, M. Retegan, F. Thomas, G. Royal, E. Saint-Aman and C. Bucher, Chem.–Eur. J., 2012, 18, 7648–7653 CrossRef CAS PubMed.
- M. R. Geraskina, A. T. Buck and A. H. Winter, J. Org. Chem., 2014, 79, 7723–7727 CrossRef CAS PubMed.
- M. J. Juetten, A. T. Buck and A. H. Winter, Chem. Commun., 2015, 51, 5516–5519 RSC.
- S. Al Shehimy, O. Baydoun, S. Denis-Quanquin, J.-C. Mulatier, L. Khrouz, D. Frath, É. Dumont, M. Murugesu, F. Chevallier and C. Bucher, J. Am. Chem. Soc., 2022, 144, 17955–17965 CrossRef CAS PubMed.
- M. Marchini, M. Baroncini, G. Bergamini, P. Ceroni, M. D'Angelantonio, P. Franchi, M. Lucarini, F. Negri, T. Szreder and M. Venturi, Chem.–Eur. J., 2017, 23, 6380–6390 CrossRef CAS PubMed.
- C. Gao, S. Silvi, X. Ma, H. Tian, M. Venturi and A. Credi, Chem. Commun., 2012, 48, 7577 RSC.
- N. Leblanc, N. Mercier, O. Toma, A. H. Kassiba, L. Zorina, P. Auban-Senzier and C. Pasquier, Chem. Commun., 2013, 49, 10272 RSC.
- Y. Jiao, H. Mao, Y. Qiu, G. Wu, H. Chen, L. Zhang, H. Han, X. Li, X. Zhao, C. Tang, X.-Y. Chen, Y. Feng, C. L. Stern, M. R. Wasielewski and J. F. Stoddart, J. Am. Chem. Soc., 2022, 144, 23168–23178 CrossRef CAS PubMed.
- J. C. Barnes, A. C. Fahrenbach, D. Cao, S. M. Dyar, M. Frasconi, M. A. Giesener, D. Benítez, E. Tkatchouk, O. Chernyashevskyy, W. H. Shin, H. Li, S. Sampath, C. L. Stern, A. A. Sarjeant, K. J. Hartlieb, Z. Liu, R. Carmieli, Y. Y. Botros, J. W. Choi, A. M. Z. Slawin, J. B. Ketterson, M. R. Wasielewski, W. A. Goddard and J. F. Stoddart, Science, 2013, 339, 429–433 CrossRef CAS PubMed.
- Y. Yang, D. Liu, M. Song, D. Shi, B. Liu, K. Cheng, Y. Lu, H. Liu, M. Yang, W. Wang, J. Li and J. Wei, Chem.–Eur. J., 2017, 23, 7409–7413 CrossRef CAS PubMed.
- C. Bao, N. Yan, T. Cao, X. Zhang, Y. Zhu, Y. Zhang, M. Maximov, S. Xiong and G. He, Dyes Pigm., 2024, 227, 112136 CrossRef CAS.
- R. L. Ward and S. I. Weissman, J. Am. Chem. Soc., 1957, 79, 2086–2090 CrossRef CAS.
- J. Hankache and O. S. Wenger, Chem. Rev., 2011, 111, 5138–5178 CrossRef CAS PubMed.
- A. Jain and S. J. George, Mater. Today, 2015, 18, 206–214 CrossRef CAS.
- F. Xing, S. Li, L. Chen, J.-S. Dang and X. He, ACS Nano, 2023, 17, 21432–21442 CrossRef PubMed.
- M. Kolek, F. Otteny, P. Schmidt, C. Mück-Lichtenfeld, C. Einholz, J. Becking, E. Schleicher, M. Winter, P. Bieker and B. Esser, Energy Environ. Sci., 2017, 10, 2334–2341 RSC.
- A. N. Davis, K. Parui, M. M. Butala and A. M. Evans, Nanoscale, 2024, 16, 10142–10154 RSC.
- P. Audebert and F. Miomandre, Chem. Sci., 2013, 4, 575–584 RSC.
- E. Quemener and M. Corvellec, Linux J., 2013, 235, 3 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5sc01361e |
| ‡ The two first authors contributed equally to this work. |
| § 19% is the best yield obtained over three attempts and the average yield over these attempts is 15%. |
| ¶ It should be noted, however, that this stabilizing effect is not sufficient to stabilize the radical state under oxygen. |
| This journal is © The Royal Society of Chemistry 2025 |