
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIcosahedron kernel defect in Pt1Agx series of bimetallic nanoclusters enhances photocatalytic hydrogen evolution†
Dong
Tan‡
a,
Tengfei
Ding‡
a,
Kaidong
Shen‡
a,
Chang
Xu
 a,
Shan
Jin
b,
Daqiao
Hu
*a,
Song
Sun
*a and
Manzhou
Zhu
*a
a,
Shan
Jin
b,
Daqiao
Hu
*a,
Song
Sun
*a and
Manzhou
Zhu
*a
aDepartment of Chemistry and Centre for Atomic Engineering of Advanced Materials, Key Laboratory of Structure and Functional Regulation of Hybrid Materials of Ministry of Education, Anhui University, Hefei, Anhui 230601, China. E-mail: hudaqiao@ahu.edu.cn; suns@ustc.edu.cn; zmz@ahu.edu.cn
bInstitutes of Physical Science and Information Technology, Anhui University, Hefei, Anhui 230601, China
First published on 25th April 2025
Abstract
Developing high-efficiency photocatalysts for photocatalytic hydrogen production and understanding the structure–property relationships is much desired. In this study, a family of Pt1Agx (x = 9, 11, 13 and 14) nanoclusters (NCs), including a new Pt1Ag11(SR)5(P(Ph-OMe)3)7 NC, were designed and synthesized via ligand engineering (SR = 2,3,5,6-tetrafluorothiophenol, P(Ph-OMe)3 = tris(4-methylphenyl)phosphine). The positive effect of the kernel structural defect on photocatalytic activity was investigated using the photocatalytic water-splitting reaction as a model, and the mechanistic relationship between the defect structure and catalytic activity was clarified. In this series of Pt1Agx bimetallic NCs, the Pt1Ag11 NC, which exhibits a distinctive defect-containing icosahedral kernel structure, displayed excellent catalytic performance for photocatalytic hydrogen evolution, with the hydrogen production rate reaching 1780 μmol g−1 h−1. The experimental results revealed that the superior catalytic activity of Pt1Ag11/g-C3N4 may originate from the formation of Z-scheme heterojunction between Pt1Ag11 and the g-C3N4, facilitating efficient electron–hole separation and charge transfer. Furthermore, density-functional theory (DFT) calculations reveal the critical role of the defect-containing icosahedron-kernel on photocatalytic activity, which is favourable for the formation of the most stable nanocomposites and the easy absorption of H* intermediates on the Ag sites in Pt1Ag11/g-C3N4. This paper provides insights into the effect that the defects have on the mechanism of the photocatalytic hydrogen evolution reaction at the atomic level and promotes the rational design of high-efficiency photocatalysts.
1 Introduction
Photocatalytic water splitting for hydrogen production has attracted extensive interest because it offers a pollution-free and sustainable route to alleviate the energy crisis and tackle environmental issues.1–3 However, achieving high conversion efficiency remains a significant challenge due to the complex processes involved in photocatalytic reactions, including photo absorption, charge separation, transport, dissociation, and recombination.4,5 Therefore, the development of high-efficiency photocatalysts for photocatalytic hydrogen production, and a detailed understanding of structure–property relationships, are desired.6Atomic scale metal nanoclusters (NCs) (1–3 nm in diameter) have shown potential for application in photocatalysis due to their unique atomic stacking modes, optical properties and abundance of catalytic active sites.7,8 Moreover, their ultrasmall size endows metal NCs with discrete molecule-like electronic energy levels, whereby they can be excited as small-band-gap semiconductors to generate electrons and holes.9 To avoid the unfavorable agglomeration of metal NCs caused by high surface energies during the photocatalytic reaction, NC-based composite photocatalysts were fabricated to improve catalytic stability and activity.10,11 In 2013, Negishi et al. reported glutathione-protected Au25 NCs loaded onto BaLa4Ti4O15 as hybrid catalysts for the water-splitting reaction; the hybrid catalysts exhibited photocatalytic activity 2.6 times higher than that of co-catalysts loaded with larger gold nanoparticles (10–30 nm).12 Subsequently, a wide range of metal NCs has been used in photocatalytic hydrogen evolution.13 For instance, Lu et al. synthesized Pt5(GSH)10 NCs and immobilized them on multi-arm CdS nanorods (NRs). The Pt NCs extracted the photoinduced electrons of the CdS NRs and enhanced charge separation, thereby ensuring the Pt5–CdS composite catalysts exhibited an improved photocatalytic H2 production rate of 13.0 mmol g−1 h−1 H2.14 Wang et al. synthesized Ag44 NCs and immobilized them on TiO2. The photocatalytic H2 production of Ag44–TiO2 was 7.4 mmol g−1 h−1. This elevated performance of Ag44–TiO2 was attributed to both the extension of the photoresponse time and the efficient separation and transport of charge carriers.15 Several methods have been proposed to utilize photogenerated carriers effectively for photocatalysis, such as heteroatom doping or Z-scheme heterojunctions,10,16–19 however, the efficient charge transfer and separation of NC-based hybrid catalysts in photocatalytic reactions still poses a challenge.20
Previous work has revealed that a NC structure that contains defects can significantly enhance catalytic performance. For example, Nematulloev et al. reported that defect-containing Cu28 NCs have structural vertex defects which cause a distortion in the framework and lower the symmetry, thus exhibiting more efficient selectivity in C–C cross-coupling reactions compared with Cu29 NCs.21 In addition, Silalahi et al. reported the discovery of hydride-containing 2-electron palladium/copper superatomic alloys, namely PdHCu11 and PdHCu12. The distinctive defects in PdHCu11 expose the central Pd atom, thus providing an active site to catalyze the reaction, resulting in better hydrogen evolution reaction activity than PdHCu12.22 A few reports have been published on the structural defects of metal NCs for catalysis; however, the effect of defects on the formation of heterojunctions or related mechanisms for enhanced photocatalysis have not yet been clarified for composite photocatalysts.
Herein, Pt1Ag11(SR)5(P(Ph-OMe)3)7 (Pt1Ag11 for short, where SR is 2,3,5,6-tetrafluorothiophenol and P(Ph-OMe)3 is tris(4-methylphenyl)phosphine) with three vertex Ag atom defects within the icosahedral kernel was synthesized to construct NCs that exhibit a defect effect. The X-ray crystal structure of Pt1Ag11 was identified and categorized as belonging to the Pt1Agx family (x = 9, 11, 13 and 14) (Scheme 1). Considering this, the relationship between the defect-containing icosahedral kernel and the catalytic activity was established using photocatalytic water splitting as a model. Nanocomposite photocatalysts were prepared by loading four Pt1Agx NCs onto two-dimensional graphite-like carbon nitride (g-C3N4). g-C3N4 has a sheet-like structure with a suitable bandgap, is easily functionalized, and exhibits excellent thermal stability, and chemical corrosion resistance.23,24 In this series of Pt1Agx bimetallic NCs, the Pt1Ag11 NC, which exhibits a distinctive defect-containing icosahedral kernel structure displayed excellent catalytic performance for photocatalytic hydrogen evolution, with the hydrogen production rate reaching 1780 μmol g−1 h−1. The superior catalytic activity of Pt1Ag11/g-C3N4 may originate from the formation of a Z-scheme heterojunction between the two materials, facilitating the extension of the photoresponse time whilst facilitating more efficient electron–hole separation and charge transfer. Theoretical calculations further demonstrated that defects within the icosahedral kernel facilitated electron transport and separation. This paper offers a detailed understanding of how defects influence the photocatalytic hydrogen evolution reaction at the atomic level, facilitating the rational design of efficient photocatalysts.
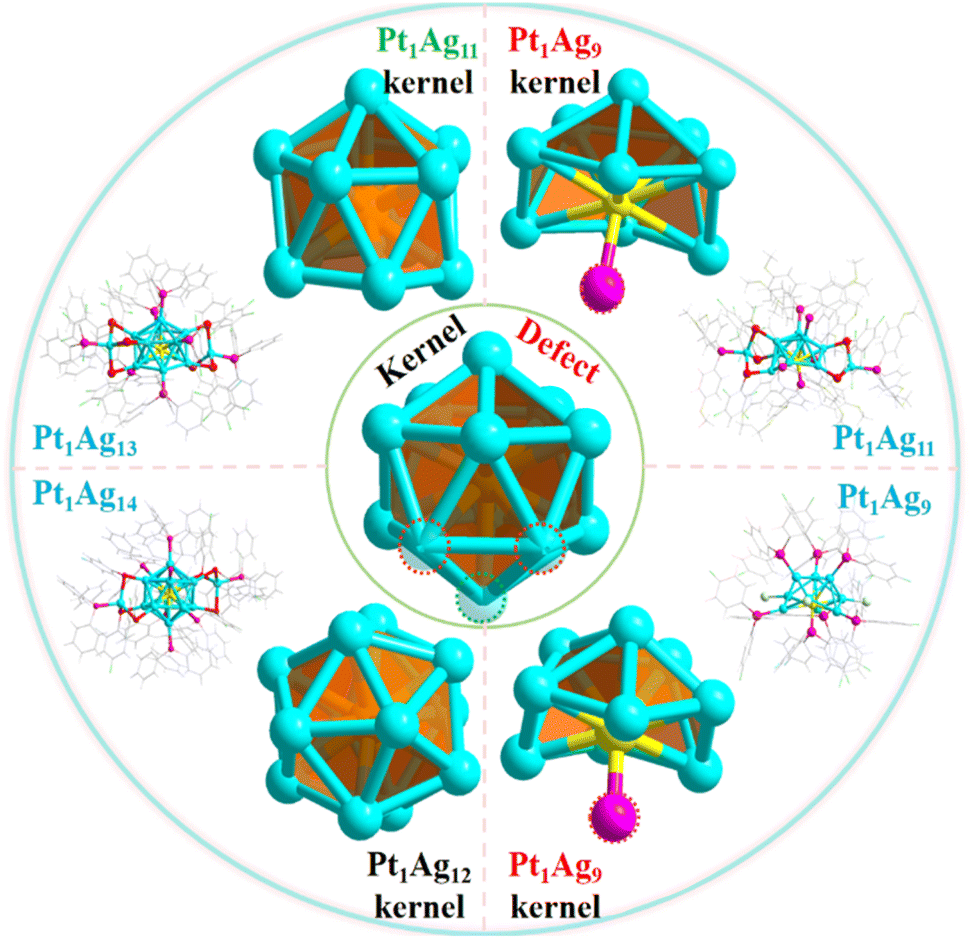 | ||
| Scheme 1 Structure of the Pt1Agx series of nanoclusters (including the kernel and the whole structure). Color code: Pt-yellow, Ag-turquoise, S-red, P-pink, F-bright green, Cl-green, O-orange, C-grey. | ||
2 Experimental section
2.1 Chemicals
Most reagents were purchased from Sigma-Aldrich and used without further purification, including hexachloroplatinic (IV) acid (H2PtCl6·6H2O, 99.99%, metals basis), potassium tetrachloroplatinate (K2PtCl4, 99.9%, metals basis), silver nitrate (AgNO3), 2,3,5,6-tetrafluorothiophenol (SR, C6H2F4S), pentafluorobenzenethiol (PFBT, C6HF5S), 2-chloro-4-fluorobenzenethiol (C6H4FClS), sodium borohydride (NaBH4), tris(4-fluorophenyl)phosphine (P(Ph-F)3), tris(4-methylphenyl)phosphine (P(Ph-OMe)3) and triphenylphosphine (PPh3). 5,5-Dimethyl-1-pyrroline-1-oxide (DMPO), tetraphenyl phosphonium bromide (PPh4Br, 98%), methylene chloride (CH2Cl2, HPLC), methanol (CH3OH, HPLC) and n-hexane (HeX, HPLC grade) were purchased from Energy Chemical.2.2 Synthesis of Pt1Ag11(SR)5(P(Ph-OMe)3)7
40 mg of AgNO3 and H2PtCl6·6H2O (12 mg, 0.0225 mmol) were dissolved into 10 mL of methanol in a 50 mL round-bottomed flask. The solution was stirred vigorously at room temperature for 10 min. The solution immediately turned brown. Subsequently, 40 μL of 2,3,5,6-tetrafluorothiophenol was added into the flask. After 5 min of reaction, 200 mg of tris(4-methylphenyl)phosphine dissolved in 10 mL of CH2Cl2 was added under vigorous stirring. The color of the solution became transparent. After half an hour, 3 mL of an aqueous solution of NaBH4 (80 mg) was added quickly to the reaction mixture under vigorous stirring. The solution color immediately changed from brown to black. The reaction was subsequently carried out for a duration of 9 hours under a N2 atmosphere at room temperature. The crystals were crystallized from CH2Cl2/hexane at room temperature in the dark to afford red block single crystals after 2 weeks.2.3 Synthesis of Pt1Ag14(SR)6(P(Ph-F)3)8
40 mg of AgNO3 and H2PtCl6·6H2O (12 mg, 0.0225 mmol) were dissolved into 10 mL of methanol in a 50 mL round-bottomed flask. The solution was stirred vigorously at room temperature for 10 min. The solution immediately turned brown. Subsequently, 40 μL of 2,3,5,6-tetrafluorothiophenol was added into the flask. After 5 min of reaction, 200 mg of tris(4-fluorophenyl)phosphine dissolved in 10 mL of CH2Cl2 was added under vigorous stirring. The color of the solution became transparent. After half an hour, 2 mL of an aqueous solution of NaBH4 (25 mg) was added quickly to the reaction mixture under vigorous stirring. The solution color immediately changed from brown to black. The reaction was subsequently carried out for a duration of 22 h under a N2 atmosphere at room temperature. The crystals were crystallized from CH2Cl2/hexane at room temperature in the dark to afford orange-red block single crystals after 5 days.2.4 Synthesis of Pt1Ag14(SR)6(PPh3)8
40 mg of AgNO3 and H2PtCl6·6H2O (12 mg, 0.0225 mmol) were dissolved into 10 mL of methanol in a 50 mL round-bottomed flask. The solution was stirred vigorously at room temperature for 10 min. The solution immediately turned brown. Subsequently, 40 μL of 2,3,5,6-tetrafluorothiophenol were added into the flask. After 5 min of reaction, 200 mg of triphenylphosphine dissolved in 10 mL of CH2Cl2 was added under vigorous stirring. The color of the solution became transparent. After half an hour, 2 mL of an aqueous solution of NaBH4 (40 mg) was added quickly to the reaction mixture under vigorous stirring. The solution color immediately changed from brown to black. The reaction was subsequently carried out for a duration of 12 h under a N2 atmosphere at room temperature. The crystals were crystallized from CH2Cl2/hexane at room temperature in the dark to afford yellow rod-like single crystals after 2 weeks.2.5 Synthesis of the g-C3N4 nanosheet
Pristine g-C3N4 was synthesized via a thermal polymerization method. 5 g of melamine was calcined at 550 °C for 4 h at a rate of 5 °C min−1 in a muffle furnace. Then, the resulting yellow sample was collected and spread in a crucible, then calcined at 500 °C for 2 h at a heating rate of 5 °C min−1 to obtain the carbon nitride nanosheet.2.6 Preparation of Pt1Agx/g-C3N4 nanocomposites
For the preparation process, 5 mg of PtAg nanoclusters were dissolved in 10 mL of methylene chloride in a round-bottom flask (RBF), which gave a medium to intense color. To the brown-colored solution, 100 mg of g-C3N4 nanosheet were added and the mixture was stirred for 12 h. The color of the supernatant became faint as the nanoclusters were successfully embedded onto the g-C3N4 nanosheet. The reaction mixture was then centrifuged at 8000 rpm for 10 min. The clear DCM layer was discarded, and the g-C3N4 precipitate was dried under vacuum for a few hours. The dry powder was characterized via TEM analyses and used for the catalytic reactions.2.7 Characterization
The data collection for the single-crystal X-ray diffraction (SC-XRD) analysis of all the nanocluster crystal samples was carried out on a Stoe Stadivari diffractometer under a nitrogen flow, using graphite-monochromatized Cu Kα radiation (λ = 1.54186 Å). Data reductions and absorption corrections were performed using the SAINT and SADABS programs, respectively. The structure was solved by direct methods and refined with full-matrix least squares on F2 using the SHELXTL software package. All non-hydrogen atoms were refined anisotropically, and all the hydrogen atoms were set in geometrically calculated positions and refined isotropically using a riding model. All crystal structures were treated with PLATON SQUEEZE. The diffuse electron densities from these residual solvent molecules were removed. The CCDC number of Pt1Ag11 is 2380779.Electrospray ionization mass spectrometry (ESI-MS) measurements were performed on a MicrOTOF-QIII high-resolution mass spectrometer. All UV-vis spectra of the nanoclusters were recorded using an Agilent 8453, and the samples were dissolved in CH2Cl2 whose background correction was made using a CH2Cl2 blank. Transmission electron microscopy (TEM, JEM-2010) was used to investigate the morphologies and energy-dispersive X-ray spectroscopy (EDS) analyses were performed on a JEOL JEM-2100F FEG TEM operated at 200 kV. Nanocluster powder samples were used for the analysis. X-ray photoelectron spectroscopy (XPS, Thermo-VG Scientific, E = 1486.60 eV, Mg Kα radiation, USA) measurements were performed to detect the elemental composition. The samples were analyzed for photoluminescence spectra using a MicroTime 200 fluorescence spectrophotometer at room temperature. The emission lifetimes were measured with nanoclusters on a HORIBA FluoroMax-4P. The nanocluster was purged with N2 for 5 min, then saturated with O2 for 5 min, respectively.
The powder X-ray diffraction (PXRD) measurements of the materials were carried out using a diffractometer operating (Smartlab 9 kW, Cu Kα radiation) at 40 kV and 200 mA, in a 2θ range of 10–80° with a step width of 0.01°. Fourier-transform infrared spectroscopy (FT-IR) was collected on a Thermo Scientific Nicolet iS50R spectrometer. UV-vis diffuse reflectance spectroscopy (DRS) was conducted on a Shimadzu UV-2600i spectrophotometer at room temperature using BaSO4 as the reference. The electron paramagnetic resonance (EPR) spectra were obtained using a JEOL JES FA200 to detect ˙O2− and ˙OH radicals using 5,5-dimethyl-1-pyrroline-1-oxide (DMPO) as a spin trap. Inductively coupled plasma-atomic emission spectrometry (ICP-AES) measurements were performed on an Atomscan advantage instrument from Thermo Jarrell Ash Corporation (USA), whereby 2.5 mg of PtAg NC/g-C3N4 nanocomposites were dissolved in 1 mL of concentrated nitric acid and 3 mL of deionized water for testing.
2.8 Photoelectrochemical measurements
The electrochemical impedance spectroscopy (EIS), photocurrent–time profiles and Mott–Schottky diagram were recorded on a CHI760E electrochemical workstation with a standard three-electrode system, where photocatalyst-coated fluorine-doped tin oxide (FTO) was used as the working electrode, Pt plate as the counter electrode, and a saturated Ag/AgCl electrode as the reference electrode. A 0.1 M Na2SO4 solution was used as the electrolyte. The as-synthesized samples (10 mg) were added into 400 μL of ethanol and a 20 μL Nafion mixed solution, and the working electrodes were prepared by dropping the suspension (200 μL) onto an FTO glass substrate before drying at room temperature. Mott–Schottky plots were measured at 500, 1000, and 1500 Hz, respectively. EIS was recorded with a bias potential of −1.4 V in the dark.2.9 Photocatalytic hydrogen evolution
The photocatalytic hydrogen production tests were performed in a Pyrex top-irradiation reaction vessel with a stationary temperature at 25 °C under full-spectrum light, which was connected to a glass closed gas system (Labsolar-6A, Perfect Light). A 300 W Xe lamp was employed to serve as the light source. 50 mg of the as-prepared photocatalyst was ultrasonically dissolved in 10 mL of triethanolamine (TEOA, sacrifice reagent) and 90 mL of H2O and then loaded into a 370 mL sealed quartz reactor and evacuated using a vacuum pump. The photocatalytic hydrogen performance of the photocatalyst was calculated using gas chromatography (GC-5190, China) equipped with a thermal-conductivity detector (TCD, with Ar as the carrier gas and a 5 Å molecular sieve column). The injection temperature and detection temperature were set to 100 °C during testing, and the column furnace temperature was set to 50 °C. The production of H2 every 30 min was monitored using a gas chromatograph. The temperature of the whole process was kept at 8 °C using circulating cooling water.2.10 Apparent quantum efficiency (AQE) measurements
The AQE measurement methods were similar with those of the photocatalytic measurements except for the wavelength of the light. Particularly, different monochromatic light (360 nm, 380 nm, 400 nm, 420 nm, 440 nm) was utilized to assess the quantum efficiency of the photocatalyst. Based on the amounts of evolved H2, the AQE is calculated via the following formula:where n (mol) refers to the amount of H2 molecules, v (mol s−1) represents the rate of H2 production, NA (6.022 × 1023 per mol) represents the Avogadro constant, ℏ (6.626 × 10−34 J S) represents the Planck constant, c (3 × 108 m s−1) is the speed of light, S is the irradiation area (cm2), P refers the intensity of the irradiation light (W cm−2), t signals the photoreaction time (s), and λ is the wavelength of the monochromatic light (m).

The simplified formula is translated as follows:

2.11 Computational method
The Vienna ab initio simulation package (VASP) was selected to perform our density-functional theory (DFT) calculations. The exchange–correlation functional was treated through the generalized gradient approximation (GGA) using the Perdew–Burke–Ernzerhof (PBE) functional. The cutoff energy was set to 570 eV for all calculations. The Brillouin zone (BZ) was sampled using a 2 × 2 × 1 gamma-centered Monkhorst–Pack grid. The energy and the force standard received on each atom were set to be 10−6 eV and 0.02 eV Å−1, respectively. To avoid the interaction between adjacent slabs, a vacuum space of 20 Å was used. The visualization for electronic and structural analysis software (VESTA 3) was utilized for visualization and plotting. The VASPKIT code was employed for the postprocessing of the calculated data.The charge density difference (Δρ) was determined by
| Δρ = ρ*M − ρM − ρ* | (1) |
In addition, Bader charges were utilized to detect charge transfer between the substrate and clusters. To evaluate the energetic stability of the g-C3N4-loaded clusters, we calculated their binding energy (Ebind), which is defined as:
| Ebind = E*M − E* − EM | (2) |
| ΔG = ΔE + ΔEzpe − TΔS | (3) |
The HER can be decomposed into two steps, and the reaction equation can be written as:
| * + H+ + e− → H* | (4) |
| H* + H+ + e− → * + H2 | (5) |
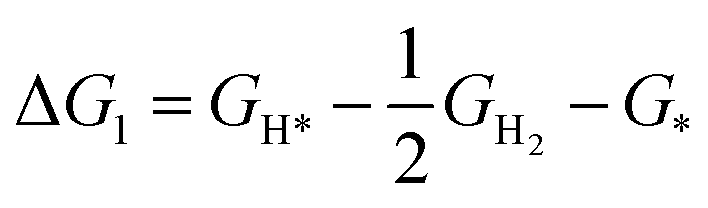 | (6) |
 | (7) |
3 Results and discussion
The icosahedral kernel structure was chosen as the model for investigating the defect effect because it is the most widely observed structure in NCs. A novel Pt1Ag11 NC was synthesized, and can be categorized as part of the Pt1Agx NC series along with three previously reported NCs, Pt1Ag9,25 Pt1Ag13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 7 and Pt1Ag14.26 Single-crystal X-ray crystallography (SC-XRD) revealed that Pt1Ag11 crystalized in the triclinic space group P
7 and Pt1Ag14.26 Single-crystal X-ray crystallography (SC-XRD) revealed that Pt1Ag11 crystalized in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) . The Pt1Ag11 NC comprised a Pt1Ag9 kernel, stabilized by five P(Ph-OMe)3 ligands, one Ag(SR)2[P(Ph-OMe)3] motif and one Ag(SR)3[P(Ph-OMe)3] motif (Fig. 1a). The chemical composition of Pt1Ag11 was definitively confirmed using electrospray ionization mass spectrometry (ESI-MS) in positive ion mode, as shown in the Fig. 1b. The spectrum contained two intense peaks at m/z of 2500.726 Da (calculated = 2500.765 Da) and 2511.792 Da (calculated = 2511.755 Da), corresponding to the characteristic peaks of Pt1Ag11(C6HF4S)5(C21H21O3P)7Ag2(CH3OH) and Pt1Ag11(C6HF4S)5(C21H21O3P)7Ag2(CH3ONa). Transmission electron microscopy (TEM) images indicated that Pt1Ag11 has a uniform appearance, with an average size of 1.32 nm (Fig. S1†). The UV-vis spectrum of Pt1Ag11 in CH2Cl2 displays one strong peak centered at 372 nm and three weak shoulder peaks at 425, 455, and 550 nm (Fig. 1c). For comparison, the UV-vis spectra of the Pt1Ag9, Pt1Ag13, and Pt1Ag14 NCs revealed different absorption peaks (Fig. S2†). Moreover, the Pt1Agx series in the solid state emitted between 600 and 700 nm (Fig. S3†).
. The Pt1Ag11 NC comprised a Pt1Ag9 kernel, stabilized by five P(Ph-OMe)3 ligands, one Ag(SR)2[P(Ph-OMe)3] motif and one Ag(SR)3[P(Ph-OMe)3] motif (Fig. 1a). The chemical composition of Pt1Ag11 was definitively confirmed using electrospray ionization mass spectrometry (ESI-MS) in positive ion mode, as shown in the Fig. 1b. The spectrum contained two intense peaks at m/z of 2500.726 Da (calculated = 2500.765 Da) and 2511.792 Da (calculated = 2511.755 Da), corresponding to the characteristic peaks of Pt1Ag11(C6HF4S)5(C21H21O3P)7Ag2(CH3OH) and Pt1Ag11(C6HF4S)5(C21H21O3P)7Ag2(CH3ONa). Transmission electron microscopy (TEM) images indicated that Pt1Ag11 has a uniform appearance, with an average size of 1.32 nm (Fig. S1†). The UV-vis spectrum of Pt1Ag11 in CH2Cl2 displays one strong peak centered at 372 nm and three weak shoulder peaks at 425, 455, and 550 nm (Fig. 1c). For comparison, the UV-vis spectra of the Pt1Ag9, Pt1Ag13, and Pt1Ag14 NCs revealed different absorption peaks (Fig. S2†). Moreover, the Pt1Agx series in the solid state emitted between 600 and 700 nm (Fig. S3†).
 | ||
| Fig. 1 (a) Structural anatomy of a Pt1Ag11 nanocluster (Pt1Ag9 kernel; Pt1Ag9@5P; Pt1Ag9@Ag1S3P1@Ag1S2P1; Pt1Ag11(C6H2F4S)5(C21H21O3P)7 overall structure), color labels: yellow, Pt; turquoise, Ag; red, S; pink, P; bright green, F; orange, O; gray, C; white, H. (b) ESI-MS of the Pt1Ag11 nanoclusters [Pt1Ag11(C6HF4S)5(C21H21O3P)7Ag2(CH3OH) (☆) and Pt1Ag11(C6HF4S)5(C21H21O3P)7Ag2(CH3ONa) (★)]; (c) UV-vis spectra of the Pt1Ag11 nanoclusters. The XPS spectra of (d) Pt 4f and (e) Ag 3d for the Pt1Agx nanoclusters (x = 9, 11, 13, 14). | ||
A cycle between enhanced emission intensity with N2 and quenched emission intensity with O2 was detected for the Pt1Ag11 NC. Meanwhile, the average PL lifetime of the Pt1Ag11 NC was 4.28 μs, which was prolonged to 5.91 μs in a N2 atmosphere, and reduced to 2.16 μs in an O2 atmosphere. Similar results were observed for the other three NCs, indicating that the Pt1Agx series of NCs (x = 9, 11, 13, and 14) were phosphorescent7 (Fig. S4–S6†). In addition, the valence states of the Pt and Ag atoms in the Pt1Agx series of bimetallic NCs were investigated and compared with the results obtained from X-ray photoelectron spectroscopy (XPS). The XPS results revealed the presence of Pt, Ag, P, F, O, C, and S in the Pt1Ag11 NC (Fig. S7†). As shown in Fig. 1d, the Pt 4f binding energies in Pt1Agx were as follows: Pt1Ag14 (71.5 eV) > Pt1Ag9 (71.4 eV) > Pt1Ag13 (71.3 eV) > Pt1Ag11 (71.1 eV). The lower binding energies of the Pt 4f peaks indicate that the Pt atoms in Pt1Ag11 were more partial to 0 valence (71.0 eV) compared with the other members of the Pt1Agx family of NCs. For the binding energies of Ag, a sequence of Pt1Ag13 (368.4 eV) ≈ Pt1Ag11 (368.4 eV) > Pt1Ag14 (368.2 eV) ≈ Pt1Ag9 (368.2 eV) can be established. This illustrates that the valence state of Ag in the Pt1Agx NCs lies between 0 and +1 (Fig. 1e).
In addition to determining the difference in the electronic structure of the Pt1Agx series of NCs, analysis of their geometric structures is crucial. SC-XRD revealed that Pt1Ag13 crystalized in the triclinic space group P similar to Pt1Ag11, whereas Pt1Ag14 crystalized in the monoclinic space group C2/c; Pt1Ag9 was found to have the trigonal space group R3 (Tables S1 and S2†). Comparison of the crystal structures of the Pt1Agx series of NCs indicated that Pt1Ag14 has a complete icosahedral Pt1Ag12 kernel, while the remaining three NCs exhibit different icosahedral-kernel defects (Fig. S8†). In Pt1Ag13, removing the vertex silver atom from the icosahedral kernel caused the loss of one Ag–P bond, whereas the peripheral Ag1S3P1 motif did not change. However, three silver atoms were absent from the icosahedral kernel in Pt1Ag11, and thus, the Ag1S3P1 motif experienced the loss of an Ag–S bond, leading to the formation of the Ag1S2P1 motif (Fig. 2a). Meanwhile, a new Pt–P bond was formed within the icosahedral kernels in both Pt1Ag11 and Pt1Ag9. Pt1Ag11 and Pt1Ag9 have the same kernel, albeit with only a P ligand included for the latter (Fig. 2b). A detailed comparison of the bond lengths and angles within the kernel structures of the Pt1Ag9 and Pt1Ag11 NCs further suggested that the kernel of Pt1Ag11 was more distorted (Tables S3 and S4†). In addition, the geometric structures were further compared by analyzing the bond lengths of the four Pt1Agx NCs. The Pt–Ag average distance within the icosahedral kernel in the Pt1Agx NCs was Pt1Ag14 (2.757 Å) > Pt1Ag9 (2.747 Å) > Pt1Ag13 (2.744 Å) > Pt1Ag11 (2.743 Å). The Ag–Ag average distance in Pt1Agx was Pt1Ag14 (2.899 Å) > Pt1Ag9 (2.891 Å) > Pt1Ag13 (2.873 Å) > Pt1Ag11 (2.861 Å). The above results also indicated that the kernel of the NCs became more compact with the loss of kernel silver atoms. Moreover, the kernel of Pt1Ag11 was the most compact (Fig. S9–S11†). The geometric defects in the icosahedral kernel of the Pt1Agx NCs were correlated with distinct electronic structures. Specifically, both Pt1Ag9 and Pt1Ag11 exhibited a free electron count of 6e, while Pt1Ag13 and Pt1Ag14 displayed counts of 7e and 8e, respectively.
 | ||
| Fig. 2 Structures of the Pt1Agx nanoclusters (x = 9, 11, 13, 14). (a) Framework structure: metallic kernel, ligand, and surface motifs; (b) metallic kernel on the Pt1Agx NCs. Color labels: yellow, Pt; turquoise, Ag; red, S; pink, P; green, Cl. All of the C and H atoms are omitted. The crystal data sources for Pt1Ag9, Pt1Ag13 and Pt1Ag14 were taken from the literature.7,25,26 | ||
The photocatalytic hydrogen evolution reaction was then used to examine the effect of the structural defects on the catalytic activity. A schematic diagram of the photocatalyst preparation process is shown in Scheme S1.†27 Lamellar g-C3N4 nanosheet was first synthesized via secondary calcination and exhibited a larger specific surface area (63.9405 m2 g−1) compared with bulk g-C3N4 (10.053 m2 g−1), as confirmed by BET measurements (Fig. S12†).28–32 Next, the Pt1Agx NCs with an optimal loading of 5 wt% were anchored onto the g-C3N4 nanosheet via impregnation (Fig. S13†). Four types of Pt1Agx/g-C3N4 nanocomposites labeled Pt1Ag9/g-C3N4, Pt1Ag11/g-C3N4, Pt1Ag13/g-C3N4, and Pt1Ag14/g-C3N4 were prepared using this method. Inductively coupled plasma-atomic emission spectroscopy (ICP-AES) was utilized for further analysis, unambiguously identifying the similar Pt and Ag contents of the four types of Pt1Agx/g-C3N4 nanocomposite (Table S5†). Moreover, TEM images and energy-dispersive X-ray spectroscopy (EDX) elemental mapping confirmed the presence and homogeneous distribution of the Pt1Agx NCs on the g-C3N4 nanosheet (Fig. S14†). The Pt1Agx/g-C3N4 nanocomposites were further analyzed using X-ray diffraction (XRD). As shown in Fig. S15a,† the two diffraction peaks at 13.0° and 27.7° that correspond to the (100) and (002) diffraction of the typical graphitic interlayer stacking structure of the pure g-C3N4 nanosheet did not change in the nanocomposites. Moreover, as shown in Fig. S15b,† the characteristic Fourier-transform infrared (FTIR) peaks located at approximately 810 cm−1, 1200–1650 cm−1, and 3000–3400 cm−1, corresponding to the vibrations of the s-triazine ring unit, C–N heterocycles, and amino groups (N–H), respectively, remained unchanged in the NCs compared with those of pure g-C3N4 nanosheet. These results indicate that the basic backbone of the g-C3N4 nanosheet was still well-maintained after the addition of the Pt1Agx NCs. As shown in Fig. S15c,† the optical absorption intensity of Pt1Agx/g-C3N4 was enhanced in the full spectrum compared with that of the pure g-C3N4 nanosheet. Moreover, according to the Kubelka–Munk function, the corresponding intrinsic bandgap of the g-C3N4 nanosheet, Pt1Ag14/g-C3N4, Pt1Ag13/g-C3N4, Pt1Ag9/g-C3N4, and Pt1Ag11/g-C3N4 were estimated to be 2.76, 2.75, 2.73, 2.71 and 2.69 eV, respectively (Fig. S15d†). The enhanced optical absorption intensity and narrowed bandgap of the as-prepared Pt1Ag11/g-C3N4 contributes to the generation of more photogenerated carriers. The hydrogen evolution performance of the four types of Pt1Agx/g-C3N4 (x = 9, 11, 13, and 14) nanocomposites were then obtained from the full solar spectrum.
As shown in Fig. 3a, when 10 wt% triethanolamine (TEOA) was used as the sacrificial agent, Pt1Ag11/g-C3N4 displayed the highest hydrogen production performance (1780 μmol g−1 h−1), which was 33.3 times that of the pure g-C3N4 nanosheet (53.4 μmol g−1 h−1) and 27.9 times that of Pt1Ag11 (63.8 μmol g−1 h−1), as well as outperforming several other corresponding counterparts (Table S6†). To exclude the reaction of the NCs with the sacrificial agent, we performed a set of light–dark contrast tests, which indicated that no hydrogen was produced in the absence of light (Fig. S16†). Moreover, the hydrogen production rate of Pt1Ag11/g-C3N4 was ∼2.83 times higher than that of Pt1Ag9/g-C3N4 (628.4 μmol g−1 h−1) and much higher than that of Pt1Ag13/g-C3N4 (448 μmol g−1 h−1) and Pt1Ag14/g-C3N4 (185.2 μmol g−1 h−1) (Fig. 3b and Table S7†), resulting in the following activity trend; Pt1Ag11/g-C3N4 > Pt1Ag9/g-C3N4 > Pt1Ag13/g-C3N4 > Pt1Ag14/g-C3N4 > g-C3N4 nanosheet. The apparent quantum efficiency of Pt1Ag11/g-C3N4 under light irradiation at 420 nm was ∼3.11% (Table S8†). In cycling experiments (Fig. 3c), the hydrogen production rate of Pt1Ag11/g-C3N4 was maintained for three photocatalytic cycles of 9 h. In the fourth cycle, a slight decrease was observed, which may be attributed to shedding of the NCs owing to constant agitation during the cycle reaction. To accurately characterize the changes in the catalysts before and after the reaction, UV-vis diffuse reflection spectroscopy was performed and the characteristic peak position did not change and the intensity decreased slightly, indicating that the overall structure of the catalyst remained unchanged and the structure of Pt1Ag11 was well maintained (Fig. S17†). In addition, the effect of the ligand on the photocatalytic performance was excluded by obtaining the time-dependent H2 production profiles for various Pt1Ag14 NCs with different ligands (Fig. S18†). As shown in Fig. S19,† the photocatalytic hydrogen production rates of Pt1Ag14-1/g-C3N4, Pt1Ag14-2/g-C3N4, and Pt1Ag14/g-C3N4 were 129.7, 149.8, and 185.2 μmol g−1 h−1, respectively. These findings indicated that the influence of the ligand on the photocatalytic performance was minimal.33
 | ||
| Fig. 3 (a) Photocatalytic H2 production of Pt1Ag11, g-C3N4 and Pt1Ag11/g-C3N4. (b) Photocatalytic H2 production of Pt1Agx/g-C3N4 (x = 9, 11, 13, and 14). (c) Cycling runs of the Pt1Ag11/g-C3N4 catalyst under light irradiation. (d) Transient photocurrent response and (e) EIS of Pt1Agx/g-C3N4 and g-C3N4. (f) EPR spectra of ˙OH and ˙O2− on Pt1Ag11/g-C3N4 under light irradiation or in the dark (x = 9, 11, 13, and 14). | ||
To better understand the superior photocatalytic hydrogen evolution performance of the Pt1Ag11/g-C3N4 catalysts, a series of photo-electrochemistry tests were performed to probe the electron–hole separation and electron transfer behavior of the nanocomposite photocatalyst.34–39 As shown in Fig. 3d, Pt1Ag11/g-C3N4 exhibited the strongest transient photocurrent response and the response displayed the following trend under light irradiation, Pt1Ag11/g-C3N4 > Pt1Ag9/g-C3N4 > Pt1Ag13/g-C3N4 > Pt1Ag14/g-C3N4 > g-C3N4 nanosheet, suggesting the promoted efficient separation of the electron–hole pairs in the nanocomposite photocatalysts. This was further confirmed by electrochemical impedance spectroscopy (EIS). The Nyquist curve shown in Fig. 3e indicated that the interfacial charge transfer resistance of Pt1Ag11/g-C3N4 was much smaller than those of g-C3N4 and the other Pt1Agx/g-C3N4 (x = 9, 13, and 14) nanocomposites. Moreover, steady-state photoluminescence (PL) spectra obtained via excitation at 373 nm revealed a broadband centered at 450 nm for the g-C3N4 nanosheet and Pt1Agx/g-C3N4 nanocomposites with significantly different PL intensities, indicating that the recombination of photogenerated carriers in Pt1Agx/g-C3N4 can be efficiently restrained (Fig. S20†). In addition, time-resolved PL spectra (Fig. S21†) were obtained and the fitting of the time-resolved PL spectra determined the PL lifetimes to be 2.089 ns (g-C3N4 nanosheet), 2.015 ns (Pt1Ag14/g-C3N4), 1.998 ns (Pt1Ag13/g-C3N4), 1.798 ns (Pt1Ag11/g-C3N4), and 1.619 ns (Pt1Ag9/g-C3N4). The reduced decay lifetime of Pt1Agx/g-C3N4 signified the promotion of efficient separation of electron–hole pairs and an accelerated charge transport process. In summary, the order of charge transfer and separation efficiency of all the photocatalysts closely aligned with the order of the photocatalytic evolution performance.
To understand the photogenerated charge transfer pathways in the Pt1Agx/g-C3N4 nanocomposites, the band structure of the g-C3N4 nanosheet and Pt1Agx NCs were evaluated. The band gap of 2.76 eV for the g-C3N4 nanosheet was determined from the UV-vis absorption spectrum; the conduction band (CB) level was estimated by Mott–Schottky measurements to be −0.31 V vs. NHE (PH = 7, Fig. S22†). Because the metal NCs can be regarded as small band gap semiconductors, the energy band structure of the Pt1Agx NCs was determined in the same way as that of the g-C3N4 nanosheet (Fig. S23 and S24†).10,13 The energy diagram showed that all the lowest unoccupied molecular orbitals (LUMO) of the Pt1Agx NCs were slightly more negative than the CB of g-C3N4, suggesting the formation of a type-II or Z-scheme heterojunction (Fig. S25†). Taking Pt1Ag11/g-C3N4 as a model, EPR analysis was performed to distinguish the exact types of heterostructures. To produce ˙O2− radicals, the potential of the photogenerated electrons in the CB should be more negative than −0.33 V. Meanwhile, the potential of the photogenerated holes in the valence band (VB) should be more positive than 2.40 V to produce ˙OH radicals. Apparently, ˙O2− radicals and ˙OH radicals could not be produced by the type-II heterojunction due to the high potential of the photogenerated electrons of −0.31 V and the low potential of the photogenerated holes of 0.95 V. However, as shown in Fig. 3f, under light irradiation, the production of ˙O2− radicals and ˙OH radicals was observed, demonstrating the transfer of the photogenerated electrons from g-C3N4 to Pt1Ag11, forming a Z-scheme heterojunction in Pt1Ag11/g-C3N4.40,41
To gain a molecular level understanding of the effect of the kernel structural defects on photocatalytic activity, density-functional theory (DFT) calculations using the Vienna ab initio simulation package (VASP) were conducted.42,43 After geometric relaxation, we concluded that the Pt1Agx NCs could maintain a stable structure on the surface of the g-C3N4 monolayer (Fig. S26†), which might be attributed to the formation of a Ag–N covalent bond and abundant π–π interactions between Pt1Agx NC and g-C3N4. The binding energy (Ebind) of the NCs with the g-C3N4 monolayer can also be obtained. The Ebind value of Pt1Ag11, Pt1Ag9, Pt1Ag13 and Pt1Ag14 were calculated as −0.95 eV, −0.72 eV, −0.65 eV and −0.68 eV, respectively, where the high Ebind (−0.95 eV) value of Pt1Ag11 enables it to exhibit the most stable loading structure on the g-C3N4 surface compared with the other NCs (Fig. 4a). The differential charge density was analyzed to determine the charge transfer between the NCs and monolayers. As shown in Fig. 4b, the g-C3N4 layer accumulated negative charge, while the NCs gathered positive charge, thus forming an internal electric field directing from the NCs to g-C3N4. Bader44 charge analysis showed that the Pt1Ag9 (0.66|e|) and Pt1Ag11 (0.82|e|) NCs transferred more charge to g-C3N4 than was transferred by Pt1Ag13 (0.48|e|) and Pt1Ag14 (0.62|e|). This difference indicates a stronger internal electric field in Pt1Ag11/g-C3N4, allowing Pt1Ag11 to interact more closely with g-C3N4. Furthermore, the photocatalytic hydrogen evolution reaction was investigated with ΔG as an indicator of the absorption of H* intermediates on the Ag sites in the Pt1Agx/g-C3N4 nanocomposites. As shown in Fig. 4c, Pt1Ag11/g-C3N4 has the lowest Gibbs free energy change (ΔG) (0.23 eV), following the trend of Pt1Ag11/g-C3N4 < Pt1Ag9/g-C3N4 (0.29 eV) < Pt1Ag13/g-C3N4 (0.33 eV) < Pt1Ag14/g-C3N4 (0.67 eV), which suggests optimal catalytic activity on the surface of the icosahedron with atomic defects. Moreover, the d-band center of the Ag atoms in Pt1Ag11 are closer to the Fermi level, which suggests a stronger binding interaction between Pt1Ag11/g-C3N4 and the H species (Fig. S27†).
 | ||
| Fig. 4 (a) Binding energies of the Pt1Agx nanoclusters loaded on a g-C3N4 monolayer. (b) Differential charge density of nanoclusters loaded on a g-C3N4 monolayer, where yellow and cyan areas represent electron accumulation and depletion, respectively. The total amount of charge transfer is given. (c) The calculated Gibbs free energy (ΔGH*) diagrams for the photocatalytic hydrogen evolution reaction on Pt1Agx/g-C3N4. The optimized structures of H* adsorption on Pt1Ag9/g-C3N4 (blue), Pt1Ag11/g-C3N4 (yellow), Pt1Ag13/g-C3N4 (green), and Pt1Ag14/g-C3N4 (purple). (d) The energy level diagrams and the interface charge transfer route in Pt1Ag11/g-C3N4. An internal electric field (IEF) is formed inside the heterojunction formed by g-C3N4 loaded with Pt1Ag11. Evacuum, Ef1, Ef2 and Ef3 represent the vacuum level and Fermi levels of g-C3N4, Pt1Ag11 and Pt1Ag11/g-C3N4, respectively. VB and CB represent the top of the valence band and the bottom of the conduction band of g-C3N4. HOMO and LUMO refer to the highest occupied molecular orbital and the lowest unoccupied molecular orbital of the electrons in the Pt1Ag11 cluster, respectively. | ||
Building on this, we explored the mechanism of photocatalytic water splitting for hydrogen production (Fig. 4d). When NCs are loaded onto the surface of g-C3N4, free electrons transfer across the interface due to the differing Fermi levels.45,46 Specifically, electrons from Pt1Ag11, which has a higher Fermi level, migrate to g-C3N4, which has a lower Fermi level, until equilibrium is reached at a common Fermi level. This observation aligns with the differential charge density results shown in Fig. 4b. Under the influence of the Coulomb force, the band edge in g-C3N4 at the interface bent downward, and the energy level of Pt1Ag11 bent upward. When illuminated, electrons in the VB of the g-C3N4 within the Pt1Ag11/g-C3N4 heterojunctions absorbs the energy and transits it to the CB, leaving behind holes in the VB. Simultaneously, the HOMO electrons of Pt1Ag11 gained energy to transition to the LUMO, resulting in a vacant HOMO. Driven by the internal electric field, the photogenerated electrons in g-C3N4 were transferred to Pt1Ag11. The band (and energy level) bending are crucial for the migration of photogenerated carriers under optical excitation.47 The downward band bending in g-C3N4 facilitates the free flow of electrons while inhibiting hole outflow. Conversely, the upward energy level bending in Pt1Ag11 suppresses electron de-excitation. Thus, electrons flowing from the CB of g-C3N4 recombine with the vacant orbitals in the HOMO of Pt1Ag11, allowing the electrons in the LUMO of Pt1Ag11 to participate in the hydrogen evolution reaction. This process enables direct Z-scheme photocatalytic water splitting for hydrogen production.
4 Conclusions
A family of Pt1Agx (x = 9, 11, 13, and 14) NCs, including the new Pt1Ag11 NC, was designed and synthesized via ligand engineering. Single-crystal X-ray diffraction revealed that the crystal structures of these NCs possessed defect-containing icosahedral kernels where one or three vertical Ag atoms are lost. Using the photocatalytic water-splitting reaction as a model, the positive effect of the kernel structural defect on the photocatalytic activity was elucidated, and the mechanism that links the defect structure and catalytic activity was well established. Importantly, Pt1Ag11/g-C3N4 has the highest hydrogen production rate reaching 1780 μmol g−1.h−1, that is, ∼2.83, ∼3.97, ∼9.61, and ∼33.3 times higher than that of Pt1Ag9/g-C3N4, Pt1Ag13/g-C3N4, Pt1Ag14/g-C3N4 and the g-C3N4 nanosheets, respectively. DFT calculations further demonstrated the importance of the defect-containing kernel structure for the activation of H2, electron–hole separation, and the charge transfer efficiency of the Pt1Ag11/g-C3N4 photocatalyst. These improved properties which might be attributed to the formation of Z-scheme heterojunctions, accounting for their very high photocatalytic H2 production rate. This study lays a strong foundation for the design of highly efficient and precise nanocomposite photocatalysts and provides new insights into the mechanism of photocatalytic hydrogen production at the atomic level.Abbreviations
| Pt1Ag11 | Pt1Ag11(SR)5(P(Ph-OMe)3)7, (SR is 2,3,5,6-tetrafluorothiophenol and P(Ph-OMe)3 is tris(4-methylphenyl)-phosphine) |
| Pt1Ag9 | Pt1Ag9(P(Ph-F)3)7Cl3, (P(Ph-F)3 is tris(4-fluorophenyl)phosphine) |
| Pt1Ag13 | Pt1Ag13(PFBT)6(PPh3)7, (PFBT is pentafluorobenzenethiol and PPh3 is triphenylphosphine) |
| Pt1Ag14 | Pt1Ag14(SR)6(PPh3)8, (SR is 2-chloro-4-fluorobenzenethiol and PPh3 is triphenylphosphine) |
| Pt1Ag14-1 | Pt1Ag14(SR)6(P(Ph-F)3)8, (SR is 2,3,5,6-tetrafluorothiophenol and P(Ph-F)3 is tris(4-fluorophenyl)phosphine) |
| Pt1Ag14-2 | Pt1Ag14(SR)6(PPh3)8, (SR is 2,3,5,6-tetrafluorothiophenol and PPh3 is triphenylphosphine) |
Data availability
CCDC 2380779 contain the supplementary crystallographic data for this paper. These data can be obtained free of charge viahttps://www.ccdc.cam.ac.uk/data_request/cif, or by emailing E-mail: data_request@ccdc.cam.ac.uk, or by contacting The Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK; fax: +44 1223 336033.Author contributions
D. Hu, S. Sun and M. Zhu conceived and designed the project. D. Tan and T. Ding carried out the synthesis and catalytic experiments. S. Jin and D. Tan carried out crystallography and mass spectrometry. K. Shen and C. Xu conducted theoretical calculations. All authors contributed to the discussion of the results and the preparation of the manuscript.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
We acknowledge the financial support provided by the National Natural Science Foundation of China (22371003, 21871001, 22103001) and the Natural Science Fund of the Education Department of Anhui Province (2023AH050108).Notes and references
- Y. Shang, H. Fan, X. Che and W. Wang, Promoted photocatalytic hydrogen evolution via double-electron migration in Ag@g-C3N4 heterojunction, Int. J. Hydrogen Energy, 2023, 48, 17370–17382 CrossRef CAS.
- Y. Han, C. Wang, R. Zhao, J. Han and L. Wang, Ni-doped CdSe/ZnSnO3 double-shell nanocubes heterojunction for efficient photocatalytic hydrogen evolution, Fuel, 2023, 353, 129247 CrossRef CAS.
- P. Zhou, I. A. Navid, Y. Ma, Y. Xiao, P. Wang, Z. Ye, B. Zhou, K. Sun and Z. Mi, Solar-to-hydrogen efficiency of more than 9% in photocatalytic water splitting, Nature, 2023, 613, 66–70 CrossRef CAS PubMed.
- A. Shu, C. Qin, M. Li, L. Zhao, Z. Shangguan, Z. Shu, X. Yuan, M. Zhu, Y. Wu and H. Wang, Electric effects reinforce charge carrier behaviour for photocatalysis, Energy Environ. Sci., 2024, 17, 4907 RSC.
- C. Xia, H. Wang, J. K. Kim and J. Wang, Rational Design of Metal Oxide-Based Heterostructure for Efficient Photocatalytic and Photoelectrochemical Systems, Adv. Funct. Mater., 2021, 31, 2008247 CrossRef CAS.
- A. Meng, W. Tian, H. Yang, X. Wang, X. Wang and Z. Li, Molybdenum sulfide-modified metal-free graphitic carbon nitride/black phosphorus photocatalyst synthesized via high-energy ball-milling for efficient hydrogen evolution and hexavalent chromium reduction, J. Hazard. Mater., 2021, 413, 2008247 Search PubMed.
- C. Fang, C. Xu, W. Zhang, M. Zhou, D. Tan, L. Qian, D. Hu, S. Jin and M. Zhu, Dual-quartet phosphorescent emission in the open-shell M1Ag13 (M = Pt, Pd) nanoclusters, Nat. Commun., 2024, 15, 5962 CrossRef CAS PubMed.
- Y. Du, H. Sheng, D. Astruc and M. Zhu, Atomically Precise Noble Metal Nanoclusters as Efficient Catalysts: A Bridge between Structure and Properties, Chem. Rev., 2020, 120, 526–622 CrossRef CAS PubMed.
- Y. Wang, X.-H. Liu, R. Wang, B. Cula, Z.-N. Chen, Q. Chen, N. Koch and N. Pinna, Secondary Phosphine Oxide Functionalized Gold Clusters and Their Application in Photoelectrocatalytic Hydrogenation Reactions, J. Am. Chem. Soc., 2021, 143, 9595–9600 CrossRef CAS PubMed.
- H. Wang, X. Zhang, W. Zhang, M. Zhou and H.-L. Jiang, Heteroatom-Doped Ag25 Nanoclusters Encapsulated in Metal-Organic Frameworks for Photocatalytic Hydrogen Production, Angew. Chem., Int. Ed., 2024, e202401443 CAS.
- C. Wang, P. Lv, D. Xue, Y. Cai, X. Yan, L. Xu, J. Fang and Y. Yang, Zero-Dimensional/Two-Dimensional Au25(Cys)18 Nanoclusters/g-C3N4 Nanosheets Composites for Enhanced Photocatalytic Hydrogen Production under Visible Light, ACS Sustain. Chem. Eng., 2018, 6, 8447–8457 CrossRef CAS.
- Y. Negishi, M. Mizuno, M. Hirayama, M. Omatoi, T. Takayama, A. Iwasea and A. Kudo, Enhanced photocatalytic water splitting by BaLa4Ti4O15 loaded with ∼1 nm gold nanoclusters using glutathione-protected Au25 clusters, Nanoscale, 2013, 5, 7188 RSC.
- Y. Du, C. Li, Y. Dai, H. Yin and M. Zhu, Recent progress in atomically precise metal nanoclusters for photocatalytic application, Nanoscale Horiz., 2024, 9, 1262 RSC.
- X. Lu, A. Tong, D. Luo, F. Jiang, J. Wei, Y. Huang, Z. Jiang, Z. Lu and Y. Ni, Confining single Pt atoms from Pt clusters on multi-armed CdS for enhanced photocatalytic hydrogen evolution, J. Mater. Chem. A, 2022, 10, 4594 RSC.
- Y. Wang, X.-H. Liu, Q. Wang, M. Quick, S. A. Kovalenko, Q.-Y. Chen, N. Koch and N. Pinna, Insights into Charge Transfer at an Atomically Precise Nanocluster/Semiconductor Interface, Angew. Chem., Int. Ed., 2020, 59, 7748–7754 CrossRef CAS PubMed.
- X. Huang, K. Yin, S. Zhang, T. Wu, Y. Yuan, X. Wang, Y. Jia, Z. Xiao, J. Gu and D. Wang, Interfacial chemical bond-modulated Z-scheme Cs2AgBiBr6/WO3 enables stable and highly efficient photocatalysis, Appl. Surf. Sci., 2023, 637, 157877 CrossRef CAS.
- J. Wang, X. Li, Y. You, X. Yang, Y. Wang and Q. Li, Interfacial coupling induced direct Z-scheme water splitting in metal-free photocatalyst: C3N/g-C3N4 heterojunctions, Nanotechnology, 2018, 29, 365401 CrossRef PubMed.
- Q. Zhu, H. Shen, C. Han, L. Huang, Y. Zhou, Y. Du, X. Kang and M. Zhu, Rationally construction of atomic-precise interfacial charge transfer channel and strong build-in electric field in nanocluster-based Z-scheme heterojunctions with enhanced photocatalytic hydrogen production, Nano Res., 2024, 17, 5002–5010 CrossRef CAS.
- Y. Du, J. Li, X. Ma and Q. Guo, Visible light driven mesoporous ag/Ag2CrO4/g-C3N4 degrades multiple organic pollutants efficiently: synthesis, mechanism, and degradation pathway, Vacuum, 2024, 226, 113323 CrossRef CAS.
- S. O. Lee, S. K. Lakhera and K. Yong, Strategies to Enhance Interfacial Spatial Charge Separation for High-Efficiency Photocatalytic Overall Water-Splitting: A Review, Adv. Energ. Sust. Res., 2003, 4, 2300130 Search PubMed.
- S. Nematulloev, A. Sagadevan, B. Alamer, A. Shkurenko, R. Huang, J. Yin, C. Dong, P. Yuan, K. E. Yorov, A. A. Karluk, W. J. Mir, B. E. Hasanov, M. N. Hedhili, N. M. Halappa, M. Eddaoudi, O. F. Mohammed, M. Rueping and O. M. Bakr, Atomically Precise Defective Copper Nanocluster Catalysts for Highly Selective C-C Cross-Coupling Reactions, Angew. Chem., Int. Ed., 2023, 62, e202303572 CrossRef CAS PubMed.
- R. P. B. Silalahi, Y. Jo, J.-H. Liao, T.-H. Chiu, E. Park, W. Choi, H. Liang, S. Kahlal, J.-Y. Saillard, D. Lee and C. W. Liu, Hydride-containing 2-Electron Pd/Cu Superatoms as Catalysts for Efficient Electrochemical Hydrogen Evolution, Angew. Chem., Int. Ed., 2023, 62, e202301272 CrossRef PubMed.
- D. Zhu and Q. Zhou, Nitrogen doped g-C3N4 with the extremely narrow band gap for excellent photocatalytic activities under visible light, Appl. Catal., B, 2021, 281, 119474 CrossRef CAS.
- Z. Yu, Y. Li, A. Torres-Pinto, A. P. LaGrow, V. M. Diaconescu, L. Simonelli, M. J. Sampaio, O. Bondarchuk, I. Amorim, A. Araujo, A. M. T. Silva, C. G. Silva, J. L. Faria and L. Liu, Single-atom Ir and Ru anchored on graphitic carbon nitride for efficient and stable electrocatalytic/photocatalytic hydrogen evolution, Appl. Catal., B, 2022, 310, 121318 CrossRef CAS.
- W. Sun, S. Jin, W. Du, X. Kang, A. Chen, S. Wang, H. Sheng and M. Zhu, Total Structure Determination of the Pt1Ag9[P(Ph-F)3]7Cl3 Nanocluster, Eur. J. Inorg. Chem., 2020, 590–594 CrossRef CAS.
- X. Lin, K. Sun, X. Fu, X. Ren, Y. Yang, C. Liu and J. Huang, Correlating Kernel−Shell Structures with Optical Properties of Pt1Ag24 and Pt1Ag14 Nanoclusters, J. Phys. Chem. C, 2021, 125, 2194–2201 CrossRef CAS.
- A. K. Das, S. Mukherjee, S. S. R, A. S. Nair, S. Bhandary, D. Chopra, D. Sanyal, B. Pathak and S. Mandal, Defects Engineering on Ceria and C−C Coupling Reactions Using [Au11(PPh3)7I3] Nanocluster: A Combined Experimental and Theoretical Study, ACS Nano, 2020, 14, 16681–16688 CrossRef CAS PubMed.
- P. Niu, L. Zhang, G. Liu and H.-M. Cheng, Graphene-Like Carbon Nitride Nanosheets for Improved Photocatalytic Activities, Adv. Funct. Mater., 2012, 22, 4763–4770 CrossRef CAS.
- W. Yan, L. Yan and C. Jing, Impact of doped metals on urea-derived g-C3N4 for photocatalytic degradation of antibiotics: structure, photoactivity and degradation mechanisms, Appl. Catal., B, 2019, 244, 475–485 CrossRef CAS.
- X. Zhang, F. Wu, G. Li, L. Wang, J. Huang, A. Song, A. Meng and Z. Li, Mechanistic insight into the synergy between platinum cluster and indium particle dual cocatalysts for enhanced photocatalytic water splitting, J. Colloid Interface Sci., 2024, 670, 774–784 CrossRef CAS PubMed.
- C. Huang, Y. Wen, J. Ma, D. Dong, Y. Shen, S. Liu, H. Ma and Y. Zhang, Unraveling fundamental active units in carbon nitride for photocatalytic oxidation reactions, Nat. Commun., 2021, 12, 320 CrossRef CAS PubMed.
- X. Zhang, F. Wu, G. Li, L. Wang, J. Huang, A. Song, A. Meng and Z. Li, Construction of intramolecular donor-acceptor type carbon nitride for photocatalytic hydrogen production, J. Colloid Interface Sci., 2024, 655, 439–450 CrossRef CAS PubMed.
- A. Ma, Y. Ren, Y. Zuo, J. Wang, S. Huang, X. Ma and S. Wang, Ligand-controlled exposure of active sites on the Pd1Ag14 nanocluster surface to boost electrocatalytic CO2 reduction, Chem. Commun., 2024, 60, 3162–3165 RSC.
- M. Xu, D. Li, K. Sun, L. Jiao, C. Xie, C. Ding and H.-L. Jiang, Interfacial Microenvironment Modulation Boosting Electron Transfer between Metal Nanoparticles and MOFs for Enhanced Photocatalysis, Angew. Chem., Int. Ed., 2021, 60, 16372–16376 CrossRef CAS PubMed.
- M. Xu, X. Ruan, D. Meng, G. Fang, D. Jiao, S. Zhao, Z. Liu, Z. Jiang, K. Ba, T. Xie, W. Zhang, J. Leng, S. Jin, S. K. Ravi and X. Cui, Modulation of Sulfur Vacancies in ZnIn2S4/MXene Schottky Heterojunction Photocatalyst Promotes Hydrogen Evolution, Adv. Funct. Mater., 2024, 34, 2402330 CrossRef CAS.
- X. Yang, Y. Zhang, J. Deng, X. Huo, Y. Wang and R. Jia, Fabrication of Porous Hydrophilic CN/PANI Heterojunction Film for High-Efficiency Photocatalytic H2 Evolution, Catalysts, 2023, 13(1), 139 CrossRef CAS.
- L. Mao, B. Zhai, J. Shi, X. Kang, B. Lu, Y. Liu, C. Cheng, H. Jin, E. Lichtfouse and L. Guo, Supercritical CH3OH-Triggered Isotype Heterojunction and Groups in g-C3N4 for Enhanced Photocatalytic H2 Evolution, ACS Nano, 2024, 18, 13939–13949 CrossRef CAS PubMed.
- L. Wang, Y. Li, Y. Ai, E. Fan, F. Zhang, W. Zhang, G. Shao and P. Zhang, Tracking Heterogeneous Interface Charge Reverse Separation in SrTiO3/NiO/NiS Nanofibers with In Situ Irradiation XPS, Adv. Funct. Mater., 2023, 33, 2306466 CrossRef CAS.
- F. Xing, C. Wang, S. Liu, S. Jin, H. Jin and J. Li, Interfacial Chemical Bond Engineering in a Direct Z-Scheme g-C3N4/MoS2 Heterojunction, ACS Appl. Mater. Interfaces, 2023, 15, 11731–11740 CrossRef CAS PubMed.
- H. Wang, Z. Chen, Y. Shang, C. Lv, X. Zhang, F. Li, Q. Huang, X. Liu, W. Liu, L. Zhao, L. Ye, H. Xie and X. Jin, Boosting Carrier Separation on a BiOBr/Bi4O5Br2 Direct Z-Scheme Heterojunction for Superior Photocatalytic Nitrogen Fixation, ACS Catal., 2024, 14, 5779–5787 CrossRef CAS.
- H. Shi, C. Li, L. Wang, W. Wang, J. Bian and X. Meng, Photocatalytic reduction of nitrate pollutants by novel Z-scheme ZnSe/BiVO4 heterostructures with high N2 selectivity, Sep. Purif. Technol., 2022, 300, 121854 CrossRef CAS.
- G. Kresse and J. Hafner, Ab initio molecular dynamics for liquid metals, Phys. Rev. B:Condens. Matter Mater. Phys., 1993, 47, 558–561 CrossRef CAS PubMed.
- G. Kresse and J. Furthmuller, Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set, Phys. Rev. B:Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed.
- W. Tang, E. Sanville and G. Henkelman, A grid-based Bader analysis algorithm without lattice bias, J. Phys. Condens. Matter, 2009, 21, 084204 CrossRef CAS PubMed.
- L. Xu, M. Yang, S. J. Wang and Y. P. Feng, Electronic and optical properties of the monolayer group-IV monochalcogenides M X (M = Ge, Sn; X = S, Se, Te), Phys. Rev. B, 2017, 95, 235434 CrossRef.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- X. Jia, J. Wang, Y. Lu, J. Sun, Y. Li, Y. Wang and J. Zhang, Designing SnS/MoS2 van der Waals heterojunction for direct Z-scheme photocatalytic overall water-splitting by DFT investigation, Phys. Chem. Chem. Phys., 2022, 24, 21321–21330 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2380779. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5sc01735a |
| ‡ D. Tan, T. Ding and K. Shen contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |