
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSolid-state temperature-dependent luminescence of C,C′-diaryl-o-carboranes based on restriction of excited-state structural relaxation†
Kazuhiro
Yuhara
a and
Kazuo
Tanaka
 *ab
*ab
aDepartment of Polymer Chemistry, Graduate School of Engineering, Kyoto University, Katsura, Nishikyo-ku, Kyoto 615-8510, Japan. E-mail: tanaka@poly.synchem.kyoto-u.ac.jp
bDepartment of Technology and Ecology, Graduate School of Global Environmental Studies, Kyoto University, Katsura, Nishikyo-ku, Kyoto 615-8510, Japan
First published on 17th March 2025
Abstract
Here, we show mechanistic analyses of solid-state temperature-dependent luminescence of C,C′-diaryl-o-carborane derivatives. Solid-state stimuli-responsiveness is a promising property to visualize environmental changes on the molecular scale. Thus, mechanistic study of responsiveness is meaningful for broadening the scope of materials. We previously reported solid-state temperature-dependent luminochromism of C-anthryl-o-carborane derivatives. Among them, we re-focused on C-anthryl-C′-phenyl-o-carborane as a prototypical compound and synthesized derivatives with electronically different substituents on aryl units. Based on extensive analyses with crystallography, variable temperature optical measurements and theoretical calculation, we show that luminochromic behaviors can be explained by partially restricted excited-state elongation of the carbon–carbon bond in the cluster. Our results indicate that thermal expansion and contraction of the crystalline lattice should play a key role in modulating the degree of excited-state structural relaxation including the bond elongation in the cluster.
Introduction
Stimuli-responsive materials change their chemical structures and physical properties in response to external stimuli. These materials can convert applied stimuli and environmental changes to observable signals. Numerous materials have been reported to have various responsiveness toward external stimuli with changing their properties.1,2 Luminescent color change is one of the important outputs for stimuli detection due to its reproducibility, repeatability, and high sensitivity. Some molecular skeletons show unique responsiveness utilizing excited-state processes such as twisted intramolecular charge transfer (TICT),3 excited-state intramolecular proton transfer (ESIPT),4 and excimer/exciplex formation.5 Basically, luminescent molecules with such processes have large Stokes shifts, which enables us to recognize luminescent color change in response to stimuli.6 Recently, excited-state structural relaxation processes of flexible π-conjugated skeletons have gained attention in the molecular science and material fields.6,7 Upon excitation, such flexible molecules structurally transform to stable structures in the excited state. In an environment where structural change freely occurs, excited molecules can adopt a completely relaxed geometry. In contrast, when the relaxation is partially restricted by the surrounding environment or external stimuli, these compounds show luminescent color change. Based on this excited-state dynamics, stimuli-responsive properties have been reported with flexible π-conjugated skeletons, mainly focusing on responsiveness in solution states.8–11 At this stage, solid-state stimuli-responsive luminescent molecules with flexible skeletons have rarely been reported.12,13 Those molecules have advantages of high sensitivity visualizing slight external environmental change based on the abundant stable structures in the excited state even though stimuli-responsiveness accompanying structural change is often cancelled by densely-packed surrounding molecules in film or crystalline materials. Therefore, the development of molecules with both flexible skeletons and stimuli-responsiveness has been regarded as a challenging research target. Additionally, when we aim for practical usages, solid-state stimuli-responsive luminescent molecules can be regarded as promising candidates because they are suitable for optoelectronic applications, such as organic light-emitting diodes, solid-state lasers and sensing devices, owing to the durability of the compound against heat and chemical stimuli without vaporization of the solvent and large volume change.14,15 This is a major difference in comparison with a conventional chromophore, which is highly emissive only in the solution state, but suffers from quenching in the solid state due to intermolecular interactions such as π–π interactions.16We have focused on the icosahedral compound o-carborane as a key skeleton of solid-state stimuli-responsive luminescent materials.17o-Carborane is representative of a class of boron cluster compounds composed of two carbon and ten boron atoms in their core units.18,19 Optical properties of C-aryl-o-carborane derivatives have attracted attention from photophysical aspects during the last decade20–25 and been analyzed in theoretical studies in recent years.26–29 One of the impressive optical properties is intramolecular charge transfer (ICT) emission.30 Owing to the electron deficiency of boron atoms when connected to π-conjugated units, o-carborane can work as an electron acceptor with orbital conjugation. After photoexcitation, a locally excited state of the π-units is formed, where molecular geometry is close to that of the Franck–Condon state. Subsequent structural relaxation forms the ICT state through electronic charge transfer from the π-units to the boron cluster. During the relaxation process, the carbon–carbon bond in the o-carborane unit (Ccage–Ccage) elongates due to the electronic conjugation from π-orbitals of aryl groups to anti-bonding σ*-orbital of the Ccage–Ccage bond.30–32 In this process, the Ccage–Ccage bonds adopt a perpendicular conformation to the π-orbitals of aryl groups and elongate to ca. 2.3 Å.26,30 Some of the C-aryl derivatives have an aggregation-induced emission (AIE)1,16 property based on ICT emission. Only trace of luminescence is observed in solution, while intense emission in aggregation and/or solid can be obtained. The origin of the AIE phenomena is often described in terms of suppression of non-radiative decay in the aggregated state.33–36 Our group experimentally demonstrated that the Ccage–Ccage bond elongation process plays a key role in the photophysical process in the excited state leading to the AIE property.26 Additionally, previous studies have discussed the mechanism based on energetic accessibility to a non-emissive dark CT state with a structure of further elongated Ccage–Ccage bonds (ca. 2.6 Å) and parallel conformation of the Ccage–Ccage bond to aryl π-orbitals.27,29 Luminescence can be quenched by structural relaxation in the solution state, while the rigid environment in the solid state partially restricts the relaxation, which should lead to AIE phenomena. Based on the above-mentioned excited-state dynamics of o-carborane derivatives and solid-state/solution-state luminescent properties, various types of stimuli-responsive materials have been reported by utilizing π-conjugated scaffolds such as anthracene,37–41 pyrene,40,42–47 carbazole,22,48,49 triphenylanime,50 and bis(arylethynyl)benzene.51,52
Here, we report mechanistic analyses of temperature-dependent luminescent properties of C,C′-diaryl-o-carborane derivatives. We previously reported solid-state temperature-dependent luminochromism of C-anthryl-o-carborane derivatives.32 Theoretical study by Su et al. suggested the critical role of the Ccage–Ccage bond elongation process.28 In this paper, we focus on the C-phenyl-C′-anthryl derivative (H–H), which we regard as a prototypical compound with C,C′-diaryl units for further functionalization. Through an organic synthetic approach, we introduced electronically different substituents on the phenyl or/and anthryl groups and clarified the mechanism of thermo-responsive luminescent properties based on the results of photophysical measurements, crystallographic data, and thermal analysis. Moreover, these optical properties were well realized by theoretical calculations. To the best of our knowledge, we firstly indicate the crucial role of solid-state partial Ccage–Ccage bond elongation in the temperature-dependent luminescent properties of C,C′-diaryl-o-carborane derivatives both experimentally and theoretically.
Results and discussion
Synthesis
The synthetic scheme of C,C′-diaryl-o-carborane derivatives is shown in Scheme 1 (see the ESI† for details). We introduced electron-donating 2-thienyl or electron-accepting bromo groups on phenyl and/or 9-anthryl moieties. The prototypical compound, H–H, was prepared by the alkyne insertion reaction53,54 with 9-(phenylethynyl)anthracene and decarborane (B10H14) according to previous literature.32 The mono- and bis-brominated derivatives Br–H and Br–Br were synthesized through nucleophilic aromatic substitution with Br2 and H–H, and alkyne insertion with 9-bromo-10-((4-bromophenyl)ethynyl)anthracene and B10H14, respectively. Afterwards, Suzuki–Miyaura coupling reactions of Br–H and Br–Br with 2-thipheneboronic acid afforded mono-thienyl derivative T–H and bis-thienyl derivative T–T, respectively. | ||
| Scheme 1 Synthetic schemes of C,C′-diaryl-o-carborane derivatives. | ||
We also performed co-oligomerization reactions with fluorene or strongly electron-donating bithiophene used as comonomer units. Sonogashira–Hagihara coupling reactions between Br–Br and corresponding diethynyl-aryl comonomers afforded alternating co-oligomers with fluorene (o-FL) and bithiophene (o-BT), respectively. Ethynylene spacers were incorporated to reduce steric hindrance of chain-ends during propagation reactions between V-shaped diaryl-o-carborane and bis-brominated monomers. The products showed good solubility in conventional organic solvents, such as chloroform (CHCl3), dichloromethane and tetrahydrofuran. According to the physical properties shown in the ESI,† we concluded that polymeric products should be long enough for evaluating the influence of extension of π-conjugation through the main-chains on electronic properties.
X-ray diffraction analysis
Crystal structures of H–H, Br–H, Br–Br, T–H, and T–T were determined using single-crystal X-ray diffraction (SCXRD) analyses (Fig. 1 and Table S1†). We collected crystallographic data from all compounds under the same conditions, although the data for H–H were previously reported. From diffraction data, molecular structures of the derivatives were unambiguously characterized. All compounds had V-shaped geometries with phenyl and anthryl groups arranged almost perpendicular to the Ccage–Ccage bonds. The bond lengths between carbon atoms were within a narrow range of 0.01 Å (1.820(4)–1.830(5) Å) and longer than that of parent o-carborane (1.62 Å).55 This is because steric repulsion should occur between phenyl and anthryl groups. Additionally, the five compounds had similar values of geometrical parameters such as the dihedral angles of the Ccage–Ccage bond with the anthryl group and the distance between phenyl and anthryl units (Table S2†). These results indicate that the derivatives have similar ground-state geometry independent of the substituents.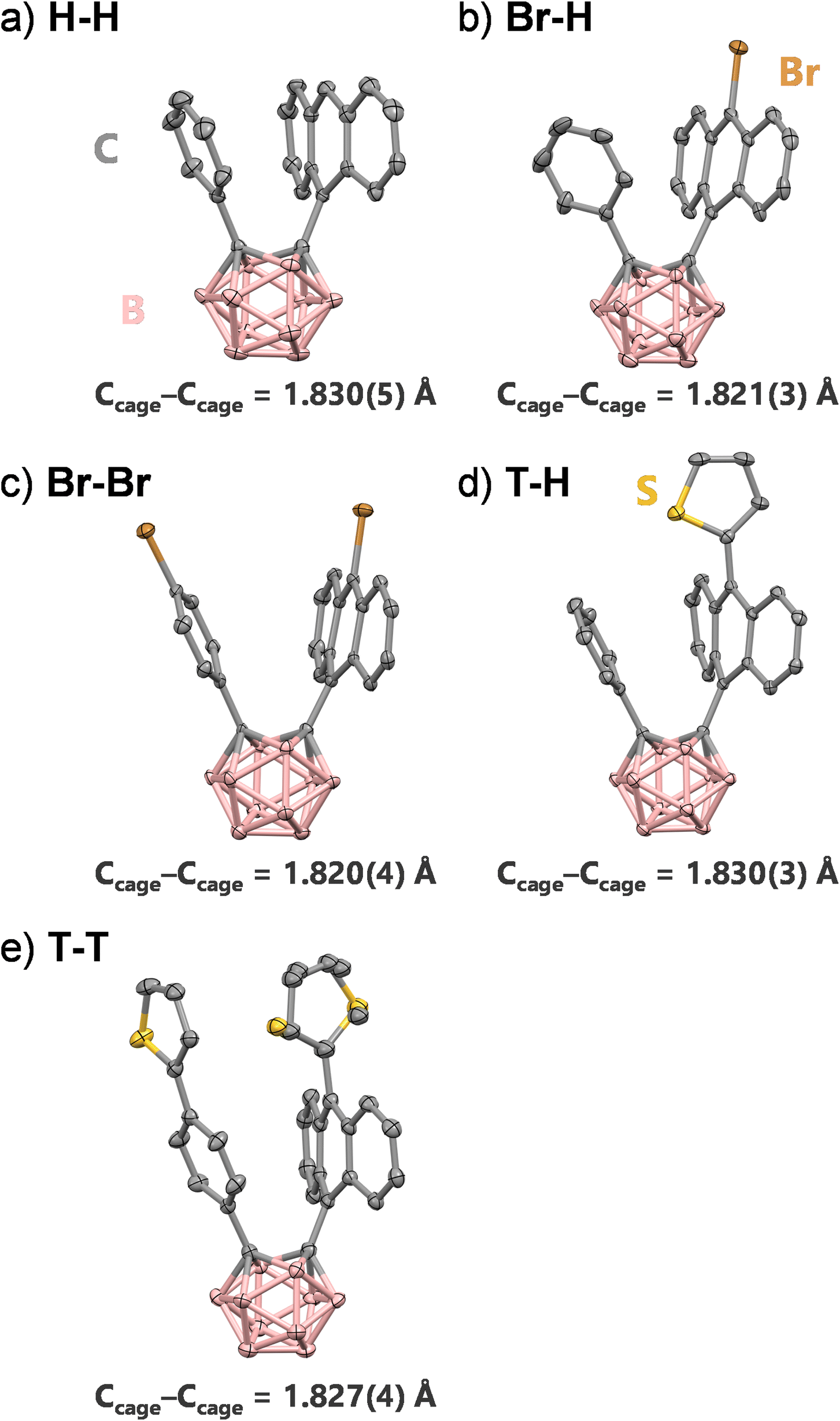 | ||
| Fig. 1 ORTEP drawings of (a) H–H, (b) Br–H, (c) Br–Br, (d) T–H and (e) T–T. Thermal ellipsoids are drawn at 50% probability level. Hydrogen atoms are omitted for clarity. The thiophene ring on the anthracene unit of T–T is refined with ring flipping disorder. Colors: B, pink; C, grey; S, yellow; Br, brown. | ||
The temperature dependency of the crystalline structures was examined for H–H (Tables S3 and S4†). Diffraction data measured at 143 K were compared with those obtained at 208 and 243 K. In all data, H–H crystallized in the same space group (P21/n) and its molecular geometries were hardly affected by temperature difference (Table S5†). However, the lattice constant was affected by temperature change. We observed a cell volume (Vcell) expansion along with temperature increasing (143 K, 2172.9(19) Å3; 208 K, 2180.6(7) Å3; 243 K, 2183.5(8) Å3). This change should originate from molecular vibration activated by external heat. The void volume in the unit cell (Vvoid) also increased linearly with temperature elevation (Fig. S1;† 143 K, 253 Å3; 208 K, 262 Å3; 243 K, 267 Å3). Additionally, the space occupied by each molecule (Vmol) was calculated by (Vcell − Vvoid)/Z, where Z denotes the number of molecules in the asymmetric unit. The Vmol values were almost the same at all temperatures (143 K, 480 Å3; 208 K, 480 Å3; 243 K, 479 Å3). These results suggest that each molecule in the crystalline lattice gains more available space for accompanying excited-state structural change in higher temperature regions.
Crystallinity of the compounds was investigated with powder X-ray diffraction (PXRD) analyses. Solid samples of H–H, Br–H, Br–Br, T–H, and T–T were prepared by recrystallization from hot CHCl3/MeOH solutions. All peak positions in PXRD patterns were well matched with those simulated from SCXRD data (Fig. S2†). This correspondence supports that the as-synthesized solid samples have sufficient crystallinity to discuss solid-state luminescent properties considering SCXRD structures.
Optical properties
Optical measurements were conducted to obtain information about the electronic structures of the compounds (Fig. 2, S3–S14, Tables 1, S6 and S7†). UV-visible absorption spectra were measured in CHCl3. Compared to H–H, it was found that the maximum absorption wavelengths (λabs) of the lowest energy band bathochromically shifted by 24 nm (T–H) and 26 nm (T–T), implying that the introduction of electron-donating thiophene units could contribute to lowering the energy gaps. o-FL and o-BT exhibited even more red-shifted absorption than small molecules. This result indicates that electronic conjugation should be developed through polymer main-chains. Photoluminescence (PL) in CHCl3 was also red-shifted with a similar tendency to absorption. Luminescent maximum wavelengths of PL bands (λPL) of o-FL and o-BT reached the NIR region (o-FL, 709 nm; o-BT, 743 nm). To examine the origin of this tendency, energy levels of frontier molecular orbitals were determined with cyclic voltammetry (CV) measurements (Fig. S18 and S19†). Accordingly, energy levels of the highest occupied molecular orbital (HOMO) were slightly elevated by the introduction of thiophene units (H–H, −5.72 eV; T–H, −5.55 eV; T–T, −5.62 eV), while those of the lowest unoccupied molecular orbital (LUMO) remained at almost the same level (H–H, −3.57 eV; T–H, −3.61 eV; T–T, −3.56 eV). CV results indicate that narrower energy gaps should originate from increases in HOMO levels by the introduction of electron-donating groups. This effect is especially observed in o-BT which has bithiophene as the strongest electron-donating group, resulting in NIR emission. To determine luminescent species, we performed optical measurements in various solvents (Fig. S3, S4 and Table S6†). For all compounds, larger Stokes shifts were observed in solvents with higher polarity, clearly meaning that all luminescent bands have ICT characters. For small molecules, very weak PL was observed (luminescent quantum yield (ΦPL) ≤ 0.01) in the solution state, similarly to typical C-aryl-substituted o-carboranes.22,30,32,56 This is because the non-radiative pathways should be opened during excited-state structural relaxation.26 | ||
| Fig. 2 Normalized UV-visible absorption (Abs.) and photoluminescence (PL) spectra of C,C′-diaryl-o-carborane derivatives (solution: chloroform, 1.0 × 10−5 M). | ||
| Compound | State | λ abs/nm | λ PL /nm | Φ PL , | τ PL /ns |
|---|---|---|---|---|---|
| a 1.0 × 10−5 M in CHCl3 solution (per repeating units for oligomers). b Prepared by a spin-coating method. c Excited at λabs. d Absolute quantum yield determined by an integrating sphere method. e Excited at 369 or 375 nm. —: not measured. n.d.: cannot be determined reliably due to weak luminescence. | |||||
| H–H | Solutiona | 408 | 643 | 0.01 | n.d. |
| Crystal | — | 592 | 0.70 | 7.7 | |
| Br–H | Solutiona | 419 | 637 | <0.01 | n.d. |
| Crystal | — | 619 | 0.26 | 2.9 | |
| Br–Br | Solutiona | 422 | 643 | <0.01 | n.d. |
| Crystal | — | 625 | 0.26 | 3.2 | |
| T–H | Solutiona | 432 | 676 | <0.01 | n.d. |
| Crystal | — | 637 | 0.66 | 7.5 | |
| T–T | Solutiona | 434 | 677 | <0.01 | n.d. |
| Crystal | — | 671 | 0.22 | 3.5 | |
| o-FL | Solutiona | 478 | 709 | 0.05 | n.d. |
| Filmb | 491 | 700 | 0.06 | 1.0 (56%), 0.2 (15%), 2.8 (29%) | |
| o-BT | Solutiona | 479 | 743 | 0.01 | n.d. |
| Filmb | 494 | 729 | 0.02 | n.d. | |
Solid-state luminescent properties were measured with crystal samples of the compounds and oligomer films (Fig. 2 and Table 1). The maximum emission wavelengths were red-shifted by the introduction of electron-donating groups as observed in the solution state (H–H, 592 nm; T–H, 637 nm; T–T, 671 nm) originating from elevation of HOMO energy levels. In all compounds, higher ΦPL values in the solid state were observed than those in solution, representing that these compounds have AIE properties. In addition, in all compounds the λPL values were lower than those in CHCl3 and greater than those in low-polarity solvent (hexane or CCl4). The degree of structural relaxation of the ICT-emissive aryl-o-carborane system can be strongly affected by the polarity around the molecule.20,26,38,56 Then depending on the polarity, the solution-state luminescent wavelength can be located at both shorter and longer wavelength regions in comparison with that in the solid state.
Temperature dependency of solid-state luminescence
The temperature dependency of luminescent properties was examined to gain deeper insight into the relaxation process in the excited state. Solid-state variable temperature PL (VTPL) properties were monitored (Fig. 3 and Table S7†). Notably, VTPL spectra of oligomers were recorded below glass transition temperatures (ca. 340 K determined by differential scanning calorimetry (DSC) (Fig. S21†)) to exclude consideration of glass transition. All compounds showed continuous red shifts of luminescent bands with increasing temperature, although wavelength regions were dependent on the substituents on aryl units as we observed for H–H previously (Table S7†).32 The maximum luminescent wavelengths progressively increased with temperature rising (e.g.H–H, 576 nm (80 K), 587 nm (230 K), and 607 nm (410 K); Br–Br, 617 nm (80 K), 622 nm (230 K), and 640 nm (410 K); T–T, 654 nm (80 K), 668 nm (230 K), and 693 nm (410 K)). As we observed from the temperature-dependent X-ray diffraction of H–H (Fig. S1, Tables S3 and S4†), solid-state organic compounds have lower density at higher temperature due to thermal expansion of the crystal lattice.57 It is suggested that this lattice expansion should form larger void volume in crystals where a larger degree of excited-state structural relaxation can be allowed. To discuss the temperature effect of solid-state molecular conditions on luminescence, thermogravimetric analysis (TGA) and DSC were performed (Fig. S20 and S21†). Within the temperature ranges for PL measurements, TGA and DSC curves indicated that significant weight losses and thermal phase transitions should hardly proceed in the solid state. Based on these results, we can exclude the possibility of decompositions and thermal phase transitions during VTPL measurements. | ||
| Fig. 3 Solid-state temperature-dependent luminescence spectra of C,C′-diaryl-o-carboranes. | ||
Additionally, we investigated the temperature dependency of PL quantum yields and lifetimes (Fig. S8–S14 and Table S7†). A downward trend of quantum yield and shortening of lifetime were observed with temperature increasing. According to these values, radiative and non-radiative decay constants (kr and knr, respectively) were calculated (Fig. S15, S16 and Table S7†). It was revealed that kr values were almost independent or lowered with temperature increasing. In contrast, knr values exponentially increased with temperature although each compound had a different extent of increase. T–T, o-FL, and o-BT had knr values one order of magnitude larger than those of other compounds, especially in higher temperature regions. Due to the narrower energy gaps, these three compounds should potentially have more non-radiative decay pathways as explained by the energy gap law.58 Therefore, emission quenching proceeds by heating. Larger knr values of brominated derivatives (Br–H and Br–Br) than H–H in the whole temperature range can be explained by the heavy atom effect of bromine atoms, which may accelerate intersystem crossing from the singlet to the triplet excited dark state.59 These kinetic analyses suggest that molecular thermal vibration in the excited state should accelerate at higher temperature, which can lead to non-radiative decay pathways. Temperature-dependent luminescent properties were also investigated in the solution state (Fig. S17†), where larger molecular structural change can be allowed than in the solid state. As we expected, drastic spectral change was observed in the 2-methyltetrahydrofuran solution of H–H, while the spectra were strongly affected by glass transition of the solvent and change in dielectric constant along with temperature changes.60 This result suggests that the surrounding environment strongly influences the luminescent properties of the derivatives.
Theoretical calculation
To gain a mechanistic insight into the photophysical properties, we performed theoretical calculation on H–H, Br–H, Br–Br, T–H, and T–T. Density functional theory (DFT) and time-dependent DFT (TD-DFT) calculations were used with the C.01 version of the Gaussian 16 package.61 After screening of the functionals and basis sets (Table S8†), calculation data were obtained by the CAM-B3LYP functional with LanL2DZ (Br atoms) and 6-31+G(d,p) (all other atoms) basis sets, which can provide the most reproduced molecular geometries close to those of crystal structures. We focused on the molecular structures in both the ground state (S0) and the first excited state (S1). The solvent effect of CHCl3 was modeled with the polarized continuum model62–64 in the solution state. Quantum mechanics and molecular mechanics (QM/MM) calculation26,30,65 was used for structural optimization in the crystalline state. The molecular coordinates for QM/MM analyses were extracted from the single-crystal structures (Fig. S23†). One central molecule was treated as a QM molecule using the TD-DFT method and the surrounding molecules were treated as frozen state by the MM method.66 Under these conditions, molecular structures were optimized in the S0 and S1 states.Firstly, from energy levels and distribution of frontier molecular orbitals, luminescent characters of the derivatives were evaluated (Fig. 4). Compared to H–H, electron-donating thiophene units induced an elevation of the HOMO level resulting in a narrower energy gap between HOMO and LUMO as calculated from the CV data. This result supports red-shifted absorption and luminescent bands of T–H and T–T compared with H–H. Indeed, TD-DFT calculation reproduced the trends of the absorption and luminescence wavelengths, and the validity of selection of our calculation level was supported (Table 2). Additionally, luminescence was driven by electronic transition between HOMO and LUMO in the S1 state. Emission species were characterized by distribution analysis of orbitals. In the S1 state, HOMOs were mainly located on anthracene and o-carborane moieties, whereas LUMOs were spread over the whole molecules. Along with the luminescent transition, the charge transfer between aryl units and o-carborane moieties should occur, supporting our characterization of ICT luminescence. The oligomerization was supposed to enhance ICT character through introduction of electron-donating groups (fluorene and bithiophene). As a result, far more red-shifted luminescence should be induced compared to other compounds.
 | ||
| Fig. 4 Calculated Kohn–Sham frontier orbital distribution and S0–S1 electronic transition energy in nm for energy-minimum geometries of S0 and S1 states (colors: boron, pink; carbon, grey; sulfur, yellow; bromine, red). Solvent effect of chloroform was considered with polarized continuum model. f denotes the oscillator strength. Isovalues are 0.02. CAM-B3LYP functional with 6-31+G(d,p) (for H, B, and C) and LanL2DZ (for Br) basis sets were used. Red and blue line plots indicate HOMO and LUMO energy levels, respectively. | ||
| Compound | Solution | Crystal | ||||
|---|---|---|---|---|---|---|
| Ccage–Ccagea/Å | λ em calc | λ em exp | Ccage–Ccagec/Å | λ em calc | λ em exp | |
| a Solvent effect of chloroform was considered with polarized continuum model. b Measured in chloroform solution (1.0 × 10−5 M). c Estimated by QM/MM analyses. | ||||||
| H–H | 2.334 | 586 | 643 | 2.216 | 517 | 592 |
| Br–H | 2.290 | 602 | 637 | 2.230 | 515 | 619 |
| Br–Br | 2.293 | 606 | 643 | 2.254 | 520 | 625 |
| T–H | 2.305 | 631 | 676 | 2.238 | 537 | 637 |
| T–T | 2.307 | 636 | 677 | 2.227 | 553 | 671 |
Next, we found stark differences in the molecular structures between ground and excited states. The most characteristic structural difference was observed in the Ccage–Ccage bond length (Table 2). In the solution state, the bond lengths were 1.778–1.791 Å in the ground state; meanwhile bond extension was observed to 2.290–2.334 Å in the excited state. In the crystalline state, the excited-state Ccage–Ccage bond elongation was also induced to 2.216–2.254 Å. These results suggest that the bond elongation process should be one of the main excited-state structural relaxation processes. The suppression of relaxation calculated in crystals can induce smaller Stokes shifts in solid than in CHCl3 solution. Additionally, these relaxation behaviors should lead to a non-radiative decay process. In the solution state, emission from C,C′-diaryl-o-carborane derivatives is quenched by free structural relaxation. However, in the crystalline state, such processes should be partially restricted by surrounding molecules, and subsequently strong AIE properties can be induced.
Finally, thermochromic luminescence was theoretically analyzed. Correlation between the Ccage–Ccage bond length and electronic states suggested the role of the Ccage–Ccage bond in progressive shifts of luminescent bands upon temperature change. The S0 and S1 potential energy curves (PESs) were calculated through the scan method with variation of the Ccage–Ccage bond length (Fig. 5 and S24†). By changing the Ccage–Ccage bond length between 1.8 Å and 2.7 Å in 0.1 Å increments, energy levels were estimated in both ground state (Sopt0) and excited state (Sopt1) and corresponding vertically de-excited state (SFC0). The energy difference between Sopt1 and SFC0 states represents luminescent energy with consideration of the Franck–Condon approximation. In the S0 state, elongation of the bond resulted in monotonic increase of potential energy, while the S1 PES had a global minimum around 2.3 Å in the solution state. Similar results were obtained in our previous system with relaxation through the Ccage–Ccage bond elongation.26 Here we applied the same method to QM/MM analyses in crystals. PES in the crystalline state showed a steeper energy change along with the lengths of the Ccage–Ccage bond compared to those in the solution state. This difference suggests that structural relaxation processes in the solid were restricted because solid-state molecules were confined in the condensed packing state. The global minimum in the crystalline state is located around 2.2 Å, reflecting restriction of relaxation. Moreover, differences of energy levels between Sopt1 and SFC0 states are decreased by increasing the Ccage–Ccage bond length. These data correspond to red shifts of calculated luminescent wavelengths along with the Ccage–Ccage bond elongation (Fig. S24†). Based on the above analyses, we propose a plausible description of temperature-dependent consecutive spectral shifts of solid-state luminescence. By heating solid-state samples, void volume increased. This structural alteration provided more void volumes as enlarged available spaces to allow a larger degree of excited-state structural relaxation mainly triggered through the Ccage–Ccage bond elongation. Then incomplete, but larger degree of elongation results in luminescence in longer wavelength regions at higher temperatures. To the best of our knowledge, this is the first paper to provide discussions combining experimental and theoretical analyses for progressive wavelength shift upon temperature change of solid-state aryl-o-carborane derivatives.
 | ||
| Fig. 5 Energy levels of the S0 and S1 states of H–H with variable Ccage–Ccage bond lengths in the (a) solution state and (b) crystalline state. Sopt0 and Sopt1 denote energy levels of optimized geometries in ground state and excited state, respectively. SFC0 denotes energy levels calculated with single-point energy calculation using S1 excited-state optimized geometries. | ||
Conclusion
In this paper, we performed mechanistic analyses of temperature-dependent luminescence of C,C′-diaryl-o-carborane derivatives in the solid states. Six novel derivatives were synthesized via bromine-substituted derivatives as key intermediates. Temperature dependency of optical properties and theoretical calculations indicate that the compounds can show Ccage–Ccage bond elongation as a key excited-state structural relaxation pathway. Thermally induced expansion or contraction of the crystalline lattice affected the crystal void volume which can be accompanied by excited-state structural changes. The consecutive shifts of luminescence bands were able to be explained by the changes in the degree of relaxation upon thermally induced micro-environmental alteration in the crystalline state. Our results are meaningful for the study of excited-state chemistry and material applications of C-aryl-o-carborane derivatives. These mechanistic analyses of excited-state photophysical dynamics and exploration of stimuli-responsiveness of aryl-o-carborane derivatives are promising for significant information for creating advanced sensing materials.Data availability
The data supporting this article have been included as part of the ESI.† Crystallographic data for H–H (measured at three different temperatures), Br–H, Br–Br, T–H and T–T have been deposited at the CCDC under 2411824–2411828, 2425738, and 2425739.Author contributions
K. Y. performed experiments, wrote the original draft and conducted the data analysis. K. T. supervised the project. Both authors discussed the results and contributed to editing the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was partially supported by the Asahi Glass Foundation and JSPS KAKENHI Grant Number 24K01570.References
- J. Mei, Y. Hong, J. W. Y. Lam, A. Qin, Y. Tang and B. Z. Tang, Aggregation-induced emission: The whole is more brilliant than the parts, Adv. Mater., 2014, 26, 5429–5479 CrossRef CAS.
- M. A. C. Stuart, W. T. S. Huck, J. Genzer, M. Müller, C. Ober, M. Stamm, G. B. Sukhorukov, I. Szleifer, V. V Tsukruk, M. Urban, F. Winnik, S. Zauscher, I. Luzinov and S. Minko, Emerging applications of stimuli-responsive polymer materials, Nat. Mater., 2010, 9, 101–113 CrossRef.
- S. Sasaki and G. P. C Drummen, Recent advances in twisted intramolecular charge transfer (TICT) fluorescence and related phenomena in materials chemistry, J. Mater. Chem. C, 2016, 4, 2731 RSC.
- P. Gayathri, M. Pannipara, A. G. Al-Sehemi and S. P. Anthony, Recent advances in excited state intramolecular proton transfer mechanism-based solid state fluorescent materials and stimuli-responsive fluorescence switching, CrystEngComm, 2021, 23, 3771–3789 RSC.
- X. Zhang, S. Wang, Z. Fu, C. Wang, Z. Yang, Y. Gao, H. Liu, S. T. Zhang, C. Gu and B. Yang, Switching Monomer-to-Excimer Fluorescence by Noncovalent Interaction Competition Strategy, Adv. Funct. Mater., 2023, 33, 2301228 CrossRef CAS.
- Z. Zhang, W. Song, J. Su and H. Tian, Vibration-Induced Emission (VIE) of N,N′-Disubstituted-Dihydribenzo[a,c]phenazines: Fundamental Understanding and Emerging Applications, Adv. Funct. Mater., 2020, 30, 1902803 CrossRef CAS.
- C. Yuan, S. Saito, C. Camacho, S. Irle, I. Hisaki and S. Yamaguchi, A π-conjugated system with flexibility and rigidity that shows environment-dependent RGB luminescence, J. Am. Chem. Soc., 2013, 135, 8842–8845 CrossRef CAS.
- R. Kotani, H. Sotome, H. Okajima, S. Yokoyama, Y. Nakaike, A. Kashiwagi, C. Mori, Y. Nakada, S. Yamaguchi, A. Osuka, A. Sakamoto, H. Miyasaka and S. Saito, Flapping viscosity probe that shows polarity-independent ratiometric fluorescence, J. Mater. Chem. C, 2017, 5, 5248 RSC.
- Z. Zhang, X. Jin, X. Sun, J. Su and D. H. Qu, Vibration-induced emission: Dynamic multiple intrinsic luminescence, Coord. Chem. Rev., 2022, 472, 214768 CrossRef CAS.
- P. Valat, V. Wintgens, J. Kossanyi, L. Biczók, A. Demeter and T. Bérces, Influence of Geometry on the Emitting Properties of 2,3-Naphthalimides, J. Am. Chem. Soc., 1992, 114, 946–953 CrossRef CAS.
- Y. Chen, J. Zhao, H. Guo and L. Xie, Geometry relaxation-induced large stokes shift in red-emitting borondipyrromethenes (BODIPY) and applications in fluorescent thiol probes, J. Org. Chem., 2012, 77, 2192–2206 CrossRef CAS.
- L. Wang, K. Wang, B. Zou, K. Ye, H. Zhang and Y. Wang, Luminescent chromism of boron diketonate crystals: Distinct responses to different stresses, Adv. Mater., 2015, 27, 2918–2922 CrossRef CAS.
- T. Nishiuchi, S. Aibara, T. Yamakado, R. Kimura, S. Saito, H. Sato and T. Kubo, Sterically Frustrated Aromatic Enes with Various Colors Originating from Multiple Folded and Twisted Conformations in Crystal Polymorphs, Chem.–Eur. J., 2022, 28, e202200286 CrossRef CAS.
- M. K. Bera, P. Pal and S. Malik, Solid-state emissive organic chromophores: design, strategy and building blocks, J. Mater. Chem. C, 2020, 8, 788–802 RSC.
- C. Pan, K. Sugiyasu, Y. Wakayama, A. Sato and M. Takeuchi, Thermoplastic fluorescent conjugated polymers: Benefits of preventing π-π Stacking, Angew. Chem., Int. Ed., 2013, 52, 10775–10779 CrossRef CAS.
- Y. Hong, J. W. Y. Lam and B. Z. Tang, Aggregation-induced emission, Chem. Soc. Rev., 2011, 40, 5361–5388 RSC.
- J. Ochi, K. Tanaka and Y. Chujo, Recent Progress in the Development of Solid-State Luminescent o-Carboranes with Stimuli Responsivity, Angew. Chem., Int. Ed., 2020, 59, 9841–9855 CrossRef CAS.
- T. L. Heying, J. W. Ager, S. L. Clark, D. J. Mangold, H. L. Goldstein, M. Hillman, R. J. Polak and J. W. Szymanski, A New Series of Organoboranes. I. Carboranes from the Reaction of Decaborane with Acetylenic Compounds, Inorg. Chem., 1963, 2, 1089–1092 CrossRef CAS.
- R. N. Grimes, Carboranes, Elsevier, 2nd edn, 2011 Search PubMed.
- L. Weber, J. Kahlert, R. Brockhinke, L. Böhling, J. Halama, A. Brockhinke, H. G. Stammler, B. Neumann, C. Nervi, R. A. Harder and M. A. Fox, C,C′-Bis(benzodiazaborolyl)dicarba-closo-dodecaboranes: Synthesis, structures, photophysics and electrochemistry, Dalton Trans., 2013, 42, 10982–10996 RSC.
- R. Huang, H. Liu, K. Liu, G. Wang, Q. Liu, Z. Wang, T. Liu, R. Miao, H. Peng and Y. Fang, Marriage of Aggregation-Induced Emission and Intramolecular Charge Transfer toward High Performance Film-Based Sensing of Phenolic Compounds in the Air, Anal. Chem., 2019, 91, 14451–14457 CrossRef CAS PubMed.
- S. H. Lee, M. S. Mun, J. H. Lee, S. Im, W. Lee, H. Hwang and K. M. Lee, Impact of the Electronic Environment in Carbazole-Appended o-Carboranyl Compounds on the Intramolecular-Charge-Transfer-Based Radiative Decay Efficiency, Organometallics, 2021, 40, 959–967 CrossRef CAS.
- F. Aniés, I. Hamilton, C. S. P. De Castro, F. Furlan, A. V. Marsh, W. Xu, V. Pirela, A. Patel, M. Pompilio, F. Cacialli, J. Martín, J. R. Durrant, F. Laquai, N. Gasparini, D. D. C. Bradley and M. Heeney, A Conjugated Carboranyl Main Chain Polymer with Aggregation-Induced Emission in the Near-Infrared, J. Am. Chem. Soc., 2024, 146, 13607–13616 CrossRef PubMed.
- N. Shida, S. Owaki, H. Eguchi, T. Nishikawa, I. Tomita and S. Inagi, Bis(pentafluorophenyl)-o-carborane and its arylthio derivatives: Synthesis, electrochemistry and optical properties, Dalton Trans., 2020, 49, 12985–12989 RSC.
- T. Lee, J. H. Jang, N. N. T. Nguyen, J. Jung, J. H. Lee and M. H. Lee, Ortho-Carborane Decorated Multi-Resonance TADF Emitters: Preserving Local Excited State and High Efficiency in OLEDs, Adv. Sci., 2024, 11, 2309016 CrossRef CAS PubMed.
- J. Ochi, K. Tanaka and Y. Chujo, Experimental proof for emission annihilation through bond elongation at the carbon-carbon bond in o-carborane with fused biphenyl-substituted compounds, Dalton Trans., 2021, 50, 1025–1033 RSC.
- L. Ji, S. Riese, A. Schmiedel, M. Holzapfel, M. Fest, J. Nitsch, B. F. E. Curchod, A. Friedrich, L. Wu, H. H. Al Mamari, S. Hammer, J. Pflaum, M. A. Fox, D. J. Tozer, M. Finze, C. Lambert and T. B. Marder, Thermodynamic equilibrium between locally excited and charge-transfer states through thermally activated charge transfer in 1-(pyren-2′-yl)-o-carborane, Chem. Sci., 2022, 13, 5205–5219 RSC.
- Y. C. Duan, Q. Q. Pan, Z. W. Zhao, Y. Gao, Y. Wu, L. Zhao, Y. Geng, M. Zhang and Z. M. Su, Theoretical Simulations of Thermochromic and Aggregation-Induced Emission Behaviors of a Series of Red-Light Anthracene-o-carborane Derivatives, Chem.–Eur. J., 2021, 27, 9571–9579 CrossRef CAS PubMed.
- D. Tahaoǧ Lu, H. Usta and F. Alkan, Revisiting the Role of Charge Transfer in the Emission Properties of Carborane-Fluorophore Systems: A TDDFT Investigation, J. Phys. Chem. A, 2022, 126, 4199–4210 CrossRef PubMed.
- H. Naito, K. Nishino, Y. Morisaki, K. Tanaka and Y. Chujo, Solid-State Emission of the Anthracene-o-Carborane Dyad from the Twisted-Intramolecular Charge Transfer in the Crystalline State, Angew. Chem., Int. Ed., 2017, 56, 254–259 CrossRef CAS PubMed.
- J. Ochi, K. Yuhara, K. Tanaka and Y. Chujo, Controlling the Dual-Emission Character of Aryl-Modified o-Carboranes by Intramolecular CH⋯O Interaction Sites, Chem.–Eur. J., 2022, 28, e202200155 CrossRef CAS PubMed.
- H. Naito, K. Nishino, Y. Morisaki, K. Tanaka and Y. Chujo, Highly-efficient solid-state emissions of anthracene–o-carborane dyads with various substituents and their thermochromic luminescence properties, J. Mater. Chem. C, 2017, 5, 10047–10054 RSC.
- R. Iwai, S. Suzuki, S. Sasaki, A. S. Sairi, K. Igawa, T. Suenobu, K. Morokuma and G. Konishi, Bridged Stilbenes: AIEgens Designed via a Simple Strategy to Control the Non-radiative Decay Pathway, Angew. Chem., Int. Ed., 2020, 59, 10566–10573 CrossRef CAS PubMed.
- S. Suzuki, S. Maeda and K. Morokuma, Exploration of Quenching Pathways of Multiluminescent Acenes Using the GRRM Method with the SF-TDDFT Method, J. Phys. Chem. A, 2015, 119, 11479–11487 CrossRef CAS PubMed.
- R. Respo-Otero, Q. Li and L. Lancafort, Exploring Potential Energy Surfaces for Aggregation-Induced Emission-From Solution to Crystal, Chem.–Asian J., 2019, 14, 700–714 Search PubMed.
- P. Zhou, P. Li, Y. Zhao and K. Han, Restriction of Flip-flop Motion as a Mechanism for Aggregation-Induced Emission, J. Phys. Chem. Lett., 2019, 10, 2024 CrossRef PubMed.
- H. Naito, Y. Morisaki and Y. Chujo, o -Carborane-Based Anthracene: A Variety of Emission Behaviors, Angew. Chem., Int. Ed., 2015, 54, 5084–5087 CrossRef CAS PubMed.
- K. Yuhara, K. Tanaka and Y. Chujo, Regulation of solid-state dual-emission properties by switching luminescence processes based on a bis-o-carborane-modified anthracene triad, Mater. Chem. Front., 2022, 6, 1414–1420 RSC.
- K. Yuhara and K. Tanaka, Direction Switching and Self-Recovering Mechanochromic Luminescence of Anthracene-Modified o-Carboranes, Chem.–Eur. J., 2023, 29, e202301189 CrossRef CAS PubMed.
- H. Yamamoto, J. Ochi, K. Yuhara, K. Tanaka and Y. Chujo, Switching between intramolecular charge transfer and excimer emissions in solids based on aryl-modified ethynyl–o-carboranes, Cell Rep. Phys. Sci., 2022, 3, 100758 CrossRef CAS.
- K. Yuhara and K. Tanaka, The Photosalient Effect and Thermochromic Luminescence Based on o-Carborane-Assisted π-Stacking in the Crystalline State, Angew. Chem., Int. Ed., 2024, 63, e202319712 CrossRef CAS PubMed.
- K. Nishino, H. Yamamoto, K. Tanaka and Y. Chujo, Development of Solid-State Emissive Materials Based on Multifunctional o-Carborane-Pyrene Dyads, Org. Lett., 2016, 18, 4064–4067 CrossRef CAS PubMed.
- T. Yanagihara and K. Tanaka, Photophysical Properties of Pyrene-Substituted nido-Carborate Anion and the Change of Luminescence Species by Counter Cations in Solution and Solid States, Adv. Opt. Mater., 2023, 11, 2300492 CrossRef CAS.
- X. Zhao, X. Zhang, X. Li, L. Wu and L. Ji, Energy Barriers and Gap Between Two Excited States in a Dual-Emissive Carborane, Chem.–Eur. J., 2024, 30, e202401246 CrossRef CAS PubMed.
- K. Nishino, H. Yamamoto, J. Ochi, K. Tanaka and Y. Chujo, Time-Dependent Emission Enhancement of the Ethynylpyrene-o-Carborane Dyad and Its Application as a Luminescent Color Sensor for Evaluating Water Contents in Organic Solvents, Chem.–Asian J., 2019, 14, 1577–1581 CrossRef CAS PubMed.
- K. Nishino, H. Yamamoto, K. Tanaka and Y. Chujo, Solid-State Thermochromic Luminescence through Twisted Intramolecular Charge Transfer and Excimer Formation of a Carborane−Pyrene Dyad with an Ethynyl Spacer, Asian J. Org. Chem., 2017, 6, 1818–1822 CrossRef CAS.
- S. Nishiyama, J. Ochi and K. Tanaka, Design for a Solid-State Luminescent Sensor Through Photo-Induced Electron Transfer Based on Pyrene-Modified o-Carborane, Asian J. Org. Chem., 2025, 14, e202400513 CrossRef CAS.
- J. Li, C. Hou, Q. Qi, L. Jiao, L. Dai, D. Chen, H. Geng, W. Y. Lai and W. Huang, Thermal Energy-Driven Solid-State Molecular Rotation Monitored by Real-Time Emissive Color Switching, CCS Chem., 2022, 4, 2711–2723 CrossRef CAS.
- X. Yu, J. Xu, M. Wang, X. Miao, Q. Fan, D. Tu, Z. Wang, C. Duan, C. Lu, H. Xu and H. Yan, A Hot Exciton Luminogen Constructed by an o-Carborane Scaffold, Adv. Opt. Mater., 2024, 12, 2302218 CrossRef CAS.
- K. Nishino, K. Tanaka and Y. Chujo, Tuning of Sensitivity in Thermochromic Luminescence by Regulating Molecular Rotation Based on Triphenylamine-Substituted o-Carboranes, Asian J. Org. Chem., 2019, 8, 2228–2232 CrossRef CAS.
- H. Mori, K. Nishino, K. Wada, Y. Morisaki, K. Tanaka and Y. Chujo, Modulation of luminescence chromic behaviors and environment-responsive intensity changes by substituents in bis-o-carborane-substituted conjugated molecules, Mater. Chem. Front., 2018, 2, 573–579 RSC.
- K. Wada, K. Hashimoto, J. Ochi, K. Tanaka and Y. Chujo, Rational design for thermochromic luminescence in amorphous polystyrene films with bis-o-carborane-substituted enhanced conjugated molecule having aggregation-induced luminochromism, Aggregate, 2021, 2, e93 CrossRef CAS.
- R. Schaeffer, A New Type Of Substituted Borane, J. Am. Chem. Soc., 1957, 79, 1006–1007 CrossRef CAS.
- A. Toppino, A. R. Genady, M. E. El-Zaria, J. Reeve, F. Mostofian, J. Kent and J. F. Valliant, High yielding preparation of dicarba-closo-dodecaboranes using a silver(I) mediated dehydrogenative alkyne-insertion reaction, Inorg. Chem., 2013, 52, 8743–8749 CrossRef CAS PubMed.
- J. Li, R. Pang, Z. Li, G. Lai, X. Q. Xiao and T. Müller, Exceptionally Long C–C Single Bonds in Diamino-o-carborane as Induced by Negative Hyperconjugation, Angew. Chem., Int. Ed., 2019, 58, 1397–1401 CrossRef CAS PubMed.
- N. Shin, S. Yu, J. H. Lee, H. Hwang and K. M. Lee, Biphenyl-and Fluorene-Based o-Carboranyl Compounds: Alteration of Photophysical Properties by Distortion of Biphenyl Rings Chart 1. Examples of o-Carborane-Based Luminophores, Organometallics, 2017, 36, 1522–1529 CrossRef CAS.
- C. S. Changquan, Thermal Expansion of Organic Crystals and Precision of Calculated Crystal Density: A Survey of Cambridge Crystal Database, J. Pharm. Sci., 2007, 96, 1043–1052 CrossRef PubMed.
- J. V. Caspar and T. J. Meyer, Application of the energy gap law to nonradiative, excited-state decay, J. Phys. Chem., 1983, 87, 952–957 CrossRef CAS.
- B. Valeur, Molecular Fluorescence Principles and Applications, Wiley-VCH Verlag, 2001 Search PubMed.
- P. D. Zoon and A. M. Brouwer, A push-pull aromatic chromophore with a touch of merocyanine, Photochem. Photobiol. Sci., 2009, 8, 345–353 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Rev. C.01, Gaussian, Inc., Wallingford CT Search PubMed.
- G. Scalmani, M. J. Frisch, B. Mennucci, J. Tomasi, R. Cammi and V. Barone, Geometries and properties of excited states in the gas phase and in solution: Theory and application of a time-dependent density functional theory polarizable continuum model, J. Chem. Phys., 2006, 124, 094107 CrossRef PubMed.
- R. Improta, V. Barone, G. Scalmani and M. J. Frisch, A state-specific polarizable continuum model time dependent density functional theory method for excited state calculations in solution, J. Chem. Phys., 2006, 125, 054103 CrossRef PubMed.
- M. Cossi and V. Barone, Time-dependent density functional theory for molecules in liquid solutions, J. Chem. Phys., 2001, 115, 4708–4717 CrossRef CAS.
- Q. Wu, T. Zhang, Q. Peng, D. Wang and Z. Shuai, Aggregation induced blue-shifted emission-the molecular picture from a QM/MM study, Phys. Chem. Chem. Phys., 2014, 16, 5545–5552 RSC.
- A. K. Rappé, C. J. Casewit, K. S. Colwell, W. A. Goddard and W. M. Skiff, UFF, a Full Periodic Table Force Field for Molecular Mechanics and Molecular Dynamics Simulations, J. Am. Chem. Soc., 1992, 114, 10024–10035 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2411824–2411828, 2425738 and 2425739. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5sc00875a |
| This journal is © The Royal Society of Chemistry 2025 |