
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceObservation of the solvent enantio-isotope effect in asymmetric ring-opening of cyclic diaryliodoniums with selenocyanate†
Yuanyuan
Li
a,
Chenyu
Tao
a,
Longhui
Duan
*a and
Zhenhua
Gu
 *ab
*ab
aDepartment of Chemistry, University of Science and Technology of China, 96 Jinzhai Road, Hefei, Anhui 230026, P. R. China. E-mail: zhgu@ustc.edu.cn; dlh1993@ustc.edu.cn
bState Key Laboratory of Coordination Chemistry, Nanjing University, Nanjing 210093, P. R. China
First published on 11th March 2025
Abstract
A Cu-catalyzed asymmetric coupling reaction between cyclic diaryliodoniums and the ambident nucleophile KSeCN was reported. Utilizing water as a co-solvent (CH2Cl2/H2O) achieves high chemoselectivity by forming a nitrogen-hydrogen-bond, thereby blocking the N-site of ambident NCSe− species, thus realizing efficient C–Se coupling. In contrast to the well-known kinetic isotope effect used to evaluate whether the C–H/D bond cleavage is rate-determining, the influence of deuterium-containing solvents on enantioselectivity remained largely unexplored. In this reaction, we observed a notable enhancement in enantioselectivity upon replacing H2O with D2O.
Introduction
Kinetic isotope effect (KIE) experiments have been widely used in modern physical organic chemistry. KIEs, measured by H/D labeling and crossover experiments, are possibly the most widely used methods to establish C–H/D bond breaking as a rate-determining step (Scheme 1a).1 Imamoto, Gridnev and co-workers observed that replacing H2 with D2 also significantly altered the enantioselectivity to various degrees in Rh-catalyzed hydrogenation of enamides (Scheme 1b).2 In 2005, Sharpless et al. reported a significant solvent isotope effect in an aqueous emulsion [2σ + 2σ + 2π] based-cycloaddition, noting a marked decrease in the reaction rate when H2O was replaced with D2O (Scheme 1c).3 McErlean et al. similarly observed that the conversion of an aromatic aza-Claisen rearrangement in D2O was only 40%, compared to full conversion in H2O over the same period.4 While deuterium kinetic isotope effects have been investigated in some aqueous reactions by substituting H2O with D2O, the literature remains devoid of any reports of the solvent enantio-isotope effect, such as D2O displaying an ability to improve enantioselectivity over H2O. | ||
| Scheme 1 Deuterium isotope effects. | ||
Selenium is among the fourteen essential trace elements and plays a vital role in human health.5 Chiral electrophilic selenium reagents have been used for the enantioselective functionalization of inactivated alkenes.6 Significant progress has recently been achieved in synthesizing optically active chiral selenium compounds through asymmetric difunctionalization of alkenes.7 Selenocyanate compounds showed broad utilities in preparing other types of organoselenium compounds.8 However, coupling with ambident nucleophiles, such as KSeCN and KSCN, usually encounters chemo-selectivity challenges. A precedent from the Reiser group demonstrated that the C–S versus C–N selectivity in Cu-catalyzed coupling involving SCN anions can be manipulated through the electronic properties of free radicals: nucleophilic radicals preferentially attack the electrophilic Cu(II) center, generating a Cu(III) species that affords a C–N coupling product upon reductive elimination (Scheme 2a),9 whereas radicals bearing an electron-withdrawing group favor direct attack at the sulfur site, forming a C–S bond directly via an outer-sphere mechanism. We reasoned that the softness and relatively larger size of the Se atom facilitate hydrogen bonding with the N atom (Scheme 2b). Considering the significance of selenocyanates and the broad utility of atropisomers,10 we herein report a Cu-catalyzed asymmetric ring-opening/C–Se coupling reaction of cyclic diaryliodoniums with KSeCN, wherein co-solvent water blocks the N-site of the SeCN anion through hydrogen bonding. Intriguingly, we observed that D2O (in contrast to H2O) unexpectedly improved the enantioselectivity of this reaction.
 | ||
| Scheme 2 Strategy for site-selective coupling of SeCN or SCN. | ||
Results and discussion
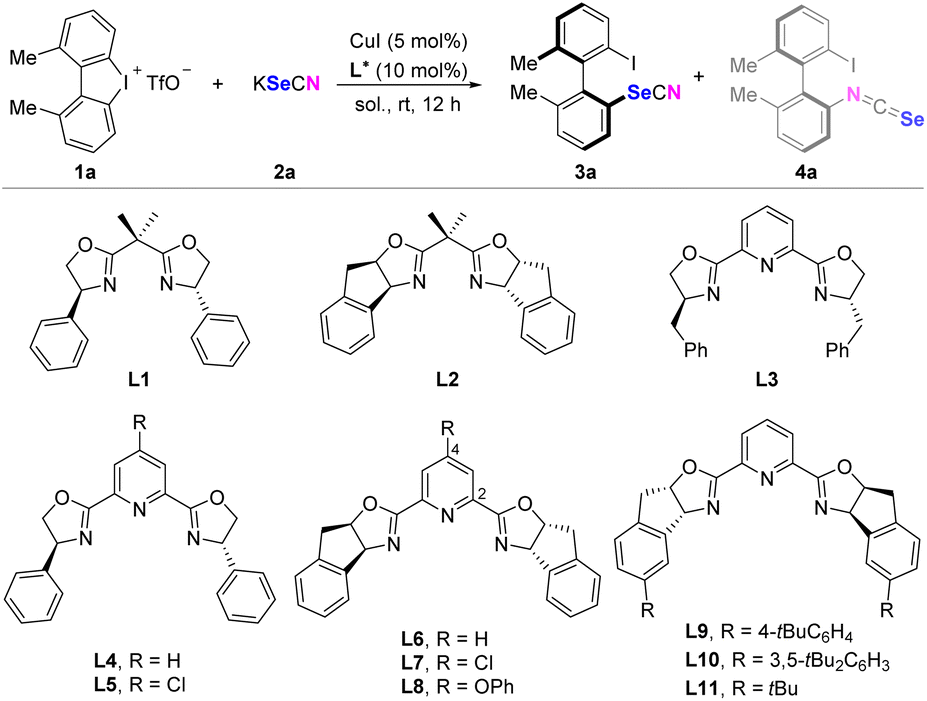
Optimization
Optimization studies were conducted using dimethyl substituted cyclic diaryliodonium 1a and KSeCN as the model substrates in dichloromethane at room temperature, with CuI as the catalyst. Chiral bisoxazoline ligands L1–L2 afforded high enantiomeric excesses of N-arylated product 4a, while the stereoselectivity of Se-arylated product 3a was below 40% ee. Moreover, the reaction exhibited poor chemoselectivity between Se-arylation and N-arylation, with 3a/4a ratios ranging from 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.5 to 2:1 (Table 1, entries 1 and 2). Introduction of benzyl substituted pyridine-2,6-bis(oxazolines) (PyBox) L3 enhanced the enantioselectivities, shifting the ratio of 3a/4a to 5/1 (entry 3). However, the PyBox derived from phenylglycinol exhibited no improvement in stereo-inductivity (entries 4 and 5). Chiral indeno-PyBox ligands L6–L8 led to excellent enantioselectivities for both 3a and 4a, while modifications of the pyridine ring's electronic properties with chloro or phenoxyl groups had minimal impact on the Se-arylation/N-arylation ratio (entries 6–8). Guided by our hypothesis that water might influence Se/N selectivity through differential hydrogen bonding affinities, we examined mixed solvent systems. Gratifyingly, employing CH2Cl2/H2O (5:1) significantly improved chemoselectivity, achieving a 16:1 ratio of 3a/4a, although the enantioselectivity of 3a decreased slightly to 86% (entry 9). An unexpected discovery was that deuterated water enhanced enantioselectivity, while maintaining a high Se/N ratio (entry 10). Further optimization efforts focused on modifying the indeno structure of PyBox, leading to the identification of the optimal ligand L11, which achieved the highest enantioselectivity (94% ee) for 3a (entries 11–13). Under these optimized conditions with L11, using CH2Cl2/H2O as the solvent system in lieu of CH2Cl2/D2O resulted in slightly reduced enantiomeric excess (92% ee) (entry 14).
:1.5 to 2:1 (Table 1, entries 1 and 2). Introduction of benzyl substituted pyridine-2,6-bis(oxazolines) (PyBox) L3 enhanced the enantioselectivities, shifting the ratio of 3a/4a to 5/1 (entry 3). However, the PyBox derived from phenylglycinol exhibited no improvement in stereo-inductivity (entries 4 and 5). Chiral indeno-PyBox ligands L6–L8 led to excellent enantioselectivities for both 3a and 4a, while modifications of the pyridine ring's electronic properties with chloro or phenoxyl groups had minimal impact on the Se-arylation/N-arylation ratio (entries 6–8). Guided by our hypothesis that water might influence Se/N selectivity through differential hydrogen bonding affinities, we examined mixed solvent systems. Gratifyingly, employing CH2Cl2/H2O (5:1) significantly improved chemoselectivity, achieving a 16:1 ratio of 3a/4a, although the enantioselectivity of 3a decreased slightly to 86% (entry 9). An unexpected discovery was that deuterated water enhanced enantioselectivity, while maintaining a high Se/N ratio (entry 10). Further optimization efforts focused on modifying the indeno structure of PyBox, leading to the identification of the optimal ligand L11, which achieved the highest enantioselectivity (94% ee) for 3a (entries 11–13). Under these optimized conditions with L11, using CH2Cl2/H2O as the solvent system in lieu of CH2Cl2/D2O resulted in slightly reduced enantiomeric excess (92% ee) (entry 14).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Ligand | Solvent | 3a/4ab | Yieldc (%)/ee (%) | |
| 3a/4a | |||||
|
a Conditions: 0.10 mmol of cyclic diaryliodoniums (1a), 0.20 mmol KSeCN (2a), CuI (5 mol%), ligand (10 mol%) in CH2Cl2 (0.04 M) [or CH2Cl2/H2O (5:1) or CH2Cl2/D2O (5:1)] at room temperature.
b NMR to determine the ratio.
c Isolated yield.
d
ent-3a was obtained.
|
|||||
| 1 | L1 | CH2Cl2 | 2/1 | 54/38 | 30/94 |
| 2 | L2 | CH2Cl2 | 1/1.5 | 38/32 | 55/94 |
| 3 | L3 | CH2Cl2 | 5/1 | 74/84 | 20/90 |
| 4 | L4 | CH2Cl2 | 3/1 | 70/48 | 22/87 |
| 5 | L5 | CH2Cl2 | 3/1 | 68/56 | 18/88 |
| 6 | L6 | CH2Cl2 | 5/1 | 76/90 | 16/95 |
| 7 | L7 | CH2Cl2 | 4/1 | 73/90 | 20/96 |
| 8 | L8 | CH2Cl2 | 4/1 | 73/90 | 19/94 |
| 9 | L6 | CH2Cl2/H2O | 16/1 | 88/86 | —/92 |
| 10 | L6 | CH2Cl2/D2O | 16/1 | 90/90 | —/94 |
| 11d | L9 | CH2Cl2/D2O | 12/1 | 83/92 | —/92 |
| 12d | L10 | CH2Cl2/D2O | 24/1 | 92/88 | —/86 |
| 13d | L11 | CH2Cl2/D2O | 20/1 | 90/94 | —/90 |
| 14d | L11 | CH2Cl2/H2O | 18/1 | 90/92 | —/— |
The addition of organic hydrogen bond donor urea did not obviously improve the Se/N selectivity (Scheme 3a). A radical pathway can be ruled out as TEMPO had a minimal effect on product formation. The most intriguing aspect of this reaction is the enhanced enantioselectivity observed with deuterated water (D2O) compared to regular water (H2O). While the isotope effect on enantioselectivity has been observed for reactions involving C–H/D bond formation or cleavage,11 our observed enhancement of enantioselectivity by D2O is distinct as it occurs without breaking any chemical bonds of D2O. Therefore, explaining such a deuterium isotope effect on stereoselectivity remains particularly challenging, as both chemical and physical properties, including viscosity, molecular polarizability, or the pKa value, may contribute to the observed outcomes. Given the pH differences [pH(H2O) = 6.9976 pD(D2O) = 7.43 at 25 °C], KHCO3 and K2CO3 were used to adjust the pH of the CH2Cl2/H2O mixture; however, no noticeable improvement in enantioselectivity was observed. We further conducted a series of experiments combining either H2O or D2O with CH2Cl2, (CH2Cl)2, CHCl3, toluene and THF as a mixed solvent (Scheme 3b). Notably, enantiomeric excess improvements can be observed in CH2Cl2, (CH2Cl)2, CHCl3, or toluene when H2O was replaced by D2O. Conversely, an opposite trend was observed in THF/H2O and THF/D2O solvent systems. A similar enhancement was noted when the reaction was conducted in CH2Cl2/CD3OD instead of CH2Cl2/CD3OH, yielding compound 3a with an ee value improved from 50% to 64%.
 | ||
| Scheme 3 Control experiments. | ||
While a definitive explanation remains elusive, from the current literature12 we reasoned three possible factors that possibly affect the enantioselectivity: (a) the previous ab initio calculations on hydrogen-bonded oligomers suggest that the bond strength in D2O is 0.2–0.3 kcal mol−1 stronger than that in H2O, suggesting a potential stronger hydrogen bond in the CH2Cl2/D2O system. (b) Studies using X-ray measurements with molecular simulations indicate that the covalent O–D bond in D2O is 3% shorter than the O–H bond in regular water. (c) The hydrogen-bond network in D2O is more tetrahedral compared to that in H2O. These distinctions may create a different three-dimensional environment compared to H2O, potentially impacting enantioselectivity.
Substrate scope
Under optimized conditions, the substrate scope of various diaryliodonium salts was explored (Scheme 4). Pleasingly, high Se/N arylation ratios (typically exceeding 15:1) were consistently observed across all tested substrates. With 2,2′-diethyl-1,1′-biphenyl-derived cyclic diaryliodonium as the substrate, the reaction proceeded smoothly, albeit with slightly reduced stereocontrol (3b). Both dichlorinated and dibrominated cyclic diaryliodoniums exhibited good reactivity, resulting in excellent enantioselectivities (3c and 3d). High enantiomeric excess (ee) values were also achieved for the ortho,ortho′-dioxygenated atropisomeric biaryls (3e and 3f). The binaphthyl cyclic diaryliodonium underwent a smooth asymmetric ring-opening reaction, yielding excellent yield and near-perfect stereoselectivity (3g). This compound also facilitated the determination of the absolute configuration through single crystal X-ray diffraction analysis (CCDC 2373878). Cyclic diaryliodonium bearing additional methyl or chloro groups at the para- or meta-positions exhibited comparable reactivity and selectivity (3h–k). However, unsymmetric cyclic diaryliodonium with a substituent adjacent to the C–I bond displayed poor regioselectivity, consistent with our previous findings in the trifluoromethylthiolation reaction.13 For instance, products 3l and 3l′ were isolated in 18% and 70% yields, respectively, though both the products exhibited excellent ee values.
 | ||
| Scheme 4 Substrate scope of C–Se coupling. Reaction conditions: cyclic diaryliodonium 1 (0.2 mmol, 1.0 equiv.), KSeCN (0.40 mmol, 2.0 equiv.), CuI (1.9 mg, 0.01 mmol, 5 mol%), L11 (10.1 mg, 0.02 mmol, 10 mol%) in CH2Cl2/D2O (5 mL/1 mL) at 25 °C for 12 h. The values in parentheses refer to the results by using CH2Cl2/H2O as the solvent. | ||
Isoselenocyanates represent valuable building blocks that were widely used in synthesizing selenium-containing compounds.14 The classic synthesis involves a two-step modification of corresponding amines via Barton's protocol.15 Although complete control of chemo-selective N-arylation remained elusive, we evaluated several substrates in anhydrous CH2Cl2 using CuI/L2 as the catalyst, with N/Se ratios ranging from 1:1 to 1.5:1 (see the ESI† for details) (Scheme 5). All substrates tested provided the products in decent isolated yields. Similar to the Se-arylation reaction, the N-arylation reaction of 2,2′-diethyl-1,1′-biphenyl-derived cyclic diaryliodonium resulted in a diminished enantioselectivity (4b). Notably, cyclic diaryliodoniums bearing dimethyl, dichloro, or dibromo substituents, as well as the binaphthyl substrate, afforded products with excellent stereoselectivities (4a and 4c–f).
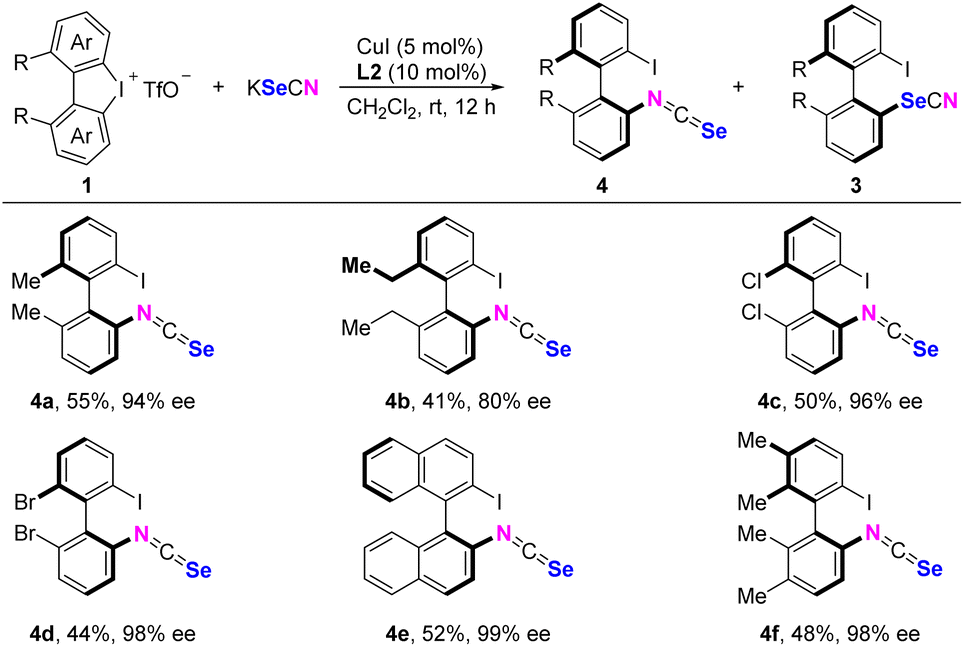 | ||
| Scheme 5 Substrate scope of C–N couplinga. aReaction conditions: cyclic diaryliodonium 1 (0.2 mmol, 1.0 equiv.), KSeCN (0.40 mmol, 2.0 equiv.), CuI (1.9 mg, 0.01 mmol, 5 mol%), L2 (7.0 mg, 0.02 mmol, 10 mol%) in CH2Cl2 (5 mL) at 25 °C for 12 h. The ratios of 4:3 range from 1:1 to 1.5:1, see the ESI† for details. | ||
The selenium atom in selenocyanate exhibits electrophilic character and readily reacts with Grignard reagents. This reactivity enabled the formation of selenanes 5a–h in yields ranging from 80–99% without erosion of enantiopurity (Scheme 6). The reaction demonstrated compatibility with vinyl and alkynyl magnesium reagents, although 2-methylalkenyl selane 5j and alkynyl selane 5k were obtained in lower yields. Additionally, cyclopropyl magnesium bromide also proved compatible, smoothly yielding 5l in 96% yield. Selenocyanate 3a served as a versatile synthetic platform for accessing diverse functionalities. Treatment of (R)-3a with TMSCF3/Cs2CO3 in MeCN yielded the atropisomeric trifluoromethyl selane 5m in 66% yield. Reduction of 3a with NaBH4 produced a diselenide compound upon workup. Subsequent reactions with SelectFluor, followed by an electrophilic cyclization with 1-methoxy-2-(phenylethynyl)benzene, yielded the bicyclic selane 5n in a moderate yield, albeit with a slight decrease in enantiopurity.
 | ||
| Scheme 6 Synthetic applications. | ||
To further demonstrate synthetic utility, we sought to convert isoselenocyanate (S)-4a into substituted selenoureas. The transformation was accomplished by simply stirring a mixture of isoselenate (S)-4a with amines in CH2Cl2 at room temperature. Primary amines, i.e. methylamine and 4-methoxyaniline readily underwent an addition reaction to produce selenoureas 6a, 6b (CCDC 2373879) and 6d in good yields without compromising stereochemical integrity. Secondary amine, N-ethylaniline, also efficiently coupled with isoselenate to provide 6c in excellent yield.
Kinetic studies
The kinetic studies were conducted via calorimetry analysis, which enabled us to continuously collect in situ data throughout the reaction process.16 Notably, both water and deuterated water were almost equally effective in accelerating the reaction, showing no obvious isotope effect on the reaction rate. Careful reaction calorimetry measurements showed that the reaction in CH2Cl2/D2O was very slightly slower than that in CH2Cl2/H2O (Scheme 7a). Furthermore, kinetic studies at 30 °C yielded two interesting observations: (i) the reaction exhibited zeroth-order kinetics with respect to the CuI/L6 catalyst (Scheme 7b). (ii) After ensuring complete dissolution of KSeCN in H2O, kinetics for varying the loadings of KSeCN were measured (Scheme 7c). The reaction exhibited pseudo-first-order kinetic dependence on KSeCN when the loading ranged from 0.50 to 1.0 equivalent [0.50 to 1.0 mol L−1 in H2O]. However, at loadings of KSeCN exceeding 1.5 equivalents, the reaction exhibited pseudo-zeroth-order kinetics dependence on the KSeCN concentration. | ||
| Scheme 7 Kinetic studies. | ||
In an [Ir(COD)py(PCy3)]PF6-catalyzed hydrogenation reaction, Rempel and co-workers observed zeroth-order kinetics with respect to catalyst concentration at high catalyst loadings.17 In our system, the kinetic behavior can be rationalized through phase-transfer considerations. The transfer of KSeCN from the aqueous phase to the organic phase is concentration-dependent only up to 1.0 equivalent of KSeCN (1.0 mol L−1 in H2O). Beyond this point, the concentration of SeCN anions in the organic phase reaches saturation, remaining relatively unchanged despite a further increase in KSeCN loading. Consequently, the high loading of KSeCN caused zeroth-order dependence on the KSeCN concentration. In contrast, the concentration of the CuI/L6 catalyst remains in excess compared to KSeCN in the organic phase, causing the reaction to exhibit zeroth-order kinetics with respect to the copper catalyst.
The role of water in accelerating aqueous reactions can be explained using the Marcus–Jung model of hydrogen bonding between the dangling –OH and the lipophilic substrates or Kobayashi's three layers model, which involves partial polarization.18 We reasoned that the catalytic cycle involves crucial deuterium bond (D–N) formation between D2O and the SeCN anion (Scheme 8).19 This biphasic reaction system presents specific concentration dynamics: (i) KSeCN distribution: the concentration of KSeCN in the aqueous phase significantly exceeds its concentration in the organic phase. (ii) SeCN species saturation: at high KSeCN loadings, the concentration of SeCN in the organic phase reaches saturation, resulting in rate independence from further KSeCN addition. (iii) Catalyst excess: the concentration of the copper catalyst remains in excess relative to the organic phase SeCN species, even at high KSeCN loadings. Mechanistically, the catalytic cycle commences with the association of copper/PyBox complex M1 with selenium cyanate, a step identified as the rate-determining step through the kinetic analysis. Upon loading 2.0 equivalents of KSeCN, the concentration of KSeCN in the organic phase is controlled by its solubility in CH2Cl2, other than its mass transfer rate across the phase boundary.20 The N-site blocking effect of the deuterium bond facilitates the formation of Cu–SeCN intermediate M2. This intermediate is capable of undergoing oxidative addition with cyclic diaryliodonium 1a to yield atropisomeric biaryl Cu(III) complex M4. Finally, reductive elimination of M4 produced the target axially chiral product 3a and a monovalent copper species M1.
 | ||
| Scheme 8 Plausible catalytic cycle. | ||
Conclusions
In summary, we have developed a Cu-catalyzed highly chemo- and atroposelective C–Se coupling reaction between cyclic diaryliodoniums and KSeCN in a CH2Cl2/D2O mixed solvent system. The distinctive role of D2O manifests in two critical ways: (a) as a deuterium bond donor that selectively blocks the N-site of NCSe− species, thereby promoting C–Se coupling over C–N coupling; (b) as an enhancer of enantioselectivity compared to regular water, D2O exhibits noticeable improvement in enantioselectivity. This enhanced stereoselectivity likely stems from either the slightly shorter covalent O–D bond of D2O than the O–H bond of regular water, or the enhanced tetrahedral ordering of the D2O hydrogen-bond network. Our findings underscore the importance of the deuterium isotope effect in enantioselectivity control, establishing it as an additional factor worthy of consideration in asymmetric catalysis.Data availability
The data that support the findings of this study are available in the ESI† or on request from the corresponding authors.Author contributions
Y. Li: methodology, investigation, data curation, writing – original draft. C. Tao: methodology, investigation. L. Duan: supervision, funding acquisition. Z. Gu: conceptualization, funding acquisition, writing – original draft, writing – review & editing. The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Supercomputing Center of the University of Science and Technology of China for providing the computational resources. The authors are grateful for financial support from the National Key Research and Development Program of China (no. 2021YFA1500100), the National Natural Science Foundation of China (22301291 and 22471254), and the Open Research Fund of State Key Laboratory of Coordination Chemistry, School of Chemistry and Chemical Engineering, Nanjing University.Notes and references
- (a) E. M. Simmons and J. F. Hartwig, Angew. Chem., Int. Ed., 2012, 51, 3066–3072 CrossRef CAS PubMed; (b) M. Gómez-Gallego and M. A. Sierra, Chem. Rev., 2011, 111, 4857–4963 CrossRef PubMed.
- T. Imamoto, T. Itoh, K. Yoshida and I. D. Gridnev, Chem.–Asian J., 2008, 3, 1636–1641 CrossRef CAS PubMed.
- S. Narayan, J. Muldoon, M. G. Finn, V. V. Fokin, H. C. Kolb and K. B. Sharpless, Angew. Chem., Int. Ed., 2005, 44, 3275–3279 CrossRef CAS PubMed.
- K. D. Beare and C. S. P. McErlean, Org. Biomol. Chem., 2013, 11, 2452–2459 RSC.
- (a) M. P. Rayman, Lancet, 2012, 379, 1256–1268 CrossRef CAS PubMed; (b) M. Roman, P. Jitaru and C. Barbante, Metallomics, 2014, 6, 25–54 Search PubMed.
- (a) Y. Kawamata, T. Hashimoto and K. Maruoka, J. Am. Chem. Soc., 2016, 138, 5206–5209 CrossRef CAS PubMed; (b) M. Tiecco, L. Testaferri, C. Santi, C. Tomassini, F. Marini, L. Bagnoli and A. Temperini, Chem.–Eur. J., 2002, 8, 1118–1124 Search PubMed.
- (a) L. Liao and X. Zhao, Acc. Chem. Res., 2022, 55, 2439–2453 CrossRef CAS PubMed; (b) F. V. Singh and T. Wirth, Catal. Sci. Technol., 2019, 9, 1073–1091 RSC; (c) V. Rathore, C. Jose and S. Kumar, New J. Chem., 2019, 43, 8852–8864 RSC; (d) S. E. Denmark, D. J. P. Kornfilt and T. Vogler, J. Am. Chem. Soc., 2011, 133, 15308–15311 CrossRef CAS PubMed.
- (a) M. C. Kozlowski, B. J. Morgan and E. C. Linton, Chem. Soc. Rev., 2009, 38, 3193–3207 RSC; (b) D. Zhang and Q. Wang, Coord. Chem. Rev., 2015, 286, 1–16 CrossRef CAS; (c) T. Hayashi, K. Hayashizaki, T. Kiyoi and Y. Ito, J. Am. Chem. Soc., 1988, 110, 8153–8156 CrossRef CAS; (d) X. Shen, G. O. Jones, D. A. Watson, B. Bhayana and S. L. Buchwald, J. Am. Chem. Soc., 2012, 132, 11278–11287 CrossRef PubMed; (e) G. Xu, W. Fu, G. Liu, C. H. Senanayake and W. Tang, J. Am. Chem. Soc., 2014, 136, 570–573 CrossRef CAS PubMed; (f) Y.-H. Chen, D.-J. Cheng, J. Zhang, Y. Wang, X.-Y. Liu and B. Tan, J. Am. Chem. Soc., 2015, 137, 15062–15065 CrossRef CAS PubMed; (g) H. Yang, J. Sun, W. Gu and W. Tang, J. Am. Chem. Soc., 2020, 142, 8036–8043 CrossRef CAS PubMed; (h) S. Yan, W. Xia, S. Li, Q. Song, S.-H. Xiang and B. Tan, J. Am. Chem. Soc., 2020, 142, 7322–7327 CrossRef CAS PubMed; (i) P. G. Karmaker and F. Huo, Asian J. Org. Chem., 2022, 11, e20220022 CrossRef; (j) J. C. Guillemin, Curr. Org. Chem., 2011, 15, 1670–1687 CrossRef CAS; (k) V. P. Reddy, A. V. Kumar, K. Swapna and K. R. Rao, Org. Lett., 2009, 11, 951–953 CrossRef CAS PubMed; (l) D. Kundu, S. Ahammed and B. C. Ranu, Green Chem., 2012, 14, 2024–2030 RSC; (m) Y.-M. Huang, J.-W. Ren, J.-A. Xiao, X.-L. Cheng, R.-F. Meng, X.-S. Qin, H. Peng, Z.-Z. Xie and J.-G. Cui, Synthesis, 2020, 53, 954–960 Search PubMed; (n) N. Mukherjee, D. Kundu and B. C. Ranu, Adv. Synth. Catal., 2017, 359, 329–338 CrossRef CAS; (o) M. Bürger, N. Ehrhardt, T. Barber, L. T. Ball, J. C. Namyslo, P. G. Jones and D. B. Werz, J. Am. Chem. Soc., 2021, 143, 16796–16803 CrossRef PubMed; (p) M. Bürger, S. H. Röttger, M. N. Loch, P. G. Jones and D. B. Werz, Org. Lett., 2020, 22, 5025–5029 CrossRef PubMed.
- Y. Abderrazak and O. Reiser, ACS Catal., 2024, 14, 4847–4855 CrossRef CAS.
- (a) Y.-B. Wang and B. Tan, Acc. Chem. Res., 2018, 51, 534–547 CrossRef CAS PubMed; (b) J. K. Cheng, S.-H. Xiang, S. Li, L. Ye and B. Tan, Chem. Rev., 2021, 121, 4805–4902 CrossRef CAS PubMed; (c) A. Link and C. Sparr, Chem. Soc. Rev., 2018, 47, 3804–3815 RSC; (d) J. Wencel-Delord, A. Panossian, F. R. Leroux and F. Colobert, Chem. Soc. Rev., 2015, 44, 3418–3430 RSC; (e) G. Liao, T. Zhang, Z.-K. Lin and B.-F. Shi, Angew. Chem., Int. Ed., 2020, 59, 19773–19786 CrossRef CAS PubMed; (f) X. Bao, J. Rodriguez and D. Bonne, Angew. Chem., Int. Ed., 2020, 59, 12623–12634 CrossRef CAS PubMed; (g) T.-Z. Li, S.-J. Liu, W. Tan and F. Shi, Chem.–Eur. J., 2020, 26, 15779–15792 CrossRef CAS PubMed; (h) C.-X. Liu, W.-W. Zhang, S.-Y. Yin, Q. Gu and S.-L. You, J. Am. Chem. Soc., 2021, 143, 14025–14040 CrossRef CAS PubMed; (i) J. A. Carmona, C. Rodríguez-Franco, R. Fer-nández, V. Hornillos and J. M. Lassaletta, Chem. Soc. Rev., 2021, 50, 2968–2983 RSC; (j) Y. Liang, J. Ji, X. Zhang, Q. Jiang, J. Luo and X. Zhao, Angew. Chem., Int. Ed., 2020, 59, 4959–4964 CrossRef CAS PubMed; (k) G. Xu, W. Fu, G. Liu, C. H. Senanayake and W. Tang, J. Am. Chem. Soc., 2014, 136, 570–573 CrossRef CAS PubMed; (l) J. M. Lassaletta, Atropisomerism and Axial Chirality, World Scientific, 2019 CrossRef.
- A. Jongejan, J. A. Jongejan and W. R. Hagen, Biochim. Biophys. Acta, 2003, 1647, 297–302 CrossRef CAS PubMed.
- (a) G. Giubertoni, M. Bonn and S. Woutersen, J. Phys. Chem. B, 2023, 127, 8086–8094 CrossRef CAS PubMed; (b) M. Cortes-Clerget, J. Yu, J. R. A. Kincaid, P. Walde, F. Gallou and B. H. Lipshutz, Chem. Sci., 2021, 12, 4237–4266 RSC; (c) J. K. Beattie, C. S. P. McErlean and C. B. W. Phippen, Chem.–Eur. J., 2010, 16, 8972–8974 CrossRef CAS PubMed.
- L. Duan, Z. Wang, K. Zhao and Z. Gu, Chem. Commun., 2021, 57, 3881–3884 RSC.
- K. Yan, M. Liu, J. Wen, W. Liu, X. Li, X. Liu, X. Sui, W. Shang and X. Wang, Org. Chem. Front., 2021, 8, 5139–5144 RSC.
- (a) D. H. Barton, S. I. Parekh, M. Tajbakhsh, E. A. Theodorakis and C.-L. Tse, Tetrahedron, 1994, 50, 639–654 CrossRef CAS; (b) J. G. Fernández-Bolaños, Ó. López, V. ı. Ulgar, I. Maya and J. Fuentes, Tetrahedron Lett., 2004, 45, 4081–4084 CrossRef.
- (a) A. C. Ferretti, J. S. Mathew and D. G. Blackmond, Ind. Eng. Chem. Res., 2007, 46, 8584–8589 CrossRef CAS; (b) J. Burés, A. Armstrong and D. G. Blackmond, Acc. Chem. Res., 2016, 49, 214–222 CrossRef PubMed.
- N. Hinchiranan, K. Charmondusit, P. Prasassarakich and G. L. Rempel, J. Appl. Polym. Sci., 2006, 100, 4219–4233 CrossRef CAS.
- (a) Y. Jung and R. A. Marcus, J. Am. Chem. Soc., 2007, 129, 5492–5502 CrossRef CAS PubMed; (b) T. Kitanosono and S. Kobayashi, Chem.–Eur. J., 2020, 26, 9408–9429 CrossRef CAS PubMed; (c) M.-Z. Lu and T.-P. Loh, Acc. Chem. Res., 2024, 57, 70–92 CrossRef CAS PubMed.
- (a) E. A. Merritt and B. D. Olofsson, Angew. Chem., Int. Ed., 2009, 48, 9052–9070 CrossRef CAS PubMed; (b) K. Zhao, L. Duan, S. Xu, J. Jiang, Y. Fu and Z. Gu, Chem, 2018, 4, 599–612 CrossRef CAS; (c) S. Xu, K. Zhao and Z. Gu, Adv. Synth. Catal., 2018, 360, 3877–3883 CrossRef CAS; (d) K. Zhu, K. Xu, Q. Fang, Y. Wang, B. Tang and F. Zhang, ACS Catal., 2019, 9, 4951–4957 CrossRef CAS.
- R. D. Baxter, Y. Liang, X. Hong, T. A. Brown, R. N. Zare, K. N. Houk, P. S. Baran and D. G. Blackmond, ACS Cent. Sci., 2015, 1, 456–462 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2373878 and 2373879. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5sc00014a |
| This journal is © The Royal Society of Chemistry 2025 |