
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElectrocatalytic hydrogenation of alkynes and alkenes using a proton conductive graphene oxide membrane†
Muhammad Sohail
Ahmad
 ab,
Imam
Sahroni
cd,
Taiga
Kodama
c,
Kazuto
Hatakeyama
c and
Tetsuya
Kida
*abc
ab,
Imam
Sahroni
cd,
Taiga
Kodama
c,
Kazuto
Hatakeyama
c and
Tetsuya
Kida
*abc
aInstitute of Industrial Nanomaterials (IINa), Kumamoto University, Kumamoto, Japan. E-mail: tetsuya@kumamoto-u.ac.jp
bInternational Research Organization for Advanced Science and Technology (IROAST), Kumamoto University, Kumamoto, Japan
cDepartment of Applied Chemistry and Biochemistry, Graduate School of Science and Technology, Kumamoto University, Kumamoto, Japan
dDepartment of Chemistry, Faculty of Mathematics and Natural Sciences, Universitas Islam Indonesia, Sleman, Yogyakarta 55584, Indonesia
First published on 3rd April 2025
Abstract
Graphene-based membranes are emerging as promising materials for energy and chemical conversion due to their exceptional proton conductivity and stability. In this study, we report a graphene oxide (GO) nanosheet membrane for electrochemical hydrogenation reactions. The GO membrane demonstrates excellent proton conductivity, confirmed through concentration cell measurement and complex impedance spectroscopy, and efficiently facilitates proton transport when integrated with active platinum catalysts as the cathode and anode. This system enables selective hydrogenation of alkynes and alkenes into their corresponding alkanes, achieving selectivities of 82% to 93%. This work highlights the potential of graphene-based membrane reactors as cost-effective, scalable, and energy-efficient alternative to traditional hydrogenation methods.
Introduction
Proton conductors,1 which enable the efficient transfer of protons, have attracted significant interest due to their critical role in advancing various fields, including biological systems and chemical filtration technologies.2,3 Achieving ultrahigh proton conductivity remains a key objective in energy-related applications such as steam electrolysis,4 fuel cells,5 and hydrogen separation,6 where proton exchange efficiency directly impacts the device's performance.7 To date, various materials such as Nafion,8 metal–organic frameworks,9 covalent organic frameworks,10 and oxidized carbon-based materials,11,12 have demonstrated promising proton conducting properties.13 However, many of these materials face challenges related to fabrication complexity and high production costs, limiting their practical applications.Cost-effective oxidized carbon allotropes are highly promising due to their excellent conductivity, ease of production, and stable, non-toxic nature. Proton transport in these materials occurs efficiently through an adsorbed water film on hydrophilic active sites, such as oxygen-containing groups, embedded within the hydrophobic carbon skeleton.14,15 Additionally, the structural characteristics of oxidized carbon allotropes enable hybridization with guest molecules, further enhancing proton conduction pathways.16 Among these materials, graphene oxide (GO) stands out as a two-dimensional (2D) oxidized carbon material with exceptional performance. Its flexible, planar structure facilitates the uniform distribution of hydrophilic sites within its interior and along the edges of its nanosheets. Its unique properties have led to the development of graphene-based proton-exchange membranes (PEMs), which are particularly promising for electrochemical applications.17 GO, a derivative of graphene, further enhances proton conductivity,18 making it an ideal candidate for use in membrane reactors for energy-efficient chemical transformations.
The ability of GO membranes to selectively conduct protons while preventing direct hydrogen interaction with substrates offers distinct advantages compared to traditional catalytic methods, which require high temperatures and hydrogen pressures (Fig. 1). Unlike conventional thermocatalytic hydrogenation, which is energy-intensive and often results in high waste generation,19 electrochemical hydrogenation using a GO-based PEM operates under ambient conditions, reducing energy consumption and minimizing environmental impact. As a proton-conducting membrane, GO offers a cost-effective and scalable alternative to conventional palladium (Pd)-based membranes.20,21 Pd membranes are widely recognized for their excellent performance in hydrogen-related applications;22 however, their reliance on rare and expensive metals limits large-scale adoption.23,24 This challenge underscores the need for sustainable, abundant, and affordable materials that can drive innovation in hydrogenation technologies. However, a major challenge of GO membranes is their low stability under humid conditions,25 which compromises their long-term performances. To address these issues several chemical modifications using dopants have been explored to enhance their durability.26,27 Among these, the incorporation of Ce ions has proven highly effective in maintaining both high conductivity and stability,28 making cerium functionalized graphene oxide (CeGO) a promising candidate for electrochemical hydrogenation applications.
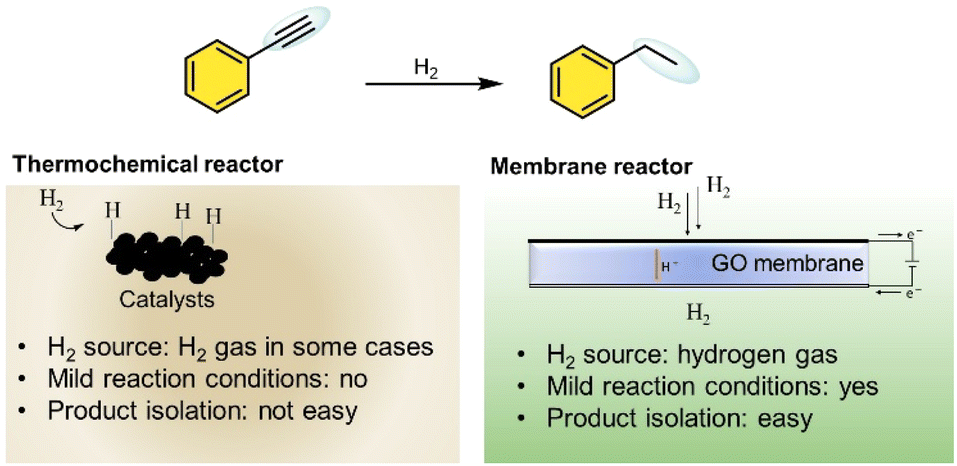 | ||
| Fig. 1 Reactions conditions and reactivities comparing thermochemical hydrogenation with the graphene oxide membrane reactor. The membrane reactor uniquely enables mild condition hydrogenation and simplified product separation. | ||
Hydrogenation, a cornerstone of chemical synthesis, is vital across industries such as petrochemicals, pharmaceuticals, and agrochemicals.29,30 However, conventional methods are energy-intensive, requiring high temperatures, and often producing significant waste.31
This study demonstrates the use of a GO-based PEM reactor for electrochemical hydrogenation. The GO membrane facilitates efficient proton conduction and selective hydrogenation of alkenes and alkynes under mild conditions, achieving up to 93% selectivity. By eliminating the need for Pd and high-pressure H2, this approach offers a sustainable and scalable pathway for hydrogenation processes.
Results and discussion
GO, synthesized through a modified Tour's method32via chemical oxidation and exfoliation of natural graphite, served as the basis for membrane fabrication. Using the vacuum filtration technique, individual GO nanosheets were assembled into a well-organized laminated structure, as confirmed by scanning electron microscopy (Fig. 2 SEM cross-section). However, a major challenge with GO membranes is their low stability, primarily due to the detachment of oxygen functional groups from the GO framework.33 This detachment reduces the interlayer spacing between stacked GO nanosheets, thereby leading to a decrease in the proton conductivity, as reported previously.26 To address this, cerium ions (3–4 nm) were controllably added to the GO dispersions before vacuum filtration. The resulting membranes, termed CeGO, were fabricated under optimized mixing conditions (Fig. S1†). | ||
| Fig. 2 Cross-sectional SEM image of the graphene oxide (GO) membrane, illustrating a well-organized laminated structure assembled via vacuum filtration. The observed stratification indicates the alignment of GO nanosheets, supporting both membrane's stability and selective proton permeability. | ||
The X-ray diffraction (XRD) patterns further supported the structural changes induced by Ce+ incorporation. While parental GO exhibited a d-spacing of 1.1 nm, the CeGO membrane showed an increased d-spacing of 1.18 nm, with a corresponding shift in the XRD peak to lower 2θ values (Fig. 3a). This increase indicates the intercalation of Ce+ within the interlayer gallery space of GO sheets.28 The chemical functionalities of GO suggest two primary interaction modes for Ce+ within the GO membrane (Scheme 1), (i) bridging the edge of the sheets via carboxyl group coordination and (ii) intercalating between the basal planes through either weak alkoxide bonds. Energy dispersive X-ray spectroscopy (EDX) mapping of the CeGO membrane cross-section confirmed the homogeneous distribution of Ce+ across the laminated structure (Fig. 2). These structural modifications significantly enhance the stability of the CeGO membrane, as intercalated Ce+ mitigates the collapse of interlayer spacing while contributing to improved proton conductivity.
 | ||
| Fig. 3 (a) XRD patterns of GO and CeGO membranes, (b) FTIR spectra of GO and CeGO, (c) high-resolution XPS survey spectrum of S 1s and (d) Ce 3d XPS spectrum of CeGO. | ||
 | ||
| Scheme 1 Schematic representation of Ce+ grafting in the graphene oxide framework. For clarity, only a few functional groups are shown for clarity. | ||
The Fourier-transform infrared (FT-IR) spectra of parental GO displayed characteristic peaks for C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, aromatic CC, carboxy C–O, and epoxy C–O stretch. In comparison, the FT-IR spectra of the CeGO membrane showed reduced intensity for the carboxy COOH and epoxy C–O–C stretches, with a noticeable shift in peak position (Fig. 3b). These changes indicate interactions between the carboxy and epoxy groups of GO and the Ce+ ions, suggesting proper integration of Ce+ into the GO framework through strong electrostatic interactions.34 To gain more insight into the oxygen functionalities in GO membranes, X-ray photoelectron spectroscopy (XPS) measurements were conducted, as shown in Fig. S2.† The C 1s XPS spectrum was deconvoluted to identify the carbon–oxygen bonds (Fig. S3†). The relative intensities of the oxygen functional groups in the CeGO membrane decreased due to the incorporation of Ce ions. Furthermore, the S 1s and Ce XPS spectra confirmed the retaining of Ce+ species in the CeGO membrane during the filtration process (Fig. 3c, d and S2†). Specifically, the Ce 3d band exhibited Ce 3d5/2 and Ce 3d3/2 spin–orbit doublet peaks, respectively. The peaks 3d5/2 at 881.9 eV and 886.3 eV: 3d3/2 at 900.5 eV and 904.2 eV are primarily attributed to the +3 oxidation state of cerium. However, the appearance of the peaks at 888.6 eV, and 908.1 eV and the weak high-binding energy peak at 917.7 eV indicate the presence of Ce4+ species.26,35–37 Ce ions have large coordination numbers and interact strongly with the oxygen functional groups of GO, thereby suppressing the desorption of the oxygen functional groups and improving stability.
O, aromatic CC, carboxy C–O, and epoxy C–O stretch. In comparison, the FT-IR spectra of the CeGO membrane showed reduced intensity for the carboxy COOH and epoxy C–O–C stretches, with a noticeable shift in peak position (Fig. 3b). These changes indicate interactions between the carboxy and epoxy groups of GO and the Ce+ ions, suggesting proper integration of Ce+ into the GO framework through strong electrostatic interactions.34 To gain more insight into the oxygen functionalities in GO membranes, X-ray photoelectron spectroscopy (XPS) measurements were conducted, as shown in Fig. S2.† The C 1s XPS spectrum was deconvoluted to identify the carbon–oxygen bonds (Fig. S3†). The relative intensities of the oxygen functional groups in the CeGO membrane decreased due to the incorporation of Ce ions. Furthermore, the S 1s and Ce XPS spectra confirmed the retaining of Ce+ species in the CeGO membrane during the filtration process (Fig. 3c, d and S2†). Specifically, the Ce 3d band exhibited Ce 3d5/2 and Ce 3d3/2 spin–orbit doublet peaks, respectively. The peaks 3d5/2 at 881.9 eV and 886.3 eV: 3d3/2 at 900.5 eV and 904.2 eV are primarily attributed to the +3 oxidation state of cerium. However, the appearance of the peaks at 888.6 eV, and 908.1 eV and the weak high-binding energy peak at 917.7 eV indicate the presence of Ce4+ species.26,35–37 Ce ions have large coordination numbers and interact strongly with the oxygen functional groups of GO, thereby suppressing the desorption of the oxygen functional groups and improving stability.
The structural and chemical modifications of the CeGO membrane, described earlier, suggested improved stability and functional properties due to the incorporation of Ce+ ions. To verify the impact of these modifications on proton conduction, we measured the transport number (t = 1) of the CeGO membrane reactor using a concentration cell setup (Fig. 4a). The electromotive force (EMF) generated between the anode and the cathode was recorded upon varying the hydrogen concentration at the anode. The time-dependent EMF response of the GO membrane fitted with Pt@graphene electrodes (synthesis method is presented in the ESI†) at room temperature demonstrated good sensitivity to hydrogen (Fig. 4a, inset), confirming equilibrium between hydrogen and protons with a reaction (n) of 2 (H2 = 2H+ + 2e−). The linear dependence of EMF on the logarithm of hydrogen concentration in Ar (Fig. 4a) showed a slope of 28.2 mV per decades, consistent with Nernst's equation n = 2 (29.5 mV per decade):

 are the hydrogen partial pressures are at the anode and cathode, respectively. The calculated t for the CeGO membrane was close to unity (0.956) with a slight deviation at higher
are the hydrogen partial pressures are at the anode and cathode, respectively. The calculated t for the CeGO membrane was close to unity (0.956) with a slight deviation at higher  suggesting slight electrical conductivity in the membrane. Furthermore, the proton conductivity of the CeGO membrane was measured at room temperature using complex impedance spectroscopy (Fig. 4b). Fitting the impedance data to an equitant circuit revealed the contribution from bulk resistance, surface resistance, and interface capacitance. The parental GO sheets exhibited a proton conductivity of 0.140 mS cm−1 at room temperature, comparable to that of commercial Nafion (approximately 0.1–0.05 mS cm−1).38
suggesting slight electrical conductivity in the membrane. Furthermore, the proton conductivity of the CeGO membrane was measured at room temperature using complex impedance spectroscopy (Fig. 4b). Fitting the impedance data to an equitant circuit revealed the contribution from bulk resistance, surface resistance, and interface capacitance. The parental GO sheets exhibited a proton conductivity of 0.140 mS cm−1 at room temperature, comparable to that of commercial Nafion (approximately 0.1–0.05 mS cm−1).38
 | ||
| Fig. 4 (a) Dependence of EMF on hydrogen concentration in Ar gas at room temperature. (b) Electrochemical impedance spectroscopy of GO and CeGO. | ||
The addition of Ce+ using a facile one-pot method led to significant enhancements in proton conduction, increasing the proton conductivity to 0.397 mS cm−1, over twice that of the unmodified GO membrane (Fig. 4b). This improvement is attributed to the presence of Ce+ ions, which facilitate stronger interlayer interactions and promote efficient proton transport pathways. This finding highlights the dual role of Ce in enhancing both the structural integrity and functional performances of GO-based membranes. To demonstrate the potential of the graphene-based membrane reactor for energy-efficient production of value-added chemicals, we explored the electrocatalytic hydrogenation of phenylacetylene to ethylbenzene as a model reaction. Hydrogenation, a fundamental chemical process, involves adding hydrogen atoms to unsaturated bonds or replacing functional groups, enabling the synthesis of more valuable molecules. This process is essential to various industries, including petrochemicals, pharmaceuticals, agrochemicals, fine chemicals, etc.
Extensive catalytic research has highlighted the efficiency of metals such as Pd, Pt, and Ni in hydrogenation reactions, prized for their high selectivity, ease of workup, and catalyst recyclability, contributing to excellent atomic efficiency.39–41 However, traditional thermocatalytic hydrogenation typically requires high temperatures and elevated H2 pressures, resulting in significant energy consumption and costs. Electrochemical hydrogenation, in contrast, eliminates the need for harsh chemical oxidants and reductants, offering a continuous, energy-efficient, and milder process. This approach is more sustainable and enhances selectivity, making it a favorable alternative to diverse applications.
The key component of the membrane reactor is a thin CeGO membrane, which serves as a hydrogen-permeable layer. A Teflon sheet with carbon paper physically separates the electrolysis compartment from the hydrogenation compartment, which contains a solution of alkenes or alkynes dissolved in isopropanol. The H2 split on the surface of the catalyst (Pt@graphene) generates protons (H+), which diffuse through the CeGO membrane28,42 and react with organic compounds in the hydrogenation chamber. This architecture enables electrochemical and hydrogenation reactions to occur in separate compartments, with the hydrogenation compartment featuring an opening to sample reaction solution during hydrogenation reactions (Fig. S4†).
For the electrocatalytic hydrogenation reaction, the hydrogenation chamber was filled with 0.03 M phenylacetylene dissolved in isopropanol.
Electrolysis was conducted for 24 hours at a constant current density of 100 mA cm−2, corresponding to the geometric surface area of one face of the Pt@graphene catalyst. The reactor operated at ambient pressure and room temperature, and the reaction progress was monitored using gas chromatography-mass spectrometry (GC-MS). The Pt@graphene hybrid was synthesized by thermal annealing of fresh nanodiamonds at 1100 °C under an argon atmosphere (details are in the ESI†).
Initially, 0.03 M phenylacetylene in IPA was tested with a bare GO membrane (entry 1b, Table 1). No ethylbenzene formation was observed after 24 hours, and the pristine GO membrane dissolved in IPA. To overcome this limitation, we employed a CeGO membrane and explored its hydrogenation chemistry with Pt@graphene, achieving an ethylbenzene yield of 83% (entry 1, Table 1). Encouraged by this result, we examined the influence of applied current density and reaction time on the hydrogenation process. Electrochemical hydrogenation was performed at current densities of 50 and 100 mA cm−2. Increasing the current density from 50 to 100 mA cm−2 enhanced the conversion of phenylacetylene to ethylbenzene from 45% to 83% over 24 hours (Fig. S10a†). Notably, at 100 mA cm−2, the conversion remained consistently higher across all time points. Time-dependent cell voltage measurements at different applied current densities (Fig. S10b†) further confirmed the durability of the CeGO membrane during the hydrogenation reaction. We then tested the broad reactant scope of hydrogenation chemistries with the Pt@graphene-coated CeGO membrane (Table 1). The products were confirmed by GC-MS analysis (Fig. S11–S15†) and quantified using decane as the internal standard. Ethylbenzene (entry 1, Table 1), methyl ethylbenzene (entry 2, Table 1), and chloroethyl benzene (entry 4, Table 1) were synthesized with high purity and selectivity. Similarly, ethyl benzene 2a was obtained from styrene in 93% yield. Substituted styrene has been reduced to the corresponding products, as shown in Table 2. Typically, the introduction of electron-withdrawing groups onto the benzene ring of the substrates (such as entries 4 and 5 Table 1, and/or 2cTable 2) reduced the electron density of the ring, thereby enhancing the reactivity in hydrogenation. The yield remained consistent, demonstrating the efficiency and adaptability of our reaction system.
|
|
|||
|---|---|---|---|
| Entryc | Reactant | Product | Yieldb/% |
| a Reaction conditions: CeGO membrane sandwiched with Pt@graphene, 28 °C, 100 mA cm−2, solvent isopropanol, reaction time 24 h, H2 5 mL min−1. b Yield calculated using decane as an internal standard. c GO membrane was used and no products were detected. | |||
| 1 |

|
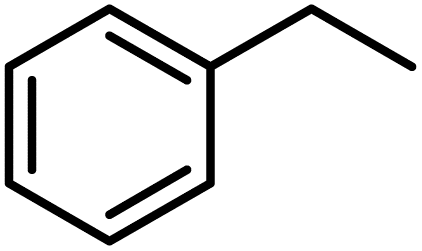
|
83 |
| 2 |

|

|
87 |
| 3 |

|

|
89 |
| 4 |

|

|
91 |
| 5 |

|

|
90 |

| a Reaction conditions: CeGO membrane sandwiched with Pt@graphene, 28 °C, 100 mA cm−2, solvent isopropanol, reaction time 24 h, H2 5 mL min−1. |
|---|

|
As previously reported, hydrogen permeation occurs through conduction in the CeGO membrane.28 First, hydrogen is electro-chemically oxidized to protons and electrons on the surface of the membrane coated with an electrocatalyst (Pt@graphene) (H2 → 2H+ + 2e−). These protons diffuse into the CeGO membrane and finally react to form the corresponding product on the other side of the membrane. The most critical advantage of this technology is the 100% selectivity toward H2 when a dense, crack-free CeGO membrane is used. Notably, the use of the Pt@graphene sandwiched CeGO membrane with IPA solvent in the hydrogenation chamber effectively minimized the formation of the byproducts over a long time, such as 24 hours. This outcome is particularly significant, as byproducts are often observed in related hydrogenation reactions.43,44
We developed a novel room temperature (25–30 °C) electrochemistry-assisted selective hydrogenation method for converting alkynes and alkenes into their corresponding products using molecular hydrogen as the hydrogen source. This process demonstrates highly efficient catalytic performance, low energy requirement, and enhanced safety, showcasing its potential for future industrial applications. Furthermore, the CeGO membrane exhibited excellent long-term stability as confirmed by the immersion test in water, where no significant structural degradation was observed (Fig. S16†). To advance this technology toward industrial relevance, further studies could focus on developing cost-effective modified graphene-based membranes, such as ultrathin self-supporting GO asymmetric membranes, with improved hydrogen permeability and catalytic efficiency.
Conclusions
In this study, we developed CeGO membranes as a cost-effective and efficient alternative for electrocatalytic hydrogenation, overcoming the limitations of traditional Pd- and Nafion-based systems. The CeGO membranes demonstrated enhanced proton conductivity, stability, and hydrogen selectivity, enabling the efficient hydrogenation of phenylacetylene to ethylbenzene under mild conditions. By integrating CeGO membranes into a reactor design that separates electrochemical and hydrogenation reactions, we achieved a sustainable process using water or H2 as a proton source. This work highlights the potential of CeGO membranes for advancing sustainable chemical processes and paves the way for broader applications in catalytic and electrochemical technologies.Data availability
All experimental procedures, analytical data and spectra are available in the ESI.†Author contributions
M. S. Ahmad; conceptualization, methodology, investigations, writing – original draft. I. Sahroni; formal analysis. T. Kodama; formal analysis, validations. K. Hatakeyama; formal analysis. T. Kida; supervision, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge financial support from the Japan Society for the Promotion of Science (JSPS) through the grant-in-Aid for Early-Career Scientists (Grant No. 24K17588). Support from the Young Faculty Members Support Program of IROAST, and the Institute of Industrial Nanomaterials (IINa) Kumamoto University, is also greatfully acknowledged.Notes and references
- K.-D. Kreuer, Chem. Mater., 1996, 8, 610–641 CrossRef CAS
.
- M. Lozada-Hidalgo, S. Hu, O. Marshall, A. Mishchenko, A. N. Grigorenko, R. A. W. Dryfe, B. Radha, I. V. Grigorieva and A. K. Geim, Science, 2016, 351, 68–70 CrossRef CAS PubMed
- D. D. Ordinario, L. Phan, W. G. Walkup, J.-M. Jocson, E. Karshalev, N. Hüsken and A. A. Gorodetsky, Nat. Chem., 2014, 6, 596–602 CrossRef CAS PubMed
- L. Bi, S. Boulfrad and E. Traversa, Chem. Soc. Rev., 2014, 43, 8255–8270 RSC
- K. Jiao, J. Xuan, Q. Du, Z. Bao, B. Xie, B. Wang, Y. Zhao, L. Fan, H. Wang, Z. Hou, S. Huo, N. P. Brandon, Y. Yin and M. D. Guiver, Nature, 2021, 595, 361–369 CrossRef CAS PubMed
- L. Mogg, G.-P. Hao, S. Zhang, C. Bacaksiz, Y.-C. Zou, S. J. Haigh, F. M. Peeters, A. K. Geim and M. Lozada-Hidalgo, Nat. Nanotechnol., 2019, 14, 962–966 CrossRef CAS PubMed
- E. Fabbri, D. Pergolesi and E. Traversa, Chem. Soc. Rev., 2010, 39, 4355–4369 RSC
- B. Liu, B. Hu, J. Du, D. Cheng, H.-Y. Zang, X. Ge, H. Tan, Y. Wang, X. Duan, Z. Jin, W. Zhang, Y. Li and Z. Su, Angew. Chem., Int. Ed., 2021, 60, 6076–6085 CrossRef CAS PubMed
- D.-W. Lim and H. Kitagawa, Chem. Soc. Rev., 2021, 50, 6349–6368 RSC
- Z. Wang, Y. Yang, Z. Zhao, P. Zhang, Y. Zhang, J. Liu, S. Ma, P. Cheng, Y. Chen and Z. Zhang, Nat. Commun., 2021, 12, 1982 CrossRef CAS PubMed
- M. R. Karim, K. Hatakeyama, M. Koinuma and S. Hayami, J. Mater. Chem. A, 2017, 5, 7243–7256 RSC
- X. Meng, H.-N. Wang, S.-Y. Song and H.-J. Zhang, Chem. Soc. Rev., 2017, 46, 464–480 RSC
- A. Karmakar, R. Illathvalappil, B. Anothumakkool, A. Sen, P. Samanta, A. V. Desai, S. Kurungot and S. K. Ghosh, Angew. Chem., Int. Ed., 2016, 55, 10667–10671 CrossRef CAS PubMed
- K. Hatakeyama, M. S. Islam, K. Michio, C. Ogata, T. Taniguchi, A. Funatsu, T. Kida, S. Hayami and Y. Matsumoto, J. Mater. Chem. A, 2015, 3, 20892–20895 RSC
- K. Hatakeyama, H. Tateishi, T. Taniguchi, M. Koinuma, T. Kida, S. Hayami, H. Yokoi and Y. Matsumoto, Chem. Mater., 2014, 26, 5598–5604 CrossRef CAS
- N. L. Hamidah, M. Shintani, A. S. Ahmad Fauzi, G. K. Putri, S. Kitamura, K. Hatakeyama, M. Sasaki, A. T. Quitain and T. Kida, ACS Appl. Nano Mater., 2020, 3, 4292–4304 CrossRef CAS
- J. Tong, Y. Fu, D. Domaretskiy, F. Della Pia, P. Dagar, L. Powell, D. Bahamon, S. Huang, B. Xin, R. N. Costa Filho, L. F. Vega, I. V. Grigorieva, F. M. Peeters, A. Michaelides and M. Lozada-Hidalgo, Nature, 2024, 630, 619–624 CrossRef CAS PubMed
- X. Pan, M. G. Debije, A. P. H. J. Schenning and C. W. M. Bastiaansen, ACS Appl. Mater. Interfaces, 2021, 13, 28864–28869 CrossRef CAS PubMed
- K. Boonpalit, C. Uthayopas and P. Surawatanawong, Organometallics, 2022, 41, 259–269 CrossRef CAS
- A. Singha Hazari, M. L. Frisch, Y. Wen, M. D. Stankovic and C. P. Berlinguette, J. Am. Chem. Soc., 2024, 146, 28153–28160 CAS
- M. D. Stankovic, N. E. LeSage, J. F. Sperryn, A. Kurimoto and C. P. Berlinguette, ACS Energy Lett., 2024, 9, 4459–4464 CrossRef CAS
- R. S. Sherbo, R. S. Delima, V. A. Chiykowski, B. P. MacLeod and C. P. Berlinguette, Nat. Catal., 2018, 1, 501–507 CrossRef CAS
- A. Fukazawa, Y. Shimizu, N. Shida and M. Atobe, Org. Biomol. Chem., 2021, 19, 7363–7368 RSC
- R. S. Delima, M. D. Stankovic, B. P. MacLeod, A. G. Fink, M. B. Rooney, A. Huang, R. P. Jansonius, D. J. Dvorak and C. P. Berlinguette, Energy Environ. Sci., 2022, 15, 215–224 RSC
- C.-N. Yeh, K. Raidongia, J. Shao, Q.-H. Yang and J. Huang, Nat. Chem., 2015, 7, 166–170 CrossRef CAS PubMed
- N. L. Hamidah, M. Shintani, A. S. Ahmad Fauzi, E. G. Mission, K. Hatakeyama, A. T. Quitain and T. Kida, SN Appl. Sci., 2019, 1, 630 CrossRef CAS
- M. Zhang, Y. Mao, G. Liu, G. Liu, Y. Fan and W. Jin, Angew. Chem., Int. Ed., 2020, 59, 1689–1695 CrossRef CAS PubMed
- S. Kitamura, G. K. Putri, T. Kodama, T. Nakahara, N. L. Hamidah, T. Shinkai, I. Sahroni, Y. Inomata, K. Hatakeyama, A. T. Quitain, M. S. Ahmad and T. Kida, Nano Lett., 2024, 24, 15226–15233 CrossRef CAS PubMed
- J. Zhang, H. Zhang, Y. Wu, C. Liu, Y. Huang, W. Zhou and B. Zhang, J. Mater. Chem. A, 2022, 10, 5743–5757 RSC
- X. Zhou, X. Yu, B. You and Y. Jing, J. Mater. Chem. A, 2024, 12, 20527–20541 RSC
- P. Lara and K. Philippot, Catal. Sci. Technol., 2014, 4, 2445–2465 RSC
- D. C. Marcano, D. V. Kosynkin, J. M. Berlin, A. Sinitskii, Z. Sun, A. Slesarev, L. B. Alemany, W. Lu and J. M. Tour, ACS Nano, 2010, 4, 4806–4814 CrossRef CAS PubMed
- K. H. Thebo, X. Qian, Q. Zhang, L. Chen, H.-M. Cheng and W. Ren, Nat. Commun., 2018, 9, 1486 CrossRef PubMed
- N. Shauloff, N. L. Teradal and R. Jelinek, ACS Sens., 2020, 5, 1573–1581 CrossRef CAS PubMed
- X. He, X. Liu, R. Li, B. Yang, K. Yu, M. Zeng and R. Yu, Sci. Rep., 2016, 6, 22238 CrossRef CAS PubMed
- D. R. Mullins, S. H. Overbury and D. R. Huntley, Surf. Sci., 1998, 409, 307–319 CrossRef CAS
- M. Veith, S. Mathur, A. Kareiva, M. Jilavi, M. Zimmer and V. Huch, J. Mater. Chem., 1999, 9, 3069–3079 RSC
- M. R. Karim, K. Hatakeyama, T. Matsui, H. Takehira, T. Taniguchi, M. Koinuma, Y. Matsumoto, T. Akutagawa, T. Nakamura, S. Noro, T. Yamada, H. Kitagawa and S. Hayami, J. Am. Chem. Soc., 2013, 135, 8097–8100 CrossRef CAS PubMed
- M. G. Avello, J. Blas Martínez, T. Romero, V. Papaefthimiou, M. J. Chetcuti, V. Ritleng, C. Pham-Huu, C. Oelschlaeger and C. Michon, ACS Sustain. Chem. Eng., 2024, 12, 10739–10751 CrossRef CAS
- M. Ashraf, M. S. Ahmad, Y. Inomata, N. Ullah, M. N. Tahir and T. Kida, Coord. Chem. Rev., 2023, 476, 214928 CrossRef CAS
- K. Murugesan, A. S. Alshammari, M. Sohail, M. Beller and R. V. Jagadeesh, J. Catal., 2019, 370, 372–377 CrossRef CAS
- Y. Jin, B. Gao, C. Bian, X. Meng, B. Meng, S. I. Wong, N. Yang, J. Sunarso, X. Tan and S. Liu, Green Chem., 2021, 23, 3374–3385 RSC
- H. Xue, H. Xu, X. Song, M. Chen, X. Wang, M. Ji and M. Wang, Inorg. Chem., 2024, 63, 16011–16017 CrossRef CAS PubMed
- O. Verho, H. Zheng, K. P. J. Gustafson, A. Nagendiran, X. Zou and J.-E. Bäckvall, ChemCatChem, 2016, 8, 773–778 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5sc00423c |
| This journal is © The Royal Society of Chemistry 2025 |