
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHierarchical chirality conversion switched by biaxial halogen bonding†
Weilong
Ma
,
Zhaozhen
Cao
,
Na
Zhang
*,
Aiyou
Hao
 and
Pengyao
Xing
*
and
Pengyao
Xing
*
Key Laboratory of Colloid and Interface Chemistry of Ministry of Education, School of Chemistry and Chemical Engineering, Shandong University, Jinan 250100, People's Republic of China. E-mail: nzhang@sdu.edu.cn; xingpengyao@sdu.edu.cn
First published on 11th April 2025
Abstract
Precise control over chirality across hierarchical levels of peptides enriches the toolbox of peptide-based chiroptical materials. In this work, we report a controlled conversion from folded molecular chirality to supramolecular chirality, which can be effectively switched by biaxial halogen bonds. Amino acid segments were conjugated to the bipyridine core as a β-turn to facilitate the formation of parallel β-sheet arrays, which enables intramolecular chirality transfer to the appended pyrenes. This allows for efficient excimer emission and boosted circularly polarized luminescence with a dissymmetry factor of up to 10−2. The folding structures are self-assembled into nanohelices where supramolecular chirality is dominated by emerged heterochirality in response to the solvent medium. The introduction of hypervalent iodine(III) species binds to the peptide via strong biaxial I⋯N halogen bonds that transform well-ordered helices into amorphous nanoparticles. The biaxial halogen bonding complexation rearranges the assembly with extremely high efficacy, suppressing the supramolecular chirality and recovering the folded molecular chirality, when the chiroptical activities are rationally manipulated. This work unveils the dynamic conversion of artificially folded peptides, which will inspire the design of protocols for peptide- or protein-based chiroptical materials.
Introduction
Peptides or proteins exhibit a hierarchical level of structures owing to the spontaneous folding and subsequent assembly procedure driven by noncovalent forces.1–3 Structural evolution from folded secondary to assembled tertiary or higher motifs could be inversely achieved by denaturation triggered by thermal, chemical, pH, mechanical and many other stimulus factors (Scheme 1a).4–8 Denaturation is sometimes reversible owing to the refolding or reassembly mechanisms.9 Synthetic short peptides or peptoids undergo spontaneous folding into parallel or anti-parallel β-sheet motifs facilitated by the β-turn.10–12 Rotatable segments, such as the bi-aromatic axis, are conducive to the formation of intramolecular folding via hydrogen bonds. For instance, Herrick's conformation represents the 1,1′-ferrocenyl peptides with parallel β-sheet motifs, whereby the efficient chirality transfer from peptides to ferrocene has been observed.13–16 | ||
| Scheme 1 (a) Schematic of the structural conversion between folded secondary and self-assembled tertiary peptide motifs, where self-assembly and reversible denaturation dominate the processes. (b) Chemical structures of the folded peptide structures rotated by a bipyridine axle. It shows the transformation process between dominated molecular and supramolecular chiralities through self-assembly and coassembly with I(III). | ||
Folded peptides with a rotatable aromatic axle are an emerging platform for investigating chirality transfer under intramolecular folding and their aggregation in the assembly state, which represent molecular- and supramolecular-level chirality. The conformational locking by the peptide folding allows for the chirality transfer to aromatic entities or the covalently attached functional substituents to afford interesting chiroptical properties and applications.17,18 The employment of kinetic and dynamic protocols to manipulate the folding scenarios shall efficiently alter and control the folded chirality; the peptide sequences, temperature, concentration, and solvent media exhibit dramatic impacts.19–22 Furthermore, the assembly and formation of ordered-packed molecular arrays generate supramolecular chirality that introduces macroscopic helicity with distinct and evolved chiroptical properties.23,24 Dynamic transformation between folded intramolecular and supramolecular chirality in the assembly state shows increased potentials in the fabrication of peptide-based chiral materials,25–27 which also are reminiscent of the reversible denaturation process depicted in Scheme 1a.9
We design a peptide sequence in which 2,2′-bipyridine provides a rotatable axis acting as a β-turn to facilitate parallel β-sheet folding. The folding allows for the chirality transfer across spacers towards the bipyridine core and the signaling pyrenes (Scheme 1b). The folding is sensitive to solvation showing tunable excimer emission, whereby the chiroptical properties, including circular dichroism and circularly polarized luminescence (CPL), could be finely tailored, and the dissymmetry factors are up to 10−2. Self-assembly by increasing the portion of methyl cyclohexane (MCH) in chloroform (TCM) is triggered by the evolution from folded molecular chirality to supramolecular chirality. Heterochiral nanohelices emerged in response to hypersensitivity to the delicate fractions of good/poor solvents. A particular 70 vol% of MCH prefers an opposite handedness compared to other systems. This reflects the disparity in chirality adaptive to the balance between kinetic and thermodynamic aggregation. Recovery from supramolecular chirality to the molecular level is achieved by halogen bonding complexation between the hypervalent I(III) and bipyridine domains. The complexation is ultrahigh, either in a pure solution or an assembly state. The in situ addition of merely 0.1 molar equiv. I(III) rapidly transforms one-dimensional nanohelices into zero-dimensional nanoparticles. Within the amorphous nanoparticles, the complexation by halogen bonding efficiently suppresses the supramolecular chirality and recovers the folded molecular chirality, similar to that of the denaturation of peptides. This work fabricates a β-sheet-like folded peptide in which the conversion between molecular and supramolecular chirality is readily achieved, expressing great potential in responsive peptide-based chiroptical materials.
Results and discussion
The synthesis route along with the characterization via1H and 13C nuclear magnetic resonance (NMR) spectroscopy, as well as mass spectrometry (MS), are detailed in the ESI.† The interaction between solute and solvent significantly influences the folding behavior, thereby potentially regulating the properties.22,28–30 We initially examined the fluorescence of PAP and PPP in five different solvents (Fig. 1a and S1–S4†). PAP and PPP exhibit excimer peaks at 480 nm and 475 nm, with monomer peaks at 375 nm and 377 nm, respectively. The excimer characteristic peak observed in the fluorescence spectra provides direct evidence of intramolecular folding. Additionally, experiments show that at concentrations of 0.02 mM for PAP and PPP, no intermolecular aggregation occurs, thus minimizing the impact of aggregation on the excimer (Fig. S35 and S36†). The ratio between these peaks elucidates the stacking scenario of the two pyrene segments, correlating with spatial arrangements and noncovalent interplay. The excimer-monomer ratios of PAP and PPP in methanol (MeOH), tetrahydrofuran (THF), dichloromethane (DCM), and TCM, ranging from 1.0 to 1.8, indicate distinct interactions between pyrenes. However, in dimethyl sulfoxide (DMSO), the excimer proportion sharply decreases, indicating a weakened interaction or elongated distance between the pyrene. This observation suggests that DMSO might engage in hydrogen bonding with the amides of PAP and PPP, which significantly reduces the formation of excimers. Thus, solvent media could modulate the opening and closure of the clamp-like structure. We conducted a solvent ratio-dependent fluorescence comparison. As shown in Fig. 1b and S5,† as the volume fraction of DMSO increases, the excimer peak of PAP at 480 nm gradually decreases. This trend aligns with the observations shown in Fig. 1a, confirming that DMSO solvation affects the opening and closure of the molecular clamp in PAP. In addition to influencing the folding behavior of molecules, solvents also significantly impact the lifetime and quantum yield (Fig. 1c, S6 and S7†). For PAP, the fluorescence lifetime was 37.61 ns in DMSO and 13.94 ns in TCM, indicating a nearly threefold difference. Similarly, the quantum yields in these two solvents differ significantly, with values of 31.72% in DMSO and 8.17% in TCM. Regarding PPP, its fluorescence lifetime is 37.90 ns in DMSO and 16.38 ns in TCM, demonstrating a significant solvent-dependent effect on fluorescence dynamics. The quantum yields also vary notably between these solvents, with values of 30.21% in DMSO and 8.28% in TCM, underscoring the distinct impact of solvents on fluorescence efficiency. Owing to the distinct polarities of DMSO and TCM, based on the excimer/monomer ratio, TCM facilitates more excimer formation than DMSO. Electron coupling in TCM between excited and ground pyrene pairs increases the nonirradiative decay pathways compared to that in DMSO, greatly declining the quantum yields although with a shorter lifetime. | ||
| Fig. 1 (a) Ratio of excimer to monomer peak in different solvents. (b) Normalized emission spectra of LPAP with different DMSO volume fractions (DMSO/TCM). (c) Luminescence lifetime of LPAP in TCM and DMSO. (d) CD spectra of L/DPAP in TCM and DMSO. (e and f) CPL spectra of L/DPAP and L/DPPP in TCM and DMSO. λex = 340 nm, c = 0.02 mM. | ||
In addition, solvent-induced changes are also evident in the circular dichroism (CD) spectra. For PAP, there is a significant disparity in CD signals between TCM and DMSO (Fig. 1d). In TCM, a distinct Cotton effect is observed at 345 nm in the pyrene absorption region, indicating mirror-image relationships between the molecules. This effect demonstrates the transfer of point chirality from the amino acid to the pyrene unit. In contrast, in DMSO, owing to solvation effects, this chirality transfer to the pyrene groups is diminished, resulting in the absence of a significant Cotton effect at 345 nm. In contrast, PPP exhibits a Cotton effect at 345 nm in both TCM and DMSO (Fig. S8†). The difference observed for PAP in DMSO can be attributed to environmental factors, which can lead to changes in the molecular conformation, causing slight shifts in peak values. Interestingly, both PAP and PPP demonstrate significant CPL in solution (Fig. 1e and f). The luminescence dissymmetry factor (glum) for PAP in DMSO and TCM reaches ±6.0 × 10−3 and ±3.0 × 10−3, respectively. PPP exhibits even stronger dissymmetric luminescence performance, with glum values of ±1.0 × 10−2 and ±4.2 × 10−3 in DMSO and TCM, respectively. It is noteworthy that the absorbance dissymmetry factor (gabs) for PAP in DMSO and TCM is ±2.8 × 10−5 and ±7.9 × 10−5, while for PPP, gabs values are approximately ±7.2 × 10−5 and ±7.0 × 10−5 in DMSO and TCM, respectively. The |glum|/|gabs| ratios for PAP in DMSO and TCM are up to 214.28 and 37.97, respectively. For PPP, the corresponding ratios are 138.89 and 60.00 in DMSO and TCM, further highlighting their pronounced CPL properties in different solvents. As summarized by Mori, there is a general relationship between gabs and glum in π–π* transitions of chiral organic fluorophores: |glum| ≈ 0.81 × |gabs|.31 However, according to Masahiko's work, it is often observed that |glum| > |gabs| for chiral pyrene excimers, indicating a stronger dissymmetry in luminescence compared to absorption.32,33 For PAP and PPP in their excimer states, where the two pyrene skeletons are closely aligned due to exciton resonance interactions, the lower |gabs| value suggests that the pyrene rings are more disordered in the ground state. In the excited state, the closely packed pyrene rings contribute to the larger |glum| value. Generally, pyrene excimers with high dissymmetric luminescence factors and CPL activity hold significant potential for the development of new chiroptical materials.
The results from the density functional theory (DFT) calculations offer theoretical insights into the structural characteristics of PAP and PPP (Fig. 2a and b). In PAP, the dihedral angle is 54.09°, which slightly distorts the molecule and facilitates the formation of double C⋯O interaction (3.271 Å). Additionally, the distance between the two pyrene rings is 3.407 Å, facilitating π–π interactions. These intramolecular forces contribute to the stability of the PAP conformation. Similar intramolecular forces are present within the PPP, collectively stabilizing its molecular conformation. Using Multiwfn and VMD programs, visualized images of non-covalent interactions (NCI) iso-surfaces were generated to represent the types and distribution of interactions (Fig. 2c and d).34 As previously mentioned, C⋯O and π–π interactions are all evident. According to DFT and NCI calculations, the stabilizing forces in the folded structures mainly involve interstrand dipole–dipole interactions, CH⋯O hydrogen bonds, and π–π stacking between the pyrene groups. These strong interactions stabilize their conformations, resembling a clamping structure.
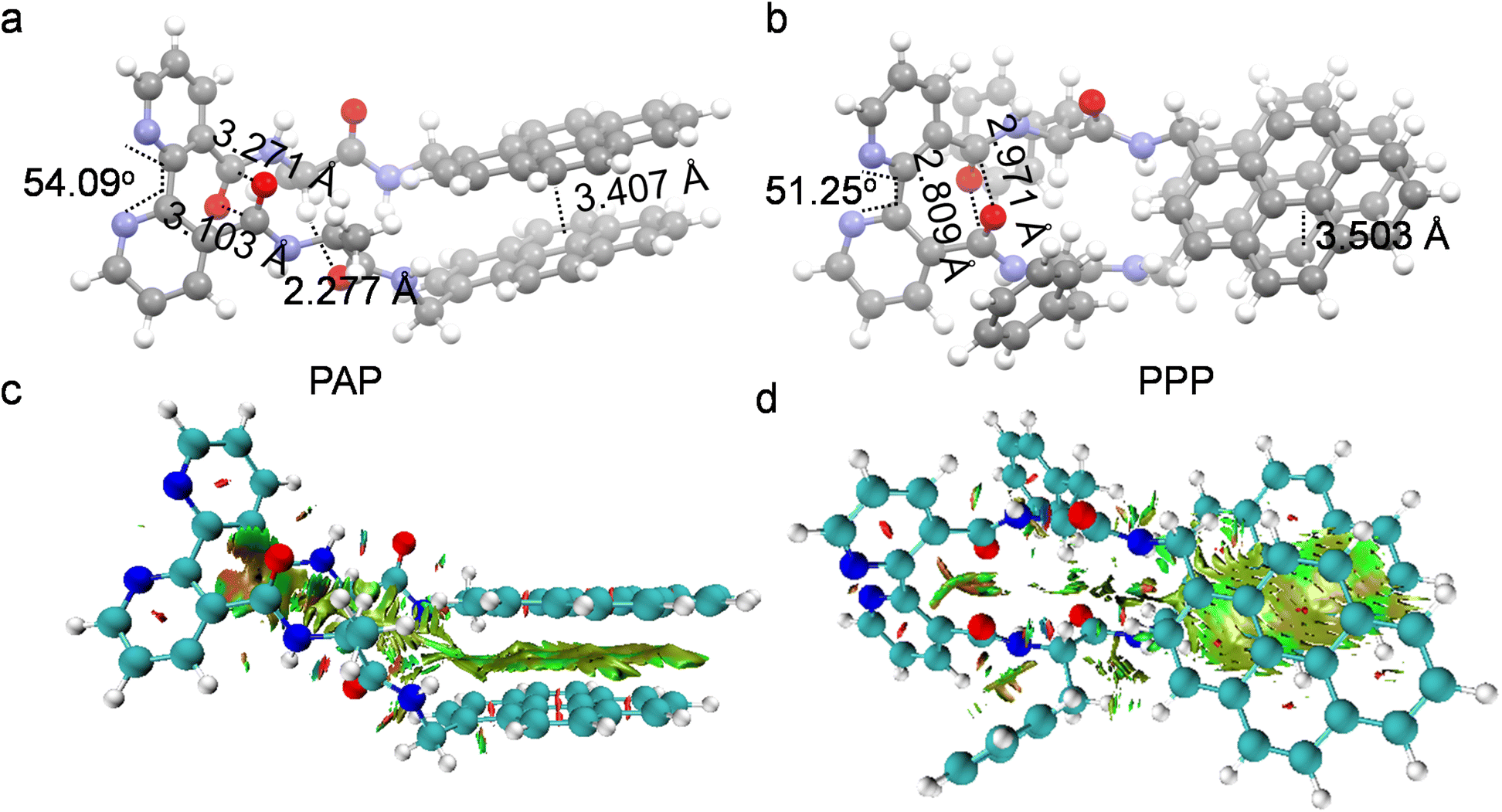 | ||
| Fig. 2 (a and b) DFT-optimized structures. (c and d) NCI analysis. The green regions represent the vdWs interaction. | ||
Self-assembly was conducted to transmit the folding molecular chirality to supramolecular chirality. The concentrations of PAP and PPP were 0.02 mM. The assembly was conducted using two solvents: TCM and MCH, with MCH comprising volume fractions of 60%, 70%, 80%, and 90%. After 24 hours of solvent exchange-initiated assembly, the morphological characteristics under different conditions were examined using scanning electron microscopy (SEM). At a 60% volume fraction of MCH, LPAP assembled into spherical structures with a diameter of approximately 500 nm. As the volume fraction of MCH increased beyond 60%, the morphology transformed into various micron-sized flower-like structures. Specifically, at 70%, 80%, and 90% MCH, LPAP formed micron-sized flowers with diameters of about 6 µm (Fig. 3a–d). In contrast, LPPP exhibited different helical chiralities under different assembly conditions. This showed M-handed chirality at MCH volume fractions of 60%, 80%, and 90%, whereas it exhibited P-handed chirality at a volume fraction of 70% (Fig. 3e–h). DPPP displayed the corresponding enantiomeric effect. Surprisingly, at a volume fraction of 70% MCH, the assembly morphology of PPP showed an opposite helical direction compared to the other three ratios (Fig. 3i–l). To further investigate this phenomenon, CD spectra were collected to examine the chiral inversion at different assembly ratios (Fig. 3m–p and S17†). At MCH volume fractions of 60%, 80%, and 90%, LPPP exhibited positive CD signals at 355 nm, indicating a consistent chiral orientation of the molecules in these conditions. However, at a volume fraction of 70%, the CD signal in this region shifted to negative, suggesting a spatial change in the aggregation orientation of the pyrene groups at this specific concentration. This observation highlights that altering the assembly conditions influences the aggregation behavior of the molecules during the assembly process. The fiber bundling effect plays a crucial role here. During the self-assembly process, when molecules aggregate via the fiber bundling effect, intermolecular interactions (such as π–π stacking and hydrogen bonding) can be strengthened, which influences the conformation and arrangement of the molecules. The bundling effect can alter the distance and alignment between molecules, thereby affecting their chiral states. When the volume fraction of MCH reaches 70%, the fiber bundling effect may accelerate the reversal of chirality through kinetic factors during the self-assembly process, leading to opposite characteristics observed in both SEM and CD.
 | ||
| Fig. 3 (a–l) SEM of LPAP, LPPP and DPPP in different solvent ratios. (m–p) CD spectra of L/DPPP in different solvent ratios. c = 0.02 mM. | ||
Next, we investigated the CPL properties of the system using MCH volume fractions of 70% and 80%, focusing particularly on the chiral optical properties of the molecules in their excited states (Fig. 4a). The CD sign in aggregated systems is most likely associated with the formation of supramolecular chirality. Clearly, the variation in the MCH fraction is accompanied by the sign inversion of the CD signal and the handedness inversion of the nanofibers. Ascribed to the inherent absolute chirality of amino acids, the changes in CD signs should be related to the asymmetric packing of pyrenes in assemblies. However, CPL reflects the chirality in the photoexcited state, and pyrene excimer pairs could, in most cases, alter their topology with structural relaxation. As previously mentioned, the molecular conformation of PPP remains compact in the excited state, leading to consistent CPL signals across different MCH volume fractions. However, differences in aggregation behavior affect the overall assembly properties. Specifically, at a 70% MCH volume fraction, the assembly is more stable, resulting in stronger fluorescence and CPL intensities compared to the 80 vol% MCH. This indicates that when the intrinsic CPL properties of PPP in their excited states are compact, the assembly conditions significantly influence the overall optical properties. Fourier transform infrared spectroscopy (FT-IR) analysis was performed on the assemblies obtained using the nanoprecipitation method with MCH volume fractions of 70% and 80%. The stretching vibration peaks of the amide N–H bonds were observed at 3158 cm−1 and 1592 cm−1. The FT-IR results revealed slight differences between the MCH volume fractions of 70% and 80% in these vibration peaks (Fig. 4b). These differences indicate that the spatial arrangement of the pyrene groups within the assemblies has changed, resulting in a modified chemical environment for the amide N–H bonds. This alteration in the environment of the amide groups is reflected in the variations observed in the stretching vibration peaks. To investigate the molecular packing patterns under assembly conditions, Grazing Incidence Wide-Angle X-ray Scattering (GIWAXS) was performed on thin film samples, as depicted in Fig. 4c. Aggregates were collected using a high-speed centrifuge and cast onto silicon wafers to air dry. During this process, chiral nanofibers with specific aspect ratios aligned parallel to silicon wafer surfaces. The 2D GIWAXS patterns and integral curves in Fig. 4c reveal distinct peaks of LPPP at Q values of 0.271 Å−1, 0.427 Å−1, and 0.534 Å−1, with a ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :√3:2. This characteristic ratio signifies a hexagonal stacking pattern, yielding a calculated lattice parameter of 2.3 nm. Importantly, the hexagonal stacking observed in GIWAXS correlates with the helical fibers observed in SEM. Moreover, aggregates precipitated at different MCH volume fractions (80%) exhibited a hexagonal stacking pattern (Fig. S9†). This consistency suggests that variations in the direction of helical fibers do not significantly alter the aggregate arrangement pattern. Overall, the GIWAXS analysis confirms that the chiral nanofibers form a hexagonal stacking structure with a lattice parameter of 2.3 nm, which remains stable under different assembly conditions.
:√3:2. This characteristic ratio signifies a hexagonal stacking pattern, yielding a calculated lattice parameter of 2.3 nm. Importantly, the hexagonal stacking observed in GIWAXS correlates with the helical fibers observed in SEM. Moreover, aggregates precipitated at different MCH volume fractions (80%) exhibited a hexagonal stacking pattern (Fig. S9†). This consistency suggests that variations in the direction of helical fibers do not significantly alter the aggregate arrangement pattern. Overall, the GIWAXS analysis confirms that the chiral nanofibers form a hexagonal stacking structure with a lattice parameter of 2.3 nm, which remains stable under different assembly conditions.
 | ||
| Fig. 4 (a) CPL spectra of PPP in different solvent ratios. c = 0.02 mM. (b) FT-IR spectra comparison of PPP in different solvent ratios. (c and d) GIWAXS patterns and corresponding integral peaks for LPPP (fMCH = 70%). | ||
We employed molecular dynamic simulations to investigate the aggregation behavior of 200 molecules deposited into a simulation box measuring 10 × 10 × 10 nm3, which was filled with water molecules. After 15 ns of equilibration, snapshots are depicted in Fig. 5a–d. They highlight the π–π interactions between pyrene planes, which dominate the aggregated system. Additionally, Radial Distribution Function (RDF) calculations were used to analyze the statistical distances between pyrene planes. By selecting atoms C1 and C2 to represent two pyrene planes, Fig. 5f shows a major distance of 3.82 Å between them, consistent with theoretical expectations for π–π interactions. To further understand the aggregated system, we plotted the solvent accessible surface (SAS) area against the equilibration time. After equilibration, the SAS areas of PAP and PPP decreased by approximately 49% and 50%, respectively. This marked decrease in the SAS area correlates closely with the hydrophobic nature of the molecules and their subsequent helical aggregation.
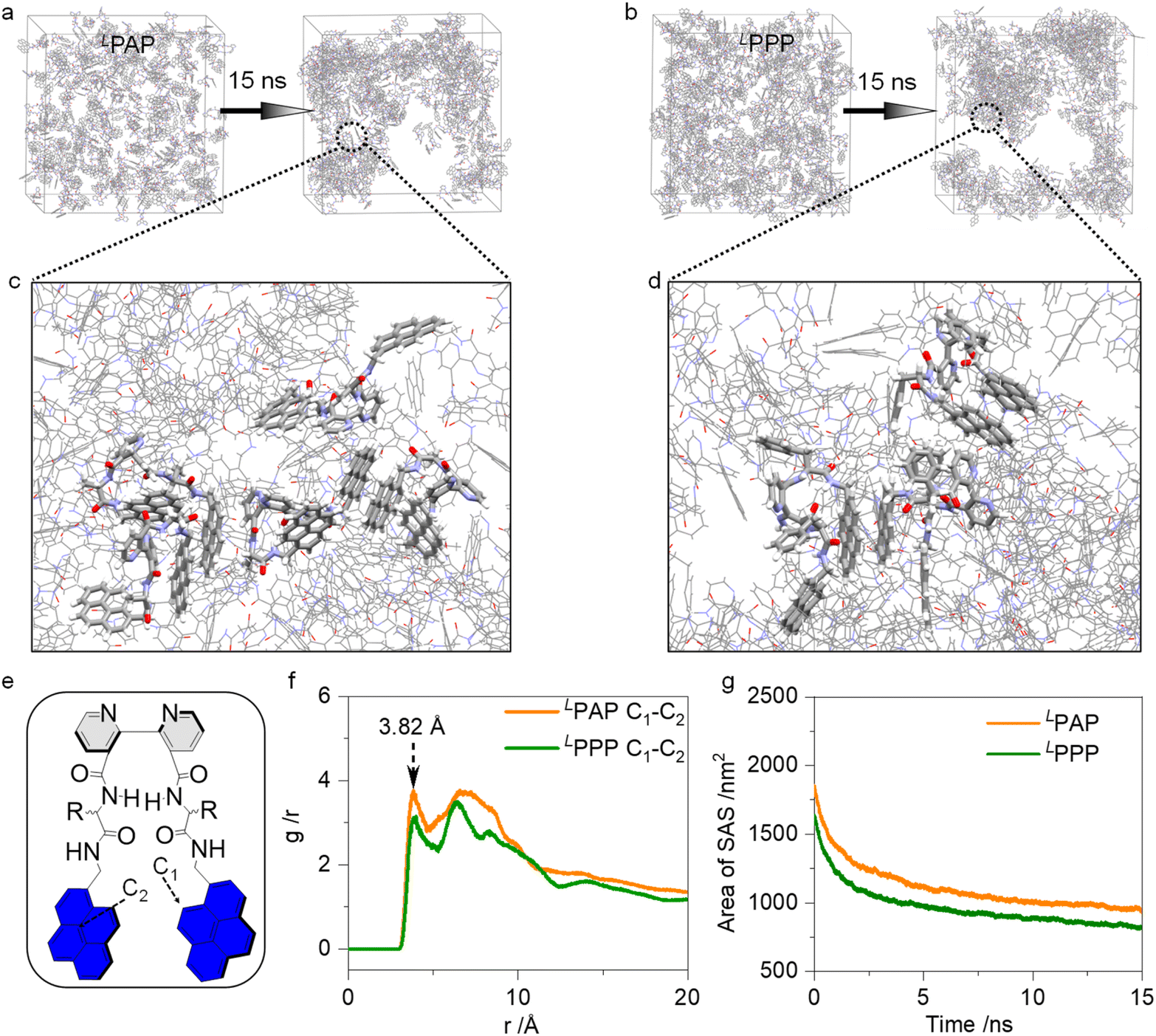 | ||
| Fig. 5 (a and b) Molecular dynamic simulation (15 ns) of LPAP and LPPP. (c and d) Partial snapshots of LPAP and LPPP systems show the molecular packing of self-assembly. (e) Atomic numbering assignment of PAP and PPP. (f) Solvent accessible surface areas of LPAP and LPPP. (g) RDF profiles of distances about π–π interaction between pyrenes. | ||
PAP and PPP are suitable for forming supramolecular complexes with guest molecules through halogen bonding due to the presence of bipyridines. However, the formation of halogen bonding is vulnerable to solvation effects.35,36 In this regard, cationic halogen bond donors are considered to enhance and stabilize halogen bonds in solution, such as iodinated imidazolium cations, iodotriazolium cations, and hypervalent iodine cations.37–40 The advantage of halogen bonding in this context lies in its stable and reversible regulatory capability. Halogen bonding demonstrates exceptionally strong binding affinity in both the solution and assembly states. We here selected I·BArF4 (referred to as I(III)) as the biaxial halogen bonding donors and confirmed strong interactions between the host and guest molecules using high-resolution mass spectrometry (HRMS) and titration experiments. In HRMS (Fig. 6a and b), m/z peaks at 1243.3341, 1244.3470, 1245.3558, and 1246.3540 are consistent with the theoretically predicted corresponding positions. These results validate the presence of stable complexes in the solution (CHCl3). As shown in Fig. 6d and f, increasing I(III) results in fluorescence quenching. Nonlinear fitting of the fluorescence intensity at 476 nm across different equivalents of I(III) yielded a binding constant of 2.9 × 104 M−1 with a 1:1 binding stoichiometry, underscoring strong non-covalent interactions. Similarly, fluorescence titration of I(III) with PAP also demonstrates quenching (Fig. 6c and e), indicating analogous charge transfer phenomena. Notably, upon the addition of I(III), the excimer peaks of both PAP and PPP gradually diminish. Simultaneously, with the addition of I(III), the UV absorbance at the pyrene regions of PAP and PPP gradually decreases, indicating the switching of pyrene moieties (Fig. S26†). This suggests that I(III) binding increases the distance between the two pyrene units, potentially disrupting their stacking interaction. These findings highlight the potential for I(III) to modulate the supramolecular structure of pyrene-based compounds by affecting their intermolecular distances.
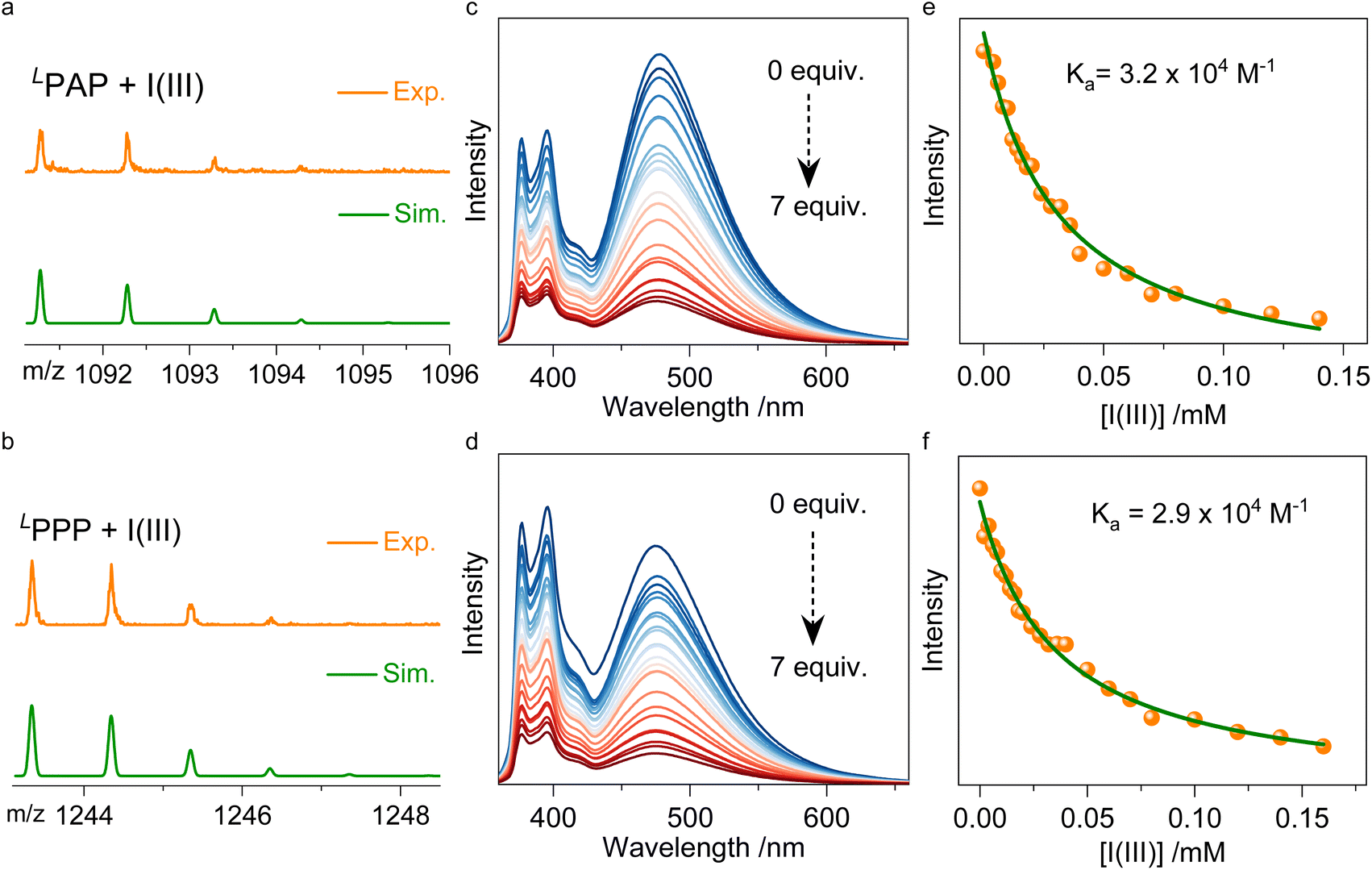 | ||
| Fig. 6 (a and b) HRMS of LPAP + I(III) and LPPP + I(III) with a 1:1 binding ratio. (c and d) Fluorescence titration experiments of LPAP and LPPP in CHCl3 with the addition of I(III). (e and f) Fitting curves of fluorescence intensity at 478 and 476 nm for LPAP and LPPP, respectively. | ||
Hypervalent iodine(III) cationic compound possesses two electrophilic regions in the opposite direction of the two C–I bonds, known as σ-holes. As depicted in Fig. 7a, these σ-holes are located at the extensions of the C–I covalent bonds in the cyclic diaryliodonium ion. Owing to their inherent cationic nature, the σ-holes exhibit a surface electrostatic potential of up to 126.79 kcal mol−1. In the case of PPP, the presence of lone pair electrons on the pyridine nitrogen atoms results in a surface electrostatic potential of −55.14 kcal mol−1 (Fig. 7b). Similarly, for PAP, the electrostatic potential in the vicinity of the pyridine nitrogen atoms is −53.82 kcal mol−1 (Fig. S10†). This distribution of electrostatic potential provides halogen bonds with distinctive directionality and contributes to their strong binding affinity. Therefore, the σ-holes generated by the hypervalent iodine(III) compound and the corresponding electrostatic potentials on PPP and PAP surfaces illustrate the directional nature and strong binding capabilities of halogen bonds in these systems. Fig. 7c illustrates the strong directionality of the halogen bond formed between I(III) and PPP, which is characterized by a bond angle of 167.02° and a bond length of 2.746 Å. Additionally, hydrogen bonds between the carbonyl oxygen and aromatic hydrogen atoms are observed at distances of 2.365 Å and 2.489 Å, further enhancing the binding capability between I(III) and PPP. Similarly, Fig. 7d shows interactions between PAP and I(III) involving both hydrogen bonds and halogen bonds. Furthermore, the binding energies of PAP and PPP with I(III) are calculated. PPP exhibits a slightly higher binding energy compared to PAP, with values of −49.07 kcal mol−1 and −45.62 kcal mol−1, respectively (Fig. 7e). These findings underscore the specific structural arrangements and energetics of halogen and hydrogen bonding interactions between I(III), PPP, and PAP, highlighting their roles in the formation of stable supramolecular complexes. We investigated solid halogen-bonded complexes using X-ray Photoelectron Spectroscopy (XPS). The shifts observed in the I d3/2 and I d5/2 peaks for PAP, from 634.70 and 623.20 eV to 632.15 and 620.65 eV, respectively, indicate a significant charge transfer from the nitrogen atom within the pyridine unit to iodine (Fig. 7j). This transfer increases the electron density around iodine, thereby reducing its nuclear attraction and resulting in a decrease in binding energy. In contrast, the N 1s peak of the pyridine nitrogen shifted slightly from 398.75 eV to 399.00 eV, indicating a change in the chemical environment of the nitrogen owing to its interaction with iodine (Fig. 7i). However, there was no observable shift in the XPS peak of the amide nitrogen. Notably, the O 1s peak remained unchanged before and after the formation of halogen-bonded complexes (Fig. 7k), strongly suggesting that I(III) predominantly interacts with the nitrogen atom on the pyridine ring and not with other oxygen-containing sites. XPS provides clear evidence of a specific interaction between I(III) and the nitrogen atom in PAP, illustrating the selective nature of halogen bonding. Further XPS analysis of PPP also supports this conclusion, confirming that I(III) specifically interacts with the nitrogen atom on the pyridine ring (Fig. 7l, S11 and S12†). It provides compelling evidence that I(III) forms halogen bonds with nitrogen atoms in pyridine-containing molecules, influencing their electronic environments and highlighting distinctive interaction patterns. Simultaneously, based on the changes in XPS and fluorescence intensity, π-stacking interactions between the cationic aromatic hypervalent iodine(III) species and the pyrene moiety can be ruled out. The C 1s peak of the pyrene group showed no significant shift before and after the addition of I(III), so we can exclude the presence of π-stacking interactions between the cationic aromatic hypervalent iodine(III) species and the pyrene group (Fig. S37†). If π-stacking interactions were present, such interactions could alter molecular interactions, change the electron distribution, and reduce the energy level difference, leading to a redshift in fluorescence. However, as shown in Fig. 6c and d, there was no shift in the monomer and excimer peaks, which further ruled out the presence of π-stacking interactions.
 | ||
| Fig. 7 (a and b) ESP of I(III) and LPPP. (c and d) DFT-optimized structure of LPPP + I(III) and LPAP + I(III), respectively. (e) Interaction energies of PAP + I(III) and PPP + I(III). (f–i) XPS patterns of LPAP, LPPP and its halogen bonding complex with the 1:1 ratio. | ||
To further investigate the properties of halogen-bonded complexes, we conducted additional characterizations to examine the behavior of PAP and PPP upon the addition of I(III) in both the solution and assembly systems. In TCM, the CD signals of both PAP and PPP increased with the addition of I(III) (Fig. 8a, b, S13 and S14†). This indicates that as the distance between the two pyrene rings increases, the point chirality at the amino acid positions is more likely to transfer to the supramolecular level. This is due to the decrease in π–π interactions between the pyrene rings within the molecule, while the π–π interactions between molecules increase, leading to closer intermolecular correlations. Consequently, the arrangement of PAP and PPP in space becomes more extended and orderly, facilitating the transfer of point chirality to the supramolecular level. In TCM, the CPL of PAP and PPP showed little change before and after the addition of I(III) (Fig. 8c, d, S15 and S16†). This phenomenon is attributed to the molecules maintaining their relatively stable conformations in the excited state, as mentioned earlier. Therefore, even after the formation of halogen-bonded complexes upon adding I(III), there is no substantial impact on the conformational stability of PAP and PPP in a pure solution system. In summary, these findings underscore how the introduction of I(III) influences the supramolecular chirality and intermolecular interactions of PAP and PPP while maintaining their stable excited-state conformations.
 | ||
| Fig. 8 (a–d) CD and CPL spectra of PAP and PPP with I(III) in TCM. (e and f) CD spectra of PAP and PPP with I(III) under different co-assembly conditions. (g and h) CPL spectra of PPP with I(III) in TCM with fMCH = 70% and 80%, respectively. (i and j) UV-vis absorption and fluorescence titration experiments of LPAP in TCM and MCH (1:9) with the addition of I(III). (k) UV absorption of PAP at 343 nm and fluorescence intensity at 474 nm under different equivalents of I(III). The inserted image shows the changes in PAP after the addition of 0.1 equivalents of I(III). (l) SEM of LPPP with I(III) in TCM with fMCH = 60%; the concentrations of PPP and I(III) are both 0.02 mM. | ||
In the assembly system, the formation of halogen bonds has a profound impact on the entire system. In contrast to the disperse system in solution, adding only 0.1 equivalents of I(III) to LPAP causes a substantial increase in both absorption and fluorescence intensities (Fig. 8i and j). After the in situ addition of 0.1 equivalents of I(III) to the PAP assembly, the solution immediately became clear (insert of Fig. 8k). This is because before the addition of I(III), PAP was arranged regularly and compactly, resulting in a high extinction coefficient of the solution. The stacking between pyrenes caused aggregation-caused quenching, leading to weak fluorescence. After the addition of I(III), the introduction of a positive charge and the bulk counterion BArF4− disrupted the regular stacking of PAP. Therefore, the halogen bond complexes formed a loose spherical arrangement without chiral stacking, greatly suppressing the supramolecular chirality and recovering the folding molecular chirality (Fig. 8l). Owing to the maintenance of molecular chirality by the halogen bond complexes in the assembly system, the chiral signals observed at different volume ratios of MCH are consistent with the same sign. LPPP + I(III) shows a positive Cotton effect in all four assembly systems, which is distinctly different from the chiral inversion observed for PPP at a 70% volume fraction of MCH (Fig. 8e and f). Moreover, when the molecular chirality recovers in the assembled system, the CPL signal of the halogen-bonded complex is enhanced compared to that of the PPP (Fig. 8g and h). Because PAP and PPP exist in their molecular states in pure TCM, this condition can serve as a highly representative reference. By comparing the CD, CPL, UV, and PL data from the pure solution and the co-assembled system in MCH/TCM (Fig. S38–S41†), we found that the spectra of the PAP and PPP co-assembled with hypervalent iodine (I(III)) species closely resemble those in the pure TCM solution. This also suggests that the introduction of hypervalent iodine effectively suppresses supramolecular chirality, leading to its conversion into molecular chirality. Although it gives large spherical aggregates in the aggregated state, the packing between building units is believed to be random without long-range packing. In this work, by constructing ultra-strong halogen bonds in the aggregated state, we successfully achieved a transition of halogen-bonded complexes from supramolecular chirality to molecular folding chirality. This multi-level chiral transition induced by halogen bonding provides new strategies and ideas for achieving controllable multidimensional chiral inversion and chiral luminescent materials.41–46
Conclusions
In summary, this work introduces a folded peptide sequence designed with 2,2′-bipyridine as a rotatable axis, covalently linking pyrene-capped amino acids. The β-turn of 2,2′-bipyridine, along with hydrogen bonding and π-stacking anchoring, facilitates the formation of parallel β-sheet folding. The folding allows for the chirality transfer across spacers towards the bipyridine core and the signaling pyrenes. Furthermore, the folding displays solvent-adaptiveness, featuring tunable excimer emission and finely tailored chiroptical activity, with an asymmetry factor of up to 10−2. The self-assembly of the peptide sequence yields a supramolecular nanohelix, exhibiting high sensitivity to the delicate fractions of good/poor solvents. A particular 70 vol% of MCH prefers an opposite handedness compared to other systems. The 2,2′-bipyridine unit serves as a halogen bond acceptor, exhibiting ultrahigh binding affinity with the biaxal halogen bond donor I(III) in both the solution and assembly states. Notably, the in situ addition of 0.1 molar equivalents of I(III) swiftly converts the one-dimensional nanohelix into zero-dimensional nanoparticles. Akin to peptide denaturation, it diminishes supramolecular chirality and re-establishes folded molecular chirality. This work reveals the dynamic transformation of synthetic folded peptides, offering new insights that could inspire design strategies for peptide or protein-based chiroptical materials.Experimental
Materials, experimental details, additional CD, and MS spectra can be found in the ESI.†Data availability
The data supporting the findings of this study are available from the authors upon reasonable request.Author contributions
W. Ma carried out the main experiments and data analysing. P. Xing and A. Hao proposed the assumption and wrote the paper.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work is also supported by the National Natural Science Foundation of China (Grant No. 22171165 and 22371170), and Natural Science Foundation of Shandong Province, No. ZR2022MB080.Notes and references
- H. Liu, R. Wang, J. Wei, C. Cheng, Y. Zheng, Y. Pan, X. He, M. Ding, H. Tan and Q. Fu, J. Am. Chem. Soc., 2018, 140, 6604–6610 CrossRef CAS PubMed.
- P. Zhou, X. Hu, J. Li, Y. Wang, H. Yu, Z. Chen, D. Wang, Y. Zhao, S. M. King, S. E. Rogers, J. Wang, J. R. Lu and H. Xu, J. Am. Chem. Soc., 2022, 144, 21544–21554 CrossRef CAS PubMed.
- Y. Jang and J. A. Champion, Acc. Chem. Res., 2016, 49, 2188–2198 CrossRef CAS PubMed.
- Y. Tang, G. Ghirlanda, W. A. Petka, T. Nakajima, W. F. DeGrado and D. A. Tirrell, Angew. Chem., Int. Ed., 2001, 40, 1494–1496 CrossRef CAS PubMed.
- J. Yoon, D. Thirumalai and C. Hyeon, J. Am. Chem. Soc., 2013, 135, 12112–12121 CrossRef CAS PubMed.
- J. J. Ewbank and T. E. Creighton, Nature, 1991, 350, 518–520 CrossRef CAS PubMed.
- B. Shan, S. McClendon, C. Rospigliosi, D. Eliezer and D. P. Raleigh, J. Am. Chem. Soc., 2010, 132, 4669–4677 CrossRef CAS PubMed.
- F. Sbordone, A. Micallef and H. Frisch, Angew. Chem., Int. Ed., 2024, 63, e202319839 CrossRef CAS PubMed.
- J. Hermans and G. Acampora, J. Am. Chem. Soc., 1967, 89, 1547–1552 CrossRef CAS PubMed.
- S. S. Bag, S. Jana, A. Yashmeen and S. De, Chem. Commun., 2015, 51, 5242–5245 RSC.
- A. D'Souza, M. Mahajan and S. Bhattacharjya, Chem. Sci., 2016, 7, 2563–2571 RSC.
- A. Perczel, I. Jákli and I. G. Csizmadia, Chem.–Eur. J., 2003, 9, 5332–5342 CrossRef CAS PubMed.
- Z. Zong, Z. Cao, A. Hao and P. Xing, J. Mater. Chem. C, 2021, 9, 12191–12200 RSC.
- S. R. Beeren and J. K. M. Sanders, Chem. Sci., 2011, 2, 1560–1567 RSC.
- T. Moriuchi and T. Hirao, Acc. Chem. Res., 2010, 43, 1040–1051 CrossRef CAS PubMed.
- R. S. Herrick, R. M. Jarret, T. P. Curran, D. R. Dragoli, M. B. Flaherty, S. E. Lindyberg, R. A. Slate and L. C. Thornton, Tetrahedron Lett., 1996, 37, 5289–5292 CrossRef CAS.
- B. Liu, J. Gao, A. Hao and P. Xing, Chem.–Eur. J., 2022, 28, e202200682 CrossRef CAS PubMed.
- J. Morimoto, J. Kim, D. Kuroda, S. Nagatoishi, K. Tsumoto and S. Sando, J. Am. Chem. Soc., 2020, 142, 2277–2284 CrossRef CAS PubMed.
- T. Fukushima, K. Tamaki, A. Isobe, T. Hirose, N. Shimizu, H. Takagi, R. Haruki, S.-i. Adachi, M. J. Hollamby and S. Yagai, J. Am. Chem. Soc., 2021, 143, 5845–5854 CrossRef CAS PubMed.
- Q. Zhang, A. Hao and P. Xing, ACS Nano, 2023, 17, 9468–9477 CrossRef CAS PubMed.
- Q. Cheng, A. Hao and P. Xing, Chem. Sci., 2024, 15, 618–628 RSC.
- X. Jiang, Y.-K. Lim, B. J. Zhang, E. A. Opsitnick, M.-H. Baik and D. Lee, J. Am. Chem. Soc., 2008, 130, 16812–16822 CrossRef CAS PubMed.
- S. Chen, Z. Li, C. Zhang, X. Wu, W. Wang, Q. Huang, W. Chen, J. Shi and D. Yuan, Small, 2023, 19, 2301063 CrossRef CAS PubMed.
- S. Adorinni, S. Gentile, O. Bellotto, S. Kralj, E. Parisi, M. C. Cringoli, C. Deganutti, G. Malloci, F. Piccirilli, P. Pengo, L. Vaccari, S. Geremia, A. V. Vargiu, R. De Zorzi and S. Marchesan, ACS Nano, 2024, 18, 3011–3022 CrossRef CAS PubMed.
- J. Li, Y. Cui, Y.-L. Lu, Y. Zhang, K. Zhang, C. Gu, K. Wang, Y. Liang and C.-S. Liu, Nat. Commun., 2023, 14, 5030 CrossRef CAS PubMed.
- Z. Wang, X. Xie, A. Hao and P. Xing, Angew. Chem., Int. Ed., 2024, 63, e202407182 CrossRef CAS PubMed.
- M. Liu, L. Zhang and T. Wang, Chem. Rev., 2015, 115, 7304–7397 CrossRef CAS PubMed.
- J. M. Maier, P. Li, E. C. Vik, C. J. Yehl, S. M. S. Strickland and K. D. Shimizu, J. Am. Chem. Soc., 2017, 139, 6550–6553 CrossRef CAS PubMed.
- B. Bhayana and C. S. Wilcox, Angew. Chem., Int. Ed., 2007, 46, 6833–6836 CrossRef CAS PubMed.
- S. L. Cockroft and C. A. Hunter, Chem. Commun., 2006, 3806–3808, 10.1039/B608165G.
- H. Tanaka, Y. Inoue and T. Mori, ChemPhotoChem, 2018, 2, 386–402 CrossRef CAS.
- Y. Ohishi and M. Inouye, Tetrahedron Lett., 2019, 60, 151232 CrossRef CAS.
- K. Takaishi, K. Iwachido, R. Takehana, M. Uchiyama and T. Ema, J. Am. Chem. Soc., 2019, 141, 6185–6190 CrossRef CAS PubMed.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- Y. Lu, H. Li, X. Zhu, W. Zhu and H. Liu, J. Phys. Chem. A, 2011, 115, 4467–4475 CrossRef CAS PubMed.
- C. J. Serpell, N. L. Kilah, P. J. Costa, V. Félix and P. D. Beer, Angew. Chem., Int. Ed., 2010, 49, 5322–5326 CrossRef CAS PubMed.
- S. H. Jungbauer and S. M. Huber, J. Am. Chem. Soc., 2015, 137, 12110–12120 CrossRef CAS PubMed.
- S. An, A. Hao and P. Xing, Adv. Mater., 2024, 36, 2402314 CrossRef CAS PubMed.
- G. Gong, S. Lv, J. Han, F. Xie, Q. Li, N. Xia, W. Zeng, Y. Chen, L. Wang, J. Wang and S. Chen, Angew. Chem., Int. Ed., 2021, 60, 14831–14835 CrossRef CAS PubMed.
- E. M. D. Allouche, E. Grinhagena and J. Waser, Angew. Chem., Int. Ed., 2022, 61, e202112287 CrossRef CAS PubMed.
- S. Li, J. Wang, M. Tian, X. Meng, J. Wang and J. Guo, Angew. Chem., Int. Ed., 2024, 63, e202405615 CrossRef CAS PubMed.
- S. Show, A. Jamadar, S. Gorai, S. Mula and A. Das, Chem. Commun., 2025, 61, 1826–1829 RSC.
- Y.-J. Wang, X.-Y. Shi, P. Xing, X.-Y. Dong and S.-Q. Zang, Sci. Adv., 2023, 9, eadj9013 CrossRef CAS PubMed.
- S. Zheng, J. Han, X. Jin, Q. Ye, J. Zhou, P. Duan and M. Liu, Angew. Chem., Int. Ed., 2021, 60, 22711–22716 CrossRef CAS PubMed.
- Z. Wang, A. Hao and P. Xing, Small, 2023, 19, 2302517 CrossRef CAS PubMed.
- Y. Xu, A. Hao and P. Xing, Angew. Chem., Int. Ed., 2022, 61, e202113786 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5sc01441g |
| This journal is © The Royal Society of Chemistry 2025 |