
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRearranging spin electrons by axial-ligand-induced hybridization state transition to boost the activity of nickel single-atom-catalysts for electrochemical CO2 reduction†
Mingxia
Peng
a,
Kai
Huang
b,
Xiuyuan
Hu
c,
Andrea
Zitolo
 d,
Honglai
Liu
ae,
Cheng
Lian
*ae and
Jingkun
Li
*a
d,
Honglai
Liu
ae,
Cheng
Lian
*ae and
Jingkun
Li
*a
aSchool of Chemistry and Molecular Engineering, East China University of Science and Technology, Shanghai 200237, P. R. China. E-mail: liancheng@ecust.edu.cn; lijingkun@ecust.edu.cn
bSchool of Pharmaceutical and Chemical Engineering, Taizhou University, Taizhou, Zhejiang 318000, P. R. China
cNo. 2 High School of East China Normal University, 555 Chenhui Rd, Pudong, Shanghai, 201203, P. R. China
dSynchrotron SOLEIL, L'Orme des Merisiers, Départementale 128, 91190 Saint-Aubin, France
eState Key Laboratory of Chemical Engineering, East China University of Science and Technology, Shanghai 200237, P. R. China
First published on 20th March 2025
Abstract
Single-atom catalysts (SACs) with M–N4 active sites show great potential to catalyze the electrochemical CO2 reduction reaction (eCO2RR) toward CO. The activity and selectivity of SACs are determined by the local coordination configuration of central metal atoms in M–N4 sites, which is readily tuned by axial ligands. In this work, we construct axial ligands in situ on two Ni–N4-type model SACs, NiPc and Ni–N–C, by adding Cl− into the electrolyte taking advantage of the strong chemisorption of Cl− over Ni–N4. Cl axial ligand lowers the energy barrier of the potential-determining step for the eCO2RR due to a hybridization state transition of Ni orbitals and the resulting rearrangement of spin electrons. Consequently, both NiPc and Ni–N–C with axial Cl exhibit superior activity for the eCO2RR toward CO. Finally, we propose the magnetic moment of Ni as a universal descriptor for the eCO2RR toward CO on Ni–N4 with various axial ligands.
Introduction
The electrochemical CO2 reduction reaction (eCO2RR) yielding value-added chemicals and fuels, coupled with renewable electricity, offers a sustainable solution to achieve carbon neutrality.1 The products of the CO2RR vary from C1 (CO, CH4, HCOOH, CH3OH, etc.) to C2+ (CH3CH2OH, C2H4, etc.).2 The high energy barrier for C–C coupling, which is the key step to form C2+ products, generally leads to high overpotential and low selectivity for the eCO2RR toward C2+.3 In contrast, C1 products with fewer electrons transferred (e.g., CO and HCOOH) are economically favorable due to their low overpotential,3 high selectivity, and enhanced energy efficiency.4 CO constitutes a crucial raw material in industrial production processes.5 Particularly, CO and H2 (the by-product of the eCO2RR) with a tunable ratio co-produced by the eCO2RR can be readily used as a feedstock for syngas.6 In addition, cascade eCO2RR systems, which integrate two consecutive steps of CO2-to-CO and CO-to-C2+ either within one CO2 electrolyzer using a bifunctional cathode or in two tandem CO2 and CO electrolyzers,7–9 have been reported to boost the current density and faradaic efficiency (FE) for the eCO2RR toward C2+. Thus, extensive efforts have been devoted to exploring catalysts for the eCO2RR toward CO with outstanding activity and tunable selectivity in the past decades.10,11Single-atom catalysts (SACs) with unique electronic and geometric structures exhibit superior activity and selectivity for the eCO2RR toward CO.12,13 The M–N4 (M = Fe, Co, Ni, Cu, etc.) moieties, in both macrocyclic metal complexes (metal porphyrins, metal phthalocyanines (MPc), etc.) and their analogous metal–nitrogen–carbon (M–N–C) materials, are widely accepted as active sites.14,15 Particularly, nickel SACs deliver extraordinary performance with a FE >90% at a wide potential range.16,17 However, nickel SACs still suffer from insufficient activity in practical CO2 electrolyzers, leading to a significantly reduced selectivity of the eCO2RR toward CO at high current densities (e.g., >200 mA cm−2).17 Thus, it is pivotal to further boost the activity of Ni SACs for the eCO2RR toward CO to promote their feasible implementation in large-scale CO2 electrolyzers. It is reported that the symmetric D4h square-planar geometry of Ni–N4 is unfavorable for adjusting the spatial distribution of electrons and, consequently impedes the electron transfer during electrochemical reactions.17–20 Hence, breaking the D4h symmetry of Ni–N4via geometric distortion modulates their electronic structures, leading to accelerated electron transfer and enhanced activity of nickel SACs for the eCO2RR toward CO.21
Regulating the planar and axial ligands are two major strategies for tuning the geometric and electronic structures of M–N4.22–25 Planar ligand regulation includes doping with heteroatoms,26,27 engineering surface vacancies,28,29 adjusting coordination numbers,30 and constructing multi-atom sites.6 For example, S. Ji et al.31 incorporated P heteroatoms in the second coordination shell of Fe–N4 to break the local symmetry of electron distribution, leading to excellent activity and stability for oxygen reduction and evolution reactions. S. Chen et al.32 doped sulfur in the second coordination shell of Fe–N4, which induces a pronounced proton-feeding effect to boost the eCO2RR. However, it remains a great challenge for the precise and site-specific manipulation of the first and/or secondary coordination shell of central metal atoms, resulting in heterogeneous structures of planar coordination particularly for M–N–Cs obtained by high-temperature pyrolysis.33,34 In contrast, M–N4 sites with axial ligands constitute a well-defined proximal coordination configuration with tunable geometric and electronic structures.22 M. Li et al.35 reported an electronic localization enhancement of Ni–N4 induced by the Cl axial ligand, accounting for the superior HER activity of Ni–N4–Cl. However, electronic-level insights into the axial effect in tuning the eCO2RR activity of M–N4 moieties are still lacking, which is crucial for designing highly efficient SACs for the eCO2RR.
Herein, we systematically investigated the electronic structures and eCO2RR activities of two Ni–N4-type model SACs, namely NiPc and Ni–N–C, with and without the axial ligand. The axial ligand is introduced in situ by adding Cl− into the electrolyte taking advantage of the strong chemisorption of Cl− on Ni–N4. The Cl axial ligand induces a transition in hybridization states of Ni orbitals and a rearrangement of spin electrons, leading to the low energy barrier for the formation of key intermediate COOH*. As a result, Ni–N4 SACs with Cl axial ligands exhibit an enhanced CO partial current density. The magnetic moment of Ni is proposed as a universal descriptor for the eCO2RR toward CO on Ni–N4 with various axial ligands. This study provides a feasible strategy for screening highly efficient M–N4-type SACs with axial ligands for the eCO2RR toward CO.
Results and discussion
There are two typical classes of SACs extensively investigated for the eCO2RR: macrocyclic metal complexes (e.g., MPc) with a well-defined square-planar M–N4 coordination configuration36 and their analogous M–N–Cs consisting of isolated metal atoms anchored on nitrogen-doped carbon.37 M–N–Cs derived from high-temperature pyrolysis generally show heterogeneous structures, with well-recognized structures of bulk-hosted M–N4–C10 moieties embedded within a graphene sheet (denoted as MN4 hereafter) and edge-hosted M–N2+2–C4+4 bridging two adjacent graphene sheets (denoted as MN2+2 hereafter).38–40 Taking into consideration the superior activity and selectivity of Ni–N4 moieties,17,36 we focus on Ni SACs and exploit NiPc, NiN4 and NiN2+2 as representative model structures (Fig. 1a–c) to investigate the effect of axial ligands on their electrocatalytic activity for the eCO2RR toward CO via density functional theory (DFT) simulations. | ||
| Fig. 1 The morphology and structure of Ni–N–C. Top views (upper panel) and side views (lower panel) of model structures for (a) NiPc, (b) NiN4 and (c) NiN2+2 with and without Cl axial ligand. The black, blue, purple, green, and white balls represent C, N, Ni, Cl, and H atoms, respectively. (d) SEM image. (e) HAADF-STEM image and the corresponding EDX mappings. (f and g) Abbreviation-corrected STEM images with different magnifications. (h) Ni K-edge XANES spectrum. The spectra for nickel foil and nickel oxide powders are also presented as references. (i) Ni 2p XPS spectrum. (j) The Fourier transform of experimental and best fit Ni K-edge EXAFS spectra. Inset is the schematic model for the best fit, with nickel and nitrogen atoms represented by purple and blue balls, respectively. No phase-shift correction was applied to the Fourier transforms. (k) N 1s XPS spectrum. (l) Ni K-edge EXAFS analysis: Ni–N, Ni–C γ(2) two-body signals and the N–Ni–N γ(3) three-body signal included in the fit, the total signal (red line) superimposed on the experimental one (black line). | ||
We then utilized NiPc and Ni–N–C as model catalysts to verify the positive effect of Cl axial ligand on the electrolytic activity of Ni–N4 for the eCO2RR toward CO. NiPc was loaded on carbon supports via ball milling to enhance the electronic conductivity, followed by a heat treatment under milder conditions (300 °C) to stabilize NiPc on carbon without altering its structure.41,42 A Ni–N–C material consisting of atomically dispersed nickel sites was synthesized according to our previous work.43 The X-ray diffraction (XRD) pattern of Ni–N–C reveals two characteristic peaks of disordered carbon (Fig. S1a†), and the defective structure of carbon is further supported by the high intensity of D band at 1350 cm−1 in the Raman spectrum (Fig. S1b†). The scanning electron microscopy (SEM) image indicates a porous structure of Ni–N–C (Fig. 1d), which consists of abundant micropores and mesopores (Fig. S1c and d†). The Brunauer–Emmett–Teller surface area of Ni–N–C is 223 m2 g−1 (Table S1†). The highly microporous and defective structure of the carbon matrix is favorable to anchor single atoms of transition metals.44 The energy-dispersive X-ray spectroscopy (EDX) mappings show a uniform distribution of Ni, N, and C elements in Ni–N–C (Fig. 1e). No metallic particle is observed after an extensive evaluation of high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) images (Fig. 1f), in good agreement with the absence of characteristic peaks arising from nickel-based agglomerates in XRD (Fig. S1a†). The isolated nickel atoms are distinctly visible (highlighted by red circles) in the abbreviation-corrected STEM image (Fig. 1g), confirming that nickel in Ni–N–C is present as single atoms.
The atomically dispersed nature of nickel in Ni–N–C is further supported by X-ray absorption spectroscopy (XAS) and X-ray photoelectron spectroscopy (XPS) (Fig. 1h–l). The Ni 2p X-ray photoelectron spectroscopy (XPS) spectrum of Ni–N–C delivers only one main peak arising from Ni2+ accompanied by several satellite peaks, and Ni0 species is not detected (Fig. 1i).45 The XANES spectrum of Ni–N–C significantly deviates from that of NiO (Fig. 1h), suggesting that the Ni2+ species detected in XPS is not in the form of oxide. Meanwhile, the Fourier transform of extended X-ray absorption fine structure (EXAFS) exhibits negligible signals at ∼2.1 and ∼2.4 Å attributed to the Ni–Ni backscattering in metallic nickel and nickel oxide (Fig. 1j and S3†), indicating the absence of nickel-based agglomerates. Thus, XPS, EXAFS, XRD and STEM characterization studies unambiguously demonstrate the atomic dispersion of nickel in Ni–N–C. The nickel content in Ni–N–C is 0.22 at% according to XPS (Table S2†).
Then we elucidate the coordination environment of atomically dispersed nickel sites in Ni–N–C with detailed analysis of N 1s XPS and EXAFS spectra. Five nitrogen species are identified in the N 1s XPS spectrum of Ni–N–C, namely, N in M–Nx, pyrrolic N (labeled as N–H), pyridinic N, graphitic N (including Ngr and N+), and N–Ox (Fig. 1k and Table S3†).43 The presence of abundant M–Nx suggests that nickel coordinates with nitrogen defects in the carbon matrix. Thus, we fit the Fourier transform of the experimental EXAFS spectrum of Ni–N–C with a variable number of in-plane nitrogen atoms binding the Ni2+ center (inset of Fig. 1j), and the best-fit result shows a coordination number of 4.3 and a Ni–N bond distance of 1.92 Å (Table S4†). The dominant contribution is given by Ni–N first shell coordination (Fig. 1l), leading to the major peak at 1.5 Å in the Fourier transform of the EXAFS spectrum (Fig. 1j). In contrast, Ni–C second shell coordination contributes to the minor peak in the range of 2–3 Å (Fig. 1j and l).40 The intensity of the N–Ni–N signal is insufficient to determine the bond angle and, consequently, the geometric structure of the Ni–N4 moiety. Therefore, the nickel single atoms in Ni–N–C are mainly present in the form of Ni–N4 moieties, with either square-planar or distorted geometry.
The eCO2RR toward CO undergoes four elementary steps (written for acidic solutions): (1) CO2(g) + * → CO2*, (2) CO2* + H+ + e− → COOH*, (3) COOH* + H+ + e− → CO* + H2O(g), and (4) CO* → CO(g) + *,46 with a debatable potential-determining step (PDS) of either COOH* formation or CO2* adsorption.46,47 The configurations of all elementary steps on NiPc, NiN4 and NiN2+2 models are shown in Fig. S3.† The free energy (G) diagrams for the eCO2RR toward CO over three models show that the Gibbs free energy change (ΔG) of COOH* formation is the highest among four elementary steps (Fig. 2a–c and Table S5†). Thus, we can infer that the first proton coupled electron transfer to form COOH* is the potential-determining step (PDS) for the eCO2RR toward CO over Ni–N4. The changing current densities with pH on an absolute scale (vs. normal hydrogen electrode (NHE)) over NiPc and Ni–N–C (with NiN4 and NiN2+2 structures) indicate that proton participates in the PDS (Fig. 2d and e), further confirming COOH* formation as PDS for the eCO2RR toward CO. Pinpointing Cl axial ligand on Ni–N4 does not alter the PDS (Fig. 2a–c), but lowers its energy barrier for all three model structures (Fig. 2f). Moreover, NiN2+2 exhibits the lowest ΔG among three models without the axial ligand due to the strong adsorption of *COOH. However, the strong adsorption of *CO over NiN2+2 as a consequence of linear scaling relationship48 leads to an uphill *CO desorption (ΔG > 0), in contrast to the spontaneous *CO desorption over Ni–N4 and NiPc (ΔG < 0). Interestingly, Cl axial ligand not only strengthens *COOH adsorption over NiN2+2, but also weakens the adsorption of *CO, leading to a reduced energy barrier of PDS and a downhill *CO desorption. Thus, Cl axial ligand breaks the linear scaling relationship over NiN2+2,48 probably due to the synergistic effect of the axial ligand and defects in the edges. Overall, the Cl axial ligand lowers the energy barrier of PDS for the eCO2RR toward CO over Ni–N4, boosting the catalytic activity of Ni single atoms.
 | ||
| Fig. 2 The effect of Cl axial ligand on the energy barrier of PDS for the eCO2RR toward CO over Ni–N4. Free energy diagrams of the eCO2RR to CO for (a) NiPc, (b) NiN4 and (c) NiN2+2 with and without the Cl axial ligand. Experimental current densities plotted against potential vs. NHE for (d) NiPc and (e) Ni–N–C. (f) Comparison of the ΔG of PDS for NiPc, NiN4 and NiN2+2 with and without the Cl axial ligand. | ||
We then measured the activity and selectivity of the eCO2RR toward CO over NiPc and Ni–N–C, and evaluated the effect of Cl axial ligand by adding KCl in the electrolyte. The metal center in the M–N4 site tends to be occupied by a H2O molecule in the backside axial position,49–51 and Cl− in the electrolyte replaces H2O due to the significantly higher binding energy of Cl than that of H2O adsorbed on Ni–N4 (including NiPc, NiN4 and NiN2+2) according to DFT simulations (Fig. 3a–c). Further charge analysis shows that Cl atoms transfer 0.14–0.15|e| charges to Ni atoms, indicative of a strong chemisorption of Cl on Ni–N4 (insets of Fig. 3a–c). In contrast, the charge transfer from H2O molecules to Ni atoms is only 0.00–0.03|e|, suggesting a physisorption of H2O on Ni–N4 (insets of Fig. 3a–c). Thus, we can infer that Ni–N4 binds with the Cl axial ligand by simply adding Cl− in the electrolyte.
 | ||
| Fig. 3 The effect of Cl axial ligand on the activity of Ni–N4 for the eCO2RR toward CO. The binding energies of Cl and H2O adsorbed on the Ni center in (a) NiPc, (b) NiN4, and (c) NiN2+2. The insets are the charge density differences of Cl and H2O on Ni–N4 sites. The cyan and yellow regions represent loss and accumulation of charge density, respectively, and the isosurface value is ±0.002 e bohr−3. CO partial current densities vs. potential (iR-corrected) for (d) NiPc and (e) Ni–N–C. H2 partial current densities vs. potential (iR-corrected) for (f) NiPc and (g) Ni–N–C. The above electrochemical characterization is measured in 1 M KHCO3 electrolyte with 0, 10 and 100 mM KCl (the data shown in (d–g) are the average of three repeated experiments). | ||
The CO and H2 partial current densities vs. potential (iR-corrected) over NiPc and Ni–N–C catalysts in electrolytes measured in 1 M KHCO3 electrolyte with 0, 10 and 100 mM KCl are presented in Fig. 3d–g. CO2 electrolysis was conducted for 30 minutes at constant current densities from 10 mA cm−2 to 60 mA cm−2 in a flow cell with Ag/AgCl as the reference electrode. The total faradaic efficiencies (FEs) of CO and H2 for NiPc and Ni–N–C are ∼100% (Fig. S4†), indicating negligible liquid products formed during the eCO2RR. Thus, we didn't measure liquid products for this study. Both NiPc and Ni–N–C exhibit enhanced CO partial current densities in a wide potential range after adding trace amount of Cl− (10 mM), and the enhancement increases with an increasing Cl− concentration (Fig. 3d and e). The improvement in the catalytic activity of Ni–N4 for the eCO2RR toward CO induced by the Cl axial ligand is consistent with the reduced energy barrier of PDS (Fig. 2a–c). In contrast, the H2 partial current densities of NiPc and Ni–N–C are not affected by the addition of Cl− (Fig. 3f and g). The above results demonstrate that the Cl axial ligand enhances the intrinsic activity of Ni–N4 for the eCO2RR toward CO without altering its HER activity, leading to an increase in the total current density (Fig. S5†). Moreover, during the 8-hour chronoamperometry (CA) tests in 1 M KHCO3 electrolyte with 100 mM KCl, both Ni–N–C and NiPc maintained stable CO faradaic efficiency (FE) and current density, as shown in Fig. S6.† The Cl 2p spectra of the Ni–N–C and NiPc after stability tests exhibit a characteristic peak at ∼198.0 eV attributed to Ni–Cl species (Fig. S7†),52 with Cl as the axial ligand considering the absence of high-temperature required to form planar coordinated Cl.24 In addition, a new peak at 858.3 eV assigned to Ni3+ emerges in the Ni 2p XPS spectra of Ni–N–C and NiPc after stability tests (Fig. S8†), further confirming the formation of Ni–N4–Cl with Cl as the axial ligand.53 The postmortem XPS studies unambiguously demonstrate the successful introduction and high stability of the Cl axial ligand. Approximately 37% and 18% of Ni3+ species are detected in Ni–N–C and NiPc after the stability tests (Fig. S8†), suggesting that Ni–N4 is partially coordinated with axial Cl ligands.
Strong chemisorption of the axial ligand on M–N4 sites pulls single metal atoms away from the carbon plane, which breaks the D4h symmetry of square-planar M–N4, and consequently promotes the electron transfer during electrochemical reactions.19,20 The distance of Ni atom from the carbon plane (dNi) and the Ni–N bond distance (dNi–N) (insets in Fig. 4a and b) describe the degree of geometric distortion of Ni–N4 sites upon adsorption of Cl axial ligand. Thus, we explore the relationship between the energy barrier of PDS (ΔGp) for the eCO2RR and dNi/dNi–N (Fig. 4a and b). For comparison, weak adsorption of H2O axial ligand on three models are considered (Fig. S9, S10 and Table S6†). In contrast to H2O molecules (dNi = 0.02–0.08 Å), Cl atom with a strong binding energy on Ni–N4 pulls Ni atom further away from the carbon plane (dNi = 0.10–0.33 Å), resulting in a larger dNi–N (Fig. 4a, b and Table S7†). There is a positive correlation between dNi and ΔGp: the further away the Ni atoms are from the carbon plane, the lower the ΔGp (Fig. 4a). The relationship between dNi–N and ΔGp falls into two linear lines for NiPc and Ni–N–C (NiN4 and NiN2+2), respectively (Fig. 4b). It is noteworthy that both the change of dNi and dNi–N of NiPc–Cl are smaller than those of NiN4–Cl and NiN2+2–Cl due to the more rigid structure of NiPc (Fig. 4a and b). Moreover, the periodic boundary conditions on the xy plane of molecular NiPc and periodic NiN4/NiN2+2 are different for DFT simulations. Thus, it is reasonable that the linear coefficient for the correlation between the geometric parameters and ΔGp of NiPc differs from that of Ni–N–C. In summary, Cl axial ligand induces geometric distortion of Ni–N4 sites, and the geometric parameters, dNi and dNi–N, are strongly correlated with the energy barrier of PDS, and consequently the electrocatalytic activity for the eCO2RR toward CO.
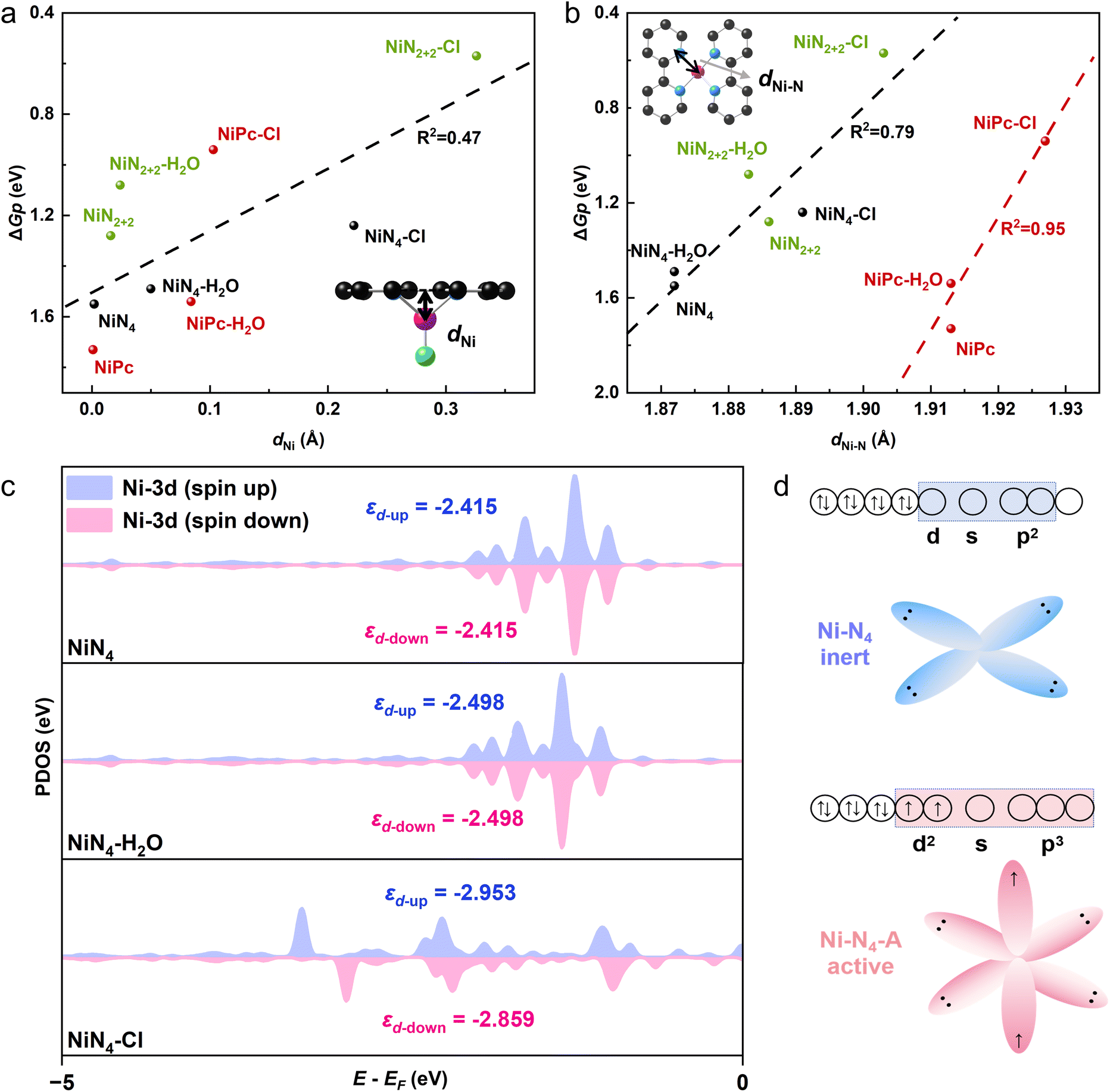 | ||
| Fig. 4 The effect of Cl axial ligand on the geometric and electronic structure of Ni–N4. The relationship between ΔGp and (a) dNi, (b) dNi–N. (c) The partial density of states and d-band center of Ni atoms in NiN4, NiN4–Cl, and NiN4–H2O. (d) Schematic diagram of orbital hybridization of Ni atoms in Ni–N4 with and without axial ligands. | ||
Then we evaluate the effect of Cl axial ligand on the electronic structures of Ni atoms in Ni–N4. The partial density of states (PDOS) show that the up and down spin images of Ni 3d orbitals of NiN4 and NiPc (with/without H2O ligands) are symmetrical (Fig. 4c and S11a†), indicating the symmetrical arrangement of the up and down spin electrons of 3d orbitals for Ni–N4 without geometric distortion (Fig. 4d). In contrast, the geometric distortion induced by Cl axial ligand results in the asymmetric up and down spin electron arrangement of Ni 3d orbitals, and thus the magnetic properties of NiN4–Cl (Fig. 4c, d and S11a†). The empty 3d, 4s and two 4p orbitals of Ni undergo dsp2 hybridization to interact with the sp2 orbitals of the four-coordinated N ligands in Ni–N4 with square planar geometry,54 whereas Ni–N4 with Cl axial ligand exhibits a d2sp3 hybridization with an octahedral coordination environment (Fig. 4d). The d2sp3 hybridization of Ni leads to the rearrangement of spin electrons, which activates Ni–N4 sites and boosts their intrinsic activity for the eCO2RR.54 Particularly, the up and down spin PDOS image of Ni 3d orbitals in NiN2+2 is asymmetric (Fig. S11b†), leading to a lower energy barrier for PDS of NiN2+2 than those of NiPc and NiN4 (Fig. 2f). The Cl axial ligand further intensifies the degree of asymmetry for the PDOS image of Ni 3d orbitals in NiN2+2 (Fig. S11b†), indicating the enhanced activity of NiN2+2–Cl for the eCO2RR toward CO.
We then analyzed the d-electron distribution of Ni atoms in NiPc, NiN4 and NiN2+2 with/without the Cl axial ligand (Fig. S12†). For all three models without Cl axial ligand, the peak of partial density of states (PDOS) below the Fermi level is mainly contributed by the d orbital components containing the z-axis direction (such as dyz, dxz, and dz2), indicating that the electrons involved in the eCO2RR are mainly provided by the d orbitals in the z-direction. Stronger the electron donating capability of d orbitals, better the eCO2RR activity we achieve. NiPc only provides electrons through dz2 and dxz, while all the dyz, dxz, and dz2 of NiN4 donate electrons. Moreover, the structural symmetry of NiN2+2 has been disrupted, enabling dxy to donate electrons in addition to dyz, dxz, and dz2. The above results are in good agreement with the superior eCO2RR activity of Ni–N–C to that of NiPc. Introducing Cl axial ligands further breaks the structural symmetry of NiPc, NiN4 and NiN2+2, which not only leads to asymmetric up and down spin images, but also brings the peak of d orbital components containing the z-axis direction closer to the Fermi level (Fig. S12†). As a result, the introduction of Cl axial ligands induces the strong electron donating capability and, consequently, enhances the eCO2RR activity of Ni–N4.
Since the asymmetric up and down spin electron arrangements of Ni 3d orbitals in Ni–N4–Cl are crucial for their enhanced eCO2RR activity, we then explore the relationship between ΔGp and Δεd (the difference in the d-band center of Ni between down and up spin). Δεd delivers a good linear relationship with ΔGp (Fig. S13a†). The change of Δεd induced by Cl axial ligands for three model structures follows the trend of NiPc (0.207 eV) > NiN2+2 (0.114 eV) > NiN4 (0.094 eV), perfectly matching the trend for the change of ΔGp induced by Cl axial ligands (NiPc > NiN2+2 > NiN4). Therefore, although the effect of axial ligands on the geometric structure of NiPc is the lowest among all three model structures due to its rigid structure, that on the electronic structures is the highest, resulting in the most significant decrease in ΔGp for NiPc induced by Cl axial ligands. Since Δεd is directly correlated with the electronic asymmetry of Ni–N4 as discussed above, we can safely conclude that the electronic symmetry breaking, which originates from the geometric symmetry breaking, is the primary factor in enhancing the eCO2RR activity.
Furthermore, the asymmetric up and down spin electron arrangements of Ni 3d orbitals will cause changes in the magnetic moment of Ni atoms (uNi), and the linear relationship between Δεd and uNi is shown in Fig. 5d. Therefore, we further investigate the relationship between ΔGp and uNi, which also exhibit a good linear correlation: larger the uNi, lower the ΔGp (Fig. S13b†). Thus, uNi and Δεd are possible descriptors of the eCO2RR activity of Ni–N4-type SACs with axial ligands. The separate linear relationships for NiPc and Ni–N–C systems are unified when using Δεd and uNi as descriptors since electronic properties circumvent the fundamentally different geometries of NiPc and Ni–N–C.
 | ||
| Fig. 5 Universal descriptors for the eCO2RR activity of Ni–N4 with various axial ligands. The relationships between ΔGp and (a) dNi, (b) dNi–N, (c) Δεd, uNi (the circular symbol represent Δεd, and the triangular symbol represents uNi). (d) The relationship between Δεd (the difference in the d-band center of Ni between down and up spin) and uNi (the magnetic moment of Ni atoms). | ||
To demonstrate the universality of the above geometric and electronic descriptors, we further extend axial ligands over NiN4 from Cl− to –NO, –SCN, and –CH3, and investigate the relationship between ΔGp and geometric/electronic parameters (dNi, dNi–N, uNi, Δεd). The configurations of all elementary steps on NiN4 models are shown in Fig. S14.† The free energy diagrams indicate that all axial ligands over NiN4 investigated exhibit reduced ΔGp, and thus promote the electrocatalytic CO2RR to CO (Fig. S15, S16 and Table S7†). There are linear correlations between ΔGp and dNi, dNi–N, uNi, Δεd for NiN4 with various ligands (Fig. S17†), which fit well in the linear lines we have described in Fig. 4a, b and S13† (Fig. 5). Therefore, uNi and Δεd are universal descriptors for evaluating the eCO2RR activity of Ni–N4-type SACs with a variety of axial ligands, and, most probably, can be extended to other M–N4-type SACs. The spin electron rearrangement in M–N4 SACs induced by axial ligands is the key for boosting eCO2RR activity.
Conclusions
In summary, we investigated the effect of axial ligand on the eCO2RR activity of two Ni–N4-type model catalysts (NiPc and Ni–N–C), and both theoretical and experimental studies demonstrate that Cl axial ligand is beneficial for the eCO2RR. The Cl axial ligand induces a hybridization state transition of nickel orbital from dsp2 to d2sp3 and, consequently, a rearrangement of spin electrons, accounting for the superior activity of Ni–N4–Cl for the eCO2RR. The low energy barrier of PDS for the eCO2RR exhibits a strong correlation with the geometric (dNi and dNi–N) and electronic (Δεd and uNi) parameters of Ni–N4. Furthermore, we propose the magnetic moment of Ni as a universal descriptor for the eCO2RR toward CO on Ni–N4 with various axial ligands (–H2O, –Cl, –NO, –SCN, and –CH3). Our work provides valuable insights on regulating the geometric configuration and electronic structure of SACs to boost their electrocatalytic activities for various energy and environmental applications.Data availability
All experimental and computational procedures and associated data are provided in the ESI.†Author contributions
M. P., K. H., and J. L. conceived the project. M. P. prepared materials, performed measurements and analysed the data. K. H. performed the DFT calculations. X. H. and A. Z. helped with some of the experiments and characterization. All authors discussed the results and commented on the manuscript. M. P., K. H., and J. L. wrote the draft, revised and finalized the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (22102114).Notes and references
- A. S. Hall, Immobilized cations boost acidic CO2 reduction, Nat. Catal., 2023, 6, 744–745 CrossRef CAS.
- M. Jiang, H. Wang, M. Zhu, X. Luo, Y. He, M. Wang, C. Wu, L. Zhang, X. Li, X. Liao, Z. Jiang and Z. Jin, Review on strategies for improving the added value and expanding the scope of CO2 electroreduction products, Chem. Soc. Rev., 2024, 53, 5149–5189 RSC.
- B. Rhimi, M. Zhou, Z. Yan, X. Cai and Z. Jiang, Cu-Based Materials for Enhanced C2+ Product Selectivity in Photo-/Electro-Catalytic CO2 Reduction: Challenges and Prospects, Nano-Micro Lett., 2024, 16, 64 CAS.
- Y. Y. Birdja, E. Pérez-Gallent, M. C. Figueiredo, A. J. Göttle, F. Calle-Vallejo and M. T. M. Koper, Advances and challenges in understanding the electrocatalytic conversion of carbon dioxide to fuels, Nat. Energy, 2019, 4, 732–745 CAS.
- N. Mohd Adli, W. Shan, S. Hwang, W. Samarakoon, S. Karakalos, Y. Li, D. A. Cullen, D. Su, Z. Feng, G. Wang and G. Wu, Engineering Atomically Dispersed FeN4 Active Sites for CO2 Electroreduction, Angew. Chem., Int. Ed., 2021, 60, 1022–1032 CrossRef CAS PubMed.
- Q. He, D. Liu, J. H. Lee, Y. Liu, Z. Xie, S. Hwang, S. Kattel, L. Song and J. G. Chen, Electrochemical Conversion of CO2 to Syngas with Controllable CO/H2 Ratios over Co and Ni Single-Atom Catalysts, Angew. Chem., Int. Ed., 2020, 59, 3033–3037 CrossRef CAS PubMed.
- P. De Luna, C. Hahn, D. Higgins, S. A. Jaffer, T. F. Jaramillo and E. H. Sargent, What would it take for renewably powered electrosynthesis to displace petrochemical processes?, Science, 2019, 364, eaav3506 CAS.
- F. Li, Y. C. Li, Z. Wang, J. Li, D. H. Nam, Y. Lum, M. Luo, X. Wang, A. Ozden, S.-F. Hung, B. Chen, Y. Wang, J. Wicks, Y. Xu, Y. Li, C. M. Gabardo, C. T. Dinh, Y. Wang, T. T. Zhuang, D. Sinton and E. H. Sargent, Cooperative CO2-to-ethanol conversion via enriched intermediates at molecule–metal catalyst interfaces, Nat. Catal., 2019, 3, 75–82 Search PubMed.
- X. Wang, J. F. De Araújo, W. Ju, A. Bagger, H. Schmies, S. Kühl, J. Rossmeisl and P. Strasser, Mechanistic reaction pathways of enhanced ethylene yields during electroreduction of CO2–CO co-feeds on Cu and Cu-tandem electrocatalysts, Nat. Nanotechnol., 2019, 14, 1063–1070 CAS.
- T. Möller, W. Ju, A. Bagger, X. Wang, F. Luo, T. Ngo Thanh, A. S. Varela, J. Rossmeisl and P. Strasser, Efficient CO2 to CO electrolysis on solid Ni–N–C catalysts at industrial current densities, Energy Environ. Sci., 2019, 12, 640–647 Search PubMed.
- S. Jin, Z. Hao, K. Zhang, Z. Yan and J. Chen, Advances and Challenges for the Electrochemical Reduction of CO2 to CO: From Fundamentals to Industrialization, Angew. Chem., Int. Ed., 2021, 60, 20627–20648 CAS.
- S. Liang, L. Huang, Y. Gao, Q. Wang and B. Liu, Electrochemical Reduction of CO2 to CO over Transition Metal/N-Doped Carbon Catalysts: The Active Sites and Reaction Mechanism, Adv. Sci., 2021, 8, 2102886 CAS.
- Y. Zhu, J. Sokolowski, X. Song, Y. He, Y. Mei and G. Wu, Engineering Local Coordination Environments of Atomically Dispersed and Heteroatom-Coordinated Single Metal Site Electrocatalysts for Clean Energy-Conversion, Adv. Energy Mater., 2020, 10, 1902844 CAS.
- S. Ren, D. Joulié, D. Salvatore, K. Torbensen, M. Wang, M. Robert and C. P. Berlinguette, Molecular electrocatalysts can mediate fast, selective CO2 reduction in a flow cell, Science, 2019, 365, 367–369 CAS.
- A. S. Varela, W. Ju, A. Bagger, P. Franco, J. Rossmeisl and P. Strasser, Electrochemical Reduction of CO2 on Metal-Nitrogen-Doped Carbon Catalysts, ACS Catal., 2019, 9, 7270–7284 CrossRef CAS.
- Y. Li, N. M. Adli, W. Shan, M. Wang, M. J. Zachman, S. Hwang, H. Tabassum, S. Karakalos, Z. Feng, G. Wang, Y. C. Li and G. Wu, Atomically dispersed single Ni site catalysts for high-efficiency CO2 electroreduction at industrial-level current densities, Energy Environ. Sci., 2022, 15, 2108–2119 RSC.
- Z. Yang, R. Chen, L. Zhang, Y. Li and C. Li, Recent progress in nickel single-atom catalysts for the electroreduction of CO2 to CO, Ind. Chem. Mater., 2024, 2, 533–555 Search PubMed.
- J. Bai, Z. Sun, H. Zhang, Y. Lian, Y. Deng, M. Xiang and Y. Su, Modulating the Local Coordination Environment of M–Nx Single-Atom Site for Enhanced Electrocatalytic Oxygen Reduction, Adv. Funct. Mater., 2024, 2417013 Search PubMed.
- R. Wang, L. Zhang, J. Shan, Y. Yang, J. F. Lee, T. Y. Chen, J. Mao, Y. Zhao, L. Yang, Z. Hu and T. Ling, Tuning Fe Spin Moment in Fe–N–C Catalysts to Climb the Activity Volcano via a Local Geometric Distortion Strategy, Adv. Sci., 2022, 9, 2203917 Search PubMed.
- X. Cheng, J. Hu, W. Shang, J. Guo, C. Xin, S. Zhang, S. Song, W. Liu and Y. Shi, Asymmetrically ligated single atomic nickel sites for efficient hydrogen peroxide electrosynthesis, Nano Res., 2024, 17, 1094–1100 CrossRef CAS.
- H. B. Yang, S. F. Hung, S. Liu, K. Yuan, S. Miao, L. Zhang, X. Huang, H. Y. Wang, W. Cai, R. Chen, J. Gao, X. Yang, W. Chen, Y. Huang, H. M. Chen, C. M. Li, T. Zhang and B. Liu, Atomically dispersed Ni(i) as the active site for electrochemical CO2 reduction, Nat. Energy, 2018, 3, 140–147 Search PubMed.
- Z. Li, R. Wu, S. Xiao, Y. Yang, L. Lai, J. S. Chen and Y. Chen, Axial chlorine coordinated iron–nitrogen–carbon single-atom catalysts for efficient electrochemical CO2 reduction, Chem. Eng. J., 2022, 430, 132882 Search PubMed.
- S. Ding, J. A. Barr, Q. Shi, Y. Zeng, P. Tieu, Z. Lyu, L. Fang, T. Li, X. Pan, S. P. Beckman, D. Du, H. Lin, J. C. Li, G. Wu and Y. Lin, Engineering Atomic Single Metal–FeN4 Cl Sites with Enhanced Oxygen-Reduction Activity for High-Performance Proton Exchange Membrane Fuel Cells, ACS Nano, 2022, 16, 15165–15174 CrossRef CAS PubMed.
- S. Wei, R. Yang, Z. Wang, J. Zhang and X. H. Bu, Planar Chlorination Engineering: A Strategy of Completely Breaking the Geometric Symmetry of Fe-N4 Site for Boosting Oxygen Electroreduction, Adv. Mater., 2024, 2404692 CrossRef CAS PubMed.
- J. X. Peng, W. Yang, Z. Jia, L. Jiao and H. L. Jiang, Axial coordination regulation of MOF-based single-atom Ni catalysts by halogen atoms for enhanced CO2 electroreduction, Nano Res., 2022, 15, 10063–10069 CrossRef CAS.
- Y. Chen, S. Ji, S. Zhao, W. Chen, J. Dong, W.-C. Cheong, R. Shen, X. Wen, L. Zheng, A. I. Rykov, S. Cai, H. Tang, Z. Zhuang, C. Chen, Q. Peng, D. Wang and Y. Li, Enhanced oxygen reduction with single-atomic-site iron catalysts for a zinc–air battery and hydrogen–air fuel cell, Nat. Commun., 2018, 9, 5422 CrossRef CAS PubMed.
- Y. Han, Y. Wang, R. Xu, W. Chen, L. Zheng, A. Han, Y. Zhu, J. Zhang, H. Zhang, J. Luo, C. Chen, Q. Peng, D. Wang and Y. Li, Electronic structure engineering to boost oxygen reduction activity by controlling the coordination of the central metal, Energy Environ. Sci., 2018, 11, 2348–2352 CAS.
- K. Mou, Z. Chen, X. Zhang, M. Jiao, X. Zhang, X. Ge, W. Zhang and L. Liu, Highly Efficient Electroreduction of CO2 on Nickel Single-Atom Catalysts: Atom Trapping and Nitrogen Anchoring, Small, 2019, 15, 1903668 CAS.
- Y. J. Sa, H. Jung, D. Shin, H. Y. Jeong, S. Ringe, H. Kim, Y. J. Hwang and S. H. Joo, Thermal Transformation of Molecular Ni2+–N4 Sites for Enhanced CO2 Electroreduction Activity, ACS Catal., 2020, 10, 10920–10931 CAS.
- J. Yang, Z. Qiu, C. Zhao, W. Wei, W. Chen, Z. Li, Y. Qu, J. Dong, J. Luo, Z. Li and Y. Wu, In Situ Thermal Atomization To Convert Supported Nickel Nanoparticles into Surface-Bound Nickel Single-Atom Catalysts, Angew. Chem., Int. Ed., 2018, 57, 14095–14100 CAS.
- S. Ji, Y. Mou, H. Liu, X. Lu, Y. Zhang, C. Guo, K. Sun, D. Liu, J. H. Horton, C. Wang, Y. Wang and Z. Li, Manipulating the Electronic Properties of an Fe Single Atom Catalyst via Secondary Coordination Sphere Engineering to Provide Enhanced Oxygen Electrocatalytic Activity in Zinc-Air Batteries, Adv. Mater., 2024, 36, 2410121 CrossRef CAS PubMed.
- S. Chen, X. Li, C. W. Kao, T. Luo, K. Chen, J. Fu, C. Ma, H. Li, M. Li, T. S. Chan and M. Liu, Unveiling the Proton-Feeding Effect in Sulfur-Doped Fe–N–C Single-Atom Catalyst for Enhanced CO2 Electroreduction, Angew. Chem., Int. Ed., 2022, 61, e202206233 CrossRef CAS PubMed.
- X. Li, T. Chen, B. Yang and Z. Xiang, Fundamental Understanding of Electronic Structure in FeN4 Site on Electrocatalytic Activity via dz2-Orbital-Driven Charge Tuning for Acidic Oxygen Reduction, Angew. Chem., Int. Ed., 2023, 62, e202215441 CAS.
- J. Li, L. Jiao, E. Wegener, L. L. Richard, E. Liu, A. Zitolo, M. T. Sougrati, S. Mukerjee, Z. Zhao, Y. Huang, F. Yang, S. Zhong, H. Xu, A. J. Kropf, F. Jaouen, D. J. Myers and Q. Jia, Evolution Pathway from Iron Compounds to Fe1(II)–N4 Sites through Gas-Phase Iron during Pyrolysis, J. Am. Chem. Soc., 2020, 142, 1417–1423 Search PubMed.
- M. Li, M. Wang, D. Liu, Y. Pan, S. Liu, K. Sun, Y. Chen, H. Zhu, W. Guo, Y. Li, Z. Cui, B. Liu, Y. Liu and C. Liu, Atomically-dispersed NiN4–Cl active sites with axial Ni–Cl coordination for accelerating electrocatalytic hydrogen evolution, J. Mater. Chem. A, 2022, 10, 6007–6015 RSC.
- S. Yang, Y. Yu, X. Gao, Z. Zhang and F. Wang, Recent advances in electrocatalysis with phthalocyanines, Chem. Soc. Rev., 2021, 50, 12985–13011 CAS.
- X. Xie, H. Peng, G. Ma, Z. Lei and Y. Xu, Recent progress in heteroatom doping to modulate the coordination environment of M–N–C catalysts for the oxygen reduction reaction, Mater. Chem. Front., 2023, 7, 2595–2619 RSC.
- Y. He, Q. Shi, W. Shan, X. Li, A. J. Kropf, E. C. Wegener, J. Wright, S. Karakalos, D. Su, D. A. Cullen, G. Wang, D. J. Myers and G. Wu, Dynamically Unveiling Metal–Nitrogen Coordination during Thermal Activation to Design High-Efficient Atomically Dispersed CoN4 Active Sites, Angew. Chem., Int. Ed., 2021, 60, 9516–9526 CrossRef CAS PubMed.
- H. Fei, J. Dong, Y. Feng, C. S. Allen, C. Wan, B. Volosskiy, M. Li, Z. Zhao, Y. Wang, H. Sun, P. An, W. Chen, Z. Guo, C. Lee, D. Chen, I. Shakir, M. Liu, T. Hu, Y. Li, A. I. Kirkland, X. Duan and Y. Huang, General synthesis and definitive structural identification of MN4C4 single-atom catalysts with tunable electrocatalytic activities, Nat. Catal., 2018, 1, 63–72 CrossRef CAS.
- F. Pan, H. Zhang, K. Liu, D. Cullen, K. More, M. Wang, Z. Feng, G. Wang, G. Wu and Y. Li, Unveiling Active Sites of CO2 Reduction on Nitrogen-Coordinated and Atomically Dispersed Iron and Cobalt Catalysts, ACS Catal., 2018, 8, 3116–3122 Search PubMed.
- L. Ding, Q. Xin, X. Zhou, J. Qiao, H. Li and H. Wang, Electrochemical behavior of nanostructured nickel phthalocyanine (NiPc/C) for oxygen reduction reaction in alkaline media, J. Appl. Electrochem., 2013, 43, 43–51 Search PubMed.
- S. A. Mirshokraee, M. Muhyuddin, N. Pianta, E. Berretti, L. Capozzoli, J. Orsilli, F. D'Acapito, R. Viscardi, A. Cosenza, P. Atanassov, C. Santoro and A. Lavacchi, Ni-Phthalocyanine Derived Electrocatalysts for Oxygen Reduction Reaction and Hydrogen Evolution Reaction: Active Sites Formation and Electrocatalytic Activity, ACS Catal., 2024, 14, 14524–14538 CrossRef CAS.
- J. Li, P. Pršlja, T. Shinagawa, A. J. Martín Fernández, F. Krumeich, K. Artyushkova, P. Atanassov, A. Zitolo, Y. Zhou, R. García-Muelas, N. López, J. Pérez-Ramírez and F. Jaouen, Volcano Trend in Electrocatalytic CO2 Reduction Activity over Atomically Dispersed Metal Sites on Nitrogen-Doped Carbon, ACS Catal., 2019, 9, 10426–10439 CrossRef CAS.
- C. C. Hou, H. F. Wang, C. Li and Q. Xu, From metal–organic frameworks to single/dual-atom and cluster metal catalysts for energy applications, Energy Environ. Sci., 2020, 13, 1658–1693 RSC.
- N. Weidler, J. Schuch, F. Knaus, P. Stenner, S. Hoch, A. Maljusch, R. Schäfer, B. Kaiser and W. Jaegermann, X-ray Photoelectron Spectroscopic Investigation of Plasma-Enhanced Chemical Vapor Deposited NiOx, NiOx(OH)y, and CoNiOx(OH)y: Influence of the Chemical Composition on the Catalytic Activity for the Oxygen Evolution Reaction, J. Phys. Chem. C., 2017, 121, 6455–6463 CrossRef CAS.
- S. Vijay, W. Ju, S. Brückner, S. C. Tsang, P. Strasser and K. Chan, Unified mechanistic understanding of CO2 reduction to CO on transition metal and single atom catalysts, Nat. Catal., 2021, 4, 1024–1031 CrossRef CAS.
- S. Liu, H. B. Yang, S. F. Hung, J. Ding, W. Cai, L. Liu, J. Gao, X. Li, X. Ren, Z. Kuang, Y. Huang, T. Zhang and B. Liu, Elucidating the Electrocatalytic CO2 Reduction Reaction over a Model Single-Atom Nickel Catalyst, Angew. Chem., Int. Ed., 2020, 59, 798–803 Search PubMed.
- Y. Tuo, W. Liu, Q. Lu, X. Zhang, X. Zhou, Y. Zhou, X. Feng, M. Wu, Z. Wang, D. Chen and J. Zhang, Breaking the scaling relationship via lattice expansion of Ag for CO2 electroreduction over a wide potential window, AIChE J., 2024, 70, e18365 CAS.
- J. W. Chen, Z. Zhang, H. M. Yan, G. J. Xia, H. Cao and Y. G. Wang, Pseudo-adsorption and long-range redox coupling during oxygen reduction reaction on single atom electrocatalyst, Nat. Commun., 2022, 13, 1734 CAS.
- S. Yu, Z. Levell, Z. Jiang, X. Zhao and Y. Liu, What Is the Rate-Limiting Step of Oxygen Reduction Reaction on Fe–N–C Catalysts?, J. Am. Chem. Soc., 2023, 145, 25352–25356 CAS.
- P. Hutchison, P. S. Rice, R. E. Warburton, S. Raugei and S. Hammes-Schiffer, Multilevel Computational Studies Reveal the Importance of Axial Ligand for Oxygen Reduction Reaction on Fe–N–C Materials, J. Am. Chem. Soc., 2022, 144, 16524–16534 CAS.
- L. Hu, C. Dai, L. Chen, Y. Zhu, Y. Hao, Q. Zhang, L. Gu, X. Feng, S. Yuan, L. Wang and B. Wang, Metal-Triazolate-Framework-Derived FeN4Cl1 Single-Atom Catalysts with Hierarchical Porosity for the Oxygen Reduction Reaction, Angew. Chem., Int. Ed., 2021, 60, 27324–27329 Search PubMed.
- J. Li, S. Ghoshal, W. Liang, M. T. Sougrati, F. Jaouen, B. Halevi, S. McKinney, G. McCool, C. Ma, X. Yuan, Z. F. Ma, S. Mukerjee and Q. Jia, Structural and mechanistic basis for the high activity of Fe–N–C catalysts toward oxygen reduction, Energy Environ. Sci., 2016, 9, 2418–2432 CAS.
- Y. Cui, C. Ren, Q. Li, C. Ling and J. Wang, Hybridization State Transition under Working Conditions: Activity Origin of Single-Atom Catalysts, J. Am. Chem. Soc., 2024, 146, 15640–15647 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc08815h |
| This journal is © The Royal Society of Chemistry 2025 |