
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA leigh syndrome mutation perturbs long-range energy coupling in respiratory complex I†
Franziska
Hoeser‡
a,
Patricia
Saura‡
b,
Caroline
Harter
a,
Ville R. I.
Kaila
 *b and
Thorsten
Friedrich
*a
*b and
Thorsten
Friedrich
*a
aInstitut für Biochemie, Albert-Ludwigs-Universität Freiburg, Germany. E-mail: Friedrich@bio.chemie.uni-freiburg.de
bDepartment of Biochemistry and Biophysics, Stockholm University, Sweden. E-mail: ville.kaila@dbb.su.se
First published on 21st March 2025
Abstract
Respiratory complex I is a central enzyme of cellular energy metabolism that couples electron transfer with proton translocation across a biological membrane. In doing so, it powers oxidative phosphorylation that drives energy consuming processes. Mutations in complex I lead to severe neurodegenerative diseases in humans. However, the biochemical consequences of these mutations remain largely unknown. Here, we use the Escherichia coli complex I as a model to biochemically characterize the F124LMT-ND5 mutation found in patients suffering from Leigh syndrome. We show that the mutation drastically perturbs proton translocation and electron transfer activities to the same extent, despite the remarkable 140 Å distance between the mutated position and the electron transfer domain. Our molecular dynamics simulations suggest that the disease-causing mutation induces conformational changes that hamper the propagation of an electric wave through an ion-paired network essential for proton translocation. Our findings imply that malfunction of the proton translocation domain is entirely transmitted to the electron transfer domain underlining the action-at-a-distance coupling in the proton-coupled electron transfer of respiratory complex I.
Introduction
The enzyme complexes of aerobic respiratory chains couple electron transfer from reducing equivalents such as nicotinamide adenine dinucleotide (NADH) to dioxygen with the translocation of protons across the membrane, generating a protonmotive force (pmf).1–3 The pmf drives the synthesis of the universal energy currency adenosine triphosphate (ATP) by ATP synthase in oxidative phosphorylation (OXPHOS).1–3 OXPHOS defects are the most common inborn errors of metabolism.4 They are genetically and clinically highly diverse, including manifestations in early-onset neurodegenerative disorders such as mitochondrial encephalomyopathy and Leigh syndrome, as well as lactic acidosis, and cardiomyopathy.5,6 OXPHOS defects are most frequently associated with a dysfunction of respiratory complex I.7–10Respiratory complex I, or the energy-converting NADH:ubiquinone oxidoreductase, is the primary electron entry point of most respiratory chains coupling NADH oxidation and ubiquinone (Q) reduction with the translocation of protons across the membrane. It comprises a peripheral arm, catalyzing electron transfer, and a membrane arm responsible for proton translocation. Mammalian complex I is composed of 45 subunits, seven of which are encoded by mitochondrial (mt) DNA.11–13 Homologues of 14 subunits, including those that are mitochondrially encoded, are found in all organisms that contain an energy-converting NADH:Q oxidoreductase. These core subunits represent the minimal structural framework necessary to carry out redox-driven proton pumping.14
Due to the dual genetic origin, mutations with an impact on the structure and function of complex I may occur in the mitochondrial and nuclear genomes. Mutations on the mtDNA are further characterized by a high level of heteroplasmy (the proportion of mutated vs. wild-type mtDNA) in different tissues.15,16 The mtT12706C mutation leads to the replacement of a conserved phenylalanine residue by a leucine residue on subunit ND5 (F124LMT-ND5, Fig. 1B and C). Patients carrying this mutation suffer from Leigh syndrome with fatal outcome. The degree of heteroplasmy varies between 33% and 60% leading to a diminished complex I activity of unknown magnitude.19–22 Measuring the activity of OXPHOS enzyme complexes in patients is hampered by the scattering of control values that might differ by as much as ±50%.23 Despite it is a known mutation, there are no experimental studies on how it affects intramolecular electron transfer, proton translocation and the coupling of both processes.
 | ||
| Fig. 1 Structure and function of respiratory complex I. (A) Model of human complex I (PDB: 5XTD17). Electron transfer from NADH to ubiquinone (Q) in the hydrophilic domain (purple) couples to proton pumping across the membrane domain. The putative proton channels are indicated by black arrows within subunits ND5 (red), ND4 (blue), ND2 (yellow) and ND1 (green) together with ND3/6/4L (orange). (B) Close-up of the homologous antiporter-like subunits NuoL (red), NuoM (blue), and NuoL (yellow) from E. coli complex I (PDB: 7Z7S18), showing the conserved axis of buried charged/polar residues involved in proton translocation. The broken transmembrane helices are highlighted in solid colors, F123L in cyan. (C) Top view of the NuoL/NuoM interface, showing the position of F123L and surrounding residues, including the conserved R175L/D178L/K229L/E144L ion-pairs, and E407M involved in proton translocation. | ||
To elucidate the effect of the F124LMT-ND5 mutation on the molecular mechanism of complex I, we use Escherichia coli as a model system. E. coli contains a minimal form of the complex with the inestimable advantage that mutations found in mtDNA can be readily introduced into bacterial DNA.24–27 The E. coli complex I comprises 13 subunits, NuoA to NuoN, adding to a molecular mass of approximately 535 kDa.14 The genes nuoC and nuoD are fused, leading to the production of a single subunit NuoCD.28 NADH is oxidized by flavin mononucleotide (FMN) at the top of the peripheral arm, and electrons are transferred by a series of seven iron–sulfur (Fe/S) clusters across a distance of about 100 Å towards the membrane,29,30 where Q is reduced by the most distal Fe/S cluster (Fig. 1A). Q is bound, reduced, and protonated in a unique cavity composed of subunits from both arms.31–34 The membrane arm contains four putative proton pathways that are connected to each other and to the Q cavity by an approximately 200 Å long central axis of buried charged residues.31–34 Three of the four proton pathways are found in NuoL, M, and N (E. coli naming) of the membrane arm that are homologous to each other and to proton/cation antiporters. Each of these subunits includes a single proton pathway established by two broken transmembrane helices (TMH) within a five-helix bundle segment (Fig. 1B).31–34 The fourth pathway is proximal to the Q binding site and composed of NuoH, A, J, and K.33,35 It was suggested that a central lysine residue (K247N, K265M and K342L; the superscript refers to the name of the subunit of E. coli complex I) of each of the antiporter-type subunits pushes a proton along the central axis of charged residues toward a distal lysine or glutamate residue (K395N, E407M and K399L) controlled by opening/closing of conserved ion-pairs at the interface between the subunits (K217/E133N, K234/E144M and R175/E144/K229/D178L; Fig. 1B).35 It was further proposed that the Q chemistry induces the propagation of an ‘electrical wave’ along the central hydrophilic axis of the membrane arm in a forward and reverse manner to drive proton translocation.35–41 Alternatively, it has been suggested that the binding of quinone leads to a transition from an ‘open’ to a ‘closed’ state.42–44 Here, quinone protonation results from a re-distribution of protons along the central axis, which in turn leads to a proton release to the cytoplasm exclusively by the most distal proton pathway on NuoL (the homologue of mitochondrial ND5).42–44
Because the F124LMT-ND5 mutation (F123LL in E. coli complex I) found in patients suffering from Leigh syndrome is located in close vicinity to the most distal proton pathway (Fig. 1C),34 it was proposed, but never experimentally tested, that the mutation directly blocks proton translocation through structurally consolidating the flexible proton channel in NuoL.23 In contrast, we show here by spectroscopy and molecular dynamics (MD) simulations that the mutation alters the ion-pair network essential for proton transport along the membrane arm. This impedes proton translocation and the alteration of the network is transmitted to the electron transfer chain up to a 140 Å distance, explaining the long-range effect of hampered proton translocation on electron transfer. In addition, we experimentally show that the mutation does not hamper the assembly of the complex and diminishes the electron and proton transfer activities in equal measures.
Results and discussion
Generation of the mutant and cell growth
F123L is located in the membrane arm of the complex, and in close proximity to the most distal proton pathway of the complex (Fig. 1A and B), near the broken TMH7 that is expected to be an essential part of that pathway. Although this residue is not directly part of the broken helix, its side chain is oriented towards K229L (Fig. 1C), which forms an ion-pair that controls the barrier for proton pumping (Fig. 1B).34 Conservation of the position was confirmed by multiple sequence alignment of both prokaryotic and eukaryotic species as also reported by others (ESI Fig. S1†).19–23We introduced the F123LL point mutation into the pBADnuoHis expression plasmid encoding the entire E. coli nuo-operon under the control of the inducible pBAD arabinose promoter. Potential recombination with the chromosomal wild-type allele during cloning was excluded by using strain DH5αΔnuo as cloning host. The expression strain BW25113Δndh/nuo:nptII_FRT was transformed with the expression plasmid either coding the original genes or those with the mutation on nuoL. The expression strain chromosomally lacks the gene of the alternative NADH dehydrogenase (ndh) and the nuo-operon encoding complex I is replaced by the nptII resistance cartridge. Thus, the NADH-induced activities of membranes solely reflect those of the complex and the variant encoded by the plasmid.
Strains producing the complex and the F123LL variant were grown in minimal medium using acetate as non-fermentable carbon source (ESI Fig. S2†). Under this condition, rapid growth of E. coli to a high OD600 depends on the presence of a functional complex I to maintain a low NADH/NAD+ ratio essential for preserving the Krebs cycle, whilst the proton translocation activity contributes to the generation of the pmf.25 As expected, the strain producing parental complex I grows fast to an OD600 of 1.2, while the strain producing the variant stops growth two hours after induction at an OD600 of 0.3 (ESI Fig. S2†). The F123LL mutation thus significantly affects the rate of NADH oxidation.
Activities of the enzymes in the membrane
Cytoplasmic membranes were obtained by differential centrifugation of disrupted cells. We estimated the amount of complex I in the membrane from the NADH/ferricyanide oxidoreductase activity mediated by NuoF that contains the flavin cofactor. This reaction is neither coupled with Q reduction nor with proton pumping. Judging by this activity, the F123LL mutant strain contains similar amounts of the protein within the measurement accuracy as the strain producing complex I (Table 1). The physiological activity of the complex and the variant was determined by measuring dioxygen consumption upon addition of NADH. In this assay, NADH is oxidized by complex I in a rate-limiting step and the produced quinol (QH2) is used by the quinol oxidases to reduce dioxygen to water. Membranes from the F123LL mutant strain exhibit only 15% of the activity of the parental strain, and the residual activity is sensitive to the complex I-specific inhibitor piericidin A (Table 1). The lesser inhibition of oxidase activity of the mutant is caused by the higher proportion of the non-enzymatic reaction contributing to the total reaction (compare Table 2). The NuoL mutant strain thus contains the same amount of complex I as the original strain, but the variant shows a substantial loss of the electron transfer activity.| Strain | NADH oxidase activity | Pic A inhibition [%] | NADH/ferricyanide oxidoreductase activity | ||
|---|---|---|---|---|---|
| [U mg−1] | [%] | [U mg−1] | [%] | ||
| WT | 0.481 ± 0.014 | 100 ± 3 | 90 ± 6 | 2.4 ± 0.2 | 100 ± 8 |
| F123LL | 0.073 ± 0.010 | 15 ± 14 | 45 ± 6 | 2.2 ± 0.2 | 92 ± 8 |
| Preparation | NADH:Q oxidoreductase activity | NADH/ferricyanide oxidoreductase activity | ||
|---|---|---|---|---|
| [U mg−1] | [%] | [U mg−1] | [%] | |
| Complex I | 40.6 ± 1.0 | 100 ± 3 | 97.3 ± 4.5 | 100 ± 5 |
| F123LL | 6.3 ± 1.1 | 15 ± 17 | 100.1 ± 4.4 | 103 ± 4 |
Assembly and stability of the variant
To test whether the diminished activity could arise from an impaired assembly of the complex, the stability of the F123LL variant was investigated by sucrose density gradient centrifugation (Fig. 2A). To this end, proteins of the cytoplasmic membrane from cells grown in rich medium were extracted with detergent. The extract was cleared by centrifugation and the supernatant was loaded onto a sucrose gradient. Solubilized membrane proteins were separated by ultra-centrifugation and the gradients were fractioned in 1 mL portions (Fig. 2A). The NADH/ferricyanide oxidoreductase activity profile of the gradient loaded with the extract from the parental strain has a maximum around fraction 17, which is typical for a stable and fully assembled complex I.45 Importantly, the activity profile of the F123LL mutant strain is virtually identical to that of the parental strain (Fig. 2A), confirming that both membranes contain a similar amount of the complex (Table 1) and that the F123LL variant is fully assembled.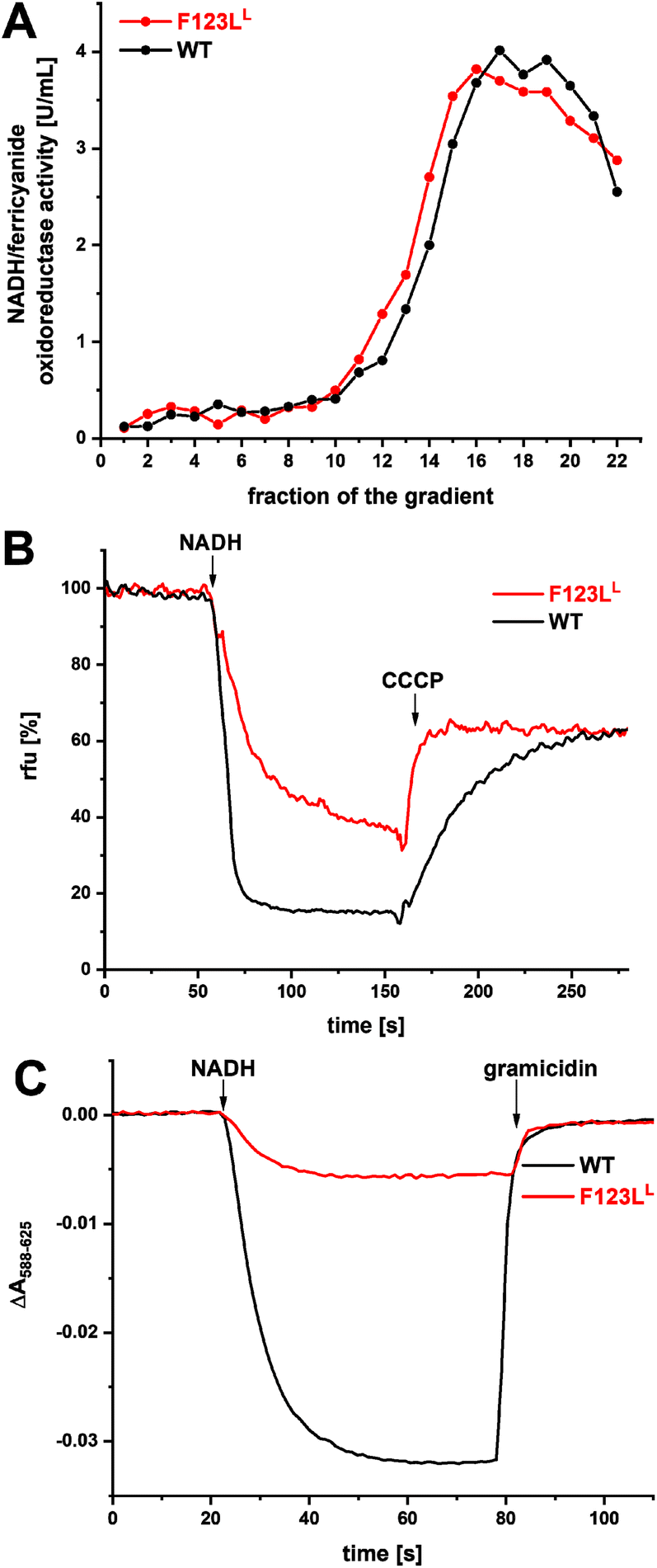 | ||
| Fig. 2 Assembly and pmf generation of complex I (black) and the F123LL variant (red). (A) Sucrose gradient centrifugation of detergent solubilized membranes from wild-type (taken from ref. 25), and the F123LL mutant strain. The NADH/ferricyanide oxidoreductase activity of each fraction normalized to 20 mg membrane protein applied on each gradient is shown. Each data point was assayed in triplicates. The standard error of the mean is ±0.12 U mL−1, which is less than the size of the individual data points. (B) Generation of a ΔpH of complex I and the variant reconstituted into liposomes determined by the ACMA assay measuring the fluorescence quench. The addition of CCCP dissipates the proton gradient. (C) Generation of a ΔΨ determined by the oxonol VI assay measuring the absorbance decrease at (588–625 nm). The membrane potential was dissipated by an addition of gramicidin. The reactions were started by an addition of 100 μM NADH. The statistics of the measurements are provided in ESI Table S1.† | ||
Biophysical characterization
The complex was isolated from the parental strain and the F123LL mutant from cells grown in rich medium. Membrane proteins were extracted with detergent and both, wild type and the variant complex were prepared to homogeneity by affinity and size-exclusion chromatography (ESI Fig. S3A and B†) as described.46 Both preparations contained all complex I subunits as judged from SDS-PAGE and the protein band pattern of both preparations is virtually identical (ESI Fig. S3C†). The EPR spectra of the variant show no differences in the number and amount of Fe/S clusters to that of complex I (ESI Fig. S3D†).Both preparations have a specific NADH/ferricyanide oxidoreductase activity of about 100 U mg−1 (Table 2), indicating a similar purity. However, the F123LL variant has only 15% of the NADH:decyl-Q oxidoreductase activity relative to complex I (Table 2), the process that triggers proton translocation. As with complex I, more than 90% of the oxidoreductase activity of the variant was inhibited by the addition of 10 μM piericidin A. These findings are consistent with the ratio of activities of the enzymes in the membrane (Table 1). Thus, the F123LL mutation does not influence NADH oxidation and electron transfer along the Fe/S clusters, but the results indicate that coupling of Q chemistry with proton translocation is disturbed by a mutation about 140 Å away from the Q active site.
The production of reactive oxygen species (ROS) by both preparations was determined using the Amplex Red assay, which measures hydrogen peroxide in a colorimetric assay as formation of resorufin.47 Complex I produces 16.6 (±1.3) nmol per min per mg resorufin in accordance with values reported in the literature.47 Resorufin production by the variant is slightly enhanced to 19.6 (±1.2) nmol min−1 mg−1. It is rather unlikely that this small increase in ROS production contributes significantly to the development of the disease in humans.
In order to quantify the NADH-driven proton translocation, both preparations were reconstituted into liposomes according to protocols established for bo3 oxidase and ATP synthase.48 The orientation of complex I in proteoliposomes was estimated by measuring the NADH/ferricyanide oxidoreductase activity before and after an addition of 0.04% detergent. 55% of the enzymes of both preparations were oriented with the NADH-binding site accessible from the outside. Only these proteins are activated in the assay as the substrate NADH is membrane impermeable. Due to the lack of a quinone regenerating system in this assay the activity of the correctly oriented enzymes in proteoliposomes is lower and amounts to 14 (±2.8) U mg−1 for complex I and 2.2 (±1.3) U mg−1 for the variant according to three technical replicates of proteoliposomes each derived from three independent protein preparations. The proteoliposomes were equilibrated with decyl-Q for one minute and proton pumping was initiated by the addition of 100 μM NADH. The NADH-driven proton translocation leads to an acidification of the liposome lumen and a positive membrane potential ΔΨ on the inside. The statistics for the measurements are provided in ESI Table S1.†
The acidification of proteoliposomes was followed using 9-amino-6-chloro-2-methoxyacridine (ACMA) fluorescence quenching method (Fig. 2C). Upon addition of NADH, the ACMA fluorescence is quenched, but the rate obtained in proteoliposomes with the F123LL variant averages 21 (±30)% relative to wild-type complex I (Fig. 2B). The signals obtained with both preparations are sensitive to an addition of the uncoupler carbonyl cyanide 3-chlorophenylhydrazone (CCCP), indicating the presence of a proton gradient.
We also followed the generation of a membrane potential ΔΨ photometrically with the potential-sensitive dye oxonol VI (Fig. 2C). The maximum rate observed with proteoliposomes containing the F123LL variant averages 20 (±24)% of the wild-type complex I (ESI Table S1†). Both signals are sensitive to the addition of the ionophore gramicidin, which dissipates the ΔΨ. The diminished rates of proton translocation are in good agreement with the decreased NADH:decyl-Q oxidoreductase activities, suggesting that the H+/e− stoichiometry is not affected by the mutation. The F123LL variant is thus capable of catalyzing redox-driven proton translocation, but with a significantly diminished activity caused by the mutation.
Perturbation of the ion-paired network
To understand the molecular impact of the F123LL mutation, we used atomistic molecular dynamics (MD) simulations based on the experimental structure of the E. coli complex I (PDB ID: 7Z7S) embedded in a lipid membrane environment. To this end, we introduced the point mutation in silico and compared the dynamics of the wild type complex to simulations of the variant (see ESI†). We find that the F123LL mutation induces local conformational changes that propagate to the ion-pair network in NuoL, altering its dynamics (Fig. 3). F123L on TMH4 forms contacts with both TMH8 (with the π-bulge at residues 254–259) and TMH5 that harbours E144L of the conserved ion-pair (Fig. 3A, ESI S1 and S4†). Our simulations suggest that the F123LL mutation alters the contacts between both of these helices, affecting adjacent polar residues (S150L, T257L, Fig. 3D) as well as hydrophobic interactions (V146L, M258L, L253L) with the surrounding helices (ESI Fig. S4†). These shifted contacts lead to a subtle shift of the conserved W143L, which forms a π-cation interaction with K229L and W238L (ESI Fig. S4†).24,49 In this regard, our simulations suggest that the F123LL substitution destabilizes these interactions, and affects the dynamics of the conserved R175/D178/K229/E144 ion-pairs (ESI Fig. S4†). The perturbed ion-pair network in turn, results in a motion of R175L towards the NuoL/NuoM interface, establishing transient interactions with the loop of the TMH12M broken helix (residues 396–397) (Fig. 3B and E). This effect is further supported by our free energy calculations, which suggest that this motion is energetically more favoured in the variant (Fig. 3E, ESI Fig. S5†), bringing the positive charge of R175L closer to the terminal proton acceptor E407M in NuoM. | ||
| Fig. 3 Dynamics of complex I and the F123LL variant. (A) Key residues around F123L in NuoL in complex I (red) and the F123LL variant (light-blue) from MD simulations. (Left) Top view, showing F/L123L, next to TMH8 (with H254L and T257L), TMH5 (with W143L and E144L), and the E144L–K229L/D178L–R175L ion-pair. (Right) Front view, showing TMH5, TMH8, and key residues undergoing conformational changes during the MD simulations. (B) Dynamics at the NuoL/NuoM interface in complex I (red) and the F123LL variant (light-blue). R175L forms hydrogen-bonded contacts with the backbone of L396M and G397M of TM12M in the F123LL, bringing the arginine closer to E407M. (C) Hydration dynamics around NuoL in complex I (red, water spheres in red) and in the F123LL variant (light-blue, water spheres in blue). (Left inset) Hydration-average at the N-side side in NuoL, showing protein residues. (Bottom inset) Hydration-average at the P-side site at the NuoM/NuoL interface, showing protein residues in the region. (D) Distances of key residues during the MD simulations of E. coli complex I. (Top) H254L–T257L distance in TMH8. (Bottom) S150L–T257L distance in TMH5 and TMH8. (E) (Top) Free energy profile of the motion of R175L from NuoL to NuoM in complex I (red) and the variant (light-blue). (Bottom) R175L–TMH12M distance at the NuoL/NuoM interface during the MD simulations. | ||
These altered contacts also show a subtle effect on the hydration of the proton exit pathway to the P-side at the NuoL/NuoM interface as well as N-side proton input channel in NuoL (Fig. 3C, ESI S6†). In this regard, we note that the substitution shifts the conformation of His254L in NuoL (located in a π-bulge in helix TMH8) that could interfere with the proton uptake and release (Fig. 3C, ESI S4 and S6†). Although these effects are rather small, they could impact the proton pumping that relay on the transient hydration of the proton pathways.24,31,34 Moreover, reverting the WT system using a relaxed structure of the F123L variant (‘L123-2-WT’, see Experimental section), results in a highly similar conformation around the TMH5 and TMH8 as observed in the WT simulations (see ESI Fig. S8†), thus further supporting our findings of the perturbed local contacts in the F123L mutation, and supporting that our results are robust.
Importantly, the disturbed contacts at the NuoL/NuoM interface leading to a hampered proton translocation and electron transfer are consistent with the major models for the mechanism of complex I. In this regard, the tightly controlled opening and closing of the ion-pairs between NuoL/M/N is essential for the ‘electrical wave’ mechanism,35,40 where the forward and backward waves propagate through the membrane arm causing proton translocation (see below). The disturbed R175L/E407M interaction results in an impeded transmission of both the forward and the reverse pulse (Fig. 3). Accordingly, this leads to a perturbation of the ‘activation signal’ between NuoL and NuoM and the energy could thus be dissipated by an uncontrolled proton release, expected to lead to a lowering of both the proton pumping and electron transfer activities, as observed in our experimental assays. On the other hand, the disruption of the balanced proton distribution in the central axis of the membrane arm is also consistent with the ‘ND5-only’ model.42,44 The mutation would disrupt the protonation pattern in the membrane arm, which is required for proton transfer to the ND5 subunit (the mitochondrial homolog of NuoL). This would interfere with the proposed de-protonation of K342L upon transition from the ‘open’ to the ‘closed’ state and the subsequently triggered de-protonation of protonated E144L.42,44 The lack of these events prevents the protonation of bound and reduced Q by conserved residues on NuoCD thus explaining long-range coupling between proton translocation and electron transfer.42,44
Formation of the putative quinol anion
Multiple Q binding positions within the substrate cavity have been proposed, initially based on MD simulations.39,50–52 and recently confirmed by cryo-electron microscopy analysis.42,53–55 However, it still remains unclear whether these sites correspond to transient resting positions or if they have a specific functional role. In this regard, it was proposed that a quinol anion (QH−) is generated within a membrane-bound Q binding site,35,40 for which there is also experimental evidence.25,56,57 The formation of such a quinol anion could induce proton translocation in the membrane arm by feeding a proton into the chain of titratable amino acid residues,25,56,57 and trigger the propagation of the forward electrical wave. The reverse wave would terminate at the Q cavity,38,40 still holding the quinol anion, which is re-protonated to form the neutral quinol species (QH2). It is important to emphasize that this model involves only an anionic and not a radical Q species.Here, complex I and the F123LL variant are reduced by a mild molar surplus of NADH and re-oxidized by dioxygen from the buffer (Fig. 4). Under these conditions, NADH reduces the cofactors of the complex including bound quinone, which leads to the physiological formation of the putative quinol anion. After all NADH is consumed, the cofactors are re-oxidized by dioxygen dissolved in the buffer in an artificial reaction. The assay therefore only provides information about the formation and stability of the quinol anion. The proposed later re-protonation to the neutral quinol form in the Q cavity cannot be detected. Our experiments show that while the NADH-reduced FMN and Fe/S cofactors are quickly re-oxidized by dioxygen, a difference absorbance remains for several seconds in the UV/vis redox-difference spectrum that can be attributed to a quinol anion (Fig. 4).25,56,57
 | ||
| Fig. 4 Reduction and re-oxidation of isolated complex I and the F123LL variant. The preparations in detergent were reduced by a twentyfold molar excess NADH. The reduced enzymes are re-oxidized by dioxygen from the buffer. After consumption of the excess NADH, the cofactors of the complex are re-oxidized one after the other and detected by UV-vis spectroscopy. (A) Absorbance of NADH at 340 nm (black) and reduction of complex I measured at 450 nm (red). (B) Absorbance of NADH at 340 nm (black) and reduction of the F123LL variant measured at 450 nm (red). (C) Re-oxidation of complex I (black) and of the F123LL variant (red) measured at 450 nm. (D) (Reduced-minus-oxidized) UV/vis difference spectrum of complex I (black) und the F123LL variant (red) observed after 90 seconds after NADH addition. Representative traces (n = 1) are shown. Experiments were made in triplicate with virtually identical kinetics and difference spectra. | ||
These findings suggest that the quinol species is trapped within the enzyme and cannot be oxidized, possibly as this would require energy provided by the membrane potential, whereas in solution, the enzyme is not exposed to a membrane potential. The signal attributed to the quinol anion is also clearly visible in the redox difference spectrum obtained with the variant (Fig. 4), and we observe no significant differences in either the rate of reduction or the re-oxidation kinetics. Hence, the F123LL mutation does not seem to hamper the formation and the stability of the putative quinol anion, but could indeed affect the steps linked to its re-protonation.
Our findings thus suggest that Q is reduced and protonated in the Q cavity in the F123LL variant. The formed QH2 moves to the second binding site in the cavity and feeds a proton into the membrane arm, initiating the electrical wave propagation (Fig. 5). Consequently, the mutation results in an energy dissipation at the interface of NuoL and NuoM due to a proton leak, which severely hampers further protonation of the quinol anion that remains in the cavity and blocks electron transfer. This model could thus explain the inhibition of electron transfer at 140 Å distance from the mutated position. Our findings, however, are also in agreement with the competing model in which Q is reduced in its binding cavity and subsequently protonated by conserved titratable residues that transfer protons from the central axis of the membrane arm to the Q cavity.42,44
 | ||
| Fig. 5 The putative catalytic mechanism of complex I. (A) Electron transfer from NADH to Q (pink arrows) couples to proton transfer from the conserved residues in the first Q-binding site (red arrows), producing quinol (QH2) that moves to a second binding site. Proton transfer from QH2 to surrounding carboxylates in the E-channel leading to the formation of a putative QH− intermediate, initiates the proton transfer cascade in the antiporter-like subunits. A transferred proton moves to NuoA/J/K, inducing a forward wave that propagates to the antiporter-like subunits NuoN, NuoM and NuoL by ion-pair opening and lateral proton transfer in each antiporter-like subunit, triggering ion-pair opening in the next subunit (red to purple arrows). NuoL ejects the terminal proton to the P-side, initiating the propagation of the backward wave by re-protonation of the central Lys, closing of the ion-pairs, and release of a proton to the P-side in the next antiporter-like subunit (blue to green arrows). This effect propagates to the quinone site, where the transient QH− is re-protonated to QH2 (green arrow) and released to the membrane. Re-protonation of the Q-cavity and binding of a new Q restores the cycle. (B) In the F123LL variant, the perturbed ion-pair dynamics in NuoL severely hampers the lateral proton transfer in NuoL and proton release across the NuoM/L interface. The flickering ion-pair leads to a broken backward signal that perturbs the propagation of the back-wave towards the quinone site, impeding the re-protonation of QH−. | ||
Conclusions
The clinically relevant complex I mutation, F124LMT-ND5 exhibits various neurological manifestations8 and is associated with an early mortality.19,23 Yet, the catalytic competence of the variant protein remained unknown due to strong variations in the degree of heteroplasmy, i.e. the number of genetic copies carrying this mutation. A study with human biopsies showed that the variant has a diminished electron transfer activity of 11–28% compared to control measurements with fibroblasts.23 As the mutation is in the close vicinity to a proton pathway, it was expected that it causes a dysfunctional proton channel.23 To elucidate the mechanistic effects of the mutation, we used here E. coli complex I as model system. Although the E. coli complex is not closely related to the human enzyme, it comprises the homologous core subunits and all redox cofactors as well as the proposed proton pathways. Our data on the assembly of the variant and its electron transfer activity are in excellent agreement with the data obtained with samples from fibroblasts.23 The mutation did not hamper assembly of the human23 and the E. coli complex (Fig. 2A). The F123LL variant exhibits about 15% of the activity of the original complex in the membrane and after isolation. In addition, we experimentally show that proton translocation is blocked to the same extent as electron transfer and that ROS production is only slightly enhanced in the variant. Furthermore, MD simulations revealed that the mutation interferes with the delicate ion-paired network in the membrane arm. The effect of the proposed proton leak in the membrane is consistent with the two major models of proton translocation by complex I.35,40,42,44 According to our favourite model, it prevents continuation of Q chemistry leading to the blockage of electron transfer activity by a remaining quinol anion. These data underscore the long-range energy-coupling in respiratory complex I, which is consistent with the proposed ‘electrical wave’ model.The MD simulations, together with free energy calculations presented here, reveal key conformational changes that could rationalize molecular principles underlying the perturbed long-range proton electron coupling process induced by the disease causing mutation. However, it should also be recognized that additional changes that are not captured by simulations can take place during the slower millisecond turnover timescales and as a result of the chemical reactions that require further QM/MM explorations.40
Based on these data, we hypothesize that the mutation in human mitochondria disrupts the electrical wave through the membrane and thus impedes proton translocation. The strong coupling of both processes causes the decrease in electron transfer. Over time, both processes could lead to a diminished ATP production and an unbalanced NAD+/NADH ratio, which could contribute to the manifestation of the disease.
Several point mutations in the membrane arm were shown to perturb electron transfer by complex I.18,58–60 However, most of these position are now known to be part of either the central hydrophilic axis or the putative proton channels. Furthermore, the effect of these mutations on proton translocation has not always been quantitatively investigated. Our data underline that each single mutation in complex I has to be investigated individually to determine their molecular consequences. For example, we recently showed that the V259AL mutation leads to the disintegration of the entire complex.25 This position is on TMH8, i.e., next to TMH4, which contains F123L, and both positions are at approximately the same level within the membrane (ESI Fig. S7†). Yet, the V259AL mutation has a structural effect by destabilizing an important π-helix, while the F123LL mutation perturbed the intricate ion-pair network leading to a functional disturbance.
Conclusively, site-directed mutagenesis in a genetically easy-to-handle bacterial system together with molecular simulations based on experimental structures, provide a powerful approach to derive a molecular understanding of the biochemical consequences of mutations in mitochondrially encoded subunits of the human respiratory complex I.
Experimental section
Chemicals
Chemicals, media ingredients and other reagents were purchased from Carl Roth GmbH & Co. KG and Sigma-Aldrich/Merck unless stated otherwise. Enzymes for molecular biology techniques, oligonucleotides and molecular weight markers for agarose gel electrophoresis and SDS-polyacrylamide gel electrophoresis were purchased from Sigma-Aldrich. Wizard SV Gel and PCR Clean-up System and Wizard Plus SV Minipreps DNA Purification System were purchased from Promega. Chromatographic materials were obtained from Cytiva, Freiburg.Strains, plasmids and oligonucleotides
A derivative of E. coli strain BW25113 (ref. 61) from which the gene ndh was deleted and the nuo operon was replaced by a resistance cartridge (nptII) by genomic replacement methods was used as host to overproduce complex I62 and its variant. In addition, E. coli strain DH5αΔnuo63 was used for site-directed mutagenesis. Oligonucleotides were obtained from Sigma-Aldrich (ESI Table S2†). Restriction enzymes were obtained from Fermentas. The plasmid pBADnuoHis64 was used to introduce the point mutations on nuoL by site-directed mutagenesis according to the QuikChange protocol (Stratagene, La Jolla, CA, USA). Silent mutations were introduced creating a new restriction site near by the point mutation to identify positive clones by restriction analysis. The primer pair nuoL_F123L_fwd/rev was used leading to plasmid the pBADnuoHis/nuoF F123LL. The PCR was performed using the KOD Hot Start DNA Polymerase (Novagen). Mutations were confirmed by DNA sequencing (GATC Eurofins, Konstanz, Germany).Cell growth and isolation of cytoplasmic membranes
Strains were grown aerobically at 37 °C while agitating at 180 rpm. Minimal medium65 with 25 mM acetate as sole carbon source was used. After 3 h, expression of the nuo operon was induced by an addition of 0.02% (w/v) L-arabinose. For the preparation of cytoplasmic membranes, cells were grown on auto-induction medium66 (1% (w/v) peptone, 0.5% (w/v) yeast extract, 0.4% glycerol, 25 mM Na2HPO4·2H2O, 25 mM KH2PO4, 50 mM NH4Cl, 5 mM Na2SO4, 2 mM MgSO4·7H2O, 0.2% (w/v) L-arabinose, 0.05% (w/v) glucose, 30 mg per L Fe-NH4-citrate, 0.5 mM L-cysteine, 50 mg per L riboflavin) containing chloramphenicol (34 μg mL−1). Cells were harvested by centrifugation (5700×g, 15 min, 4 °C; Avanti J-26 XP, Beckman Coulter; Rotor JLA 8.1000) in the exponential phase yielding 6 to 7 g cells per L. All further steps were carried out at 4 °C. The cell sediment was suspended in a fivefold volume buffer A (50 mM MES/NaOH, 50 mM NaCl, pH 6.0) containing 0.1 mM PMSF supplemented with a few grains of DNaseI and disrupted by passing three times through an EmulsiFlex-C5 (1000–1500 bar). Cell debris was removed by centrifugation (9500×g, 20 min, 4 °C; RC-5 Superspeed Refrigerated Centrifuge, Sorvall Instruments; Rotor A 8.24). Cytoplasmic membranes were obtained from the supernatant by ultra-centrifugation (160![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000×g, 60 min, 4 °C; LE-80K Ultrafuge, Beckman; Rotor 60 Ti). The sedimented membranes were suspended in an equal volume (1:1, w/v) buffer A* (buffer A with 5 mM MgCl2) containing 0.1 mM PMSF. For protein preparation, the membrane sediment was suspended in buffer A*, pH 6.8 (50 mM MES/NaOH, 50 mM NaCl, 5 mM MgCl2, pH 6.8).
000×g, 60 min, 4 °C; LE-80K Ultrafuge, Beckman; Rotor 60 Ti). The sedimented membranes were suspended in an equal volume (1:1, w/v) buffer A* (buffer A with 5 mM MgCl2) containing 0.1 mM PMSF. For protein preparation, the membrane sediment was suspended in buffer A*, pH 6.8 (50 mM MES/NaOH, 50 mM NaCl, 5 mM MgCl2, pH 6.8).
Activity measurements
Activity assays were performed at 30 °C. The NADH oxidase activity of cytoplasmic membranes was determined by a Clarke-type oxygen electrode (DW1; Hansatech) to monitor the decrease in oxygen concentration in the buffer. The electrode was calibrated by an addition of a few grains sodium dithionite to air-saturated water. The difference of the read-out before and after reduction was attributed to 237 μM oxygen.62 The assay contained 5 μL cytoplasmic membranes (80–90 mg mL−1) in 2 mL buffer A*. After equilibration, the reaction was started by an addition of 1.25 mM NADH. The NADH/ferricyanide oxidoreductase activity was determined as decrease in the absorbance of ferricyanide at 410 nm with a diode-array spectrometer (QS cuvette, d = 1 cm, Hellma; TIDAS II, J&M Aalen) using an ε of 1 mM−1 cm−1.67 The assay contained 10 μL membrane suspension or 0.1 μL complex I and 1 mM K3[Fe(CN)6] in buffer A*. The reaction was started with NADH (0.2 mM, final concentration). The NADH:decyl-ubiquinone oxidoreductase activity was measured as decrease of the NADH concentration at 340 nm using an ε of 6.3 mM−1 cm−1 (QS cuvette, d = 1 cm, Hellma; TIDAS II, J&M Aalen).27 The assay contained 60 μM decyl-ubiquinone, 2 μg complex I and a tenfold molar excess (5 μg) E. coli cytochrome bo3 oxidase in buffer A* MNG (buffer A* with 10% (v/v) glycerol, 0.005% (w/v) LMNG (2,2-didecylpropane-1,3-bis-β-D-maltopyranoside; Anatrace)). The reaction was started by an addition of 150 μM NADH. It is known that E. coli complex I preferentially produces hydrogen peroxide as ROS.68 The formation of hydrogen peroxide was determined by the Amplex Red assay as described.47 The assay contained 5 μg complex I and 30 μM NADH. Triplicates of two biological samples were measured.Sucrose gradient centrifugation
Membrane proteins were extracted by an addition of 1% (w/v) DDM (n-dodecyl-β-D-maltoside; Anatrace) to a membrane suspension (80–90 mg mL−1) in buffer A*. After incubation for 1 h at 4 °C the suspension was centrifuged for 20 min at 160000×g and 4 °C (Rotor 60Ti, L8-M Ultrafuge, Beckman). 1 mL of the supernatant was loaded onto 24 mL gradients of 5–30% (w/v) sucrose in A* DDM (buffer A* with 0.1% (w/v) DDM) and centrifuged for 16 h at 140000×g (4 °C, Rotor SW28, L8-M Ultrafuge, Beckman). The gradients were fractionated into 1 mL aliquots and the NADH/ferricyanide oxidoreductase activities were determined as described above.
Preparation of complex I and the variant
All steps were carried out at 4 °C. A final concentration of 2% (w/v) LMNG (Anatrace) was added to a membrane suspension (∼70 mg mL−1) in buffer A* pH 6.8. After incubation for 1 h at room temperature with gentle stirring, the suspension was centrifuged for 20 min at 160000×g and 4 °C (Rotor 60Ti, L8-M Ultrafuge, Beckman). The supernatant was diluted to approximately 150 mL, adjusted to 20 mM imidazole and applied to a 35 mL ProBond Ni2+-IDA column (Invitrogen) equilibrated in 20 mM imidazole, 50 mM MES/NaOH, 50 mM NaCl, 5 mM MgCl2, 10% (v/v) glycerol, 0.005% LMNG, pH 6.8 at a flow rate of 2 mL min−1. The column was first washed with the same buffer at a flow rate of 3 mL min−1 and subsequently with the same buffer containing 116 mM imidazole until the absorbance at 280 nm dropped below 500 mAu. Bound proteins were eluted with the same buffer containing 308 mM imidazole at a flow rate of 2 mL min−1. Fractions containing NADH/ferricyanide oxidoreductase activity were pooled and concentrated by ultrafiltration in 100 kDa MWCO Amicon Ultra-15 centrifugal filter (Millipore). The concentrate was applied onto a Superose 6 size exclusion chromatography column (300 mL, GE Healthcare) equilibrated in buffer A* MNG at a flow rate of 0.3 mL min−1. The fractions with highest NADH/ferricyanide oxidoreductase activity were pooled and concentrated as described above. The protein was either directly used or stored at −80 °C.
Reconstitution of complex I and its variant into liposomes
E. coli polar lipids (25 mg mL−1 in CHCl3; Avanti) were evaporated and dissolved in the fivefold volume lipid buffer (5 mM MES/NaOH, 50 mM NaCl, pH 6.7). The suspension was frozen in liquid nitrogen and thawed at 29 °C seven times to get unilamellar vesicles. Liposomes were extruded by at least 21 passes through an extruder (0.1 μM polycarbonate membrane, Mini Extruder; Avanti). For reconstitution, 0.5 to 1 mg complex I was mixed with reconstitution buffer (1:3 (v/v), 20 mM HEPES, 200 mM KCl, 73 mM sucrose, 5 mM MgSO4, 0.05% (w/v) L-α-phosphatidylcholine, 1.1% (w/v) n-octylglucoside, 0.6% (w/v) sodium deoxycholate, 0.6% (w/v) sodium cholate, pH 7.5) and incubated for 5 min on ice. 250 μL liposomes were mixed with 8 μL sodium cholate (20%, w/v) and the liposomes and complex I in reconstitution buffer were mixed and incubated for 15 min at room temperature. The solution was applied onto a size exclusion column (PD-10 Desalting Column, 8.3 mL, Sephadex G-25; GE Healthcare) equilibrated in lipid buffer in order to remove the excess detergent. The eluate was centrifuged (4 °C, 2 bar air pressure, 30 min, 150000×g, Rotor A-100, Beckman Airfuge) and sedimented proteoliposomes were gently re-suspended in 500 μL lipid buffer.
Proton translocation activity
The proton gradient (ΔpH) was determined by monitoring the fluorescence quenching of the pH sensitive dye 9-amino-6-chloro-2-methoxyacridine (ACMA; Sigma). The assay contained 100 μM decyl-ubiquinone (Sigma), 0.2 μM ACMA and 50 μL proteoliposomes in ACMA buffer (5 mM KH2PO4/KOH, 50 mM KCl and 1 mM MgCl2, pH 7.5). The assay was performed at 30 °C and the reaction was started by an addition of 100 μM NADH. The fluorescence was detected with an LS 55 fluorescence spectrometer (PerkinElmer) at an excitation wavelength of 430 nm and an emission wavelength of 480 nm. An addition of 10 μM carbonyl cyanide 3-chlorophenylhydrazone (CCCP) was used to dissipate the ΔpH. The generation of ΔΨ was determined by monitoring the changes in absorption of the potential-sensitive dye oxonol VI (Sigma). The assay contained 0.5 μM oxonol VI, 50 μM Q0 and proteoliposomes in oxonol buffer (200 mM MES/KOH, pH 6.75, 1 mM MgSO4, 300 mM mannitol). The NADH/ferricyanide oxidoreductase activity of the proteoliposomes was used to adjust the complex I content in the assays. The reaction was performed at 30 °C and started by an addition of 100 μM NADH. The absorbance at 588–625 nm was measured with a diode-array spectrometer (QS cuvette, d = 1 cm; Hellma; TIDAS II, J&M Aalen). An addition of 2 μg mL−1 gramicidin was used to dissipate ΔΨ. Protein concentration of proteoliposomes was determined by a modified Lowry method.69 Proteins were extracted from proteoliposomes by an addition of 1% LMNG (final concentration) and incubation for 30 min at room temperature. The suspension was centrifuged (4 °C, 2 bar air pressure, 10 min, 150000×g, Rotor A-100, Beckman Airfuge) and the protein concentration of the supernatant was determined.
Other analytical procedures
Protein concentration was determined by the biuret method using BSA as a standard.70 The concentration of purified complex I was determined by the difference of absorbance at 208–310 nm (TIDAS II; J&M Aalen) using an ε of 781 mM−1 cm−1 as derived from the amino acid sequence.71 SDS-PAGE (sodium dodecylsulfate polyacrylamide gel electrophoresis) was performed according to Schägger with a 10% separating gel and a 3.9% stacking gel.72 Multiple sequence alignments were performed using ClustalX 2.1.73Molecular dynamics simulations
Classical molecular dynamics (MD) simulations were performed to probe the dynamics of the wild type and F123LL variant. To this end, the coordinates of the experimental structure of complex I from E. coli (PDB ID: 7Z7S)44 were embedded in a POPE:POPG:CL (7:2:1) lipid membrane, and solvated with TIP3P water molecules and a NaCl concentration of 150 mM. Resolved water molecules and lipids (modeled as POPE) in the cryoEM structure were included in the simulation setup. Missing loops were modeled with Modeller,74 resulting in a system comprising ca. 820000 atoms, with dimensions of 285 Å × 142 Å × 200 Å. The L123L residue in the F123LL variant was introduced by modeling the Leu side-chain at the position using the UCSF Chimera rotamer plugin and the Dunbrack 2010 rotamer library,75,76 and by selecting the highest probability Leu-rotamer (χ1 = −68.3°, χ2 = 173.0°, probability = 0.67). A third set of MD simulations was performed, where the structure of the relaxed F123L variant (after 900 ns MD) was reverted to the WT system, with the L123F mutation based on the WT system. The reverted-WT system was propagated for 400 ns in two replicas (see ESI Table S3†), using the same conditions as in all the unbiased MD simulations. Protonation states of titratable residues were calculated based on PropKa3 (ref. 77) (see ESI Table S4† for non-standard protonation states). Protein, lipids, water, and ions were modeled by the CHARMM36 force field,78–80 together with DFT parameters for the cofactors.31,38 Simulations were performed at constant temperature of T = 310 K and constant pressure of p = 1 bar in an NPT ensemble, using a 2 fs integration time step. The Langevin thermostat (damping coefficient = 1/ps) and the Nosé–Hoover–Langevin piston (damping coefficient = 50 fs) were employed for constant temperature and pressure controls, respectively. Long-range electrostatics were treated by the Particle Mesh Ewald (PME) approach, with an integration grid of 1 Å. All MD simulations were performed with NAMD 2.14 and NAMD 3.0.81
Proton pathway tunnels along the NuoM P-side exit and NuoN N-side entry sites were identified with CAVER.82 The water occupancy (hydration %) along the tunnels was calculated by measuring water molecules around 5 Å radius of the selected tunnels, and dividing the number of MD snapshots with water molecules in the selection (Noccupied) by the total number of analysed snapshots (Ntotal):
Water occupancy (hydration %) = (Noccupied/Ntotal) × 100. The last 100 ns of the MD simulations, with 1 frame per ns, were considered for the hydration analysis.
Free energy calculations were performed to probe the energetics of the Arg175L flip towards NuoM in the WT and the variant using umbrella sampling (US) simulations. The reaction coordinate was defined as the difference of distances of Arg175L–D187L (atoms N–O) to Arg175L–G397M (atoms N–O). For each system, a total of 16 windows were placed spanning the whole range of the reaction coordinate, with a harmonic restraint of 20 kcal mol−1 Å−2 and using the colvars module of NAMD.83 Each window was propagated for 20 ns at T = 310 K. The free energy profile was calculated using the weighted histogram analysis method (WHAM), with statistical errors estimated by Monte Carlo bootstrap analysis.84 See ESI Table S3† for a list of all classical MD simulations, and ESI Fig. S5† for details of the free energy calculations.
Data availability
The data supporting this article have been included as part of the ESI.† Other kinetic and spectroscopic data as well as strains and plasmids generated in this study are available from the corresponding authors upon reasonable request.Author contributions
F. H.: investigation, formal analysis, visualization, writing – review and editing. P. S.: investigation, formal analysis, visualization, writing – review and editing. C. H.: investigation, formal analysis. V. R. I. K.: conceptualization, visualization, formal analysis, supervision, writing – original draft, funding acquisition. T. F.: conceptualization, visualization, formal analysis, supervision, project administration, funding acquisition, writing – original draft.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Dörte Thiel and Rebecca Mößner-Hornberger for excellent technical assistance. This work was supported by grants from the Deutsche Forschungsgemeinschaft to TF (DFG GRK2202; 235777276/RTG & SPP1927; FR 1140/11-2) and the European Research Council (ERC, grant 715311), the Knut and Alice Wallenberg foundation (grant 2019.0251), and Swedish Research Council to VRIK. We are thankful to the National Academic Infrastructure for Supercomputing in Sweden (NAISS, 2023/6-128) and the Swedish National Infrastructure for Computing (SNIC2022/1-29, 2021/1-40, 2022/13-14, LUMI project 46500017) and the PRACE (projects: pr1emi00, pr127)/MareNostrum at Barcelona Supercomputing Center (BSC) and Piz Daint hosted by the Swiss National Supercomputing Center (CSCS) for providing computing time.References
- P. Mitchell, Coupling of phosphorylation to electron and hydrogen transfer by a chemi-osmotic type of mechanism, Nature, 1961, 191, 144–148 CAS.
- P. Mitchell and J. Moyle, Chemiosmotic hypothesis of oxidative phosphorylation, Nature, 1967, 213, 137–139 CrossRef CAS.
- V. R. I. Kaila and M. Wikström, Architecture of bacterial respiratory chains, Nat. Rev. Microbiol., 2021, 19, 319–330 CrossRef CAS PubMed.
- R. Rodenburg, Mitochondrial complex I-linked disease, Biochim. Biophys. Acta, 2016, 1857, 938–945 CrossRef CAS.
- J. Finsterer, Leigh and Leigh-like syndrome in children and adults, Pediatr. Neurol., 2008, 39, 223–235 CrossRef PubMed.
- T. M. Dawson and V. L. Dawson, Molecular pathways of neurodegeneration in Parkinson's disease, Science, 2003, 302, 819–822 CrossRef CAS.
- A. Y. Abramov and P. R. Angelova, Cellular mechanisms of complex I-associated pathology, Biochem. Soc. Trans., 2019, 47, 1963–1969 CrossRef CAS PubMed.
- M. Bugiani, F. Invernizzi, S. Alberio, E. Briem, E. Lamantea, F. Carrara, I. Moroni, L. Farina, M. Spada, M. A. Donati, G. Uziel and M. Zeviani, Clinical and molecular findings in children with complex I deficiency, Biochim. Biophys. Acta, 2004, 1659, 136–147 CAS.
- S. Pitkänen, A. Feigenbaum, R. Laframboise and B. H. Robinson, NADH-coenzyme Q reductase (complex I) deficiency: heterogeneity in phenotype and biochemical findings, J. Inherit. Metab. Dis., 1996, 19, 675–686 Search PubMed.
- E. J. Tucker, G. A. Compton, S. E. Calvo and D. R. Thorburn, The molecular basis of human complex I deficiency, IUBMB Life, 2011, 63, 669–677 CAS.
- J. Hirst, Mitochondrial Complex I, Annu. Rev. Biochem., 2013, 82, 551–575 CAS.
- U. Brandt, Energy converting NADH:quinone oxidoreductase (complex I), Annu. Rev. Biochem., 2006, 75, 69–92 CAS.
- K. Fiedorczuk and L. A. Sazanov, Mammalian Mitochondrial Complex I Structure and Disease-Causing Mutations, Trends Cell Biol., 2018, 28, 835–867 CAS.
- T. Friedrich, On the mechanism of respiratory complex I, J. Bioenerg. Biomembr., 2014, 46, 255–269 CAS.
- D. C. Wallace and D. Chalkia, Mitochondrial DNA Genetics and the Heteroplasmy Conundrum in Evolution and Disease, Cold Spring Harb. Perspect. Biol., 2013, 5, a021220 Search PubMed.
- J. B. Stewart and P. F. Chinnery, The dynamics of mitochondrial DNA heteroplasmy: implications for human health and disease, Nat. Rev. Genet., 2015, 16, 530–542 CAS.
- R. Guo, S. Zong, M. Wu, J. Gu and M. Yang, Architecture of Human Mitochondrial Respiratory Megacomplex I2III2IV2, Cell, 2017, 170, 1247–1257 CAS.
- R. G. Efremov and L. A. Sazanov, Structure of the membrane domain of respiratory complex I, Nature, 2011, 476, 414–420 CAS.
- R. W. Taylor, A. A. Morris, M. Hutchinson and D. M. Turnbull, Leigh disease associated with a novel mitochondrial DNA ND5 mutation, Eur. J. Hum. Genet., 2002, 10, 141–144 CAS.
- S. Lebon, M. Chol, P. Benit, D. Mugnier, I. Chretien, I. Giurgea, I. Kern, E. Girardin, L. Hertz-Pannier, P. de Lonlay, A. Rötig, P. Rustin and A. Munnich, Recurrent de novo mitochondrial DNA mutations in respiratory chain deficiency, J. Med. Genet., 2003, 40, 896–899 CAS.
- S. I. Zhadanov, E. Y. Grechanina, Y. B. Grechanina, V. A. Gusar, N. P. Fedoseeva, S. Lebon, A. Münnich and T. G. Schurr, Fatal manifestation of a de novo ND5 mutation: Insights into the pathogenetic mechanisms of mtDNA ND5 gene defects, Mitochondrion, 2007, 7, 260–266 CAS.
- A. Kashani, I. Thiffault, M. E. Dilenge, C. Saint-Martin, K. Guerrero, L. T. Tran, E. Shoubridge, M. S. van der Knaap, N. Braverman and G. Bernard, A homozygous mutation in the NDUFS1 gene presents with a mild cavitating leukoencephalopathy, Neuorogenetics, 2014, 15, 161–164 Search PubMed.
- Y. Ni, M. A. Hagras, V. Konstantopoulou, J. A. Mayr, A. A. Stuchebrukhov and D. Meierhofer, Mutations in NDUFS1 Cause Metabolic Reprogramming and Disruption of the Electron Transfer, Cells, 2019, 8, 1149 CAS.
- M. E. Mühlbauer, P. Saura, F. Nuber, A. Di Luca, T. Friedrich and V. R. I. Kaila, Water-Gated Proton Transfer Dynamics in Respiratory Complex I, J. Am. Chem. Soc., 2020, 142, 13718–13728 Search PubMed.
- F. Nuber, J. Schimpf, J. P. di Rago, D. Tribouillard-Tanvier, V. Procaccio, M. L. Martin-Negrier, A. Trimouille, O. Biner, C. von Ballmoos and T. Friedrich, Biochemical consequences of two clinically relevant ND-gene mutations in Escherichia coli respiratory complex I, Sci. Rep., 2021, 11, 12641 CAS.
- F. Hoeser, M. Weiß and T. Friedrich, The clinically relevant triple mutation in the mtND1 gene inactivates Escherichia coli complex I, FEBS Lett., 2022, 596, 1124–1132 CAS.
- F. Hoeser, H. Tausend, S. Götz, D. Wohlwend, O. Einsle, S. Günther and T. Friedrich, Respiratory complex I with charge symmetry in the membrane arm pumps protons, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, e2123090119 CrossRef CAS.
- M. Braun, S. Bungert and T. Friedrich, Characterization of the overproduced NADH dehydrogenase fragment of the NADH:ubiquinone oxidoreductase (complex I) from Escherichia coli, Biochemistry, 1998, 37, 1861–1867 CrossRef CAS.
- S. de Vries, K. Dörner, M. J. Strampraad and T. Friedrich, Electron tunneling rates in respiratory complex I are tuned for efficient energy conversion, Angew Chem. Int. Ed. Engl., 2015, 54, 2844–2848 CAS.
- M. L. Verkhovskaya, N. Belevich, L. Euro and M. Wikström, Real-time electron transfer in respiratory complex I, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 3763–3767 CAS.
- A. Di Luca, A. P. Gamiz-Hernandez and V. R. I. Kaila, Symmetry-related proton transfer pathways in respiratory complex I, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, E6314–E6321 CrossRef CAS PubMed.
- R. Baradaran, J. M. Berrisford, G. S. Minhas and L. A. Sazanov, Crystal structure of the entire respiratory complex I, Nature, 2013, 494, 443–448 CAS.
- M. Röpke, D. Riepl, P. Saura, A. Di Luca, M. E. Mühlbauer, A. Jussupow, A. P. Gamiz-Hernandez and V. R. I. Kaila, Deactivation blocks proton pathways in the mitochondrial complex I, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2019498118 CrossRef PubMed.
- M. Röpke, P. Saura, D. Riepl, M. C. Pöverlein and V. R. I. Kaila, Functional Water Wires Catalyze Long-Range Proton Pumping in the Mammalian Respiratory Complex I, J. Am. Chem. Soc., 2020, 142, 21758–21766 CrossRef PubMed.
- V. R. I. Kaila, Long-range proton-coupled electron transfer in biological energy conversion: towards mechanistic understanding of respiratory complex I, J. R. Soc. Interface, 2018, 15, 20170916 CrossRef PubMed.
- M. Wikström, V. Sharma, V. R. I. Kaila, J. P. Hosler and G. Hummer, New perspectives on proton pumping in cellular respiration, Chem. Rev., 2015, 115, 2196–2221 Search PubMed.
- M. Verkhovskaya and M. Wikström, Oxidoreduction properties of bound ubiquinone in Complex I from Escherichia coli, Biochim. Biophys. Acta, 2014, 1837, 246–250 CAS.
- A. P. Gamiz-Hernandez, A. Jussupow, M. P. Johansson and V. R. I. Kaila, Terminal Electron-Proton Transfer Dynamics in the Quinone Reduction of Respiratory Complex I, J. Am. Chem. Soc., 2017, 139, 16282–16288 CAS.
- J. Warnau, V. Sharma, A. P. Gamiz-Hernandez, A. Di Luca, O. Haapanen, I. Vattulainen, M. Wikström, G. Hummer and V. R. I. Kaila, Redox-coupled quinone dynamics in the respiratory complex I, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, E8413–E8420 CAS.
- V. R. I. Kaila, Resolving Chemical Dynamics in Biological Energy Conversion: Long-Range Proton-Coupled Electron Transfer in Respiratory Complex I, Acc. Chem. Res., 2021, 54, 4462–4473 CrossRef CAS PubMed.
- H. Kim, P. Saura, M. C. Pöverlein, A. P. Gamiz-Hernandez and V. R. I. Kaila, Quinone Catalysis Modulates Proton Transfer Reactions in the Membrane Domain of Respiratory Complex I, J. Am. Chem. Soc., 2023, 145, 17075–17086 CrossRef CAS.
- D. Kampjut and L. Sazanov, The coupling mechanism of mammalian respiratory complex I, Science, 2020, 370, eabc4209 CrossRef CAS.
- E. Laube, J. Meier-Credo, J. D. Langer and W. Kühlbrandt, Conformational changes in mitochondrial complex I of the thermophilic eukaryote Chaetomium thermophilum, Sci. Adv., 2022, 8, eadc9952 CrossRef CAS.
- V. Kravchuk, O. Petrova, D. Kampjut, A. Wojciechowska-Bason, Z. Breese and L. Sazanov, A universal coupling mechanism of respiratory complex I, Nature, 2022, 609, 808–814 CrossRef CAS PubMed.
- H. Leif, V. D. Sled, T. Ohnishi, H. Weiss and T. Friedrich, Isolation and characterization of the proton-translocating NADH: ubiquinone oxidoreductase from Escherichia coli, Eur. J. Biochem., 1995, 230, 538–548 CrossRef CAS.
- J. Schimpf, S. Oppermann, T. Gerasimova, A. F. Santos Seica, P. Hellwig, I. Grishkovskaya, D. Wohlwend, D. Haselbach and T. Friedrich, Structure of the peripheral arm of a minimalistic respiratory complex I, Structure, 2022, 30, 80–94 CrossRef CAS.
- L. Kussmaul and J. Hirst, The mechanism of superoxide production by NADH:ubiquinone oxidoreductase (complex I) from bovine heart mitochondria, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 7607–7612 CrossRef CAS.
- C. von Ballmoos, O. Biner, T. Nilsson and P. Brzezinski, Mimicking respiratory phosphorylation using purified enzymes, Biochim. Biophys. Acta, 2016, 1857, 321–331 CrossRef CAS PubMed.
- A. Beghiah, P. Saura, S. Badolato, H. Kim, J. Zipf, D. Aumann, A. Gamiz-Hernandez, J. Berg, G. Kemp and V. R. I. Kaila, Dissected antiporter modules establish minimal proton-conduction elements of the respiratory complex I, Nat. Commun., 2024, 15, 9098 CrossRef CAS.
- O. Haapanen, A. Djurabekova and V. Sharma, Role of Second Quinone Binding Site in Proton Pumping by Respiratory Complex I, Front. Chem., 2019, 7, 221 CrossRef CAS PubMed.
- M. H. Teixeira and G. M. Arantes, Balanced internal hydration discriminates substrate binding to respiratory complex I, Biochim. Biophys. Acta, 2019, 1860, 541–548 CrossRef PubMed.
- O. Haapanen, M. Reidelbach and V. Sharma, Coupling of quinone dynamics to proton pumping in respiratory complex I, Biochim. Biophys. Acta, 2020, 1861, 148287 CrossRef CAS PubMed.
- K. Parey, O. Haapanen, V. Sharma, H. Köfeler, T. Züllig, S. Prinz, K. Siegmund, I. Wittig, D. J. Mills, J. Vonck, W. Kühlbrandt and V. Zickermann, High-resolution cryo-EM structures of respiratory complex I: Mechanism, assembly, and disease, Sci. Adv., 2019, 5, eaax9484 CrossRef CAS.
- H. R. Bridges, J. G. Fedor, N. J. Blaza, A. Di Luca, A. Jussupow, O. D. Jarman, J. J. Wright, A. A. Agip, A. P. Gamiz-Hernandez, M. M. Roessler, V. R. I. Kaila and J. Hirst, Structure of inhibitor-bound mammalian complex, Nat. Commun., 2020, 11, 5261 CrossRef CAS PubMed.
- W. Zheng, P. Chai, J. Zhu and K. Zhang, High-resolution in situ structures of mammalian respiratory supercomplexes, Nature, 2024, 631, 232–239 CrossRef CAS PubMed.
- F. Nuber, L. Mérono, S. Oppermann, J. Schimpf, D. Wohlwend and T. Friedrich, A Quinol Anion as Catalytic Intermediate Coupling Proton Translocation With Electron Transfer in E. coli Respiratory Complex I, Front. Chem., 2021, 9, 672969 CAS.
- T. Friedrich, B. Brors, P. Hellwig, L. Kintscher, T. Rasmussen, D. Scheide, U. Schulte, W. Mäntele and H. Weiss, Characterization of two novel redox groups in the respiratory NADH:ubiquinone oxidoreductase (complex I), Biochim. Biophys. Acta, 2000, 1459, 305–309 CAS.
- E. Nakamaru-Ogiso, M. C. Kao, H. Chen, S. C. Sinha, T. Yagi and T. Ohnishi, The membrane subunit NuoL(ND5) is involved in the indirect proton pumping mechanism of Escherichia coli complex I, J. Biol. Chem., 2010, 285, 39070–39078 CrossRef CAS PubMed.
- J. Michel, J. DeLeon-Rangel, S. Zhu, K. Van Ree and S. Vik, Mutagenesis of the L, M, and N subunits of Complex I from Escherichia coli indicates a common role in function, PLoS One, 2011, 6, e17420 CAS.
- M. Sato, P. K. Sinha, J. Torres-Bacete, A. Matsuno-Yagi and T. Yagi, Energy transducing roles of antiporter-like subunits in Escherichia coli NDH-1 with main focus on subunit NuoN (ND2), J. Biol. Chem., 2013, 288, 24705–24716 CAS.
- K. A. Datsenko and B. L. Wanner, One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 6640–6645 CAS.
- S. Burschel, D. Kreuzer Decovic, F. Nuber, M. Stiller, M. Hofmann, A. Zupok, B. Siemiatkowska, M. Gorka, S. Leimkühler and T. Friedrich, Iron-sulfur cluster carrier proteins involved in the assembly of Escherichia coli NADH:ubiquinone oxidoreductase (complex I), Mol. Microbiol., 2019, 111, 31–45 CAS.
- T. Pohl, T. Bauer, K. Dörner, S. Stolpe, P. Sell, G. Zocher and T. Friedrich, Iron-sulfur cluster N7 of the NADH:ubiquinone oxidoreductase (complex I) is essential for stability but not involved in electron transfer, Biochemistry, 2007, 46, 6588–6596 CAS.
- T. Pohl, M. Uhlmann, M. Kaufenstein and T. Friedrich, Lambda Red-mediated mutagenesis and efficient large scale affinity purification of the Escherichia coli NADH:ubiquinone oxidoreductase (complex I), Biochemistry, 2007, 46, 10694–10702 CAS.
- J. H. Miller, A Short Course in Bacterial Genetics – A Laboratory Manual and Handbook for Escherichia coli and Related Bacteria, J. Basic Microbiol., 1992, 33, 278 Search PubMed.
- F. W. Studier, Stable expression clones and auto-induction for protein production in E. coli, Methods Mol. Biol., 2014, 1091, 17–32 CAS.
- T. Friedrich, G. Hofhaus, W. Ise, U. Nehls, B. Schmitz and H. Weiss, A small isoform of NADH:ubiquinone oxidoreductase (complex I) without mitochondrially encoded subunits is made in chloramphenicol-treated Neurospora crassa, Eur. J. Biochem., 1998, 180, 173–180 Search PubMed.
- D. Esterházy, M. S. King, G. Yakovlev and J. Hirst, Production of reactive oxygen species by complex I (NADH:ubiquinone oxidoreductase) from Escherichia coli and comparison to the enzyme from mitochondria, Biochemistry, 2008, 47, 3964–3971 Search PubMed.
- C. M. Pomoroy, Color development time of the Lowry protein assay, Anal. Biochem., 2008, 378, 216–217 Search PubMed.
- A. G. Gornall, J. C. Bardwill and M. M. David, Determination of serum proteins by means of the biuret reaction, J. Biol. Chem., 1949, 177, 751–766 CrossRef CAS.
- S. C. Gill and P. H. von Hippel, Calculation of protein extinction coefficients from amino acid sequence data, Anal. Biochem., 1989, 182, 319–326 CrossRef CAS PubMed.
- H. Schägger and G. von Jagow, Tricine-sodium dodecyl sulfate-polyacrylamide gel electrophoresis for the separation of proteins in the range from 1 to 100 kDa, Anal. Biochem., 1987, 166, 368–379 CrossRef PubMed.
- M. A. Larkin, G. Blackshields, N. P. Brown, R. Chenna, P. A. McGettigan, H. McWilliam, F. Valentin, I. M. Wallace, A. Wilm, R. Lopez, J. D. Thompson, T. J. Gibson and D. G. Higgins, Clustal W and Clustal X version 2.0, Bioinformatics, 2007, 23, 2947–2948 CrossRef CAS PubMed.
- B. Webb and A. Sali, Comparative Protein Structure Modeling Using Modeller, Curr. Protoc. Bioinform., 2016, 54, 1–37 CrossRef PubMed.
- E. F. Pettersen, et al., UCSF Chimera-a visualization system for exploratory research and analysis, J. Comput. Chem., 2004, 25, 1605–1612 CAS.
- M. S. Shapovalov and R. L. Dunbrack Jr, A smoothed backbone-dependent rotamer library for proteins derived from Adaptive Kernel density estimates and Regressions, Structure, 2010, 19, 844–858 Search PubMed.
- M. H. Olsson, C. R. Søndergaard, M. Rostkowski and J. H. Jensen, PROPKA3: Consistent Treatment of Internal and Surface Residues in Empirical pKa Predictions, J. Chem. Theory Comput., 2011, 7, 525–537 Search PubMed.
- R. Best, X. Zhu, J. Shim, P. Lopes, J. Mittal, P. Feig and A. MacKerell, Optimization of the additive CHARMM all-atom protein force field targeting improved sampling of the backbone φ, ψ and side-chain χ(1) and χ(2) dihedral angles, J. Chem. Theory Comput., 2012, 8, 3257–3273 Search PubMed.
- N. A. Baker, D. Sept, S. Joseph, M. J. Holst and J. McCammon, Electrostatics of nanosystems: application to microtubules and the ribosome, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 10037–10041 CAS.
- G. Kieseritzky and E.-W. Knapp, Optimizing pKa computation in proteins with pH adapted conformations, Proteins, 2008, 71, 1335–1348 CAS.
- J. C. Phillips, D. J. Hardy, J. D. Maia, J. E. Stone, J. V. Ribeiro, R. C. Bernardi, R. Buch, G. Fiorin, J. Hénin, W. Jiang, R. McGreevy, M. C. Melo, B. K. Radak, R. D. Skeel, A. Singharoy, Y. Wang, B. Roux, A. Aksimentiev, Z. Luthey-Schulten, L. V. Kalé, K. Schulten, C. Chipot and E. Tajkhorshid, Scalable molecular dynamics on CPU and GPU architectures with NAMD, J. Chem. Phys., 2020, 153, 044130 CAS.
- E. Chovancová, et al., CAVER 3.0: A tool for the analysis of transport pathways in dynamic protein structures, PLoS Comput. Biol., 2012, 8, e1002708 Search PubMed.
- G. Fiorin, M. L. Klein and J. Hénin, Using collective variables to drive molecular dynamics simulations, Mol. Phys., 2013, 111(22–23), 3345–3362 CAS.
- A. Grossfield, WHAM: the weighted histogram analysis method, https://membrane.urmc.rochester.edu/wordpress/?page_id=126.
Footnotes |
| † Electronic supplementary information (ESI) available: The multiple sequence alignment, growth curves, elution profiles of chromatographic steps, SDS-PAGE, EPR spectra, dynamics of key residues in E. coli complex I and the variant, analysis of free energy calculations, hydration dynamics, disease-specific mutations, statistics on ΔpH and ΔΨ measurements, oligonucleotides, the list of molecular dynamics simulations and the list of non-standard protonation states. See DOI: https://doi.org/10.1039/d4sc04036h |
| ‡ Contributed equally. |
| This journal is © The Royal Society of Chemistry 2025 |