
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Reactivity of an arsanyl-phosphagallene: decarbonylation of CO2 and COS to form phosphaketenes†‡
Lilian S.
Szych
 ab,
Jonas
Bresien
c,
Lukas
Fischer
b,
Moritz J.
Ernst
b and
Jose M.
Goicoechea
*d
ab,
Jonas
Bresien
c,
Lukas
Fischer
b,
Moritz J.
Ernst
b and
Jose M.
Goicoechea
*d
aDepartment of Chemistry, University of Oxford, Chemistry Research Laboratory, 12 Mansfield Road, Oxford, OX1 3TA, UK
bInstitut für Chemie und Biochemie, Freie Universität Berlin, Fabeckstraße 34/36, Berlin, 14195, Germany
cInstitut für Chemie, Universität Rostock, Albert-Einstein-Straße 3a, Rostock, 18059, Germany
dDepartment of Chemistry, Indiana University, Bloomington, 800 E. Kirkwood Ave., Bloomington, IN 47405-7102, USA. E-mail: jgoicoec@iu.edu
First published on 17th March 2025
Abstract
The synthesis of an arsanyl-phosphagallene [H2CN(Dipp)]2AsP![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Ga(NacNac) (NacNac = HC[C(Me)N(Dipp)]2; Dipp = diisopropylphenyl) and its reactivity towards heterocumulenes and ketones is described. Reactions with azides, carbodiimides, isocyanates and ketones give rise to heterocycles via cyclization reactions involving the GaP π-bond (with the Ga–P σ-bond remaining unperturbed in the final products). By contrast, reactions with CO2, CS2 and COS are more intriguing, revealing a reactivity profile in which the phosphorus atom can abstract carbon monoxide from the oxygen-containing heterocumulenes. These reactions result in the formation of gallium phosphaethynolate compounds. Such reactivity is enabled by the presence of a weakly Lewis basic arsanyl moiety which, in contrast to other related compounds featuring phosphanyl groups, is insufficiently nucleophilic to play a role in frustrated Lewis-pair like reactivity.
Ga(NacNac) (NacNac = HC[C(Me)N(Dipp)]2; Dipp = diisopropylphenyl) and its reactivity towards heterocumulenes and ketones is described. Reactions with azides, carbodiimides, isocyanates and ketones give rise to heterocycles via cyclization reactions involving the GaP π-bond (with the Ga–P σ-bond remaining unperturbed in the final products). By contrast, reactions with CO2, CS2 and COS are more intriguing, revealing a reactivity profile in which the phosphorus atom can abstract carbon monoxide from the oxygen-containing heterocumulenes. These reactions result in the formation of gallium phosphaethynolate compounds. Such reactivity is enabled by the presence of a weakly Lewis basic arsanyl moiety which, in contrast to other related compounds featuring phosphanyl groups, is insufficiently nucleophilic to play a role in frustrated Lewis-pair like reactivity.
Introduction
Compounds containing multiple bonds between the heavier main group elements (n > 2) are of fundamental interest due to their relationship with unsaturated organic molecules such as alkenes and alkynes. Recently, such species have attracted attention due to their ability to activate small molecule substrates.1–3 Pioneering research in the early 1980s by West and Yoshifuji, on disilenes and diphosphenes, respectively,4 paved the way for the successful isolation of a variety of stable homo-diatomic compounds.5,6 Since then, various hetero-diatomic compounds have also been reported.6 These include species with double bonds between the following element combinations: G13G14,7,8 G13G16,9 G14G15,10–15 and G15G16.16–18 Compounds with double bonds between elements of the group 13 and 15 are of special interest due to their valence isoelectronic relationship with CC bonds. These species can be challenging to access due to the proximity of Lewis basic and Lewis acidic sites. Nonetheless, due to their increased G13G15 bond polarity, the reactivity of these compounds is expected to differ significantly from their carbon-based analogues, making them interesting synthetic targets. Even though a handful of stable G13G15 compounds have been synthesized recently,19–27 the reactivity of these compounds towards small molecules, especially towards heterocumulenes, remains largely unexplored. A recent review article published by Hering-Junghans and co-workers provides a detailed overview of compounds with G13G15 double bonds, their syntheses and reactivity.28
We recently reported that the phosphanyl-substituted phosphaalumene (A) and phosphagallene (B) can be accessed by reaction of a phosphaketene [H2CN(Dipp)]2PPCO (Dipp = 2,6-iPr2C6H3) with group 13 carbenoids E(NacNac) (E = Al, Ga; NacNac = HC[C(Me)N(Dipp)]2) (Fig. 1, top).29,30 These compounds react with small molecules in frustrated Lewis pair (FLP)-type manner (e.g. H2, CO2, amines) or in hydroelementation reactions (e.g. silanes). Due to its highly polarized AlP multiple bond compound A also undergoes intramolecular C–H activation reactions. To date, the only reported reactions of heterocumulenes with such compounds involve the reactions of A and B with CO2. This was found to selectively and irreversibly afford the five-membered ring systems C and D, respectively,29,30 as previously reported for other FLPs.31–40 Schulz and co-workers also recently reported the synthesis of a gallium-substituted phosphagallene (E) by reaction of the gallium phosphaketene (NacNac)Ga(Cl)PCO with Ga(NacNac) (Fig. 1, bottom).41,42 In contrast to A and B, the reactivity of E is mainly dominated by (partly reversible) reactions across the GaP multiple bond, involving 1,2-addition reactions (e.g. amines, alcohols, thioles, selenoles)41 and cycloaddition reactions (with heterocumulenes, ketones).42,43 Reaction of E with CO2 yields F, which readily loses CO2 upon heating to 95 °C (Fig. 1, bottom). Compound E reacts with isocyanates RNCO (R = Et, iPr, Cy) in (2 + 2) cycloaddition reactions with the CN bonds yielding the four-membered heterocycles Ga–c. Carbodiimides also undergo cycloaddition reactions yielding the products Ha and Hb. When the heterocyclic products Ga–c were reacted with CO2 the formation of six-membered ring compounds Ia–c analogous to compound F was observed.
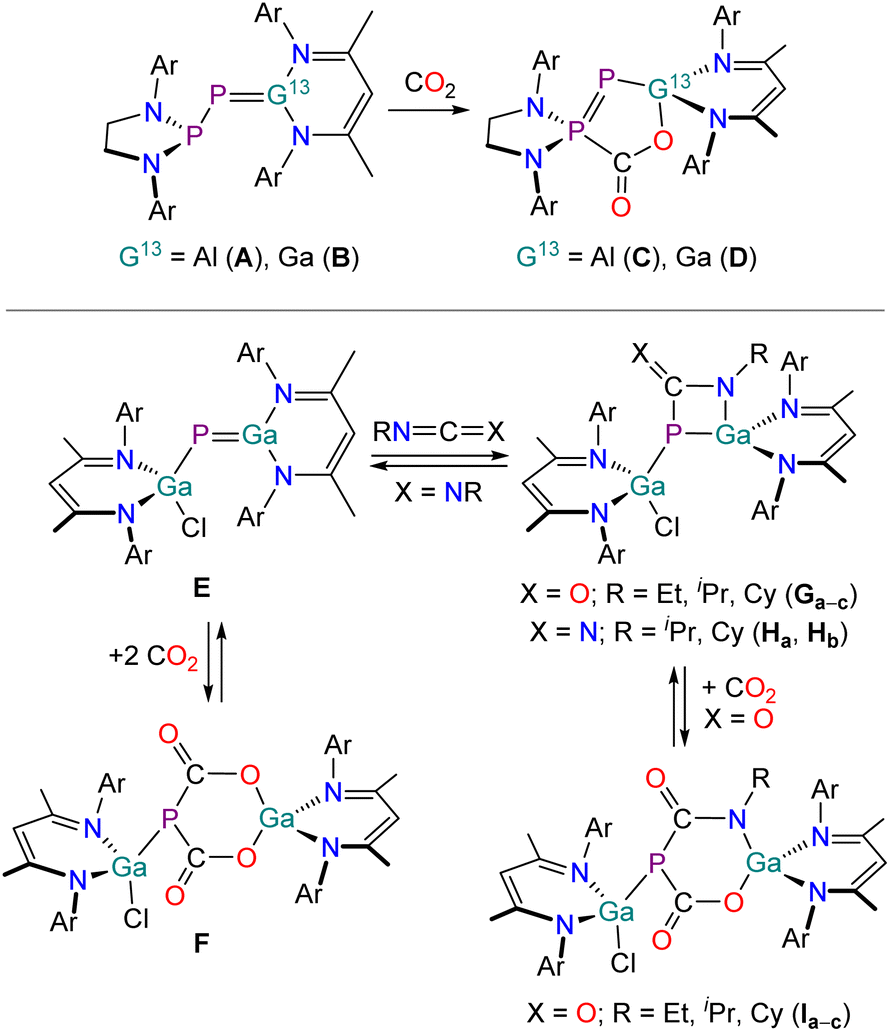 | ||
| Fig. 1 Previously reported phosphagallenes (B and E) and their reactivity towards heterocumulenes. Ar = 2,6-diisopropylphenyl (Dipp). | ||
In relation to the aforementioned studies, the compounds K[Al(NONDipp)(NMes)] and K[In(NONAr)(NMes)] (NONAr = [O(SiMe2NAr)2]2−, Ar = Dipp; Mes = 2,4,6-Me3C6H2) which exhibit G13N (G13 = Al, In) double bonds also exhibit interesting reactivity towards heterocumulenes. The aluminum complex reacts with CO2 resulting in the formation of a dimeric carbamate dianion in a (2 + 2)-addition reaction.44 The reaction of the corresponding indium derivative with azides RN3 (R = Mes, SiMe3) affords tetrazole analogues.45
The reactivity of phosphagallenes appears to be sensitive to substitution effects, as proven by the differing reactivity of B and E (vide supra). Thus, we were interested in how exchanging the adjacent basic phosphanyl group in B with a less basic arsanyl moiety affects the reactivity of the resulting species towards small molecules. These studies are described herein.
Results and discussion
Synthesis of [H2CN(Dipp)]2AsP![[double bond, length as m-dash]](https://www.rsc.org/images/entities/h3_char_e001.gif) Ga(NacNac) (1)
Ga(NacNac) (1)
Dissolution of an equimolar ratio of [H2CN(Dipp)]2AsPCO46 and Ga(NacNac) in toluene at RT resulted in rapid effervescence and an immediate color change of the reaction mixture from yellow to deep red (Scheme 1). The 31P{1H} NMR spectrum of the solution shows the quantitative formation of a product with a chemical shift of −72.2 ppm, which is significantly deshielded relative to the starting material (cf. [H2CN(Dipp)]2AsPCO: −256.8 ppm). A comparable shift was observed for the formation of B (cf. [H2CN(Dipp)]2PPCO: −245.6 ppm; [P]P[Ga] −61.3 ppm).30 Product 1 crystallizes in form of deep red single crystals from saturated n-hexane or n-pentane solution at RT in the monoclinic space group P21/c (Fig. 2). The unit cell parameters of 1 are very similar to the parameters observed for B (see Table S2‡). The crystal structure of 1 reveals a Ga1–P1 bond length of 2.1734(4) Å which suggests the presence of a double bond (∑rcov (Ga–P) = 2.35 Å; ∑rcov (GaP) = 2.19 Å).47,48 The P1–As1 bond length of 2.3166(4) Å is in the range of a single bond (∑rcov (P–As) = 2.32 Å; ∑rcov (PAs) = 2.16 Å).47,48
 | ||
| Scheme 1 Synthesis of 1. Ar = 2,6-diisopropylphenyl (Dipp). | ||
 | ||
| Fig. 2 Single crystal X-ray structure of 1. Thermal ellipsoids set at 50% probability level; hydrogen atoms and solvent of crystallization omitted for clarity. Carbon atoms are depicted as spheres of arbitrary radius. Selected interatomic distances [Å] and angles [°]: As1–P1 2.3166(4), P1–Ga1 2.1734(4), As1–P1–Ga1 99.703(15). | ||
No subsequent reactivity was observed on heating 1 in C6D6 solution to 80 °C over a period of five days. Dissolution of 1 in THF reveals no evidence of adduct formation, as evidenced by the fact that the 31P{1H} NMR chemical shift does not change.
Additions involving the GaP bond
Reaction of 1 towards azides
Upon addition of tert-butyl azide to a red solution of 1 in C6H6 at room temperature the reaction mixture instantly turns orange (Scheme 2, top). The 31P{1H} NMR spectrum suggests quantitative conversion to a single species, 2, which appears as a singlet at −31.6 ppm. Compound 2 crystallizes in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) from a concentrated n-hexane/benzene solution in form of light orange crystals suitable for X-ray crystallography (Fig. 3). The crystal structure reveals the formation of a (2 + 3) addition product exhibiting a five-membered GaPN3 ring system. The reaction proceeds regio-selectively with the functionalized nitrogen atom of the azide selectively bonded to the gallium atom. The Ga–N7, Ga1–P1 and P1–N5 bond legnths are as expected for single bonds. The N6–N7 bond is slighty longer than the N5–N6 bond, but both lie in between what is expected for a N–N single bond and a NN double bond. The GaPN3 ring system in 2 is not planar but slightly twisted. When evaporating a light orange, crystalline sample of 2 for one hour at room temperature, the orange color slowly intensifies/darkens. The same color change can be observed when heating a sample of 2 in C6D6 to 80 °C for one hour. The 31P{1H} NMR spectrum of this heated solution shows that 1 is reformed, indicating that the addition of the azide is reversible. This makes the isolation of compositionally pure samples of 2 challenging as they are often contaminated with the phosphagallene starting material.
from a concentrated n-hexane/benzene solution in form of light orange crystals suitable for X-ray crystallography (Fig. 3). The crystal structure reveals the formation of a (2 + 3) addition product exhibiting a five-membered GaPN3 ring system. The reaction proceeds regio-selectively with the functionalized nitrogen atom of the azide selectively bonded to the gallium atom. The Ga–N7, Ga1–P1 and P1–N5 bond legnths are as expected for single bonds. The N6–N7 bond is slighty longer than the N5–N6 bond, but both lie in between what is expected for a N–N single bond and a NN double bond. The GaPN3 ring system in 2 is not planar but slightly twisted. When evaporating a light orange, crystalline sample of 2 for one hour at room temperature, the orange color slowly intensifies/darkens. The same color change can be observed when heating a sample of 2 in C6D6 to 80 °C for one hour. The 31P{1H} NMR spectrum of this heated solution shows that 1 is reformed, indicating that the addition of the azide is reversible. This makes the isolation of compositionally pure samples of 2 challenging as they are often contaminated with the phosphagallene starting material.
 | ||
| Scheme 2 Synthesis of 2 (top) and 3 (bottom). Ar = 2,6-diisopropylphenyl (Dipp). | ||
 | ||
| Fig. 3 Single crystal X-ray structure of 2. Thermal ellipsoids set at 50% probability level; hydrogen atoms and solvent of crystallization omitted for clarity. Carbon atoms are depicted as spheres of arbitrary radius. Selected interatomic distances [Å] and angles [°]: As1–P1 2.4232(4), P1–Ga1 2.2782(3), P1–N5 1.7794(12), N5–N6 1.2738(17), N6–N7 1.3456(16), N7–Ga1 1.9843(11), As1–P1–Ga1 109.576(14). | ||
Reactivity of 1 towards carbodiimides
Schulz and co-workers have previously reported that E undergoes (2 + 2) cycloaddition reactions with carbodiimides yielding four-membered heterocycles Ha and Hb (vide supra).42 The same reactivity was observed on reaction of 1 with N,N′-dicyclohexylcarbodiimide C(NCy)2. Upon the addition of the carbodiimide to a red solution of 1 in C6H6 at room temperature the mixture quickly turns yellow (Scheme 2, bottom). The product, 3, exhibits a singlet in its 31P{1H} NMR spectrum at −27.6 ppm, and the resonance of the NCN atom appears as a doublet in the 13C{1H} NMR spectrum at 164.0 ppm (1JP–C = 68.1 Hz). 3 crystallizes from benzene at room temperature and X-ray crystallography confirms the formation of a slightly puckered four-membered PCNGa heterocycle (Fig. 4). The P1–Ga1 (2.3188(7) Å; ∑rcov (Ga–P) = 2.35 Å), Ga1–N6 (1.918(2) Å; ∑rcov (Ga–N) = 1.95 Å) and the P1–C1 bond lengths (1.916(3) Å; ∑rcov (P–C) = 1.86 Å) are in the range of single bonds. The C1–N5 bond length (1.273(3) Å) is as expected for an average double bond, while the C1–N6 bond (1.386(3) Å) is notably shorter than expected for a single bond (∑rcov (C–N) = 1.46 Å; ∑rcov (CN) = 1.27 Å). This shortened bond length was also observed in an analogous product from the reaction of E with carbodiimides.42
 | ||
| Fig. 4 Single crystal X-ray structure of 3. Thermal ellipsoids set at 50% probability level; hydrogen atoms and solvent of crystallization omitted for clarity. Carbon atoms are depicted as spheres of arbitrary radius. Selected interatomic distances [Å] and angles [°]: As1–P1 2.3887(7), P1–Ga1 2.3188(7), P1–C1 1.916(3), C1–N6 1.386(3), N6–Ga1 1.918(2), C1–N5 1.273(3), As1–P1–Ga1 106.21(3). | ||
Reactivity of 1 towards isocyanates
Reaction of 1 with a slight excess of mesityl isocyanate MesNCO results in a slow color change of the reaction mixture overnight from red to bright yellow (Scheme 3, top). The 31P{1H} NMR spectrum of the solution shows selective conversion to a species exhibiting a singlet resonance at −34.5 ppm, indicating the formation of a new compound 4. This is further supported by the 13C{1H} NMR spectrum which exhibits a doublet resonance at 139.9 ppm (1JC–P = 46.3 Hz) arising from the heteroallenic isocyanate carbon atom. Crystals suitable for X-ray crystallography were grown from benzene solution at room temperature (Fig. 5). The crystal structure confirms the formation of a (2 + 2) addition product as observed for reactions with carbodiimides. In contrast to the reactivity reported by Schulz and co-workers for E, the addition of MesNCO to 1 selectively proceeds at the CO bond of the isocyanate, yielding a four-membered ring system consisting of Ga1, P1, C1 and O1. The Ga1–P1 bond (2.3209(6) Å), the Ga1–O1 bond (1.9089(15) Å; ∑rcov (Ga–O) = 1.87 Å), the P1–C1 bond (1.853(2) Å) and the C1–O1 bond (1.343(3) Å; ∑rcov (C–O) = 1.38 Å) are all in the range of corresponding single bonds. The four-membered GaPCO ring system is also slightly puckered, the dihedral angle between the P1/C1/O1 plane and P1/Ga1/O1 plane is 162.59(10)°.
 | ||
| Scheme 3 Synthesis of 4 (top) and 5 (bottom). Ar = 2,6-diisopropylphenyl (Dipp). | ||
 | ||
| Fig. 5 Single crystal X-ray structure of 4. Thermal ellipsoids set at 50% probability level; hydrogen atoms and solvent of crystallization omitted for clarity. Carbon atoms are depicted as spheres of arbitrary radius. Selected interatomic distances [Å] and angles [°]: As1–P1 2.3811(6), P1–Ga1 2.3209(6), P1–C1 1.853(2), C1–O1 1.343(3), O1–Ga1 1.9089(15), As1–P1–Ga1 123.94(2). | ||
Reactivity of 1 towards benzaldehyde
Given the contrasting reactivity of 1 and E towards isocyanates, we were intrigued to investigate the reactivity of 1 towards other carbonyl-containing compounds. For this purpose, we chose to investigate its reactivity towards benzaldehyde. In addition to its ability to undergo (2 + 2) addition reactions, benzaldehyde also has a relatively protic C(sp2)–H bond that could react with the GaP bond.
Addition of benzaldehyde to a red solution of 1 in C6H6 at room temperature the reaction solution instantly turns yellow (Scheme 3, bottom). The 31P{1H} NMR spectrum of the reaction mixture indicates selective formation of a new product, 5, which shows a singlet resonance at 67.6 ppm. The 13C{1H} NMR spectrum reveals a doublet at 79.3 ppm (1JC–P = 27.3 Hz) which can be assigned to the PhCHO atom. Crystals suitable for X-ray crystallography were grown from a saturated benzene/n-hexane solution. 5 is the product of a (2 + 2) addition reaction along the CO bond, yielding a four-membered GaPCO heterocycle (Fig. 6). This heterocycle and its molecular parameters are similar to those observed for compound 4. The Ga1–O1 bond length (1.8633(15) Å) is in the range expected for a single bond, as are the Ga1–P1 (2.3457(5) Å), the P1–C56 (1.929(2) Å) and the C56–O1 bond lengths (1.423(2) Å). The ring system is again not planar but slighty puckered, the dihedral angle between the P1–C56–O1 plane and P1–Ga1–O1 plane is 166.35(11)°.
 | ||
| Fig. 6 Single crystal X-ray structure of 5. Thermal ellipsoids set at 50% probability level; hydrogen atoms and solvent of crystallization omitted for clarity. Carbon atoms are depicted as spheres of arbitrary radius. Selected interatomic distances [Å] and angles [°]: As1–P1 2.3821(5), P1–Ga1 2.3457(5), P1–C56 1.929(2), C56–O1 1.423(2), O1–Ga1 1.8633(15), As1–P1–Ga1 124.17(2). | ||
GaP bond insertion reactions and phosphorus atom extrusion
Reactivity of 1 towards CO2
The aforementioned reactions involve the formation of addition products of heterocumulenes or ketones with the GaP bond of 1. Given that closely related compounds such as A and B have been shown to exhibit FLP-like reactivity towards other heterocumulenes, e.g. CO2, we were interested to explore the role played by the pendant phosphanyl/arsanyl moiety in these reactions. The arsanyl moiety in 1 is notably less Lewis basic than the phosphanyl moiety present in B, and thus the reactivity of this compound should differ significantly.49
Reaction of a C6D6 solution of B with 2 bar of CO2 at room temperature was reported to selectively yield the colourless five-membered ring system D which exhibits two doublet resonances in its 31P{1H} NMR spectrum at 80.7 ppm and −291.0 ppm (1JP–P = 588 Hz).30 Contrastingly, the analogous reaction of 1 with CO2 yields an orange reaction mixture containing multiple products (Scheme 4).
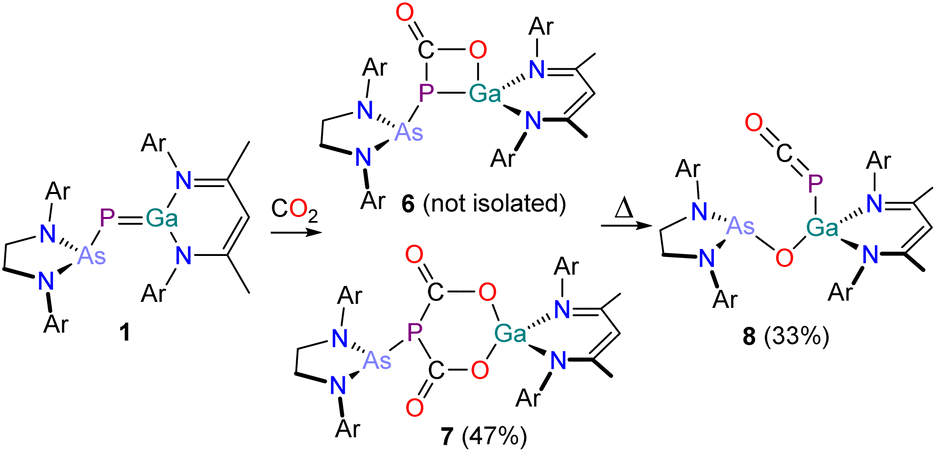 | ||
| Scheme 4 Synthesis of 6, 7 and 8. Ar = 2,6-diisopropylphenyl (Dipp). | ||
Directly after addition, the 31P{1H} NMR spectrum of the reaction mixture exhibits three singlets at −72.2 (unreacted 1), −29.5 (6) and −9.0 ppm (7). After one day at room temperature, the reaction solution decolorizes and exclusively exhibits one singlet at −9.0 ppm (see Fig. S11‡). The corresponding product, 7, can be crystallized from n-pentane at −30 °C in form of thin, colourless needles. X-ray crystallography reveals that 7 is the formal insertion product of two equivalents of CO2 into the GaP bond of 1 (Fig. 7). This motif has been reported previously by Schulz and co-workers on reaction of E with CO2.43 The six-membered GaO2C2P-heterocycle is almost planar, with the phosphorus atom protruding from the ring plane (Σangles = 291.91°). The Ga–O bonds (Ga–O1 1.852(3) Å; Ga–O3 1.831(3) Å) are nearly identical and in the range of typical single bonds, as are the C–O bonds in the heterocycle (C1–O1 1.319(5) Å; C2–O3 1.321(4) Å) and the P–C bonds (P1–C1 1.874(4) Å; P1–C2 1.8743(4) Å; ∑rcov (P–C) = 1.86 Å).
 | ||
| Fig. 7 Single crystal X-ray structure of 7. Thermal ellipsoids set at 50% probability level; hydrogen atoms and solvent of crystallization omitted for clarity. Carbon atoms (except for C1 and C2) are depicted as spheres of arbitrary radius. Selected interatomic distances [Å] and angles [°]: As1–P1 2.3985(10), P1–C1 1.874(4), P1–C2 1.873(4), Ga–O1 1.852(3), Ga1–O3 1.831(3), C1–O1 1.319(5), C1–O2 1.204(5), C2–O3 1.321(4), C2–O4 1.210(5), As1–P1–C1 98.29(15), As1–P1–C2 92.63(11), C1–P1–C2 100.99(18). | ||
Given that E has been shown to reversibly bind CO2,43 we were curious to probe the thermal stability of 7. Heating a colorless solution of 7 in C6D6 to 80 °C for one-hour results in an intense orange solution. This solution exhibits traces of 6 and the starting material 1 in its 31P{1H} NMR spectrum, indicating that the CO2 binding is thermally reversible. Upon prolonged heating, another small singlet with a chemical shift of −371.1 ppm corresponding to a new compound 8 arises in the 31P{1H} NMR spectrum. Heating a solution of 7 for three days at 80 °C cleanly affords 8. The 13C{1H} NMR spectrum of 8 shows a doublet at 181.2 ppm with a 1JC–P coupling constant of 92.9 Hz. The IR spectrum of 8 reveals an absorption band at 1922 cm−1. These data are in line with those previously reported for gallium phosphaketenes (31P{1H} NMR shifts ranging from −354.9 ppm to −394.6 ppm; 13C{1H} NMR shifts in the range of 180.8 ppm to 186.4 ppm and 1JC–P coupling constants ranging from 85 Hz to 101 Hz; PCO IR stretching bands between 1910 cm−1 and 1936 cm−1).43,50,51 To the best of our knowledge, there are only a handful of examples of deoxygenation of CO2 by compounds of the main group elements,52–55 while examples involving transition metals are more numerous.56–60
Compound 8 can be crystallized from a saturated n-hexane/benzene mixture at room temperature and X-ray elucidation confirms the formation of a gallium-phosphaketene with a Ga1–P1 bond length of 2.3512(4) Å (Fig. 8). Additionally, the Ga1 atom in 8 is bound to the arsanyl moiety via a bridging oxygen atom which originates from the carbon dioxide (Ga1–O1 1.8220(10) Å). The Ga1–P1–C1 angle of 92.98(6)° is typical for gallium phosphaketenes, as well as the almost linear P1–C1–O2 moiety (174.38(17)°).43,50,51
 | ||
| Fig. 8 Single crystal X-ray structure of 8. Thermal ellipsoids set at 50% probability level; hydrogen atoms and solvent of crystallization omitted for clarity. Carbon atoms (except for C1) are depicted as spheres of arbitrary radius. Selected interatomic distances [Å] and angles [°]: As1–O2 1.7826(10), O2–Ga1 1.8220(10), Ga1–P1 2.3512(4), P1–C1 1.644(2), C1–O1 1.160(3), As1–O2–Ga1 125.12(6), Ga1–P1–C1 92.98(6), P1–C1–O1 174.38(17). | ||
Having isolated and characterized species 7 and 8 from the reaction of 1 with CO2 [31P NMR: −9.0 (7) and −371.1 ppm (8)] we were intrigued by the nature of the unidentified intermediate observed in crude reaction mixtures which exhibits a singlet resonance at −29.5 ppm. Given that the chemical shift of this species is close to that of 3 and 4, we suspect that this compound arises from the (2 + 2) addition of CO2 and 1. This hypothesis is further supported by the observation that upon reaction of 1 with 13CO2, this resonance appears in the 31P{1H} NMR spectrum as a doublet with a 1JC–P coupling of 37.5 Hz. Furthermore, the 13C{1H} NMR spectrum of the reaction mixture exhibits a doublet resonance for the P13CO2 moiety at 179.7 ppm (1JC–P = 37.5 Hz; see ESI,‡ chapter 3).
In an attempt to characterize this compound, we halted the reaction 30 min after the addition of CO2 by removing the solvent and all volatiles under a dynamic vacuum. Redissolving the resulting orange residue in C6D6 shows the presence of 1 and of 7 by 31P{1H} NMR spectroscopy, with no evidence of 6. This suggests that the formation of 6 is reversible. Unfortunately, we were unable to isolate compound 6 from mixtures of CO2 and 1 despite repeated attempts. Varying the pressure of CO2 was attempted, giving rise to varying ratios of 1, 6, 7 and 8 at different stages of the reaction, however the reactions always contained product mixtures.
Reactivity of 1 with CS2
Due to the significant differences in the reactivity of B and 1 towards CO2 we investigated the thus far unexplored reactivity of both species towards carbon disulfide CS2. Reaction of B with CS2 in C6H6 resulted in a color change of the solution from red to a dark red/purple (Scheme 5, top). The 31P{1H} NMR spectrum of the reaction exhibits two doublets at 102.7 and −287.3 ppm (1JP–P = 614.0 Hz). The product, 9, crystallizes from n-hexane at −30 °C in form of purple needles. X-ray crystallography confirms the formation of a formal FLP-type activation product, analogous to the CO2 activation product D (Fig. 9). 9 exhibits a significantly contracted P1–P2 bond (2.0630(6) Å; ∑rcov (PP) = 2.04 Å) which is in the range of a corresponding double bond.48 The Ga1–P1 bond (2.2874(5) Å) is enlogated in comparison to B and in the range of a single bond.47,48 Adduct formation is not reversible under mild conditions (e.g. evaporation at room temperature or heating in C6D6 solution to 80 °C overnight).
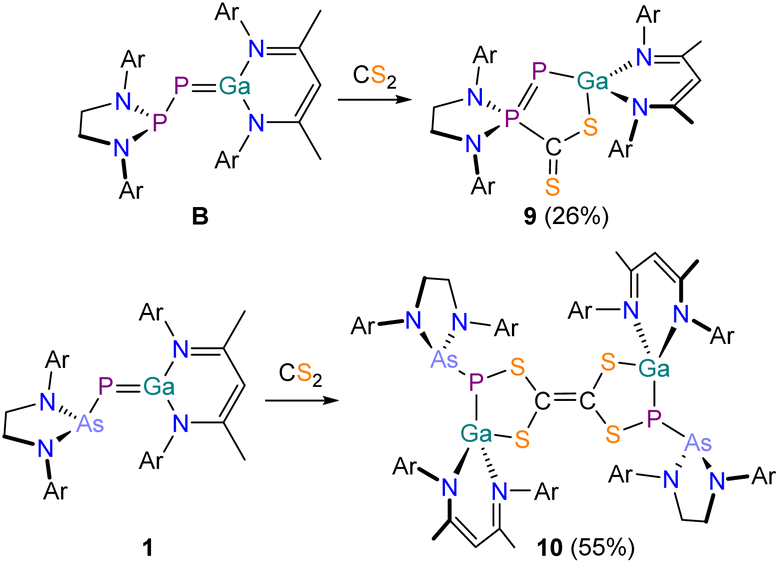 | ||
| Scheme 5 Contrasting reactivity of B (top) and 1 (bottom) with CS2. Ar = 2,6-diisopropylphenyl (Dipp). | ||
 | ||
| Fig. 9 Single crystal X-ray structure of 9. Thermal ellipsoids set at 50% probability level; hydrogen atoms and solvent of crystallization omitted for clarity. Carbon atoms (with the exception of C1) are depicted as spheres of arbitrary radius. Selected interatomic distances [Å] and angles [°]: P1–P2 2.0630(6), P1–Ga1 2.2874(5), P2–C1 1.877(2), C1–S1 1.715(2), C1–S2 1.6477(19), S1–Ga1 2.2935(5), P2–P1–Ga1 95.89(2). | ||
By contrast, the 31P{1H} NMR spectrum of a C6D6 solution of 1 after treatment with excess CS2 shows two singlets at −25.9 and −52.5 ppm in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio. The 1H NMR spectrum displays two singlets at 4.80 and 4.67 ppm stemming from two inequivalent NacNac γ-H protons, also in a 1:1 ratio. Single crystals of the product 10 were grown from n-pentane at room temperature (Fig. 10). X-ray elucidation reveals the formation of a dimeric compound, in which two identical moieties are connected via a C1C2 bond (1.350(3) Å; ∑rcov (CC) = 1.34 Å).47 This suggests that a carbene-like intermediates arising from a (2 + 3) addition reaction of 1 and CS2 dimerizes in the reaction solution to form 10. Mayer and coworkers reported on a similar product formed in the reaction of a disilene with CS2.61 A similar C2S42− moiety was recently accessed by reaction of an anionic gallium(I) compound with CS2.62 The two hereocycles in 10 are crystallographically inequivalent, but their bond metric parameters are very similar. The P–Ga bonds (P1–Ga1 2.3544(7) Å; P2–Ga2 2.3103(7) Å) are in the range expected for single bonds, as are the Ga–S bonds (Ga1–S2 2.2349(7) Å; Ga2–S3 2.2574(7) Å; ∑rcov (Ga–S) = 2.27 Å) and the C–S bonds (C1–S1 1.787(3) Å; C1–S2 1.756(2) Å; C2–S3 1.760(2) Å; C2–S4 1.804(2) Å; ∑rcov (C–S) = 1.78 Å). Compound 10 is stable at room temperature and does not undergo any rearrangement reactions even when heating a solution to 80 °C in C6D6. Given the structural similarities of 10 to tetrathiafulvalene, this product may exhibit interesting redox behavior and electrochemical properties. The formation of 10 rather than of an FLP-type activation product analogous to 9 can be rationalized due to the less nucleophilic arsanyl moiety of 1. However, to rationalize the differing reactivity of 1 towards CO2 and CS2, we performed more detailed calculations (vide infra).
:1 ratio. The 1H NMR spectrum displays two singlets at 4.80 and 4.67 ppm stemming from two inequivalent NacNac γ-H protons, also in a 1:1 ratio. Single crystals of the product 10 were grown from n-pentane at room temperature (Fig. 10). X-ray elucidation reveals the formation of a dimeric compound, in which two identical moieties are connected via a C1C2 bond (1.350(3) Å; ∑rcov (CC) = 1.34 Å).47 This suggests that a carbene-like intermediates arising from a (2 + 3) addition reaction of 1 and CS2 dimerizes in the reaction solution to form 10. Mayer and coworkers reported on a similar product formed in the reaction of a disilene with CS2.61 A similar C2S42− moiety was recently accessed by reaction of an anionic gallium(I) compound with CS2.62 The two hereocycles in 10 are crystallographically inequivalent, but their bond metric parameters are very similar. The P–Ga bonds (P1–Ga1 2.3544(7) Å; P2–Ga2 2.3103(7) Å) are in the range expected for single bonds, as are the Ga–S bonds (Ga1–S2 2.2349(7) Å; Ga2–S3 2.2574(7) Å; ∑rcov (Ga–S) = 2.27 Å) and the C–S bonds (C1–S1 1.787(3) Å; C1–S2 1.756(2) Å; C2–S3 1.760(2) Å; C2–S4 1.804(2) Å; ∑rcov (C–S) = 1.78 Å). Compound 10 is stable at room temperature and does not undergo any rearrangement reactions even when heating a solution to 80 °C in C6D6. Given the structural similarities of 10 to tetrathiafulvalene, this product may exhibit interesting redox behavior and electrochemical properties. The formation of 10 rather than of an FLP-type activation product analogous to 9 can be rationalized due to the less nucleophilic arsanyl moiety of 1. However, to rationalize the differing reactivity of 1 towards CO2 and CS2, we performed more detailed calculations (vide infra).
 | ||
| Fig. 10 Single crystal X-ray structure of 10. Thermal ellipsoids set at 50% probability level; hydrogen atoms and solvent of crystallization omitted for clarity. Carbon atoms (with the exception of C1 and C2) are depicted as spheres of arbitrary radius. Selected interatomic distances [Å] and angles [°]: As1–P1 2.4056(7), P1–Ga1 2.3544(7), P1–S1 2.1175(9), S1–C1 1.787(3), C1–S2 1.756(2), S2–Ga1 2.2349(7), C1–C2 1.350(3), As2–P2 2.3994(6), P2–Ga2 2.3103(7), P2–S4 2.0994(8), S4–C2 1.804(2), C2–S3 1.760(2), S3–Ga2 2.2574(7). | ||
Reactivity of 1 towards COS
To better understand the differing reactivity of 1 towards CO2 and CS2, we reacted 1 with carbonyl sulfide COS (Scheme 6). On addition of COS, the color of the solution changed from deep red to light yellow within a minute. The 31P{1H} NMR spectrum of the reaction mixture revealed the formation of a single species, 11, exhibiting a singlet resonance at −365.4 ppm. In contrast to the reactions between 1 with CO2 (vide supra) which required elevated temperatures, 11 is formed immediately upon addition of COS at room temperature, and no intermediate species could be observed spectroscopically. 11 crystallizes from saturated n-pentane/toluene solution at room temperature in form of colorless blocks in the space group Pna21 with two inequivalent molecules per unit cell (for clarity bond metric data for only one crystallographically independent molecule is discussed). Single-crystal X-ray analysis confirms the formation of a phosphaketene-containing compound analogous to 7 (Fig. 11). The Ga–P bond lengths are in the range of a corresponding double bond (2.3877(8) Å). In contrast to 7, the Ga1 atom in 11 is bound to the arsanyl moiety via a bridging sulfur atom (Ga1–S1 2.2261(7) Å). Compound 11 exhibits an almost linear phosphaketenyl moiety (P–C–O 173.6(3)°). In the IR spectrum the absorption band of the PCO stretching vibration can be found at 1907 cm−1. | ||
| Scheme 6 Synthesis of 11. Ar = 2,6-diisopropylphenyl (Dipp). | ||
 | ||
| Fig. 11 Single crystal X-ray structure of 11. Thermal ellipsoids set at 50% probability level; hydrogen atoms and solvent of crystallization omitted for clarity. Carbon atoms (with the exception of C1) are depicted as spheres of arbitrary radius. Selected interatomic distances [Å] and angles [°]: As1–S1 2.3216(7), S1–Ga1 2.2261(7), Ga1–P1 2.3877(8), P1–C1 1.642(4), C1–O1 1.165(5), As1–S1–Ga1 99.77(3), Ga1–P1–C1 95.25(11), P1–C1–O1 173.6(3). | ||
Computations
Electronic structure
To gain further structural insights, density functional theory (DFT) calculations63 and natural bond orbital (NBO)64–66 analyses were performed using Gaussian09 (ref. 67) at the PBE level of theory68–70 using the def2-TZVP basis set71 with dispersion correction (D3(BJ);72,73 notation: PBE-D3/def2-TZVP). The bond parameters of 1DFT were found to be similar to the experimentally observed ones (Ga–P: 2.1688 Å and P–As: 2.3175 Å). The natural population analysis (NPA) of 1DFT reveals that the Ga1–P1 π-bond in 1DFT has an occupancy of 1.87 e, (indicating a slight delocalization to the neighboring Ga–N(NacNac) bonds). This bond is almost exclusively formed from p atomic orbitals (Ga1: 99%, P1: 99% p-character) and is significantly polarized towards the P1 atom (Ga1: 17%, P1: 83%). The Ga1 atom (+1.32) and the As1 atom (+1.03) are positively charged, whilst the P1 atom is negatively charged (−0.86). Natural bond analysis (NBO) reveals a Wiberg bond index of 1.45 for the Ga–P bond in 1DFT and of 1.03 for the respective P–As bond.A comparison of the parameters calculated for 1DFT and the analogues ADFT and BDFT is well-suited to explain the experimentally observed differences in reactivity of these species. In both ADFT and BDFT the HOMO is mainly located on the lone pairs of the P1 and P2 atoms, whereas the HOMO−1 reflects the respective π-bonding interations along the G13–P1 bond (see Fig. S15‡). In accordance with these findings, A and B show FLP-type reaction behavior upon the activation of small molecules such as e.g. H2, CO2, CS2 and amines.30,74 Exchanging the [Ga] moiety in B with an [Al] moiety results in an increased reactivity, e.g. towards CH bonds, and also reduces the stability of A in comparison to B. This can be rationalized with the higher polarization of the G13P multiple bond in A.
In contrast, the HOMO in 1DFT reflects the π-bonding interations along the Ga1–P1 bond and the HOMO−1 is located at the lone pairs of the P1 and the As1 atoms. Thus, the reactivity of 1 is mainly dominated by its GaP multiple bond (vide infra). Exchanging the pendant phosphanyl [P] moiety with an arsanyl [As] moiety not only affects the energies of relevant orbitals, but also orbital shape and hybridization. According to NBO analysis, the lone pair at the As1 atom in 1DFT exhibits a significantly higher s-character (69%) than the analogous lone pair at the P2 atom in BDFT (58%). Additionally, the LP at the As1 atom in 1DFT is more diffuse. Both factors result in a less localized electron charge at the As1 atom, and thus in a decreased basicity of the As1 atom in 1DFT, in comparison to the basicity of the P2 atom in BDFT. These findings are suitable to rationalize why 1DFT does not exhibit FLP-type reactivity, as proven experimentally (vide ante).
Reaction mechanisms
Compound B was experimentally found to react with CO2 and CS2 in a FLP-type fashion, yielding analogous products for both CO2 and CS2. However, we were surprised by the differing reactivity of 1 towards CO2, CS2, and COS. Using ORCA 5.0.475 and the NEB method (nudged elastic band),76–79 we calculated the Gibbs free energies of potential reaction products and reaction mechanisms of formal (2 + 2) and (3 + 2) addition reactions of 1 and CO2, CS2 or COS, respectively. Regarding the molecular frontier orbitals of CO2, CS2 and 1DFT (see Fig. S16‡) we would expect a formal (2 + 2) addition reaction to proceed in a stepwise manner, as opposed to a concerted (3 + 2) addition reaction.
The reaction of 1 and CO2 proceeds initially via a metastable van-der-Waals adduct (ΔG° = +37.7 kJ mol−1, see Fig. 12). The experimentally observed, formal (2 + 2) addition product is the thermodynamically favored reaction product (−6.6 kJ mol−1), with an activation barrier of +75.2 kJ mol−1 with respect to the starting materials. The transition state resembles a R–PCO2− formate-like structure as the product of a nucleophilic attack of the P1 lone pair at the C1 atom of the CO2 molecule. The formation of a (3 + 2) carbene-type intermediate of 1 and CO2 was calculated to be significantly energetically uphill (+130.6 kJ mol−1). This is in accordance with the experimental observation of the formation of the (2 + 2) product 6 in solution (vide ante), which then further reacts to yield either product 7 or product 8.
 | ||
| Fig. 12 Schematic view of the energy diagram of the (2 + 2) or (2 + 3) addition reactions of 1 and CO2 or CS2, respectively, at the PBE-D3/def2-TZVP level of theory. *For computational reasons, the dimerization energy ΔGdim° formally yielding 0.5 eq. 10 was calculated for a truncated model system 10_Ph (for computational details, see ESI‡). | ||
The reaction of 1 and CS2 also leads to a metastable van-der-Waals adduct (+30.8 kJ mol−1) in the first instance. However, even though the formal (2 + 2) addition product would be the thermodynamically favored product (−91.9 kJ mol−1), the R–PCS2− thioformate-like transition state (+80.5 kJ mol−1, see Scheme S17‡) is energetically unfavorable. The formation of a carbene-type product via a (3 + 2) addition has a significantly lower activation barrier (+54.3 kJ mol−1) and is therefore the dominant reaction pathway, eventually leading to the experimentally observed dimerization product. We calculated the dimerization process of the (3 + 2) carbene-type intermediate yielding compound 10 by using a model system with phenyl-substituents instead of Dipp substituents. These calculations suggest that the dimerization reaction is a highly exergonic process (ΔGdim° = −115.3 kJ mol−1 for the reaction (3 + 2)_CS2 → ½ 10). Thus, the dimerization reaction yielding 10 is an irreversible process.
For the conversion of 1 with COS, one potential product of a (3 + 2) addition reactions was calculated to be thermodynamically uphill (65.5 kJ mol−1), whilst the formation of the formal (2 + 2) addition products was found to be exergonic (−15.9 kJ mol−1 and −64.6 kJ mol−1). This indicates that the formation of experimentally characterized compound 11 proceeds via a (2 + 2) intermediate, as analogously observed for the conversion of 1 and CO2. In this case, no further mechanistic studies were conducted.
Conclusion
We show that altering the pendant functionality of a phosphagallene has a profound effect on the reactivity of the PGa double bond. The previously reported phosphanyl-phosphagallene exhibits frustrated Lewis pair reactivity towards heteroallenes. By contrast, tempering the reactivity of the pendant Lewis base by replacement of the phosphanyl group for an arsanyl group unlocks the reactivity of the PGa bond. We have shown this through cyclization reactions with species such as azides, carbodiimides and isocyanates. More interesting are reactions occur with CO2, CS2 and COS. In the case of carbon monoxide, a deoxygenation reaction was observed cleaving one of the CO bonds. Such reactivity is very rare amongst compounds of the main-group elements. COS reacts in an analogous manner, however in this case, the weaker CS bond is cleaved. The reactivity towards CS2 is, by contrast, much more different, proceeding via a carbene intermediate that dimerizes to give a molecule with a tetrathio-oxalate core. These studies serve to expand the known reactivity of a burgeoning class of molecules possessing heteroatomic double bonds between the heavier main-group elements.
Experimental
All manipulations were carried out under oxygen- and moisture free conditions under argon or nitrogen atmosphere using standard Schlenk or drybox techniques. The reported products are (mostly very) sensitive towards oxygen and moisture. All starting materials were produced or purified as stated in the ESI.‡ Unless stated otherwise, the experiments were carried out at room temperature (approx. 23 °C) and the removal of solvents was realized in vacuo (1 × 10−3 mbar) at room temperature. Further information on experimental procedures, data acquisition and processing, purification of solvents and starting materials, on computational investigations, additional spectroscopic data and a full set of analytical data for each compound can be found in the ESI.‡Synthesis of 1
Toluene (7 mL) was added to a mixture of Ga(NacNac) (1.05 eq., 80.0 mg, 0.164 mmol) and [As]PCO (80.0 mg, 0.156 mmol) at ambient temperature (25 °C). The reaction mixture turned dark red within seconds and rapid effervescence was observed (caution: the reaction evolves of CO). The mixture was stirred at ambient temperature for one hour, after which all volatile components were removed in vacuo (1 × 10−3 mbar, 25 °C). The dark red solid was extracted with n-pentane (10 mL) and the resulting slightly turbid solution filtered. The clear, red filtrate was concentrated to a volume of approx. 3 mL in vacuo using a warm water bath (1 × 10−3 mbar, 35 °C). The product crystallizes in form of large red blocks from a concentrated n-pentane solution overnight (these crystals are also suitable for X-ray diffraction). The supernatant was removed with a syringe and discarded. The resulting crystalline product 1 was dried under a dynamic vacuum using a warm water bath for three hours (1 × 10−3 mbar, 45 °C). Yield: 92.0 mg, 0.0947 mmol, 60.7%.EA for C55H79AsGaN4P (M.W. = 971.88 g mol−1) calcd (found) in %: C 67.97 (67.50), H 8.19 (8.69), N 5.76 (5.66). 31P{1H} NMR (C6D6, 298.0 K, 202.37 MHz): δ (ppm) −72.2 (m; AsPGa). 1H NMR (C6D6, 298.0 K, 499.93 MHz): δ (ppm) 7.16–7.25 (m, 8H; ArCH), 7.05 (d, 3JH–H = 7.6 Hz, 4H; ArCH), 4.86 (s, 1H; NacNac γ-H), 4.32–4.36 (m, 2H; [SP]Dipp{CH(CH3)2}), 3.95–4.03 (m, 2H; (NCH2)2), 3.56 (sept, 3JH–H = 6.8 Hz, 2H; [SP]{CH(CH3)2}), 3.27–3.36 (m, 2H; NCH2), 2.89 (sept, 3JH–H = 6.8 Hz, 4H, NacnNac{CH(CH3)2}), 1.35–1.42 (m, 18H; NacNacCH3 and Dipp{CH(CH3)2}), 1.31 (d, J = 6.9 Hz, 6H; Dipp{CH(CH3)2}), 1.29 (d, J = 6.9 Hz, 6H; Dipp{CH(CH3)2}), 1.02 (d, 3JH–H = 6.7 Hz, 12H; Dipp{CH(CH3)2}), 0.98 (d, 3JH–H = 6.9 Hz, 12H; Dipp{CH(CH3)2}).
Synthesis of 2
1 (35 mg, 0.036 mmol) was dissolved in C6H6 (0.7 mL) in a J. Young NMR tube. Tert-butyl azide (1.05 eq., 3.70 mg, 0.0370 mmol) was added to the NMR tube causing the solution to quickly turn orange. All volatile components were removed in vacuo (1 × 10−3 mbar, 25 °C) and the orange solid extracted with n-hexane. The solution was filtered into a small vial and kept at −30 °C overnight resulting in the formation of orange crystals of 2. The supernatant was removed, discarded and the product dried under a dynamic vacuum for three hours (1 × 10−3 mbar, 25 °C). Yield: 23.2 mg, 0.0217 mmol, 61.0%. Crystals suitable for X-ray diffraction were grown from a mixture of n-hexane/benzene (1:1) at room temperature. Due to the (partly) reversible adduct formation with tBuN3 at high temperatures, it was not possible to obtain an accurate elemental analysis of 2.
EA for C59H88AsGaN7P (M.W. = 1071.01 g mol−1) calcd (found) in %: C 66.17 (67.85), H 8.28 (8.00), N 7.00 (7.20). 31P{1H} NMR (C6D6, 298.0 K, 202.38 MHz): δ (ppm) −31.6 (s; AsPGa). 1H NMR (C6D6, 298.0 K, 499.98 MHz): δ (ppm) 7.16–7.20 (m, 2H; ArCH), 7.12–7.15 (m, 2H; ArCH), 7.02–7.08 (m, 4H; ArCH), 6.86–6.97 (m, 4H; ArCH), 4.88 (s, 1H; NacNac γ-H), 4.50–4.65 (m, 2H; Dipp{CH(CH3)2}), 4.15–4.30 (m, 2H; NCH2), 3.30–3.50 (m, 4H; NCH2 and Dipp{CH(CH3)2}), 3.15–3.38 (m, 2H; Dipp{CH(CH3)2}), 2.80–2.95 (m, 2H; Dipp{CH(CH3)2}), 1.60 (s, 9H; tBuCH3), 1.29 (s, 6H; NacNacCH3), 1.25 (d, 3JH–H = 6.9 Hz, 6H; Dipp{CH(CH3)2}), 1.18–1.23 (m, 12H; Dipp{CH(CH3)2}), 1.13–1.17 (m, 6H; Dipp{CH(CH3)2}), 1.02 (br. d, 3JH–H = 6.7 Hz, 12H; Dipp{CH(CH3)2}), 0.74 (br. s, 6H; Dipp{CH(CH3)2}), 0.60 (br. d, 3JH–H = 6.3 Hz, 6H; Dipp{CH(CH3)2}).
Synthesis of 3
1 (34.0 mg, 0.0350 mmol) was dissolved in C6H6 (0.7 mL) in a J. Young NMR tube. N,N′-Dicyclohexylcarbodiimide (1.05 eq., 7.57 mg, 0.0370 mmol) was added to the NMR tube, after which the solution quickly turned yellow. The solution was filtered into a small vial affording colourless crystals of 3 within minutes. The filtration needs to be carried out quickly, otherwise the product can precipitate in the filter. The supernatant was removed, discarded and the resulting colourless crystalline solid was washed with small amounts of cold n-hexane (3 × 0.5 mL, −30 °C). The resulting crystalline solid was dried in vacuo for three hours (1 × 10−3 mbar, 25 °C). Yield: 9.70 mg, 0.008 mmol, 23.5%.EA for C68H101AsGaN6P (M.W. = 1178.21 g mol−1) calcd (found) in %: C 69.32 (70.27), H 8.64 (8.18), N 5.92 (6.79). 31P{1H} NMR (THF-d8, 298.0 K, 243.05 MHz): δ (ppm) −27.6 (s; AsPGa). 1H NMR (THF-d8, 298.0 K, 600.42 MHz): δ (ppm) 7.08–7.13 (m, 6H; ArCH), 7.03–7.07 (m, 4H; ArCH), 7.00–7.03 (br. s, 2H; ArCH), 6.93–6.98 (m, 4H; ArCH), 5.32 (s, 1H; NacNac γ-H), 3.90–3.98 (m, 2H; NCH2), 3.70–3.78 (m, 2H; Dipp{CH(CH3)2}), 3.40–3.47 (m, 1H; CyCH), 3.25–3.35 (m, 6H; NCH2 and Dipp{CH(CH3)2}), 3.18–3.25 (m, 1H; CyCH), 2.89 (sept, 3JH–H = 6.9 Hz, 2H; Dipp{CH(CH3)2}), 2.42 (br. d, 3JH–H = 10.9 Hz, 2H; CyCH2), 1.88 (br. d, 3JH–H = 12.9 Hz, 2H; CyCH2), 1.65 (br. s, 8H; CyCH2 and NacNacCH3), 1.53–1.62 (m, 2H; CyCH2), 1.25–1.38 (m, 10H; CyCH2), 1.09 (br. d, 3JH–H = 6.8 Hz, 3JH–H = 2.3 Hz, 12H; Dipp{CH(CH3)2}), 1.05 (br. s, 3H; Dipp{CH(CH3)2}), 1.04 (br. d, 3JH–H = 3.8 Hz, 6H; Dipp{CH(CH3)2}), 1.02 (br. d, 3JH–H = 4.5 Hz, 6H; Dipp{CH(CH3)2}), 1.00 (br. s, 6H; Dipp{CH(CH3)2}), 0.99 (br. s, 3H; Dipp{CH(CH3)2}), 0.93–0.98 (m, 2H; CyCH2), 0.85 (br. d, 3JH–H = 6.7 Hz, 6H; Dipp{CH(CH3)2}), 0.68 (br. d, 3JH–H = 6.7 Hz, 6H; Dipp{CH(CH3)2}).
Synthesis of 4
1 (52.0 mg, 0.0540 mmol) was dissolved in C6H6 (0.7 mL) in a J. Young NMR tube. 2,4,6-Trimethylphenyl isocyanate (1.06 eq., 9.10 mg, 0.0560 mmol) was added to the NMR tube, and the resulting reaction mixture turned yellow. After the addition, all volatile components were removed in vacuo (1 × 10−3 mbar, 25 °C) and the yellow solid was extracted with n-hexane. The solution was filtered into a small vial. Storage of the solution at −30 °C overnight afforded yellow crystals of 4. The supernatant was removed and discarded, and the solid washed with small amounts of cold n-hexane (3 × 0.3 mL, −30 °C). The resulting yellow product was dried under a dynamic vacuum for three hours (1 × 10−3 mbar, 25 °C). Yield: 33.7 mg, 0.030 mmol, 55.5%. Crystals suitable for X-ray diffraction were grown from a mixture of n-hexane/benzene (1:1) at room temperature.
EA for C65H90AsGaN5OP (M.W. = 1133.08 g mol−1) calcd (found) in %: C 68.90 (69.55), H 8.01 (7.91), N 6.18 (6.29). 31P{1H} NMR (C6D6, 298.0 K, 243.05 MHz): δ (ppm) −34.5 (s; AsPGa). 1H NMR (C6D6, 298.0 K, 600.42 MHz): δ (ppm) 7.20–7.35 (m, 6H; ArCH), 7.02–7.15 (m, 3H; ArCH), 6.95–7.01 (br. s, 2H; Mes ArCH), 6.85–6.90 (br. s, 1H; Mes ArCH), 6.59 (s, 1H; Mes ArCH), 4.66 (s, 1H; NacNac γ-H), 4.08 (br. s, 1H; Dipp{CH(CH3)2}), 3.94 (br. s, 1H; NCH2), 3.85 (br. s, 1H; Dipp{CH(CH3)2}), 3.74 (br. s, 2H; Dipp{CH(CH3)2}), 3.65 (br. s, 1H; NCH2), 3.48 (br. s, 2H; NCH2 and Dipp{CH(CH3)2}), 3.24 (br. s, 1H; NCH2), 2.87–3.05 (m, 2H; Dipp{CH(CH3)2}), 2.79 (br. s, 1H; Dipp{CH(CH3)2}), 2.14 (s, 3H; MesCH3); 1.88 (br. s, 3H; MesCH3), 1.15–1.53 (m, 42H; MesCH3 and/or NacNacCH3 and/or Dipp{CH(CH3)2}), 0.90–1.05 (m, 9H; MesCH3 and/or NacNacCH3 and/or Dipp{CH(CH3)2}), 0.73 (br. s, 3H; MesCH3 and/or NacNacCH3 and/or Dipp{CH(CH3)2}), 0.50 (br. s, 3H; MesCH3 and/or NacNacCH3 and/or Dipp{CH(CH3)2}).
Synthesis of 5
1 (58.0 mg, 0.060 mmol) was dissolved in C6H6 (0.7 mL) in a J. Young NMR tube. Benzaldehyde (1.35 eq., 8.60 mg, 0.0810 mmol) was added to the NMR tube causing the solution to quickly turn yellow. After addition, all volatile components were removed in vacuo (1 × 10−3 mbar, 25 °C) and the yellow solid extracted with n-hexane. The solution was filtered into a small vial. After storage at −30 °C overnight, yellow crystals of 5 were obtained. The supernatant was removed and discarded, and the product was dried in vacuo for three hours (1 × 10−3 mbar, 25 °C). Yield: 22.5 mg, 0.0210 mmol, 35.0%. Crystals suitable for X-ray diffraction were grown from a mixture of n-hexane/benzene (1:1) at room temperature.
EA for C62H85AsGaN4OP (M.W. = 1078.00 g mol−1) calcd (found) in %: C 69.08 (69.27), H 7.95 (7.63), N 5.20 (5.28). 31P{1H} NMR (C6D6, 298.0 K, 202.38 MHz): δ (ppm) 67.6 (s, AsPGa). 1H NMR (C6D6, 298.0 K, 499.93 MHz): δ (ppm) 7.32–7.37 (m, 2H; ArCH), 7.20–7.28 (m, 3H; ArCH), 7.17–7.19 (m, 2H; ArCH), 7.08–7.14 (m, 5H; ArCH), 6.70–6.75 (m, 1H; Ph ArCH), 6.61 (t, 3JH–H = 7.6 Hz, 2H; Ph ArCH), 5.96 (br. d, 3J = 7.1 Hz, 2H; Ph ArCH); 5.59 (d, 3JP–H = 34 Hz, 1H; PhCHO); 4.81 (s, 1H; NacNac γ-H), 4.40 (dq, 3JH–H = 12.8 Hz, J = 6.6 Hz, 1H; Dipp{CH(CH3)2}), 4.23 (td, 3JH–H = 9.7 Hz, J = 5.7 Hz, 1H; NCH2), 3.91 (sept, 3JH–H = 6.9, 1H; Dipp{CH(CH3)2}), 3.57 (sept, 3JH–H = 6.7, 1H; Dipp{CH(CH3)2}), 3.30–3.50 (m, 5H; NCH2 and Dipp{CH(CH3)2}), 3.20 (sept, 3JH–H = 6.8, 1H; Dipp{CH(CH3)2}), 3.09–3.17 (m, 1H; NCH2); 2.89 (sept, 3JH–H = 6.7, 1H; Dipp{CH(CH3)2}), 1.94 (d, 3JH–H = 6.7, 3H; Dipp{CH(CH3)2}), 1.59 (s, 3H; Dipp{CH(CH3)2}), 1.52 (s, 3H; Dipp{CH(CH3)2}), 1.45–1.49 (m, 6H; Dipp{CH(CH3)2}), 1.32–1.40 (m, 12H; Dipp{CH(CH3)2}), 1.20–1.28 (m, 12H; NacNacCH3 and Dipp{CH(CH3)2}), 1.19 (d, 3JH–H = 6.9, 3H; Dipp{CH(CH3)2}), 1.10–1.16 (m, 6H; Dipp{CH(CH3)2}), 0.93 (d, 3JH–H = 6.7, 3H; Dipp{CH(CH3)2}), 0.39 (d, 3JH–H = 6.7, 3H; Dipp{CH(CH3)2}).
Synthesis of 7
1 (50.0 mg, 0.0520 mmol) was dissolved in C6H6 (0.7 ml) in a J. Young NMR tube. Dry CO2 (2 bar) was added to the NMR tube, which was then sealed and stored at ambient temperature overnight (approx. 16 hours). Reaction progress was monitored using 31P{1H} NMR spectroscopy. When conversion to the main product in the 31P NMR spectrum (−9.0 ppm) was complete, all volatiles were removed in vacuo (1 × 10−3 mbar, 25 °C). The resulting colourless precipitate was extracted with n-pentane (0.5 mL). The resulting solution was filtered into a small vial and kept at −30 °C overnight affording small, colourless crystals of 7. The supernatant was removed with a syringe and discarded. The resulting colourless crystals were dried in vacuo for three hours (1 × 10−3 mbar, 25 °C). Yield: 25.6 mg, 0.0240 mmol, 47.0%.EA for C57H79AsGaN4O4P (M.W. = 1059.9 g mol−1) calcd (found) in %: C 64.59 (65.64), H 7.51 (6.85), N 5.29 (5.07). 31P{1H} NMR (C6D6, 298.0 K, 202.37 MHz): δ (ppm) −9.0 (s; AsP(CO2)2). 1H NMR (C6D6, 298.0 K, 499.93 MHz): δ (ppm) 7.17–7.19 (m, 1H; ArCH), 6.96–7.00 (m, 7H; ArCH), 7.03–7.14 (m, 4H; ArCH), 4.79 (s, 1H; NacNac γ-H), 3.88–3.98 (m, 2H; NCH2), 3.65–3.73 (m, 2H; Dipp{CH(CH3)2}), 3.52–3.62 (m, 2H; Dipp{CH(CH3)2}), 3.15–3.25 (m, 2H; NCH2), 3.65–3.73 (sept, 3JH–H = 6.8 Hz, 4H; Dipp{CH(CH3)2}), 1.44 (s, 6H; NacNacCH3), 1.32 (d, 3JH–H = 6.7 Hz, 12H; Dipp{CH(CH3)2}), 1.24 (d, 3JH–H = 6.9 Hz, 12H; Dipp{CH(CH3)2}), 1.22 (br. d, 3JH–H = 6.9 Hz, 6H; Dipp{CH(CH3)2}), 1.04 (d, 3JH–H = 6.7 Hz, 6H; Dipp{CH(CH3)2}), 0.96 (d, 3JH–H = 6.9 Hz, 12H; Dipp{CH(CH3)2}).
Synthesis of 8
1 (55.0 mg, 0.0570 mmol) was dissolved in C6H6 (0.7 ml) in a J. Young NMR tube. Dry CO2 (2 bar) was added to the NMR tube, which was sealed and heated to 80 °C for approx. 3 days in an oil bath. The reaction progress was monitored by 31P NMR spectroscopy. When the major species formed corresponded to a singlet at −371.1 ppm in the 31P{1H} NMR spectrum, all volatiles were removed in vacuo (1 × 10−3 mbar, 25 °C) and the colourless precipitate was extracted with n-hexane (0.5 mL). The resulting solution was filtered into a small vial and kept at −30 °C overnight affording small, colourless crystals of 8. The supernatant was removed with a syringe and discarded. The resulting colourless crystals were dried in vacuo for three hours (1 × 10−3 mbar, 25 °C). Yield: 18.9 mg, 0.0190 mmol, 32.9%. Notice: due to very broad 1H NMR signals at 298 K (see Fig. S7‡), the 1H NMR spectrum was recollected at 343 K, where the signals are sharper. In the 31P NMR spectrum at 298 K, no signal broadening was observed.EA for C56H79AsGaN4O2P (M.W. = 1015.89 g mol−1) calcd (found) in %: C 66.21 (66.00), H 7.84 (7.34), N 5.52 (5.08). 31P{1H} NMR (C6D6, 298.0 K, 243.05 MHz): δ (ppm) −371.1 (s; GaPCO). 1H NMR (C6D6, 343.0 K, 499.93 MHz): δ (ppm) 6.98–7.15 (m, 12H; ArCH), 4.89 (s, 1H; NacNac γ-H), 4.03–4.25 (m, 4H; Dipp{CH(CH3)2}), 3.71–3.85 (m, 2H; NCH2), 3.37–3.51 (m, 4H; Dipp{CH(CH3)2}), 3.05–3.19 (m, 2H; NCH2), 2.14 (br. s, 3H; Dipp{CH(CH3)2} and NacNacCH3), 1.53 (br. s; 6H; Dipp{CH(CH3)2} and NacNacCH3), 0.98–1.50 (45H; Dipp{CH(CH3)2} and NacNacCH3).
Synthesis of 9
Compound B (33 mg, 0.036 mmol) was dissolved in C6H6 (0.7 ml) in a J. Young NMR tube. CS2 (2.1 μL, 2.7 mg, 0.036 mmol) was added to the NMR tube at ambient temperature (25 °C) resulting in an immediate colour change of the reaction mixture from red to dark purple. The tube was kept at ambient temperature for one hour after which all volatile components were removed in vacuo (1 × 10−3 mbar, 25 °C). The purple solid was extracted with n-hexane (0.5 mL) and the resulting solution was filtered into a small vial and kept at −30 °C overnight to give purple crystals of 9. The supernatant was removed with a syringe and discarded. The resulting purple crystals were dried in vacuo for three hours (1 × 10−3 mbar, 25 °C). Yield: 9.2 mg, 0.0090 mmol, 26%.EA for C56H79GaN4P2S2 (M.W. = 1004.07 g mol−1) calcd (found) in %: C 66.99 (65.81), H 7.93 (6.99), N 5.58 (4.77). 31P NMR (C6D6, 298.0 K, 243.05 MHz): δ (ppm) 102.7 (d, 1JP–P = 614.0 Hz; PPGa), −287.3 (d, 1JP–P = 614.0 Hz; PPGa). 1H NMR (C6D6, 298.0 K, 600.42 MHz): δ (ppm) 7.14–7.19 (m, 2H; ArCH), 7.04–7.10 (m, 4H; ArCH), 6.93–6.98 (m, 4H; ArCH), 6.88–6.92 (m, 2H; ArCH), 4.66 (s, 1H; NacNac γ-H), 3.85–3.93 (m, 2H; NCH2), 3.78 (sept, 3JH–H = 6.9 Hz, 2H; Dipp{CH(CH3)2}), 3.72 (sept, 3JH–H = 6.8 Hz, 2H; Dipp{CH(CH3)2}), 3.20–3.30 (m, 2H; NCH2), 3.15 (sept, 3JH–H = 6.8 Hz, 2H; Dipp{CH(CH3)2}), 3.05 (sept, 3JH–H = 6.8 Hz, 2H; Dipp{CH(CH3)2}), 1.38 (s, 6H; NacNacCH3), 1.36 (d, 3JH–H = 6.7 Hz, 6H; Dipp{CH(CH3)2}); 1.12–1.17 (m, 18H; Dipp{CH(CH3)2}), 1.04–1.10 (m, 12H; Dipp{CH(CH3)2}), 0.99 (d, 3JH–H = 6.9 Hz, 6H; Dipp{CH(CH3)2}), 0.92 (d, 3JH–H = 6.7 Hz, 6H; Dipp{CH(CH3)2}).
Synthesis of 10
1 (50.0 mg, 0.0520 mmol) was dissolved in C6H6 (0.7 ml) in a J. Young NMR tube. An excess of CS2 (1 drop) was added to the NMR tube at ambient temperature (25 °C) after which the colour of the reaction mixture changed from dark red to orange. The tube was kept at ambient temperature for one hour and afterwards all volatile components were removed in vacuo (1 × 10−3 mbar, 25 °C). The orange precipitate was extracted with n-hexane (0.5 mL) and the resulting slightly turbid solution was filtered into a small vial. Storage of this solution at ambient temperature overnight afforded orange crystals of 10 (suitable for X-ray diffraction). The supernatant was removed with a syringe and discarded. The resulting orange crystals were dried in vacuo for three hours (1 × 10−3 mbar, 25 °C). Yield: 29.4 mg, 0.0140 mmol, 54.5% (of dimer, max. yield is 100%).EA for C112H158As2Ga2N8P2S4 (M.W. = 2096.04 g mol−1) calcd (found) in %: C 64.18 (64.42), H 7.60 (8.23), N 5.35 (4.93). 31P NMR (C6D6, 298.0 K, 243.05 MHz): δ (ppm) −25.9 (s; AsP(S)Ga), −52.5 (s; AsP(S)Ga). 1H NMR (C6D6, 298.0 K, 600.42 MHz): δ (ppm) 7.25–7.38 (m, 13H; ArCH), 7.19 (3JH–H = 7.5 Hz, 1H; ArCH), 7.12–7.15 (m, 2H; ArCH), 7.08 (3JH–H = 7.5 Hz, 1H; ArCH), 6.94–7.05 (m, 6H; ArCH), 6.79 (3JH–H = 7.3 Hz, 1H; ArCH), 4.80 (s, 1H; NacNac γ-H), 4.67 (s, 1H; NacNac γ-H), 4.30–4.40 (m, 1H; NCH2), 4.20–4.28 (m, 1H; NCH2), 4.02–4.11 (m, 2H; Dipp{CH(CH3)2}), 3.85–4.01 (m, 4H; Dipp{CH(CH3)2} and NCH2), 3.60–3.65 (m, 1H; Dipp{CH(CH3)2}), 3.53–3.58 (m, 2H; Dipp{CH(CH3)2} and NCH2), 3.40–3.50 (m, 4H; Dipp{CH(CH3)2} and NCH2), 3.30–3.40 (m, 1H; NCH2), 3.20–3.30 (m, 2H; Dipp{CH(CH3)2}), 3.05–3.10 (m, 3H; Dipp{CH(CH3)2}), 2.90–3.00 (m, 3H; Dipp{CH(CH3)2} and NCH2), 1.86 (d, 3JH–H = 6.5 Hz, 3H; NacNacCH3), 1.81 (d, 3JH–H = 6.5 Hz, 3H; NacNacCH3), 1.68 (d, 3JH–H = 6.7 Hz, 3H; Dipp{CH(CH3)2}), 1.60–1.64 (m, 6H; NacNacCH3), 1.59 (d, 3JH–H = 6.9 Hz, 3H; Dipp{CH(CH3)2}), 1.52–1.56 (m, 6H; Dipp{CH(CH3)2}), 1.44–1.48 (m, 21H; Dipp{CH(CH3)2}), 1.40 (d, 3JH–H = 6.7 Hz, 3H; Dipp{CH(CH3)2}), 1.32–1.37 (m, 6H; Dipp{CH(CH3)2}), 1.30 (d, 3JH–H = 6.9 Hz, 3H; Dipp{CH(CH3)2}), 1.27 (d, 3JH–H = 6.7 Hz, 3H; Dipp{CH(CH3)2}), 1.52–1.56 (m, 9H; Dipp{CH(CH3)2}), 1.14–1.18 (m, 6H; Dipp{CH(CH3)2}), 1.08–1.12 (m, 9H; Dipp{CH(CH3)2}), 0.99 (d, 3JH–H = 6.7 Hz, 3H; Dipp{CH(CH3)2}), 0.97 (d, 3JH–H = 6.7 Hz, 3H; Dipp{CH(CH3)2}), 0.94 (d, 3JH–H = 6.7 Hz, 3H; Dipp{CH(CH3)2}), 0.92 (d, 3JH–H = 6.7 Hz, 3H; Dipp{CH(CH3)2}), 0.61 (d, 3JH–H = 6.7 Hz, 3H; Dipp{CH(CH3)2}), 0.40–0.47 (m, 6H; Dipp{CH(CH3)2}), 0.21 (d, 3JH–H = 6.5 Hz, 3H; Dipp{CH(CH3)2}).
Synthesis of 11
1 (60.0 mg, 0.0610 mmol) was dissolved in C6H6 (0.7 mL) in a J. Young NMR tube. Dry COS (1 bar) was added to the NMR tube, which was then sealed and stored at room temperature for approx. 1 hour. Reaction progress was monitored using 31P{1H} NMR spectroscopy. When the spectrum revealed a singlet at −371.1 ppm, the volatile components were removed in vacuo (1 × 10−3 mbar, 25 °C) and the yellow precipitate was extracted with n-pentane/toluene (0.3 mL/0.3 mL). The resulting solution was filtered into a small vial and kept at room temperature overnight affording small, colourless crystals of 11. The supernatant was removed with a syringe, discarded and the resulting colourless crystals were dried in vacuo for three hours (1 × 10−3 mbar, 25 °C). Yield: 27.3 mg, 0.0270 mmol, 43.4%.EA for C56H79AsGaN4OSP (M.W. = 1031.95 g mol−1) calcd (found) in %: C 65.18 (65.63), H 7.72 (7.76), N 5.43 (5.14). 31P NMR (C6D6, 298.2 K, 242.95 MHz): δ (ppm) −365.4 (s, GaPCO). 1H NMR (C6D6, 298.2 K, 600.16 MHz): δ (ppm) 7.17–7.21 (m, 2H; ArCH), 7.08–7.11 (m, 3H; ArCH), 7.00–7.05 (m, 5H; ArCH), 6.96–6.99 (m, 2H; ArCH), 4.83 (s, 1H; NacNac γ-H), 3.81–3.90 (m, 2H; Dipp{CH(CH3)2}), 3.65–3.75 (m, 4H; Dipp{CH(CH3)2} and NCH2), 3.50–3.60 (m, 2H; Dipp{CH(CH3)2}), 3.15–3.25 (m, 4H; Dipp{CH(CH3)2} and NCH2); 1.54 (d, 6H, 3JH–H = 6.9 Hz; Dipp{CH(CH3)2}), 1.39 (s, 6H; NacNacCH3), 1.54 (d, 6H, 3JH–H = 6.9 Hz; Dipp{CH(CH3)2}), 1.42 (d, 6H, 3JH–H = 6.9 Hz; Dipp{CH(CH3)2}), 1.26 (d, 6H, 3JH–H = 6.5 Hz, Dipp{CH(CH3)2}), 1.23 (d, 6H, 3JH–H = 6.9 Hz, Dipp{CH(CH3)2}), 1.11 (d, 6H, 3JH–H = 6.9 Hz; Dipp{CH(CH3)2}), 1.06 (d, 6H, 3JH–H = 6.9 Hz; Dipp{CH(CH3)2}), 0.58 (d, 6H, 3JH–H = 6.9 Hz; Dipp{CH(CH3)2}), 0.53 (d, 6H, 3JH–H = 6.9 Hz; Dipp{CH(CH3)2}).
Data availability
Further information on experimental procedures, data acquisition and processing, purification of solvents and starting materials, on computational investigations, additional spectroscopic data and all full set of analytical data for each compound can be found in the ESI.‡80–85
Author contributions
L. S. S. carried out the experimental work and recorded the SCXRD data. L. S. S. and J. B. performed the computational studies. L. F. assisted with the experiments involving COS and 13CO2. M. Ernst assisted with the SCXRD measurement on compound 11. J. M. G solved the SCXRD structures. L. S. S. and J. M. G. conceptualized the project and wrote the manuscript. All authors contributed for further revision of the ESI‡ and manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the German National Academy of Sciences Leopoldina for funding (LSS; Leopoldina Fellowship Program, LPDS 2022-10). The University of Oxford and the Freie Universität Berlin are acknowledged for access to chemical crystallography facilities. The University of Rostock and M. Willert are acknowledged for access to the cluster computer and support with software installations.Notes and references
- W. E. Dasent, J. Chem. Educ., 1963, 40, 130 Search PubMed.
- K. S. Pitzer, J. Am. Chem. Soc., 1948, 70, 2140–2145 CrossRef CAS.
- R. S. Mulliken, J. Am. Chem. Soc., 1950, 72, 4493–4503 CrossRef CAS.
- (a) M. Yoshifuji, I. Shima, N. Inamoto, K. Hirotsu and T. Higuchi, J. Am. Chem. Soc., 1981, 103, 4587–4589 Search PubMed; (b) R. West, M. J. Fink and J. Michl, Science, 1981, 214, 1343–1344 CrossRef CAS PubMed.
- L. Weber, F. Ebeler and R. S. Ghadwal, Coord. Chem. Rev., 2022, 461, 214499 CrossRef CAS.
- R. C. Fischer and P. P. Power, Chem. Rev., 2010, 110, 3877–3923 Search PubMed.
- D. Raiser, C. P. Sindlinger, H. Schubert and L. Wesemann, Angew. Chem., Int. Ed., 2020, 59, 3151–3155 CrossRef CAS PubMed.
- B. Rao and R. Kinjo, Angew. Chem., Int. Ed., 2020, 59, 3147–3150 CrossRef CAS PubMed.
- D. Franz and S. Inoue, Dalton Trans., 2016, 45, 9385–9397 CAS.
- Y. Heider, P. Willmes, D. Mühlhausen, L. Klemmer, M. Zimmer, V. Huch and D. Scheschkewitz, Angew. Chem., Int. Ed., 2019, 58, 1939–1944 CAS.
- M. J. Reveley, J. Feld, D. Temerova, E. S. Yang and J. M. Goicoechea, Chem.–Eur. J., 2023, e202301542 CrossRef CAS PubMed.
- V. Y. Lee, M. Kawai, A. Sekiguchi, H. Ranaivonjatovo and J. Escudié, Organometallics, 2009, 28, 4262–4265 CrossRef CAS.
- V. Y. Lee, M. Kawai, O. A. Gapurenko, V. I. Minkin, H. Gornitzka and A. Sekiguchi, Chem. Commun., 2018, 54, 10947–10949 RSC.
- J. Feld, E. Yang, S. Urwin and J. M. Goicoechea, Chem.–Eur. J., 2024, e202401736 CrossRef CAS PubMed.
- Z. F. Zhang and M. Der Su, Inorg. Chem., 2024, 63, 19687–19700 CrossRef CAS PubMed.
- M. A. Wünsche, T. Witteler and F. Dielmann, Angew. Chem., Int. Ed., 2018, 57, 7234–7239 CrossRef PubMed.
- A. G. Baradzenka, S. F. Vyboishchikov, M. Pilkington and G. I. Nikonov, Chem.–Eur. J., 2023, e202301842 CrossRef CAS PubMed.
- C. Hu, N. Rees, M. Pink and J. M. Goicoechea, Nat. Chem., 2024, 16, 1855–1860 CrossRef CAS PubMed.
- G. Linti, H. Nöth, K. Polborn and R. T. Paine, Angew. Chem., Int. Ed. Engl., 1990, 29, 682–684 CrossRef.
- E. Rivard, W. A. Merrill, J. C. Fettinger and P. P. Power, Chem. Commun., 2006, 3800–3802 RSC.
- N. J. Hardman, C. Cui, H. W. Roesky, W. H. Fink and P. P. Power, Angew. Chem., Int. Ed., 2001, 40, 2172–2174 CrossRef CAS PubMed.
- R. J. Wright, A. D. Phillips, T. L. Allen, W. H. Fink and P. P. Power, J. Am. Chem. Soc., 2003, 125, 1694–1695 CrossRef CAS PubMed.
- J. Li, Z. Lu and L. L. Liu, J. Am. Chem. Soc., 2022, 144, 23691–23697 CrossRef CAS PubMed.
- E. A. LaPierre, B. O. Patrick and I. Manners, J. Am. Chem. Soc., 2023, 145, 7107–7112 CrossRef CAS PubMed.
- M. D. Anker, R. J. Schwamm and M. P. Coles, Chem. Commun., 2020, 56, 2288–2291 RSC.
- A. Heilmann, J. Hicks, P. Vasko, J. M. Goicoechea and S. Aldridge, Angew. Chem., Int. Ed., 2020, 59, 4897–4901 CrossRef CAS PubMed.
- A. García-Romero, C. Hu, M. Pink and J. M. Goicoechea, J. Am. Chem. Soc., 2025, 147, 1231–1239 CrossRef PubMed.
- F. Dankert and C. Hering-Junghans, Chem. Commun., 2022, 58, 1242–1262 RSC.
- L. S. Szych, L. Denker, J. Feld and J. M. Goicoechea, Chem.–Eur. J., 2024, e202401326 CAS.
- D. W. N. Wilson, J. Feld and J. M. Goicoechea, Angew. Chem., Int. Ed., 2020, 59, 20914–20918 CrossRef CAS PubMed.
- F. Bertini, V. Lyaskovskyy, B. J. J. Timmer, F. J. J. de Kanter, M. Lutz, A. W. Ehlers, J. C. Slootweg and K. Lammertsma, J. Am. Chem. Soc., 2012, 134, 201–204 CAS.
- C. Appelt, H. Westenberg, F. Bertini, A. W. Ehlers, J. C. Slootweg, K. Lammertsma and W. Uhl, Angew. Chem., Int. Ed., 2011, 50, 3925–3928 Search PubMed.
- H. S. Zijlstra, J. Pahl, J. Penafiel and S. Harder, Dalton Trans., 2017, 46, 3601–3610 CAS.
- P. Federmann, T. Bosse, S. Wolff, B. Cula, C. Herwig and C. Limberg, Chem. Commun., 2022, 58, 13451–13454 Search PubMed.
- M. N. Khan, Y. van Ingen, T. Boruah, A. McLauchlan, T. Wirth and R. L. Melen, Chem. Sci., 2023, 14, 13661–13695 CAS.
- T. W. Yokley, H. Tupkar, N. D. Schley, N. J. DeYonker and T. P. Brewster, Eur. J. Inorg. Chem., 2020, 2958–2967 Search PubMed.
- M. Devillard, R. Declercq, E. Nicolas, A. W. Ehlers, J. Backs, N. Saffon-Merceron, G. Bouhadir, J. C. Slootweg, W. Uhl and D. Bourissou, J. Am. Chem. Soc., 2016, 138, 4917–4926 CAS.
- P. Holtkamp, F. Friedrich, E. Stratmann, A. Mix, B. Neumann, H. Stammler and N. W. Mitzel, Angew. Chem., Int. Ed., 2019, 58, 5114–5118 Search PubMed.
- X. Xu, G. Kehr, C. G. Daniliuc and G. Erker, J. Am. Chem. Soc., 2013, 135, 6465–6476 Search PubMed.
- Z. Jian, G. Kehr, C. G. Daniliuc, B. Wibbeling and G. Erker, Dalton Trans., 2017, 46, 11715–11721 Search PubMed.
- M. K. Sharma, C. Wölper and S. Schulz, Dalton Trans., 2022, 51, 1612–1616 CAS.
- M. K. Sharma, C. Wölper, G. Haberhauer and S. Schulz, Angew. Chem., Int. Ed., 2021, 60, 21784–21788 CrossRef CAS PubMed.
- M. K. Sharma, C. Wölper, G. Haberhauer and S. Schulz, Angew. Chem., Int. Ed., 2021, 60, 6784–6790 CrossRef CAS PubMed.
- M. D. Anker, M. Lein and M. P. Coles, Chem. Sci., 2019, 10, 1212–1218 RSC.
- M. D. Anker, R. J. Schwamm and M. P. Coles, Chem. Commun., 2020, 56, 2288–2291 RSC.
- M. Mehta, J. E. McGrady and J. M. Goicoechea, Chem.–Eur. J., 2019, 25, 5445–5450 CrossRef CAS PubMed.
- P. Pyykkö and M. Atsumi, Chem.–Eur. J., 2008, 15, 186–197 Search PubMed.
- P. Pyykkö and M. Atsumi, Chem.–Eur. J., 2009, 15, 12770–12779 Search PubMed.
- Z. F. Zhang, M. C. Yang and M. Der Su, Inorg. Chem., 2021, 60, 15253–15269 CrossRef CAS PubMed.
- D. W. N. Wilson, W. K. Myers and J. M. Goicoechea, Dalton Trans., 2020, 49, 15249–15255 RSC.
- Y. Mei, J. E. Borger, D. Wu and H. Grützmacher, Dalton Trans., 2019, 48, 4370–4374 RSC.
- R. Becerra, J. P. Cannady and R. Walsh, J. Phys. Chem. A, 2002, 106, 4922–4927 CrossRef CAS.
- (a) C. Schulten, G. von Frantzius, G. Schnakenburg, A. Espinosa and R. Streubel, Chem. Sci., 2012, 3, 3526–3533 Search PubMed; (b) T. Taeufer, F. Dankert, D. Michalik, J. Pospech, J. Bresien and C. Hering-Junghans, Chem. Sci., 2023, 14, 3018–3023 RSC.
- L. Gu and Y. Zhang, J. Am. Chem. Soc., 2010, 132, 914–915 CrossRef CAS PubMed.
- E. Ballestero-Martínez, T. Szilvási, T. J. Hadlington and M. Driess, Angew. Chem., Int. Ed., 2019, 58, 3382–3386 CrossRef PubMed.
- I. Castro-Rodríguez and K. Meyer, Chem. Commun., 2006, 1343–1352 Search PubMed.
- U. Jayarathne, P. Chandrasekaran, H. Jacobsen, J. T. Mague and J. P. Donahue, Dalton Trans., 2010, 39, 9662–9671 Search PubMed.
- G. Fachinetti, C. Floriani, A. Chiesi-Villa and C. Guastini, J. Am. Chem. Soc., 1979, 101, 1767–1775 CrossRef CAS.
- C. H. Lee, D. S. Laitar, P. Mueller and J. P. Sadighi, J. Am. Chem. Soc., 2007, 129, 13802–13803 Search PubMed.
- J. Li and Z. Lin, Organometallics, 2009, 28, 4231–4234 CrossRef CAS.
- N. Wiberg, W. Niedermayer, K. Polborn and P. Mayer, Chem.–Eur. J., 2002, 8, 2730–2739 CrossRef CAS PubMed.
- M. Guo, B. Dong, Y. Qu, Z. Sun, L. Yang, Y. Wang, I. L. Fedushkin and X.-J. Yang, Chem.–Eur. J., 2024, e202403652 Search PubMed.
- (a) J. Bresien, SLURM interface for ORCA and Gaussian, University of Rostock, 2020 Search PubMed; (b) F. Weigend, Phys. Chem. Chem. Phys., 2006, 8, 1057–1065 RSC.
- (a) F. E. D. Glendening, J. K. Badenhoop, A. E. Reed, J. E. Carpenter, J. A. Bohmann, C. M. Morales, C. R. Landis and F. Weinhold, NBO 6.0. Theoretical Chemistry Institute, University of Wisconsin, Madison 2013 Search PubMed; (b) J. E. Carpenter and F. Weinhold, J. Mol. Struct.: THEOCHEM, 1988, 169, 41–62 CrossRef.
- F. Weinhold and C. R. Landis, in Valency and Bonding. A Natural Bond Orbital Donor-Acceptor Perspective, Cambridge University Press, 2005 Search PubMed.
- F. Weinhold and J. Carpenter, in The Structure of Small Molecules and Ions, ed. R. Naaman and Z. Vager, Springer US, Boston, MA, 1988, pp. 227–236 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision E.01, Gaussian, Inc., Wallingford CT, 2013 Search PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 Search PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1997, 78, 1396 CrossRef CAS.
- C. Adamo and V. Barone, J. Chem. Phys., 1999, 110, 6158–6170 CrossRef CAS.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297 RSC.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- J. Feld, D. W. N. Wilson and J. M. Goicoechea, Angew. Chem., Int. Ed., 2021, 60, 22057–22061 CrossRef CAS PubMed.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2022, 12, 1–15 Search PubMed.
- (a) G. Mills, H. Jónsson and G. K. Schenter, Surf. Sci., 1995, 324, 305–337 CrossRef CAS; (b) H. Jónsson, G. Mills and K. W. Jacobsen, in Classical and Quantum Dynamics in Condensed Phase Simulations, WORLD SCIENTIFIC, 1998, pp. 385–404 Search PubMed.
- G. Henkelman and H. Jónsson, J. Chem. Phys., 2000, 113, 9978–9985 CrossRef CAS.
- G. Henkelman, B. P. Uberuaga and H. Jónsson, J. Chem. Phys., 2000, 113, 9901–9904 CrossRef CAS.
- E. Maras, O. Trushin, A. Stukowski, T. Ala-Nissila and H. Jónsson, Comput. Phys. Commun., 2016, 205, 13–21 CrossRef CAS.
- O. Kysliak, H. Görls and R. Kretschmer, Dalton Trans., 2020, 49, 6377–6383 RSC.
- W. L. F. Armarego and C. L. L. Chai, Purification of Laboratory Chemicals, Elsevier, 2013 Search PubMed.
- M. Swetha, P. Venkata Ramana and S. G. Shirodkar, Org. Prep. Proced. Int., 2011, 43, 348–353 CrossRef CAS.
- CrysAlisPro, Agilent Technologies, Version 1.171.42.72a Search PubMed.
- SAINT V8.40A, Bruker AXS, Madison, WI, 2020 Search PubMed.
- (a) G. M. Sheldrick in SHELXL97, Programs for Crystal Structure Analysis (Release 97-2), Institut für Anorganische Chemie der Universität, Göttingen, Germany, 1998 Search PubMed; (b) G. M. Sheldrick, Acta Crystallogr., Sect. A, 1990, 46, 467–473 CrossRef; (c) G. M. Sheldrick, Acta Crystallogr., Sect. A, 2008, 64, 112–122 CrossRef CAS PubMed.
Footnotes |
| † Dedicated to Dr Gisela Boeck on occasion of her 70th birthday. |
| ‡ Electronic supplementary information (ESI) available. CCDC 2409889–2409898. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5sc00295h |
| This journal is © The Royal Society of Chemistry 2025 |