
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA mechanism-guided descriptor for the hydrogen evolution reaction in 2D ordered double transition-metal carbide MXenes†
Junmei
Du
 ,
Yifan
Yan
,
Xiumei
Li
,
Jiao
Chen
,
Chunsheng
Guo
,
Yuanzheng
Chen
* and
Hongyan
Wang
*
,
Yifan
Yan
,
Xiumei
Li
,
Jiao
Chen
,
Chunsheng
Guo
,
Yuanzheng
Chen
* and
Hongyan
Wang
*
School of Physical Science and Technology, Key Laboratory of Advanced Technology of Materials, Southwest Jiaotong University, Chengdu, Sichuan 610031, China. E-mail: hongyanw@swjtu.edu.cn; cyz@swjtu.edu.cn
First published on 24th April 2025
Abstract
Selecting effective catalysts for the hydrogen evolution reaction (HER) among MXenes remains a complex challenge. While machine learning (ML) paired with density functional theory (DFT) can streamline this search, issues with training data quality, model accuracy, and descriptor selection limit its effectiveness. These hurdles often arise from an incomplete understanding of the catalytic mechanisms. Here, we introduce a mechanism-guided descriptor (δ) for the HER, designed to enhance catalyst screening among ordered transition metal carbide MXenes. This descriptor integrates structural and energetic characteristics, derived from an in-depth analysis of orbital interactions and the relationship between Gibbs free energy of hydrogen adsorption (ΔGH) and structural features. The proposed model (ΔGH = −0.49δ – 2.18) not only clarifies structure–activity links but also supports efficient, resource-effective identification of promising catalysts. Our approach offers a new framework for developing descriptors and advancing catalyst screening.
1. Introduction
Hydrogen generated through clean and sustainable methods is vital for transitioning away from carbon-based fuels to renewable energy.1–3 Although noble metals such as platinum (Pt) are effective catalysts for the hydrogen evolution reaction (HER), their scarcity and high cost limit large-scale applications.4,5 Thus, developing cost-effective electrocatalysts with high efficiency and low overpotential is essential, highlighting the need for non-precious alternatives.6–9This study focuses on 2D ordered double transition-metal carbide (DTM) MXenes, derived from 312 and 413 MAX phases, respectively, which show strong potential for electrocatalytic water splitting due to their tunable structures and properties.10,11 By incorporating distinct transition metals and various surface functional groups, these MXenes offer a broad range of candidates for HER catalysis, necessitating efficient screening methods to identify optimal combinations.12
To date, most researchers have relied on high-throughput density functional theory (DFT) calculations to evaluate the HER performance of these MXenes.13–17 Although DFT provides accurate results, its computational expense limits its scalability for large datasets. This dilemma has evoked increasing interest in developing simpler descriptors that correlate with HER performance, allowing for more efficient predictions.18 Several structural and electronic descriptors have been proposed, including metal–oxygen bond strength in the outer layer of MXenes,15 which is closely related to HER activity, and the d-band center has also been shown to provide some insight into HER performance.11 Other descriptors, such as oxygen vacancy formation energy (Ef), have also been suggested, although they are supported by data from only a limited number of DTM MXenes.19
Machine learning (ML)-based approaches have also been explored.20,21 For example, B. Abraham et al. applied the Gradient Boosting Regressor (GBR) algorithm to predict GH of MM'XT2-type MXenes with a low mean absolute error (MAE) and high coefficient of determination (R2) of 0.358 eV and 0.826, respectively.22 Besides, interpretable ML models are built based on the GBR with recursive feature elimination (RFE), hyperparameter optimization (HO) and the leave-one-out (LOO) approach to identify the number of valence electrons (VT), electron affinity (EAT) of the terminating groups and d-band center variance with respect to the average (dbcσ2) as the strong key predictors of the Gibbs free energy. Anand et al. proposed the descriptor dav × χM′ for DTM MXenes, which combines the average distance of the seven nearest neighbors (dav) to the active site and the electronegativity of the outer-layer metal (χM′).11 Although ML methods such as the Symbolic Regression-Sure Independence Screening and Sparsifying Operator (SISSO) can generate reliable descriptors and exact relationships between descriptors and catalytic activity, they primarily identify mathematical patterns rather than providing a deep mechanistic understanding of catalytic activity. This focus on statistical relationships limits their ability to predict HER activity from the necessary physical and chemical insights. Besides, ML relies on large, high-quality datasets and careful descriptor selection as well as a proper optimization algorithm. Considering these challenges, it is crucial to accurately predict HER activity and gain a more comprehensive understanding from the physical/chemical insights of the structure–activity relation simultaneously.
In this study, we propose a reliable strategy to explore ordered DTM MXenes as promising non-precious HER catalysts, as illustrated in Fig. 1. We first evaluate the synthesizability, stability, and conductivity of MXenes. Then, the promising candidates for the HER are identified by using DFT. Subsequently, their structure–catalytic activity relationships are analyzed based on both microscopic and macroscopic perspectives, and a correlative descriptor (δ) is developed based on it for deeper insights. Finally, the descriptor is validated as a reliable and efficient tool for screening additional potential MXene catalysts. Consequently, we proposed a mechanism-guided universal screening descriptor for the HER based on ordered DTM MXenes.
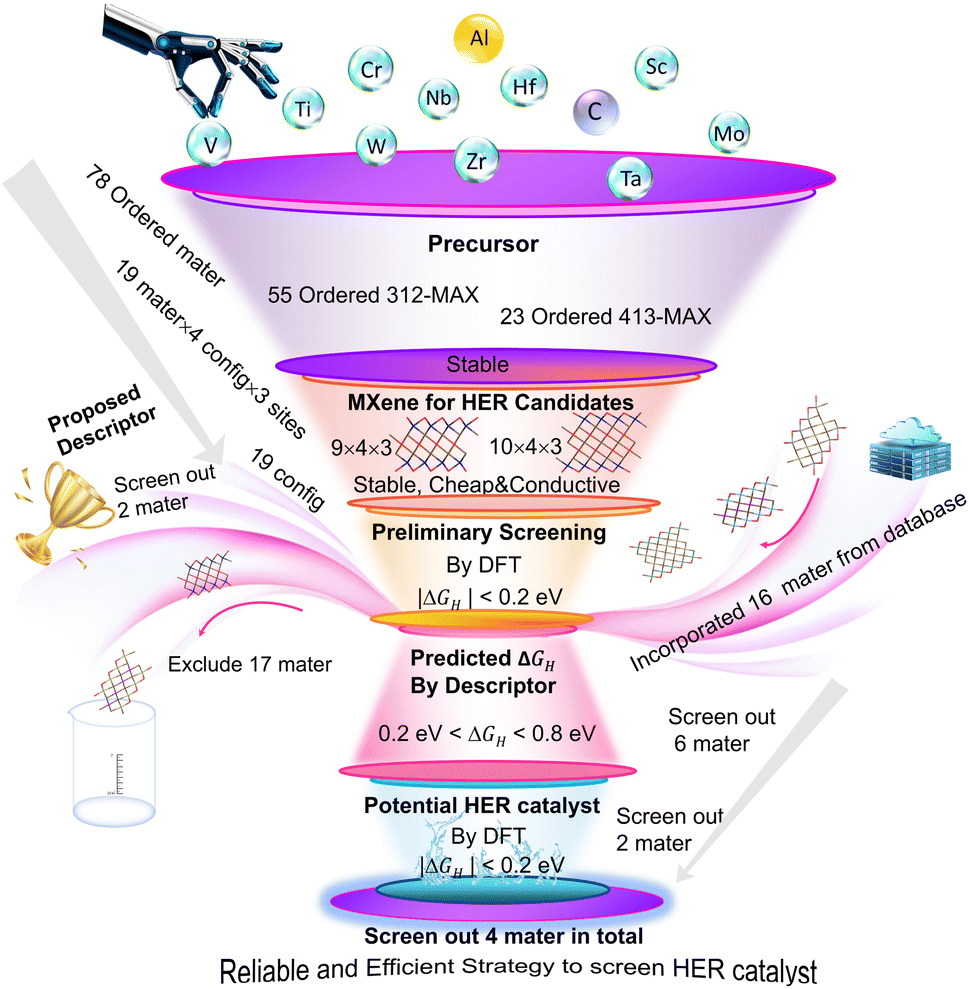 | ||
| Fig. 1 A reliable and efficient flow diagram for screening ideal HER catalysts among ordered double transition-metal carbide MXenes. ‘Config’ denotes configuration, and ‘Mater’ denotes material. | ||
2. Computational details
2.1. DFT calculations
The Vienna Ab initio Simulation Package (VASP) code23 was utilized based on spin-polarized DFT. The Perdew–Burke–Ernzerhof (PBE) functional within the generalized gradient approximation (GGA) was applied to model exchange–correlation interactions,24 and the projector augmented wave (PAW)25 method was used for core and valence electron interactions. For valence electrons, configurations included Ti[3d34s1], V[3p63d44s1], Cr[3d54s1], Zr[4s14p15s14d1], Nb[4p15s14d1], Mo[4p15s14d1], Hf[5p16s15d1], Ta[5p16s15d1], W[5p16s15d1], O[2s22p4], C[2s22p2], and H[1s1]. A kinetic energy cut-off of 500 eV was used, with convergence criteria of 1 × 10−5 eV for energy and 0.01 eV Å−1 for force. The Monkhorst–Pack scheme was employed to sample the Brillouin zone with k-point meshes of 11 × 11 × 1 for unit cells and 5 × 5 × 1 for 2 × 2 × 1 supercells. Detailed information of the coverage test of the k-point mesh and energy cutoff for accuracy is provided in Fig. S1.† A 20 Å vacuum along the z-direction minimized layer interactions, and Grimme's DFT + D3 was employed for van der Waals corrections.26,27 Dipole corrections and standard conditions (U = 0, pH = 0, p = 1 bar, and T = 298 K) were applied. Implicit solvation using the GLSSA13 solvent model28 was implemented via the VASPsol extension.29,30 The pCOHP analysis was performed using the LOBSTER 4.1.0 package.31,322.2. Gibbs free energy of hydrogen adsorption
The overall HER pathway consists of two reaction steps, which can be summarized as follows:| H+(aq) + e− + * → H* → 1/2H2(g) ΔG0 = 0 eV | (1) |
The Volmer reaction begins with an adsorbed state involving a proton (H+) in aqueous solution, an electron (e−), and pristine MXene at an active site. The adsorbed hydrogen forms an intermediate (H*). The subsequent desorption of hydrogen yields hydrogen gas (H2). The Gibbs free energy (ΔG) of the intermediate hydrogen adsorption is critical for evaluating the catalyst's hydrogen evolution reaction (HER) activity. According to the Sabatier principle, optimal HER catalysts have ΔG close to zero, as shown in the computational hydrogen electrode model (CHEM) by Nørskov et al.,33,34 under standard conditions (U = 0, pH = 0, p = 1 bar, and T = 298.15 K), which can be expressed as:
ΔGH = ΔEH + ΔEZPE + ΔU![[0 with combining right harpoon above (vector)]](https://www.rsc.org/images/entities/char_0030_20d1.gif) T − TΔS T − TΔS | (2) |
 | (3) |
 | (4) |
3. Results and discussion
3.1. Constructing ordered double transition metal carbide O-terminated MXenes
This study examines synthesizable ordered DTM MXenes, specifically M′2M′′C2T2 and M′2M′′2C3T2, derived from the 312 and 413 MAX phases, respectively.10 The 312 MAX phase has a metal-to-carbon composition ratio of M′![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :M′′:M:C as 2:1:1:2, while the 413 MAX phase follows a 2:2:1:3 ratio. We evaluate the synthesis, stability, and conductivity of these MXenes.
:M′′:M:C as 2:1:1:2, while the 413 MAX phase follows a 2:2:1:3 ratio. We evaluate the synthesis, stability, and conductivity of these MXenes.
Synthesis potential is assessed by identifying stable precursor MAX phases, with formation energies below 20 eV per atom at 0 K considered stable (details in Table S1†).36 Among 55 chemically ordered 312 MAX phases, nine are stable, while ten stable structures are found among 23413 phases. These results align with findings by Martin Dahlqvist et al., who noted similar stability trends under typical synthesis conditions (ΔHcp < 0 or 0 ≤ ΔHcp ≤ 50 at 1773 K).37 To enhance synthesis feasibility and cost-effectiveness, we excluded phases containing rare or expensive elements, such as Sc. Ultimately, 19 stable MAX phases are identified as viable precursors for M′2M′′C2T2 and M′2M′′2C3T2 MXenes, which will be explored for their potential in HER applications.
Specifically, under practical HER conditions, characterizing electrocatalyst surface structures is essential for screening high-performance candidates.38–41 The surfaces of bare MXenes fully terminated by O* or OH* functional groups under the relevant USHE and pH values are investigated by calculating their Pourbaix diagrams, as shown in Fig. 2. The results indicate that fully O*-terminated surfaces are the most thermodynamically stable for Cr2TiC2. In contrast, for other MXenes, mixed OH*/O* terminations are more stable under acidic conditions, whereas full O*-termination becomes increasingly favorable as the pH increases. These trends are consistent with prior theoretical and experimental reports.42,43 Based on these insights, we focus our subsequent HER analysis on the O*-terminated MXene surfaces. The corresponding optimized surface structures are shown in Fig. 3a. Single-point energy calculations (Table S2†) were performed at high-symmetry adsorption sites for oxygen, including hexagonal close-packed (hcp) and face-centered cubic (fcc) configurations, yielding four distinct O-terminated structures: hcp–fcc, fcc–hcp, fcc–fcc, and hcp–hcp, as shown in Fig. 3b and c. The results demonstrate that hcp–hcp sites are generally energetically favorable in MXenes, except for Cr2Ti2C3O2, Cr2V2C3O2 and Cr2Ta2C3O2, where fcc–fcc configurations are preferred. Band structure calculations demonstrate excellent electrical conductivity for these 19 configurations (Fig. S2†), enhancing charge transfer performance and improving HER efficiency.
 | ||
| Fig. 2 Surface Pourbaix diagrams of (a) Cr2TiC2, (b) Mo2TiC2, (c) Ti2Ta2C3 and (d) W2Hf3C2. The most thermodynamically stable states of the surface under relevant USHE conditions and pH values are labeled by the terminations. | ||
 | ||
| Fig. 3 Crystal structures. (a) List of 9 M′2M′′C2O2 and 10 M′2M′′2C3O2, demonstrated to be synthesizable, stable and conductive, are investigated in this work for the HER. (b and c) Atomic structures of M′2M′′C2O2 in hcp–fcc and fcc–hcp types and M′2M′′2C3O2 in fcc–fcc and hcp–hcp types viewed from the top and side, respectively. (d) Top view of three distinct adsorption structures for 1/4 hydrogen coverage on MXenes. | ||
3.2. Simulating Gibbs free energy of hydrogen adsorption
|ΔGH| serves as a key descriptor for evaluating catalytic activity in HER catalysts. For these calculations, we utilized a 2 × 2 × 1 supercell configuration, examining 19 O-terminated MXenes with 25% hydrogen coverage. The choice of 1/4 hydrogen coverage is based on the calculation results that, in most cases, DTM MXenes exhibit stable hydrogen adsorption at this level within a potential window of −1 V to −1 V, as supported by Pourbaix diagram analyses (as shown in Fig. 4). Hydrogen atoms are positioned at the top of surface functional groups (O), hcp, and fcc sites, respectively, as illustrated in the left panel of Fig. 3d. Our results indicate that hydrogen is more stable when adsorbed onto the top O site, aligning with previous studies.11,13 | ||
| Fig. 4 The surface Pourbaix diagrams as a function of pH and potential under different hydrogen coverage. Surface Pourbaix diagrams of (a) Cr2TiC2O2, (b) Mo2TiC2O2, (c) W2Hf2C3O2 and (d) W2Zr2C3O2. | ||
The Gibbs free energies for the adsorption of atomic hydrogen, ΔGH, on the 9 M′2M′′C2O2 and 10 M′2M′′2C3O2 structures have been calculated, as shown in Fig. 5a and 2b. Our results indicate that 4 M′2M′′C2O2 and 4 M′2M′′2C3O2 structures exhibit theoretical |ΔGH| values below 0.2 eV, indicating their potential as HER electrocatalysts.6 The most promising candidate is Mo2Nb2C3O2, which shows the lowest theoretical overpotential (−0.004 eV). Notably, all identified HER electrocatalysts are Mo-based MXenes, including Mo2Nb2C3O2, Mo2VC2O2, Mo2NbC2O2, Mo2Ti2C3O2, Mo2TaC2O2, Mo2Ta2C3O2, Mo2TiC2O2 and Mo2Zr2C3O2. Our DFT calculations have identified Mo-based ordered DTM MXene structures as promising HER electrocatalysts. With the increasing synthesis of Mo-based MXenes in laboratory settings, our work is poised to contribute to their expanding applications in catalysis.42,44
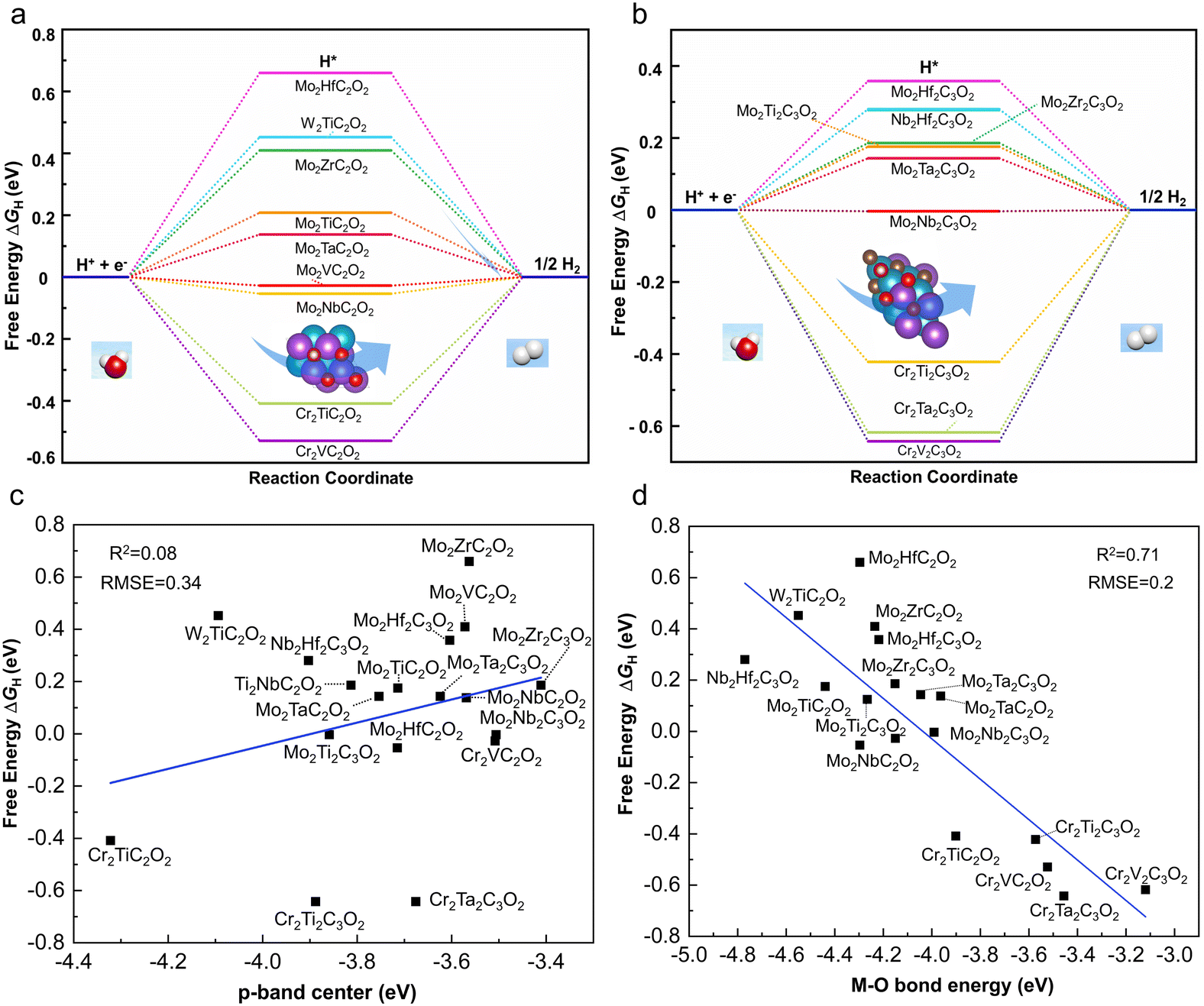 | ||
| Fig. 5 (a and b) HER free energy diagrams of M′2M′′C2O2, and M′2M′′2C3O2 under standard conditions, respectively. (c and d) Relationships between ΔGH and descriptors including (c) the p-band center of the oxygen functional group and (d) the bond energy between the first-layer transition metal and oxygen of different MXene systems, respectively. | ||
3.3. Proposing a descriptor for the HER activity
While ΔGH evaluates the activity of the HER, it fails to account for intrinsic structural characteristics. Furthermore, DFT calculations can be resource-intensive. To address this issue, several descriptors were tested to correlate with the HER activity of MXenes. Based on correlation coefficients and error metrics, we found that none of the descriptors, including the p-band center of oxygen (R2 = 0.08) (as shown in Fig. 5c) and the d-band center of the first-layer transition metal (R2 = 0.01) (as shown in Fig. S3†), could adequately describe the HER activity of MXenes. Only the bond energy between the first-layer transition metal and oxygen exhibited a slightly higher correlation (R2 = 0.71), as detailed in Fig. 5d. The reason for this is that the MXene system involves multi-layer atomic interactions, including various early transition metals interacting with carbon and oxygen atoms. Thus, a single descriptor cannot fully capture its hydrogen evolution activity, and a more comprehensive descriptor is required to properly describe the structure–activity relationship of MXenes.To address this issue, we aim to propose a direct and cost-effective structural descriptor specifically for ordered DTM MXenes.
Taking Mo2TiC2O2 as a representative example, the orbital interactions of M′–O bonding are analyzed by the projected density of states (pDOS) and the pCOHP (Fig. 6a), and it is illustrated in Fig. 6b, with two bonding and antibonding orbitals, respectively. This analysis involved a detailed examination of the overlap peaks in the pDOS for both Mo and O, combined with the bonding and anti-bonding characteristics of the Mo–O bond as well as the wave function image shown in Fig. 6c, ranging from −7 eV to 3 eV. Furthermore, our results show that the O(py)–Mo(dx2−y2) component mainly contributes to the Mo–O bond formation (supported by COHP in Fig. 6d). Additionally, the pCOHP analysis for other MXenes further supports the above conclusion, as shown in Fig. S6.†
 | ||
| Fig. 6 (a) The projected density of states (pDOS) analysis (red curve: M′(Mo) and purple curve: O) as well as pCOHP analysis of the M′(Mo)–O bond. (b) Spin-down and spin-up interactions of the Mo orbital with the O orbital, with (c) post-interaction corresponding wave functions. (d) pCOHP of the Mo(p)–O(d) bond and its O (py)–Mo (dx2−y2) component (red curve: Mo(p)–O(d) and blue curve: py-dx2−y2). (e) pDOS plots of the dx2−y2 orbital for eight distinct MXenes materials, respectively. | ||
To investigate the role of dx2−y2 orbitals in outer transition metals, we examined it across eight types of Mo- and Cr-based MXenes to ensure a diverse range of systems (Fig. 6e). The results reveal that Cr-based MXenes exhibit significantly localized dx2−y2 orbitals at the Fermi level, indicating stronger interactions with O (py) orbitals forming a σ bond. Meanwhile, this σ (Opy–M′dx2−y2) state would strongly interact with the σ (Hs–Opz) state, leading to stronger H adsorption with reduced HER activity. These findings suggest that the dx2−y2 orbital of outer transition metals plays a critical role in the M′–O bond formation and HER performance across various MXenes.
| ΔGH = ΔHH − TΔS | (5) |
| ΔHH = BDE(MXene) + BDE(H) − BDE(MXene–H) = −BDE(MXene–H) ≈ −BDE(‘O’–H) | (6) |
 | ||
| Fig. 7 (a) The top panel is the charge density difference plots for H adsorbed on Mo2Ti2C3O2 and Mo2TiC2O2 with an isosurface value of 0.0004 e bohr−3, respectively, and blue and yellow represent charge loss and charge accumulation, respectively. Bottom panel is the sliced electron localization function (ELF) of H adsorbed on Mo2Ti2C3O2 and Mo2TiC2O2 monolayers along the (001) plane, respectively. (b) The transfer of charges from hydrogen to the oxygen functional group on MXene varies depending on the material, as evaluated by Bader charge analysis. (c) Relationships between ΔGH and descriptor δstruct of different MXenes. (d) Relationships between ΔGH and descriptor δ of different MXene systems. | ||
Based on the relationship between BDE and electronegativity in the Pauling electronegativity formula (eqn (S1)†),45,46 the BDE of the O–H bond is expanded as follows:
 | (7) |
In eqn (7), an approximation has been made by replacing the BDE (O–O) with BDE (M′–O). This substitution is essential as it takes the complex structural features of 2D MXene multilayers into account. Given that M′ is chemically bonded to O, this approximation provides a more accurate representation of the system's behavior and is thus a necessary step in our analysis. It has been well-established, both in our current work and prior studies,11,15 that the outer-layer metals (M′) in MXenes are significantly involved in catalytic activity. X′ acts as a correction factor in the ΔHH calculations to account for other structural influences of MXenes. Using this expanded formula, a descriptor (δ1) is developed for HER catalytic activity, incorporating an additional correction factor (X), as shown in eqn (8):
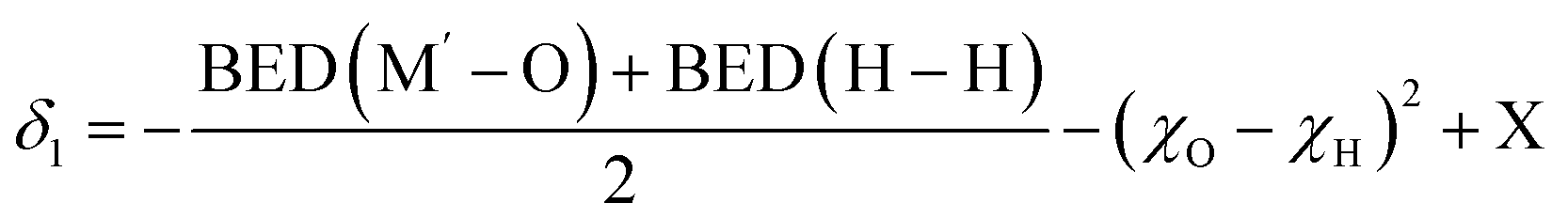 | (8) |
As a result, the outer-layer metals determine the BDE(M′–O) values, therefore, influencing δ1 values and HER activities, and the values of BDE(M′–O) are listed in Table S3.† The finding is consistent with the previous conclusion that the outer metal layer (dx2−y2 orbital) plays an important role in the HER. Therefore, the crucial role of outer-layer metals in modulating HER performance explains why all identified HER electrocatalysts in this study are Mo-based MXenes, both from microscopic and macroscopic perspectives.
Our calculations confirm that ΔGH varies with different inner transition metal layers for the same outer transition metal layer MXenes. Specifically, ΔGH decreases with the increasing electronegativity of the inner transition metal and varies with the number of inner transition metal layers, as illustrated in Fig. S7.† This indicates that both the electronegativity of the inner transition metal and the number of inner transition metal layers should be incorporated into the descriptor. Therefore, we propose the correction factor (χM′′ − χC)2 × (2.05 − 2n) as δcorrect. The term (χM′′ − χC)2 characterizes the bonding influence of the inner transition metal M′′ on the carbon atoms, employing an expression analogous to that in eqn (8). Besides, (2.05 − 2n) serves as the layer influence factor corresponding to its structural characteristics, where 2.05 is derived from the approximate axial ratio coefficient (c/2a), and n represents the number of layers of the inner transition metal. The lattice constants of the considered MXenes are illustrated in Table S4.† Thus, the descriptor δstruct is expressed as δ1 + δcorrect, which is defined as,
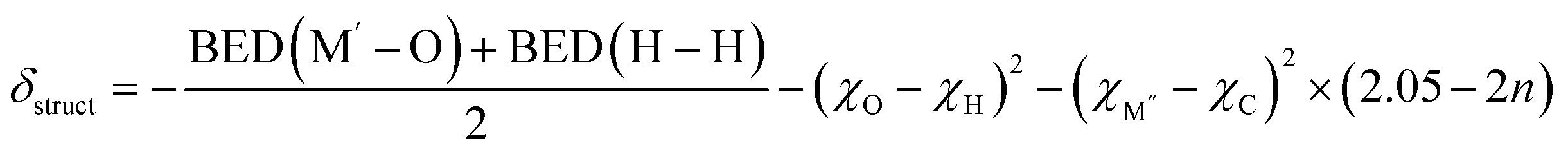 | (9) |
The scaling relationship between ΔGH and the structural descriptor δstruct is shown in Fig. 7c, characterized by a coefficient of determination (R2 = 0.82), which underscores the effectiveness of the descriptor in representing HER activity in oxygen-functionalized MXenes.
 | (10) |
This descriptor captures the sequential steps in electron transfer, offering insight into energy dynamics. The linear correlation between δ and ΔGH (Fig. 7d), expressed as ΔGH = −0.48δ − 2.12, shows improved accuracy with an R2 increase from 0.825 to 0.895 and RMSE reduction from 0.157 to 0.123.
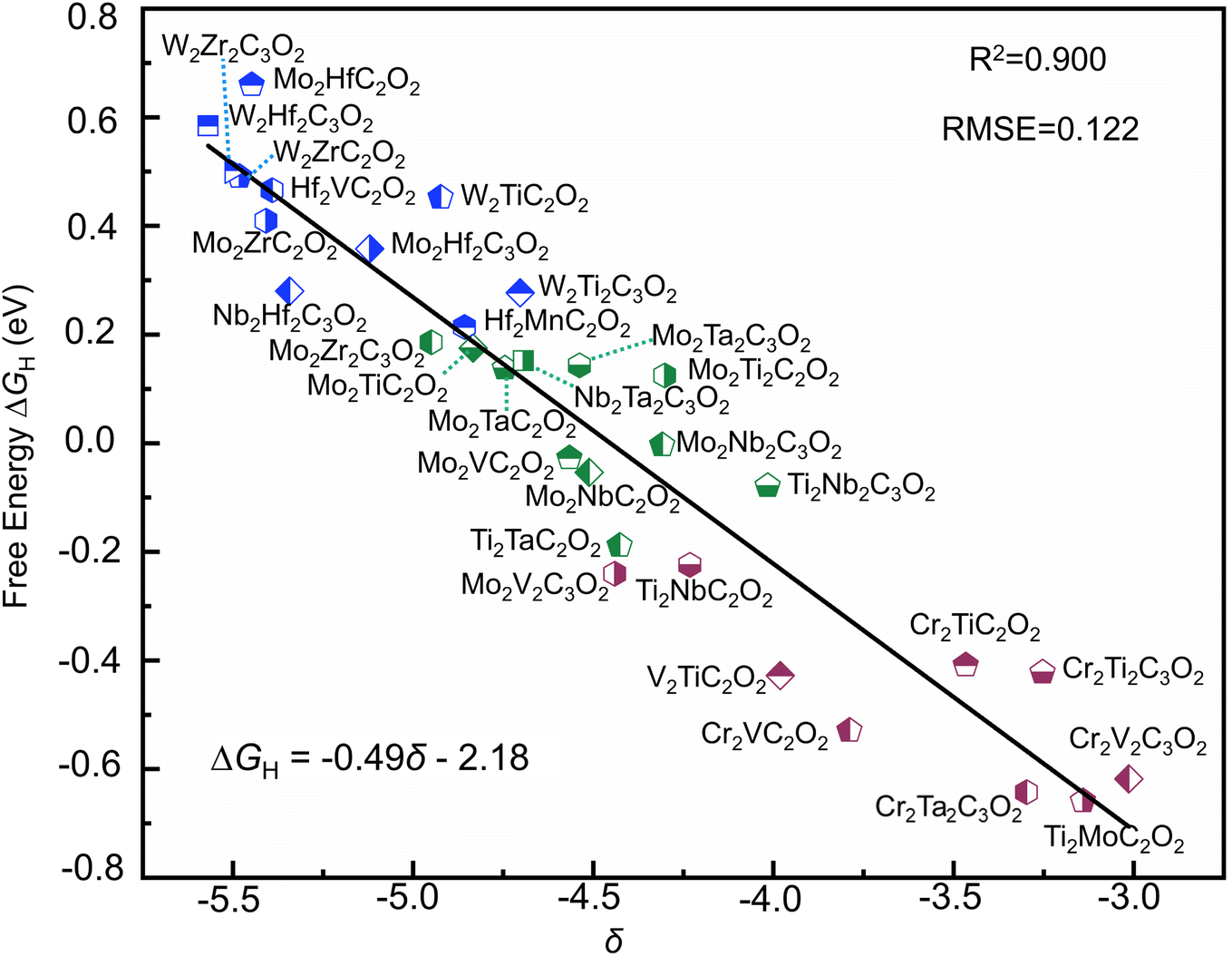 | ||
| Fig. 8 Relationships between ΔGH and descriptor δ in the 31 different MXene systems. | ||
To evaluate the generalizability of the proposed descriptor, we conducted an extension study in the application of the descriptor, especially in the M′2M′′N2O2 system, where C electronegativity is substituted with N electronegativity and the lattice coefficient is adjusted to 2.5, while keeping all other variables constant. This modification makes the descriptor more accurate for predicting ΔGH in the M′2M′′N2O2 system. We randomly selected and tested nearly 20 materials in the M′2M′′N2O2, which were later validated using DFT. The results revealed a relatively strong linear correlation between the descriptor and ΔGH, with an R2 of 0.81, as shown in Fig. S8.† This finding not only validates the effectiveness of the descriptor but also demonstrates its applicability across different systems, highlighting its potential for broader use in similar MXene structures.
3.4. Optimizing catalysis with implicit solvation models
Surface electrocatalysis at solid–liquid interfaces is inherently complex, necessitating consideration of liquid electrolyte species within the finite volume between periodically repeating slabs.50 To address this issue, the implicit and explicit solvation models are always considered in the calculation of ΔGH, and the explicit solvation model is always more accurate for the representation of solvation effects. However, in this study, we use an implicit solvation model to correct the adsorption free energies based on accuracy tests, with detailed information provided in Fig. S9 and Table S5.† The theoretical overpotentials for Mo2TiC2TX in solutions with pH values of 0, 0.3, 1, and 2 are approximately −0.39 V, −0.41 V, −0.45 V, and −0.51 V, respectively. These values align well with experimentally reported overpotentials of −0.39 V, −0.40 V, −0.45 V, and −0.47 V, underscoring the necessity of incorporating the implicit solvation model.37The results show that the solvent has a significant impact on the adsorption free energies; detailed data are shown in Table S6.† The electrostatic screening of the solvent at the surface leads to a reduction of about 0.5 eV in adsorption energy across most ordered DTM MXene systems during hydrogen adsorption. In particular, Mo2HfC2O2 exhibits a significant energy reduction of 1.338 eV. This surface, in particular, has a higher proportion of ionic bonds in the formation of polar covalent H–O bonds, which leads to a notable energy reduction.27 The calculated value of ΔGH is implied by the equation ΔGH = ΔEH + ΔEZPE + ΔUT − TΔS + kBTln10 × PH in our work, where κB is the Boltzmann constant, and T is set to 298.15 K. The dielectric constant εbulk in the implicit solvation model is 78.4, the experimental value for liquid water at 298 K.
The solvent environment significantly impacts the calculated ΔGH, thus requiring a rescreening for |ΔGH| < 0.2 eV via an implicit solvation model. Given that ΔGH generally diminishes by approximately 0.5 eV with the implicit solvation model, we elected to recompute materials whose initial ΔGH values lay within the range of 0.2 eV to 0.8 eV, as certain materials were prone to fall outside the requisite value range. This resulted in four promising candidates: Mo2Nb2C3O2, Mo2Hf2C3O2, W2Hf2C3O2, and W2Zr2C3O2,each exhibiting |ΔGH| values below 0.2 eV. Ab initio molecular dynamics (AIMD) simulations at 300 K, along with phonon band structure analysis, confirmed their mechanical and thermal stability (Fig. S10†). Additionally, the synthesizability of these materials is evaluated by demonstrating the stability of the precursor MAX phase as detailed in Table S1.† In a word, these four compounds are considered promising candidates for experimental synthesis.
4. Conclusions
This study developed a screening strategy to identify promising catalysts among ordered DTM MXenes, with four Mo- and W-based systems emerging as strong HER candidates for experimental validation. This approach considers key factors such as synthesizability, stability, and conductivity of MXenes, which are essential for practical applications. A universal screening descriptor is introduced, and its linear relationship with ΔGH (ΔGH = −0.49δ – 2.18) demonstrates its broad applicability, facilitating efficient screening of MXene catalysts for the HER. The descriptor not only deepens the understanding of structure–activity relationships, particularly highlighting the critical role of M′(dx2−y2)–O(py) orbital interactions in governing catalytic performance, but also bridges to accessible structural and electronic features such as bond dissociation energy, electronegativity, lattice parameters, ionization energy, and work function. It simultaneously enables efficient screening of MXene-based catalysts and elucidates the underlying mechanisms of their catalytic behavior. The framework and methodology behind this descriptor provide a strategic roadmap for the descriptor development and efficient catalyst screening across diverse material systems.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Junmei Du designed the study, performed the research, analyzed the data, and wrote the manuscript. Yifan Yan, Xiumei Li and Jiao Chen analyzed the data. Chunsheng Guo revised the manuscript. Yuanzheng Chen and Hongyan Wang designed the study and revised the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (12164009) and the Sichuan Science and Technology program (2022ZYD0024 and 2021YFG0228).References
- J. A. Turner, Sustainable Hydrogen Production, Science, 2004, 305, 972–974 CrossRef CAS PubMed.
- I. Roger, M. A. Shipman and M. D. Symes, Earth-abundant catalysts for electrochemical and photoelectrochemical water splitting, Nat. Rev. Chem., 2017, 1, 0003 CrossRef CAS.
- M. Ball and M. Weeda, The hydrogen economy - Vision or reality?, Int. J. Hydrogen Energy, 2015, 40, 7903–7919 CrossRef CAS.
- X. Zhao, Z. H. Levell, S. Yu and Y. Liu, Atomistic understanding of two-dimensional electrocatalysts from first principles, Chem. Rev., 2022, 122, 10675–10709 CrossRef CAS PubMed.
- C. G. Morales-Guio, L.-A. Stern and X. Hu, Nanostructured hydrotreating catalysts for electrochemical hydrogen evolution, Chem. Soc. Rev., 2014, 43, 6555–6569 RSC.
- J. Greeley, T. F. Jaramillo, J. Bonde, I. Chorkendorff and J. K. Nørskov, Computational high-throughput screening of electrocatalytic materials for hydrogen evolution, Nat. Mater., 2006, 5, 909–913 CrossRef CAS PubMed.
- X. Zou and Y. Zhang, Noble metal-free hydrogen evolution catalysts for water splitting, Chem. Soc. Rev., 2015, 44, 5148–5180 RSC.
- N. Ramaswamy, U. Tylus, Q. Jia and S. Mukerjee, Activity descriptor identification for oxygen reduction on nonprecious electrocatalysts: linking surface science to coordination chemistry, J. Am. Chem. Soc., 2013, 135, 15443–15449 CrossRef CAS PubMed.
- Z. W. Seh, J. Kibsgaard, C. F. Dickens, I. Chorkendorff, J. K. Nørskov and T. F. Jaramillo, Combining theory and experiment in electrocatalysis: Insights into materials design, Science, 2017, 355, eaad4998 CrossRef PubMed.
- B. Anasori, Y. Xie, M. Beidaghi, J. Lu, B. C. Hosler, L. Hultman, P. R. C. Kent, Y. Gogotsi and M. W. Barsoum, Two-dimensional, ordered, double transition metals carbides (MXenes), ACS Nano, 2015, 9, 9507–9516 CrossRef CAS PubMed.
- R. Anand, A. S. Nissimagoudar, M. Umer, M. Ha, M. Zafari, S. Umer, G. Lee and K. S. Kim, Late transition metal doped MXenes showing superb bifunctional electrocatalytic activities for water splitting via distinctive mechanistic pathways, Adv. Energy Mater., 2021, 11, 2102388 CrossRef CAS.
- I. Hussain, U. Sajjad, O. J. Kewate, U. Amara, F. Bibi, A. Hanan, D. Potphode, M. Ahmad, M. S. Javed, P. Rosaiah, S. Hussain, K. Khan, Z. Ajmal, S. Punniyakoti, S. S. Alarfaji, J.-H. Kang, W. Al Zoubi, S. Sahoo and K. Zhang, Double transition-metal MXenes: classification, properties, machine learning, artificial intelligence, and energy storage applications, Mater. Today Phys., 2024, 42, 101382 CrossRef CAS.
- Z. Zeng, X. Chen, K. Weng, Y. Wu, P. Zhang, J. Jiang and N. Li, Computational screening study of double transition metal carbonitrides M′2M′′CNO2-MXene as catalysts for hydrogen evolution reaction, npj Comput. Mater., 2021, 7, 80 CrossRef CAS.
- Y.-W. Cheng, J.-H. Dai, Y.-M. Zhang and Y. Song, Two-dimensional, ordered, double transition metal carbides (MXenes): A new family of promising catalysts for the hydrogen evolution reaction, J. Phys. Chem. C, 2018, 122, 28113–28122 CrossRef CAS.
- D. Jin, L. R. Johnson, A. S. Raman, X. Ming, Y. Gao, F. Du, Y. Wei, G. Chen, A. Vojvodic, Y. Gogotsi and X. Meng, Computational screening of 2D ordered double transition-metal carbides (MXenes) as electrocatalysts for hydrogen evolution reaction, J. Phys. Chem. C, 2020, 124, 10584–10592 CrossRef CAS.
- X. Wang, C. Wang, S. Ci, Y. Ma, T. Liu, L. Gao, P. Qian, C. Ji and Y. Su, Accelerating 2D MXene catalyst discovery for the hydrogen evolution reaction by computer-driven workflow and an ensemble learning strategy, J. Mater. Chem. A, 2020, 8, 23488–23497 RSC.
- J. Zheng, X. Sun, C. Qiu, Y. Yan, Z. Yao, S. Deng, X. Zhong, G. Zhuang, Z. Wei and J. Wang, High-throughput screening of hydrogen evolution reaction catalysts in MXene materials, J. Phys. Chem. C, 2020, 124, 13695–13705 CrossRef CAS.
- N. Dubouis and A. Grimaud, The hydrogen evolution reaction: from material to interfacial descriptors, Chem. Sci., 2019, 10, 9165–9181 RSC.
- W. Jiang, X. Zou, H. Du, L. Gan, C. Xu, F. Kang, W. Duan and J. Li, Universal descriptor for large-scale screening of high-performance MXene-based materials for energy storage and conversion, Chem. Mater., 2018, 30, 2687–2693 CrossRef CAS.
- L. Chen, Y. Tian, X. Hu, S. Yao, Z. Lu, S. Chen, X. Zhang and Z. Zhou, A Universal machine learning framework for electrocatalyst innovation: A case study of discovering Alloys for hydrogen evolution reaction, Adv. Funct. Mater., 2022, 32, 2208418 CrossRef CAS.
- L. Chen, X. Zhang, A. Chen, S. Yao, X. Hu and Z. Zhou, Targeted design of advanced electrocatalysts by machine learning, Chin. J. Catal., 2022, 43, 11–32 CrossRef CAS.
- B. M. Abraham, P. Sinha, P. Halder and J. K. Singh, Fusing a machine learning strategy with density functional theory to hasten the discovery of 2D MXene-based catalysts for hydrogen generation, J. Mater. Chem. A, 2023, 11, 8091–8100 RSC.
- G. Kresse and J. Furthmüller, Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set, Phys. Rev. B:Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized gradient approximation made simple, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS.
- G. Kresse and D. Joubert, From ultrasoft pseudopotentials to the projector augmented-wave method, Phys. Rev. B:Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS.
- S. Grimme, Semiempirical GGA-type density functional constructed with a long-range dispersion correction, J. Comput. Chem., 2006, 27, 1787–1799 CrossRef CAS PubMed.
- S. Grimme, S. Ehrlich and L. Goerigk, Effect of the damping function in dispersion corrected density functional theory, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- D. Gunceler, K. Letchworth-Weaver, R. Sundararaman, K. A. Schwarz and T. A. Arias, The importance of nonlinear fluid response in joint density-functional theory studies of battery systems, Modell. Simul. Mater. Sci. Eng., 2013, 21, 074005 CrossRef.
- K. Mathew, V. S. C. Kolluru, S. Mula, S. N. Steinmann and R. G. Hennig, Implicit self-consistent electrolyte model in plane-wave density-functional theory, J. Chem. Phys., 2019, 151, 234101 CrossRef PubMed.
- K. Mathew, R. Sundararaman, K. Letchworth-Weaver, T. A. Arias and R. G. Hennig, Implicit solvation model for density-functional study of nanocrystal surfaces and reaction pathways, J. Chem. Phys., 2014, 140, 084106 CrossRef PubMed.
- R. Nelson, C. Ertural, J. George, V. L. Deringer, G. Hautier and R. Dronskowski, LOBSTER: Local orbital projections, atomic charges, and chemical-bonding analysis from projector-augmented-wave-based density-functional theory, J. Comput. Chem., 2020, 41, 1931–1940 CrossRef CAS PubMed.
- S. Maintz, V. L. Deringer, A. L. Tchougréeff and R. Dronskowski, LOBSTER: A tool to extract chemical bonding from plane-wave based DFT, J. Comput. Chem., 2016, 37, 1030–1035 CrossRef CAS PubMed.
- J. K. Nørskov, T. Bligaard, A. Logadottir, J. R. Kitchin, J. G. Chen, S. Pandelov and U. Stimming, Trends in the exchange current for hydrogen evolution, J. Electrochem. Soc., 2005, 152, J23 CrossRef.
- B. Hinnemann, P. G. Moses, J. Bonde, K. P. Jørgensen, J. H. Nielsen, S. Horch, I. Chorkendorff and J. K. Nørskov, Biomimetic hydrogen evolution: MoS2 nanoparticles as catalyst for hydrogen evolution, J. Am. Chem. Soc., 2005, 127, 5308–5309 CrossRef CAS.
- V. Wang, N. Xu, J.-C. Liu, G. Tang and W.-T. Geng, VASPKIT: A user-friendly interface facilitating high-throughput computing and analysis using VASP code, Comput. Phys. Commun., 2021, 267, 108033 CrossRef CAS.
- M. Naguib, V. N. Mochalin, M. W. Barsoum and Y. Gogotsi, 25th anniversary article: MXenes: a new family of two-dimensional materials, Adv. Mater., 2014, 26, 992–1005 CrossRef CAS PubMed.
- M. Dahlqvist and J. Rosen, Predictive theoretical screening of phase stability for chemical order and disorder in quaternary 312 and 413 MAX phases, Nanoscale, 2020, 12, 785–794 RSC.
- H. Liu, X. Jia, A. Cao, L. Wei, C. D'agostino and H. Li, The surface states of transition metal X-ides under electrocatalytic conditions, J. Chem. Phys., 2023, 158, 124705 CrossRef CAS PubMed.
- W. Yang, Z. Jia, B. Zhou, L. Wei, Z. Gao and H. Li, Surface states of dual-atom catalysts should be considered for analysis of electrocatalytic activity, Commun. Chem., 2023, 6, 6 CrossRef CAS PubMed.
- Z. Guo, T. Wang, J. Xu, A. Cao and H. Li, Surface coverage and reconstruction analyses bridge the correlation between structure and activity for electrocatalysis, Chem. Commun., 2024, 60, 14346–14359 RSC.
- H. Liu, D. Zhang, S. M. Holmes, C. D'Agostino and H. Li, Origin of the superior oxygen reduction activity of zirconium nitride in alkaline media, Chem. Sci., 2023, 14, 9000–9009 RSC.
- J. Zhang, Y. Zhao, X. Guo, C. Chen, C.-L. Dong, R.-S. Liu, C.-P. Han, Y. Li, Y. Gogotsi and G. Wang, Single platinum atoms immobilized on an MXene as an efficient catalyst for the hydrogen evolution reaction, Nat. Catal., 2018, 1, 985–992 CrossRef CAS.
- G. Gao, A. P. O'Mullane and A. Du, 2D MXenes: A new family of promising catalysts for the hydrogen evolution reaction, ACS Catal., 2017, 7, 494–500 CrossRef CAS.
- B. C. Wyatt, A. Thakur, K. Nykiel, Z. D. Hood, S. P. Adhikari, K. K. Pulley, W. J. Highland, A. Strachan and B. Anasori, Design of atomic ordering in Mo2Nb2C3Tx MXenes for hydrogen evolution electrocatalysis, Nano Lett., 2023, 23, 931–938 CrossRef CAS PubMed.
- S. Trasatti, Electronegativity, work function, and heat of adsorption of hydrogen on metals, J. Chem. Soc., Faraday Trans., 1972, 1(68), 229–236 RSC.
- A. L. Allred, Electronegativity values from thermochemical data, J. Inorg. Nucl. Chem., 1961, 17, 215–221 CrossRef CAS.
- Y. Gogotsi and B. Anasori, The Rise of MXenes, ACS Nano, 2019, 13, 8491–8494 CrossRef CAS PubMed.
- H. Kwon, J. Park, J. E. Shin and B. Koo, Optimal investment strategy analysis of on-site hydrogen production based on the hydrogen demand prediction using machine learning, Int. J. Energy Res., 2024, 17, 6313421 CrossRef.
- M. V. Jyothirmai, R. Dantuluri, P. Sinha, B. M. Abraham and J. K. Singh, Machine-learning-driven high-throughput screening of transition-metal atom intercalated g-C3N4/MX2 (M = Mo, W; X = S, Se, Te) heterostructures for the hydrogen evolution reaction, ACS Appl. Mater. Interfaces, 2024, 2024, 12437–12445 CrossRef PubMed.
- S. Ringe, N. G. Hörmann, H. Oberhofer and K. Reuter, Implicit solvation methods for catalysis at electrified interfaces, Chem. Rev., 2022, 122, 10777–10820 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc08725a |
| This journal is © The Royal Society of Chemistry 2025 |