
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of bicyclo[3.2.0]heptane lactones via a ligand-enabled Pd-catalyzed C(sp3)–H activation cascade†
Zhoulong
Fan
,
Xinpei
Cai
,
Tao
Sheng
and
Jin-Quan
Yu
 *
*
Department of Chemistry, The Scripps Research Institute, 10550 North Torrey Pines Road, La Jolla, CA 92037, USA. E-mail: yu200@scripps.edu
First published on 29th April 2025
Abstract
Bicyclo[3.2.0]heptane lactones represent an important scaffold in bioactive molecules. Herein, we report a diastereoselective synthetic disconnection to access bicyclo[3.2.0]heptane lactones from bicyclo[1.1.1]pentane carboxylic acids, which proceeds through palladium-catalyzed C–H activation and C–C cleavage processes. By using two different classes of ligands, MPAA and pyridone-amine, either all-syn arylated bicyclo[3.2.0]heptane lactones or non-arylated ones can be synthesized. The bicyclo[3.2.0]heptane lactone products were converted into multiple substituted cyclobutane, γ-lactone, and oxobicyclo[3.2.0]heptane derivatives to showcase the synthetic versatility of this method.
Introduction
The bicyclo[3.2.0]heptane scaffold is frequently encountered in natural products and biologically active molecules (Fig. 1A).1–5 Its unique three-dimensional structure enhances its efficiency in binding to protein targets. Consequently, accessing diverse substitution patterns is crucial for exploring the chemical space availability of such a scaffold. The most direct method for accessing the highly functionalized bicyclo[3.2.0]heptane motif involves an intramolecular [2 + 2] cycloaddition reaction (Fig. 1B(i)), as reported by Chirik,6–8 Yoon,9,10 Burns,11 Roşca,12 You,13 Toste,14 Mascareñas,15 and Yu16via transition metal-catalysis. Additionally, Leonori, Ruffoni, Merino and coworkers have reported a dearomatization strategy for constructing 2-azabicyclo[3.2.0]heptane motifs from nitroarenes (Fig. 1B(ii)).17 Furthermore, direct functionalizations on the bicyclo[3.2.0]heptane scaffold have been achieved through transition metal-catalyzed addition of cyclobutene (Fig. 1B(iii)).18,19 However, these methods often do not offer control over diastereoselectivity. Therefore, there is a pressing need for the development of a strategy capable of accessing a functionalized bicyclic scaffold with exclusive diastereoselectivity.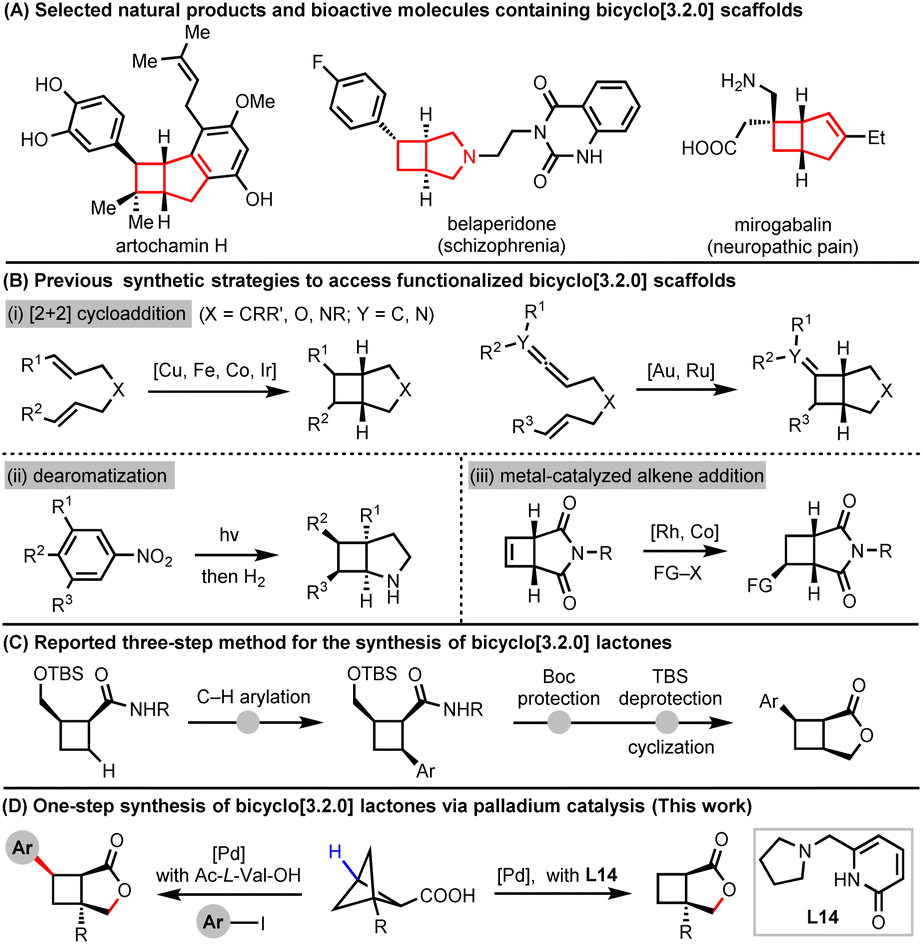 | ||
| Fig. 1 Importance and synthetic approaches of functionalized bicyclo[3.2.0]heptane scaffolds. | ||
Bicyclo[3.2.0]heptane lactones are not only bioactive but also versatile precursors for diverse bicyclo[3.2.0]heptane scaffolds, such as azabicyclo[3.2.0]heptanes and oxobicyclo[3.2.0]heptanes. The previous method involves three steps starting from pre-assembled cyclobutane derivatives, including C–H arylation, Boc protection, and TBS deprotection (Fig. 1C).20 Inspired by our recent work on palladium-catalyzed C–C cleavage of bicyclo[1.1.1]pentanol to form functionalized cyclobutenes diastereoselectively,21 we began to develop a palladium-catalyzed C–H arylation and sequential C–C cleavage/lactonization of bicyclo[1.1.1]pentane carboxylic acids for the construction of arylated bicyclo[3.2.0]heptane lactones in a diastereoselective manner (Fig. 1D). This process is realized using a mono-N-protected amino acid (MPAA) ligand. Interestingly, in the absence of aryl iodides, the different products, bicyclo[3.2.0]heptane lactones, were also obtained by switching the bidentate ligands from Ac-L-Val-OH to pyridone-amine L14. Ring opening of such motifs to generate trisubstituted cyclobutanes and γ-lactones, as well as reduction to form arylated oxobicyclo[3.2.0]heptane demonstrates the synthetic utility of these scaffolds.
Results and discussion
Our investigation commenced with the evaluation of ligand effects using bicyclo[1.1.1]pentane carboxylic acid 1a as a model substrate (Table 1). In the absence of a ligand, the desired arylated bicyclo[3.2.0]heptane lactone product 3a was observed in a very low 18% yield (entry 2). Considering the known capability of bidentate MPAA ligands to promote the C(sp3)–H activation of free carboxylic acids,22,23 we explored a series of readily available MPAAs and related derivatives (entry 1, entries 3–7). Gratifyingly, Ac-L-Val-OH, identified as an optimal ligand, delivered product 3a in a yield of 66%, while other ligands gave lower yields (30–39%). Next, we investigated additional monodentate ligand scaffolds,24–27 such as pyridines L3 and L4, pyridone L5 and quinoline L6 (entries 8–11). However, these ligands did not enhance the reaction yields (21–54%). In the absence of palladium, no product was detected (entry 12). A significantly decreased yield was obtained when replacing Pd(OAc)2 with Pd(TFA)2 (entry 13). Control experiments involving silver salts indicated that AgTFA is indispensable, suggesting that the trifluoroacetate anion plays a crucial role in the catalytic system (entries 14–16). Furthermore, several bases were also examined (entries 18–19). The use of Na2HPO4 substantially increased the yield of 3a from 25% to 66% (entry 17 vs. entry 1) while the starting material was not detected. This enhancement is attributed to the coordination of Na+ with carboxylate in a κ2 mode, allowing palladium to activate the target C–H bond in the correct orientation28 (see the ESI†).|
|
||
|---|---|---|
| Entry | Deviation from standard conditions | 3a, yield (%) |
| a Reaction conditions: 1a (0.1 mmol), 2a (0.3 mmol), Pd(OAc)2 (10 mol%), Ac-L-Val-OH (20 mol%), AgTFA (4 equiv.), Na2HPO4 (1 equiv.), HFIP (2 mL), 110 °C, 20 h. b The yield of 3a was determined by 1H NMR analysis using 1,3,5-trimethoxybenzene as an internal standard. | ||
| 1 | None | 66 |
| 2 | w/o Ac-L-Val-OH | 18 |
| 3 | Ac-Gly-OH instead of Ac-L-Val-OH | 31 |
| 4 | Ac-L-Phe-OH instead of Ac-L-Val-OH | 39 |
| 5 | Boc-L-Val-OH instead of Ac-L-Val-OH | 30 |
| 6 | L1 instead of Ac-L-Val-OH | 31 |
| 7 | L2 instead of Ac-L-Val-OH | 38 |
| 8 | L3 instead of Ac-L-Val-OH | 54 |
| 9 | L4 instead of Ac-L-Val-OH | 46 |
| 10 | L5 instead of Ac-L-Val-OH | 21 |
| 11 | L6 instead of Ac-L-Val-OH | 33 |
| 12 | w/o Pd(OAc)2 | n.d. |
| 13 | Pd(TFA)2 instead of Pd(OAc)2 | 33 |
| 14 | w/o AgTFA | n.d. |
| 15 | AgOAc instead of AgTFA | 10 |
| 16 | Ag2CO3 instead of AgTFA | 9 |
| 17 | w/o Na2HPO4 | 25 |
| 18 | Na2CO3 instead of Na2HPO4 | 27 |
| 19 | NaOAc instead of Na2HPO4 | 45 |
|
||

With the optimal reaction conditions in hand, we explored the reaction scope concerning aryl and heteroaryl iodides, and bicyclo[1.1.1]pentanes (Scheme 1). When treating 1a with a variety of phenyl iodides featuring both electron-rich and electron-poor functional groups, the corresponding syn-arylated bicyclo[3.2.0]heptane lactone products 3a–3n were obtained in moderate to good yields. To determine whether the chiral ligand produces an enantiopure product, we carried out the reaction using 4-iodobiphenyl (2d) as the coupling partner with either nonchiral Ac-β-Ala-OH or chiral Ac-L-Val-OH as the ligand. Chiral HPLC analysis of the arylated product 3d revealed a 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 ratio of the two enantiomers, indicating that the chiral ligand does not induce enantioselectivity (see the ESI†). For 6-iododihydrobenzodioxine and 2-iodonaphthalene, increasing the concentration of HFIP from 0.05 M to 0.3 M led to the desired products 3o and 3p in moderate yields. Typically, heterocycle motifs pose challenges in C–H activation reactions due to the undesired coordination of heteroatoms with palladium, deactivating the catalyst. However, under the current reaction conditions, heteroaryl iodides could serve as coupling partners. Various substituted pyridines, thiophenes, and furans were introduced at the β position of bicyclo[3.2.0]heptane lactones in a syn fashion. Additionally, three substituted bicyclo[1.1.1]pentane carboxylic acids were subjected to the standard conditions, yielding highly functionalized bicyclo[3.2.0]heptane lactone derivatives 3x to 3z.
:50 ratio of the two enantiomers, indicating that the chiral ligand does not induce enantioselectivity (see the ESI†). For 6-iododihydrobenzodioxine and 2-iodonaphthalene, increasing the concentration of HFIP from 0.05 M to 0.3 M led to the desired products 3o and 3p in moderate yields. Typically, heterocycle motifs pose challenges in C–H activation reactions due to the undesired coordination of heteroatoms with palladium, deactivating the catalyst. However, under the current reaction conditions, heteroaryl iodides could serve as coupling partners. Various substituted pyridines, thiophenes, and furans were introduced at the β position of bicyclo[3.2.0]heptane lactones in a syn fashion. Additionally, three substituted bicyclo[1.1.1]pentane carboxylic acids were subjected to the standard conditions, yielding highly functionalized bicyclo[3.2.0]heptane lactone derivatives 3x to 3z.
 | ||
| Scheme 1 Reaction scope for the synthesis of arylated bicyclo[3.2.0]heptane lactones. Reaction conditions: unless stated otherwise, 1 (0.1 mmol), 2 (0.3 mmol), Pd(OAc)2 (10 mol%), Ac-L-Val-OH (20 mol%), AgTFA (4 equiv.), Na2HPO4 (1 equiv.), HFIP (2 mL), 110 °C, 20 h. Isolated yields are reported. aUsing HFIP (0.3 M). | ||
Having established the synthetic methods for accessing arylated bicyclo[3.2.0]heptane lactones, we then sought to investigate the possibility of generating bicyclo[3.2.0]heptane lactone scaffolds without the arylation step (Scheme 2). Initially, we conducted the reaction using Ac-L-Val-OH as a ligand under the standard conditions without aryl iodide. However, the starting material 1a was recovered, and no product 4a was observed, indicating that the MPAA ligand might be applicable for the cleavage of C–C bonds at benzylic positions. Considering the effectiveness of our recently developed bidentate pyridone ligands in palladium catalysis,29,30 we tested X,L-type quinoline–pyridone ligand L9 and X,X-type pyridone–pyridone ligand L10. Although these ligands provided product 4a, the yield was low. Encouraged by these results, we examined several imine–pyridone and amine–pyridone ligands (L11–L15).31 Significantly, when L14 was employed as the ligand, the product 4a was obtained in 68% yield. These conditions were identified as the optimal ones and used to explore the substrate scope. Various 1-substituted bicyclo[1.1.1]pentane-2-carboxylic acids were subjected to the conditions, leading to the corresponding bicyclo[3.2.0]heptane lactone derivatives in yields ranging from 39% to 65%. These previously inaccessible substitution patterns on bicylco[3.2.0]heptane lactones could prove beneficial in exploring the available chemical space in pharmaceutical campaign.
 | ||
| Scheme 2 Reaction scope for the synthesis of bicyclo[3.2.0]heptane lactones. Reaction conditions: 1 (0.1 mmol), Pd(OAc)2 (10 mol%), ligand (20 mol%), AgTFA (4 equiv.), Na2HPO4 (1 equiv.), HFIP (2 mL), 110 °C, 20 h. Unless stated otherwise, isolated yields are reported. a1H NMR yield using 1,3,5-trimethoxybenzene as an internal standard. | ||
To elucidate the possible mechanism, we carried out the reaction in the presence of acetic acid-d4 and HFIP-d1 without the aryl iodide. The partially deuterated carboxylic acid [D]-1a was recovered in 53% yield, with 20% and 25% deuterium incorporation on the α-position and β-position, respectively (Scheme 3A). In addition, to rule out the possibility of non-directed C–H bond cleavage, we performed the reaction under standard conditions using 4-methylbenzyl bicyclo[1.1.1]pentane-2-carboxylate (1f) as the substrate. However, no deuteration was observed at the α- or β-positions, further verifying that the carboxylic acid directed C–H bond cleavage step occurred in the process (see the ESI†). A plausible mechanism is depicted in Scheme 3B. The β-C–H palladation intermediate II is first formed, followed by an oxidative addition and reductive elimination sequence to deliver the β-arylation intermediate IV. Subsequently, a strain-release C–C bond cleavage event takes place, generating the palladium species V. Then, this species undergoes lactonization and protodepalladation to afford product 3. In addition, we also proposed the mechanism in the absence of aryl iodides. The C–C cleavage occurs directly with the assistance of the pyridone–amine ligand, providing the intermediate III′. Then, the sequential lactonization and protodepalladation processes produce the non-arylated lactone 4.
 | ||
| Scheme 3 Mechanistic studies. | ||
To further illustrate the application of the current methods, we conducted extensive transformations of bicyclo[3.2.0]heptane lactone 3a. The ring-opening process of γ-lactone yielded the tri-substituted cyclobutane derivative 5 under sequential hydrolysis and substitution conditions. Notably, the cyclobutane ring of 3a was unexpectedly cleaved to produce γ-lactone 6 in the presence of BBr3, and the detailed mechanism remains unclear. In addition, the oxobicyclo[3.2.0]heptane scaffold 7 could be easily obtained through the sequential reduction of the ester and recyclization (Scheme 4A).20 To explore the potential utility of this bicyclic γ-lactone in chemical biology, we removed the Bn protecting group of 3f and coupled it with propargylamine, resulting in the generation of product 8 with an alkynyl group for protein labeling (Scheme 4B).
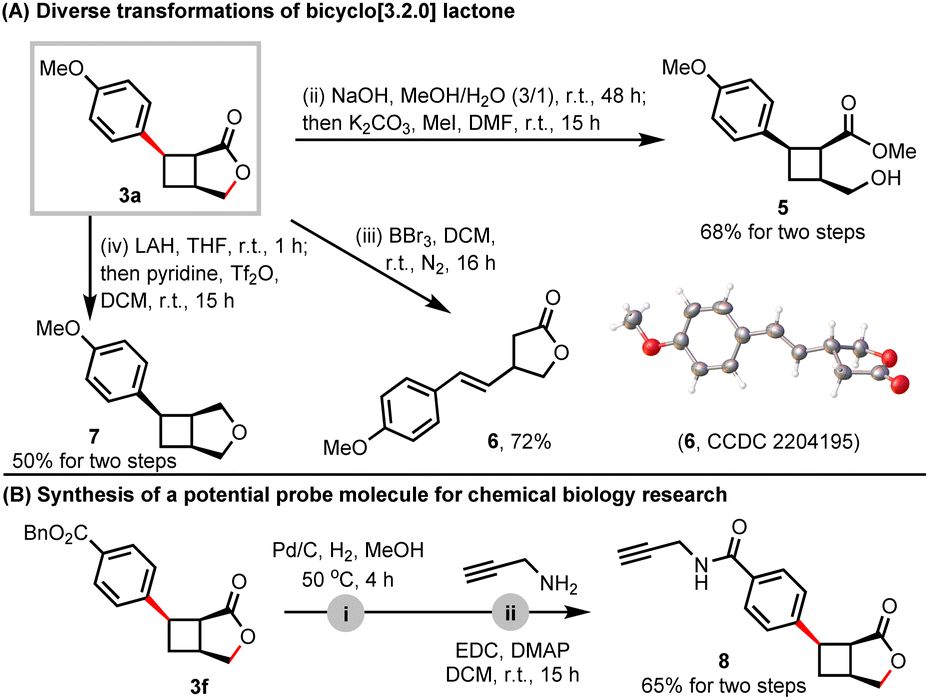 | ||
| Scheme 4 Synthetic applications. | ||
Conclusions
In summary, we developed diastereoselective methods for the synthesis of arylated or non-arylated bicyclo[3.2.0]heptane lactones from bicyclo[1.1.1]pentane carboxylic acids under palladium catalysis. The utilization of MPAA or pyridone–amine ligands is crucial for the formation of either arylated or non-arylated lactones respectively. Additionally, this lactone scaffold can effectively undergo transformation to afford a diverse range of highly functionalized cyclobutane, γ-lactone, and oxobicyclo[3.2.0]heptane derivatives. We anticipate that this approach holds potential for constructing a scaffold-based novel compound library, contributing to the identification of hit or lead compounds in drug discovery.Data availability
The data supporting this article have been included as part of the ESI,† including detailed experimental procedures and characterization data for new compounds. Crystallographic data have been deposited with the CCDC with deposition numbers: 2252736 (3e) and 2204195 (6).Author contributions
Z. F. developed the reaction, optimized the conditions, and investigated the substrate scope. X. C. prepared part of the substrates. T. S. provided the imine-pyridone and amine-pyridone ligands. Z. F. and J.-Q. Y. wrote the manuscript. J.-Q. Y. directed the project.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge The Scripps Research Institute and the NIH (National Institute of General Medical Sciences grant R01 GM084019). We thank the Scripps Automated Synthesis Facility and Scripps Center for Metabolomics and Mass Spectrometry for assistance with HRMS. We also thank Dr M. Gembicky, Dr Jake Bailey, and the University of California San Diego Crystallography Facility for X-ray crystallographic analysis. We thank Dr Kevin Wu for proofreading and providing helpful suggestions in preparing the manuscript.Notes and references
- G. Steiner, L. Unger, B. Behl, H.-J. Teschendorf and R. Munschauer, WO1994000458A1, 1994.
- K. C. Nicolaou, T. Lister, R. M. Denton and C. F. Gelin, Angew. Chem., Int. Ed., 2007, 46, 7501 CrossRef CAS PubMed.
- C. M. Marson, Chem. Soc. Rev., 2011, 40, 5514 RSC.
- A. V. Denisenko, T. Druzhenko, Y. Skalenko, M. Samoilenko, O. O. Grygorenko, S. Zozulya and P. K. Mykhailiuk, J. Org. Chem., 2017, 82, 9627 CrossRef CAS PubMed.
- E. D. Deeks, Drugs, 2019, 79, 463 CrossRef CAS PubMed.
- M. W. Bouwkamp, A. C. Bowman, E. Lobkovsky and P. J. Chirik, J. Am. Chem. Soc., 2006, 128, 13340 CrossRef CAS PubMed.
- J. M. Hoyt, K. T. Sylvester, S. P. Semproni and P. J. Chirik, J. Am. Chem. Soc., 2013, 135, 4862 CrossRef CAS PubMed.
- V. A. Schmidt, J. M. Hoyt, G. W. Margulieux and P. J. Chirik, J. Am. Chem. Soc., 2015, 137, 7903 CrossRef CAS PubMed.
- C. S. Gravatt, L. Melecio-Zambrano and T. P. Yoon, Angew. Chem., Int. Ed., 2021, 60, 3989 CrossRef CAS PubMed.
- S. O. Scholz, J. B. Kidd, L. Capaldo, N. E. Flikweert, R. M. Littlefield and T. P. Yoon, Org. Lett., 2021, 23, 3496 CrossRef CAS PubMed.
- C. M. F. Mansson and N. Z. Burns, J. Am. Chem. Soc., 2022, 144, 19689 CrossRef CAS PubMed.
- L. E. Hertwig, T. Bender, F. J. Becker, P. Jäger, S. Demeshko, S. J. Gross, J. Ballmann, D.-A. Roşca and P. Jäger, ACS Catal., 2023, 13, 6416 CrossRef CAS.
- P. Yang, R.-X. Wang, X.-L. Huang, Y.-Z. Cheng, S.-L. You and P. Yang, J. Am. Chem. Soc., 2023, 145, 21752 CrossRef CAS PubMed.
- M. R. Luzung, P. Mauleón and F. D. Toste, J. Am. Chem. Soc., 2007, 129, 12402 CrossRef CAS PubMed.
- M. Gulías, A. Collado, B. Trillo, F. López, E. Oñate, M. A. Esteruelas and J. L. Mascareñas, J. Am. Chem. Soc., 2011, 133, 7660 CrossRef PubMed.
- P. Zhang and Z.-X. Yu, J. Am. Chem. Soc., 2023, 145, 9634 CrossRef CAS PubMed.
- E. Matador, M. J. Tilby, I. Saridakis, M. Pedrón, D. Tomczak, J. Llaveria, I. Atodiresei, P. Merino, A. Ruffoni and D. Leonori, J. Am. Chem. Soc., 2023, 145, 27810 CrossRef CAS PubMed.
- F. W. Goetzke, A. M. L. Hell, L. van Dijk and S. P. Fletcher, Nat. Chem., 2021, 13, 880 CrossRef CAS PubMed.
- Z. Liang, L. Wang, Y. Wang, L. Wang, Q. Chong and F. Meng, J. Am. Chem. Soc., 2023, 145, 3588 CrossRef CAS PubMed.
- T. J. Osberger, S. L. Kidd, T. A. King and D. S. Spring, Chem. Commun., 2020, 56, 7423 RSC.
- Z. Fan, D. A. Strassfeld, H. S. Park, K. Wu and J.-Q. Yu, Angew. Chem., Int. Ed., 2023, 62, e202303948 CrossRef CAS PubMed.
- B.-F. Shi, N. Maugel, Y.-H. Zhang and J.-Q. Yu, Angew. Chem., Int. Ed., 2008, 47, 4882 CrossRef CAS PubMed.
- Q. Shao, K. Wu, Z. Zhuang, S. Qian and J.-Q. Yu, Acc. Chem. Res., 2020, 53, 833 CrossRef CAS PubMed.
- J. He, S. Li, Y. Deng, H. Fu, B. N. Laforteza, J. E. Spangler, A. Homs and J.-Q. Yu, Science, 2014, 343, 1216 CrossRef CAS PubMed.
- P. Wang, M. E. Farmer, X. Huo, P. Jain, P.-X. Shen, M. Ishoey, J. E. Bradner, S. R. Wisniewski, M. D. Eastgate and J.-Q. Yu, J. Am. Chem. Soc., 2016, 138, 9269 CrossRef CAS PubMed.
- H. Park, N. Chekshin, P.-X. Shen and J.-Q. Yu, ACS Catal., 2018, 8, 9292 CrossRef CAS PubMed.
- Z. Li, Z. Wang, N. Chekshin, S. Qian, J. X. Qiao, P. T. Cheng, K.-S. Yeung, W. R. Ewing and J.-Q. Yu, Science, 2021, 372, 1452 CrossRef CAS PubMed.
- K. M. Engle, T.-S. Mei, M. Wasa and J.-Q. Yu, Acc. Chem. Res., 2012, 45, 788 CrossRef CAS PubMed.
- Z. Wang, L. Hu, N. Chekshin, Z. Zhuang, S. Qian, J. X. Qiao and J.-Q. Yu, Science, 2021, 374, 1281 CrossRef CAS PubMed.
- H. S. S. C. Chan, J.-M. Yang and J.-Q. Yu, Science, 2022, 376, 1481 CrossRef CAS PubMed.
- T. Sheng, Z. Zhuang, Z. Wang, L. Hu, A. N. Herron, J. X. Qiao and J.-Q. Yu, J. Am. Chem. Soc., 2022, 144, 12924 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2204195 and 2252736. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5sc00711a |
| This journal is © The Royal Society of Chemistry 2025 |